


Abstract
A simple, direct method for the detection of DNA–protein interaction was developed with electrochemical methods. Single-stranded DNA (ss-DNA) probes were prepared through the chemical bonding of an oligonucleotide to a polymer film bearing carboxylic acid groups, and double-stranded DNA (ds-DNA) probes were prepared through hybridization of the complementary sequence DNA on the ss-DNA probe. Impedance spectroscopy and differential pulse voltammetry (DPV) distinguished the interaction between the DNA probes with mouse Purβ (mPurβ), an ss-DNA binding protein, and with Escherichia coli MutH, a ds-DNA binding protein. Impedance spectra obtained before and after the interaction of DNA probes with these proteins clearly showed the sequence-specific ss-DNA preference of mPurβ and the sequence-specific ds-DNA preference of MutH. The concentration dependence of proteins on the response of the DNA probes was also investigated, and the detection limits of MutH and mPurβ were 25 and 3 μg/ml, respectively. To confirm the impedance results, the variation of the current oxidation peak of adenine of the DNA probe was monitored with DPV. The formation constants of the complexes formed between the probe DNA and the proteins were estimated based on the DPV results.
INTRODUCTION
Revealing the genomic sequences of many different organisms promises unprecedented potential for new discoveries in biomedical research. One of the key questions in post-genomic fields is how a specific gene demonstrates the cellular functions necessary in a certain physiological environment. It is widely known that various intra/inter-molecular interactions, such as protein–DNA and protein–protein interactions, perform crucial roles in cellular processes in particular ways. Protein–DNA interactions are a common feature in cellular processes such as mismatched DNA repair (1) and gene regulation (2). Great advances have been made in the detection of protein–DNA interactions using labour-intensive and lengthy traditional methods, such as the gel-shifting assay (3) and the DNA foot-printing assay (4). Thus, much attention is focussed on the creation of a new, expedient method to detect the specific DNA binding activity of a protein, which would have a wide applicability in the rapidly developing biological research field.
Various methods for the detection of DNA hybridization have been developed with optical, electrochemical, surface acoustic wave and microgravimetric techniques. Of these, electrochemical techniques offer a sensitive, selective, low-cost and miniaturized device for the detection of biological molecules. Electrochemical transducers monitor the signals of current, potential, conductivity and impedance during the interaction between the probe and the target (5,6). Earlier, the possibility of direct detection of DNA hybridization using impedance measurements was introduced by the shift of the out-of-phase impedance after hybridization (7). Recently, we developed a biosensor using an electro-active polyterthiophene-functionalized DNA probe, and showed that the specific hybridization of an ss-DNA sequence with its complementary target ss-DNA in a solution induced a significant change in impedance (8). Based on this technique, a DNA probe chemically bonded to the conducting polymer was used for the detection of the DNA–protein interaction employing impedance spectroscopy and DPV (Figure 1). In the potential range of DPV with the polymer coated electrode, guanine and adenine residues of DNA are oxidized, and the oxidation currents are found to be directly proportional to the concentration of DNA. The DPV is capable of detecting the trace amount of analytes present in the solution (9). On the other hand, impedance spectroscopy is used to monitor the variation of impedance value before and after the interaction of the probe with analytes (10). The impedance changes reflect the degree of interaction between the protein and the DNA probes.
Figure 1.
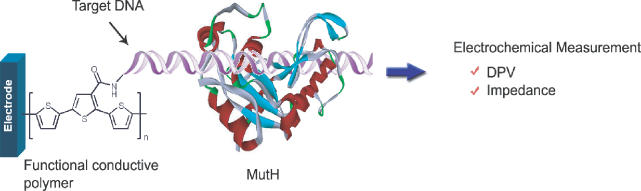
Schematic representation of the experiment. The glassy carbon electrode was coated by functional conductive polymer (polyTTCA). The probes immobilized with ss-DNA or ds-DNA probes on the polyTTCA film-coated electrode were reacted with the different concentrations of both proteins, MutH (Protein Data Bank code 2AZO) and mPurβ, for the electrochemical analysis.
The anodic peak of adenine in the DNA probe in DPV decreased and the impedance also changed, due to the interaction between DNA and the protein. Thus, this simple electrochemical method can directly detect the activity of a protein to specific ds-DNA as well as ss-DNA without any additives or radioactive labelling. In the present study, we examined two specific DNA binding proteins, MutH, a ds-DNA binding protein and mPurβ, an ss-DNA binding protein, with variable oligonucleotides for the possible application of this method to the detection of DNA–protein interactions. In addition, in order to test the chain length effect of the interaction between probe DNA and the proteins, different sizes of base DNA were used.
METERIALS AND METHODS
Materials
Acetate buffer (pH 5.2), sodium phosphate dibasic, sodium chloride, sodium dihydrogenphosphate, 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDAC) and dichloromethane (99.8%, anhydrous) were obtained from Sigma (USA). Tetrabutylammonium perchlorate (TBAP, electrochemical grade) was obtained from Fluka (USA) and purified. All chemicals were prepared and used under Dnase-, Rnase- and protease- free conditions.
Synthesis of DNA oligonucleotide probes
Oligonucleotides possessing a modified 5′ base [NH2(CH2)6] were purchased from Genemed Synthesis, Inc (USA) (Table 1). Of these, the 30mer contains an inverted core MCAT enhancer element (AGGAATG) showing a specific binding interaction to mPurβ. The 22mer-me1 has a specific binding sequence (GA*TC; A* = methylated A) to MutH. Other sequences were designed to compare the sequence specificity of both proteins. Of these probes, complementary oligonucleotides were synthesized by the Applied Biosystems Model 394 DNA/RNA Synthesizer. All nucleotides were purified by gel filtration and reverse-phase chromatography using an Invitrogen RP-C18 column.
Table 1. DNA oligonucleotide probes used in this study.
| Designation | Sequence |
|---|---|
| 36mer | 5′-NH2(CH2)6GCATACGGAAGTTAAAGTGCGGATCAT-CTCTAGCCA-3′ |
| 32mer | 5′-NH2(CH2)6 CACGTCCTAGACGGGATCCCGTCTAGG-ACGTG-3′ |
| 30mer | 5′- NH2(CH2)6 GGAGCAGAACAGAGGAATGCAGTGGA-AGAG-3′ |
| 28mer | 5′- NH2(CH2)6 GGAAGTTAAAGTGCGGATCATCTC-TAGC-3′ |
| 28mer-me1 | 5′-NH2(CH2)6 GGA AGT TAA AGT GCG GAmT CAT CTC TACC-3′ |
| 28mer-me2 | 5′- NH2(CH2)6 GGA AGT TAA AGT GCG GGmT CAT CTC TACC-3′ |
| 19mer | 5′- NH2(CH2)6 CTCCTGTGGAGAAGTCTGC-3′ |
Modification of an electrode
A terthiophene monomer bearing a carboxyl group, 3′-carboxyl-5,2′:5′,2-terthiophene was synthesized by treatment of 3′-bromothiophene with CuCN in DMF followed by hydrolysis in KOH–ethoxyethanol (11). The synthesized 5,2′;5′,2-terthiophene-3′-carboxylic acid (TTCA) was polymerized on a glassy carbon electrode (12) (area: 0.07 cm2, Kosentech Model KSGC-100 (South Korea)] in a 0.1 M Bt4NClO4/CH2Cl2 solution containing a 1.0 mM monomer by potential cycling five times from 0.0 V to 1.5 V versus Ag/AgCl (11). During the first anodic scan from 0.0V to 1.5 V in a monomer-containing solution, the oxidation peak of the monomer appeared at +1.3 V and the reverse scan towards the positive direction showed a small cathodic peak at +0.9 V, which corresponded to the reduction of the polymer film immediately formed on the electrode surface. The peak potentials of the polymer itself shifted to a more positive direction as the number of potential cycles increased. The peak then overlapped with the redox peaks of the monomer oxidation peak, which appeared at +1.3 V. The resulting reddish-brown-coloured polymer film revealed a broad redox peak at around +1.1 V during the reverse cathodic scan.
Immobilization of ss-DNA
A thoroughly washed electrode coated with poly-terthiophene-bearing carboxylic acid groups was used to attach an oligodeoxynucleotide (ODN), as shown in the previous study (8). The conducting polymer (polyTTCA)-coated electrode was immersed in a 30 mM acetic acid/acetate buffer (pH 5.2) containing 4.0 mM EDAC for 1 h. The EDAC-attached conducting polymer-coated electrode was rinsed with a buffer solution and subsequently incubated in a 11 μM probe ODN/acetate buffer solution for 6 h at 25°C. In this process, amino-linked C6-modified ODN was immobilized on the polyTTCA film by the formation of covalent bonds with carboxyl groups on the polymer.
Preparation of ds-DNA through hybridization
To prepare double-stranded DNA probes through hybridization, the ss-DNA probe-immobilized electrode was subsequently incubated for 1 h in a complementary matched target ODN solution (11 μM target ODN, 10 mM phosphate buffer pH 7.4, 0.137 M NaCl). The incubation was performed in a shaking incubator (Model: NB-205, N-Biote INC, South Korea) with a rotation speed of 80 r.p.m. Incubation temperature was different from each different sequence (60°C for 36mer, 45°C for 32mer, 30°C for 19mer, and 40°C for 28mer ODN). After the incubation, the temperature was allowed to cool down to room temperature in the incubator; subsequently the electrode was taken out and washed well with the same buffer solution.
Expression and purification of proteins
An expression vector, pQE-9, with the mouse mPurβ gene, was kindly provided by Dr R. J. Kelm of the Mayo Clinic/Foundation (13). In this construction, there is no protease site between the N-terminal His-tag and the protein. To introduce a protease site, mouse mPurβ was cloned again into a modified pET15b vector with a TEV cleavage site subsequent to the N-terminal His-tag. To over-express mPurβ, the plasmid was transformed into the E.coli strain BL21(DE3) and the protein expression levels were tested by varying the conditions of induction. mPurβ is highly expressed under conditions of a short induction time (∼1.5 h) and a small amount of IPTG (<0.15 mM). Under these conditions, the expressed mPurβ protein is highly soluble in both Tris (pH 8.0) and phosphate buffers (pH 7.0). The harvested cells were resuspended in a cell lysis buffer with 0.1 mg/ml lysozyme. The supernatant of the cell lysate was injected onto a Hi-trap Ni-column (Pharmacia Co.), and pre-equilibrated by the binding buffer. The column was washed with a 20% elution buffer and eluted with 300 mM imidazole. The fluent from the Ni-column was directly injected onto a desalting G-25 column pre-equilibrated with 20 mM Tris (pH 8.0), 1 mM EDTA, 1 mM DTT and 100 mM NaCl to adjust the buffer condition for the TEV digestion. The TEV digestion was performed at 4°C overnight (14). The TEV-digested mPurβ was further purified using a Mono-Q column. The proteins were eluted by a salt gradient from 100 to 500 mM NaCl. The target protein was eluted with ∼220 mM NaCl.
E.coli MutH with a N-terminal His-tag was cloned and over-expressed as described (15,16). His-tagged MutH was purified over a Ni-column, and the His-tag was removed by thrombin cleavage at room temperature. MutH was then purified over a Mono-Q column and concentrated to 2 mg/ml in 20 mM Tris (pH 8.0), 60 mM KCl, 1 mM DTT, 0.5 mM EDTA and 5% glycerol.
Impedance, DPV and EQCM measurements
ODN probe-modified electrodes (area: 0.07 cm2) were used as working electrodes; A Ag/AgCl electrode, as a reference electrode, and a Pt wire as a counter electrode. DPVs were recorded using a Potentiostat/Galvanostat [PAR EG&G Model 273A (USA) and Kosentech Model KST-P1A (South Korea)]. An impedance experiment was carried out using the potentiostat coupled with a Lock-in amplifier (PAR EG&G, MODEL 5210) at an open circuit voltage from 100 KHz down to 10 Hz at a sampling rate of 5 points per decade (AC Amplitude: 10 mV). An Ag/AgCl (in saturated KCl) electrode with a non-aqueous solution bridge was used for the polymer growth in a 0.1 M TBAP/CH2Cl2 solution. DPVs were recorded in a phosphate buffer solution of pH 7.4 in the potential range between 0 and 1.6 V. The DPV measurement conditions were as follows: scan rate, 5 mV/s; pulse height, 50 mV; pulse width, 50 ms. The experiment employing the electrochemical quartz microbalance (EQCM) was performed using a SEIKO EG&G model QCA917. The EG&G PARC software package (M270/250 Electrochemical analysis Software) and an Au working electrode (area, 0.196 cm2; 9 MHz; AT-cut quartz crystal) were used for the EQCM experiment.
Analytical procedures
After anodic polymerization of TTCA on a glassy carbon electrode, ss-DNA was immobilized to the carboxylic acid functional group of polyTTCA using catalyst (EDAC). Then, the ss-DNA-immobilized polyTTCA-coated electrode was immersed in a solution containing complementary ss-DNA to make a ds-DNA-modified probe. The ss-DNA and ds-DNA immobilized probes were soaked in the solution containing MutH or mPurβ. To evaluate the hybridization of DNA and the interaction of probe DNA with proteins (MutH, mPurβ), the DPV and impedance spectra were recorded in a blank phosphate buffer solution before and after hybridization and interaction reactions.
RESULTS AND DISCUSSION
Detection of DNA hybridization
To evaluate the attachment of probe ODN (ss-DNA), the amount of the probe ODN (32mer) immobilized on the polymer-coated electrode was determined using the quartz crystal microbalance technique. During immobilization of the probe ODN, the frequency gradually decreased and reached a steady state after 1 h, indicating that the immobilization time was ∼1 h. After immobilization for 1 h, the mass change due to immobilization of the probe ODN on the electrode surface was determined to be 530 ± 18 ng.
Impedance spectra (frequency range, 100 kHz to 10 Hz; AC amplitude, 10 mV) were separately obtained for the electrode modified with the polymer and the ss-DNA probe before and after hybridization in a phosphate buffer medium at room temperature (shown in Figure 2A). The ss-DNA probe decreases the impedance somewhat compared with that of the polymer-modified electrode. However, the hybridization causes a strong decrease of the impedance values, indicating that the ds-DNA is more conductive than ss-DNA immobilized on the conducting polymer coated electrode (8). To examine the chain size effect on the hybridization of DNA, 28mer, 30mer, 32mer and 36mer ODNs were hybridized using the same method. Of these, the largest reproducible difference of logarithmic impedance values before and after hybridization for the complementary sequence was around 1 kHz, which was the same to 28mer, 30mer, 32mer and 36mer ODNs. Table 2 shows the difference of logarithmic impedance (ΔlZl) values before and after hybridization for the complementary sequence ∼1 kHz. After hybridization, all impedance values decreased which indicated increasing conductivity due to the formation of double-stranded DNA.
Figure 2.
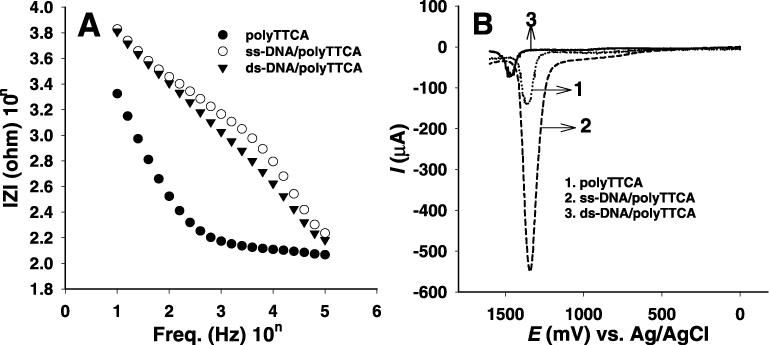
(A) Impedance spectra and (B) differential pulse voltammograms recorded for (1) polyTTCA, (2) ss-DNA/polyTTCA and (3) ds-DNA/polyTTCA-modified GCEs. Impedance spectra and differential pulse voltammograms were recorded after 1 h hybridization in a phosphate buffer solution of pH 7.4. The impedance and current changes reveal the degree of hybridization.
Table 2. The difference of logarithmic impedance values (ΔlZl) before and after hybridization of the complementary sequence according to the different ODN length at 1 KHz.
| ODN length | ΔlZl (10nΩ) |
|---|---|
| 28mer | 1.04 ± 0.05 |
| 30mer | 0.61 ± 0.05 |
| 32mer | 0.78 ± 0.05 |
| 36mer | 0.98 ± 0.05 |
To confirm the impedance results, DPVs were recorded in a blank phosphate buffer (pH 7.4/0.137 M NaCl) solution before and after the reaction, and the DPV results were employed to estimate the formation constant between the probe ds-DNAs and proteins. Figure 2B shows the DPVs recorded for (i) the polyTTCA-modified electrode, (ii) the ss-DNA/polyTTCA-modified probe and (iii) the ds-DNA/polyTTCA-modified probe in a buffer solution. The DPV recorded for polyterthiophene bearing carboxylic acid groups shows an anodic peak around 1.3V versus Ag/AgCl, which comes from the oxidation of the polymer. Meanwhile, in the DPV recorded for the modified electrode after immobilization with the ss-DNA and ds-DNA probes, two peaks appeared at +1.0 and +1.4 V, which corresponded to the oxidation of guanine and adenine on the ODN, respectively (5,17). The guanine oxidation peak is smaller than that of adenine that was observed even at a low concentration of ODN. The anodic peak current of ss-DNA is larger than that of the polymer-modified electrode itself, which appears around the adenine peak position. In contrast, the oxidation peak of ds-DNA is much smaller than that of ss-DNA (15), because the magnitude of oxidation peaks relates to the amount of guanine and adenine residues in the close proximity to the electrode surface. In the case of the ss-DNA probe, it has the largest amount of free adenine and guanine bases. After duplex formation with ds-DNA, the primary oxidation sites of adenine and guanine form a part of a hydrogen bond of the DNA's double helix, which prevent the oxidation of the base residues on the electrode surface. Thus, ds-DNA yields a smaller oxidation signal compared with that of ss-DNA.
Direct detection of the ss-DNA binding affinity of mPurβ
mPurβ is known to be an ss-DNA binding protein that is involved in transcriptional regulation of the mouse vascular smooth muscle (VSM) α-actin gene in fibroblasts (13,18). This protein interacts with the purine-rich ss-DNA in a highly conserved polypurine–polypyrimidine region in the VSM α-actin gene promoters to negatively regulate transcription. mPurβ also shows a strong sequence specificity for d(AGGAATG), an essential MCAT enhancer element in polypurine ss-DNA (19,20).
To detect the interaction between DNA and mPurβ, ss-ODN and ds-ODN-modified probes were subsequently incubated for 10 min in an mPurβ solution (450 μg/ml protein, phosphate buffer pH 7.4, 137 mM NaCl) at room temperature (25°C). After incubation, these probes were washed with the same buffer solution. Impedance spectra were separately obtained for the probe before and after binding with mPurβ. Figure 3A shows the plots of the impedance spectra obtained for 30mer ss-ODN and ds-ODN-modified probes before and after binding with mPurβ. The interaction between 30mer ss-DNA and mPurβ causes the decrease of the impedance values. The differences of the impedance values of the ss-ODN and ds-ODN-modified probes before and after binding with mPurβ in Figure 3A are shown in Figure 3B. The largest reproducible differences of logarithmic impedance values for the ss-ODN-modified electrode before and after binding with mPurβ was obtained at ∼1 kHz, which indicates that ss-DNA interacted with mPurβ. Whereas, in the case of the ds-ODN-modified electrode, there was no difference in logarithmic impedance values, indicating that there was no interaction between ds-ODN and mPurβ.
Figure 3.
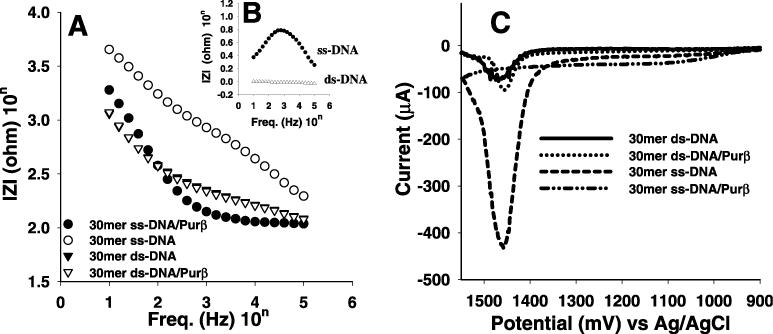
(A) Impedance spectra, (B) the difference in the impedance for 30mer ss-DNAs and ds-DNAs obtained before and after binding with mPurβ and (C) voltammograms for 30mer ss-DNAs and ds-DNAs obtained before and after binding to mPurβ. Impedance spectra and differential pulse voltammograms were recorded in a phosphate buffer solution of pH 7.4. The impedance and current changes reveal the degree of interaction between protein and DNA probes.
To elucidate the impedance result, DPVs were recorded for the DNA probes in a blank phosphate buffer solution before and after binding with mPurβ. As shown in Figure 3C, the adenine oxidation peak for the 30mer ss-ODN probe disappeared after binding with mPurβ. Such disappearance of the DNA intrinsic anodic peak is attributed to the accessibility of adenine moiety to the DNA binding pocket of the protein. On the other hand, the ds-ODN-modified electrode after binding to mPurβ still showed a big anodic peak at 1.4 V similar to that of before binding with mPurβ. These results reconfirmed the impedance results indicating that mPurβ interacted only with ss-DNA. The same experiments repeated for 32mer and 36mer, which do not have the specific sequence for mPurβ, resulted in no such differences in impedance spectra and DPV obtained before and after binding with mPurβ, clearly indicating that mPurβ is interacting with ss-DNA in a sequence-specific manner.
Direct detection of the ds-DNA binding affinity of MutH
MutH is one of three essential proteins (including MutL and MutS) for the initiation of methyl-directed DNA mismatch repair to correct mistakes made during DNA replication in E.coli (21–23). MutH has both strong sequence specificity and methylation specificity (GAmTC) (21,23). The crystal structure as well as modelling studies showed that ds-DNA with the recognition sequence is sited in a deep DNA-binding cleft composed of the N- and C-terminal arms of MutH (16).
To examine the interaction between DNA and MutH, ss-ODN and ds-ODN-modified probes were subsequently incubated for 10 min in a 450 μg/ml MutH solution (phosphate buffer of pH 7.4 and 0.137 M NaCl) at room temperature. After incubation, these probes were washed with the same buffer solution. Impedance spectra were obtained for the electrode modified with the ss-ODNs and ds-ODNs before and after binding with MutH. Figure 4A shows the plot of impedance spectra obtained for the 28mer ss-ODN and ds-ODN-modified electrodes before and after the reaction with MutH. The difference in the impedance values of the ss-ODN and the ds-ODN probes before and after the reaction with MutH are shown in Figure 4B. The largest reproducible difference in logarithmic impedance values was obtained using the ds-ODN probe, indicating the presence of an interaction between ds-ODN and MutH. However, with regard to the ss-ODN probe, there was no difference in logarithmic impedance values before and after the reaction with MutH. This indicates that there was no interaction between the ss-ODN and MutH. Unlike mPurβ, MutH is well known as a ds-DNA binding protein. The present method easily elucidates the fact that MutH interacts only with ds-ODN, due to the structural changes in immobilized ODNs after binding with MutH.
Figure 4.
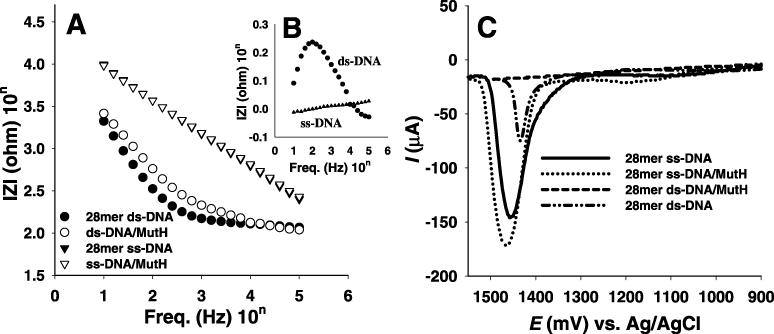
(A) Impedance spectra, (B) the difference in the impedance spectra for 28mer ss-DNAs and ds-DNAs between before and after binding with MutH and (C) the difference in the pulse voltammograms for 30mer ss-DNAs and ds-DNAs before and after binding MutH.
To confirm the impedance result, DPVs were recorded in a blank phosphate buffer solution before and after the reaction with MutH. As shown in Figure 4C, the adenine oxidation peak disappeared in CV for the 28mer ds-ODN after the reaction with MutH. This is comparable with that of the ds-ODN probe before the reaction. Such disappearance of the DNA intrinsic response is attributed by the digging of the bases including adenine and guanine moieties in the pocket of the active site of MutH through the interaction with ds-ODN, whereas DPV recorded for the ss-ODN-modified electrode after incubation in a MutH solution still showed a big anodic peak for ss-ODN at 1.4 V, similar to that before incubation with in a MutH solution. This indicates that there is no interaction between the ss-ODN and MutH. All 36mer, 30mer and 19mer ss-DNA probes showed similar results compared to those for 28mer ss-DNA probe (data not shown), indicating no length effect of ss-DNA probes. In addition, impedance spectra and DPV of the 36mer and 32mer ds-DNAs with the specific sequence d(GATC) for MutH show similar results compared with those of 28mer ds-DNA, while 19mer ds-DNA without the GATC sequence shows that MutH does not interact with the ds-DNA probe.
The interaction between the hemimethylated ODN probe and MutH
To elucidate the sequence specificity of the DNA interaction with MutH, 28mer-me1 and 28mer-me2 were used to compare their interactions with MutH. The hemimethylated ODN-modified probes were subsequently incubated for 10 min in a MutH solution at 25°C. After incubation, the probes were taken from the solution and the impedance spectra were obtained in a blank buffer solution. Figure 5A shows the impedance spectra and their differences (inset) for the 28mer-me1 ds-ODN-modified probe before and after the reaction with MutH. In this case, the interaction between the hemimethylated ds-ODN and MutH causes the decrease of the impedance values. The largest reproducible difference of logarithmic impedance values before and after binding with MutH was ∼0.12 Ω. This is 48% smaller than the 0.23 Ω of ds-28mer that has the unmethylated d(GATC) sequence. It is known that MutH has diverse interactions with the methylated and unmethylated ds-d(GATC) sequence (21,24–26). Our results show that the binding affinity of MutH to the unmethylated sequence is stronger than the methylated sequence.
Figure 5.
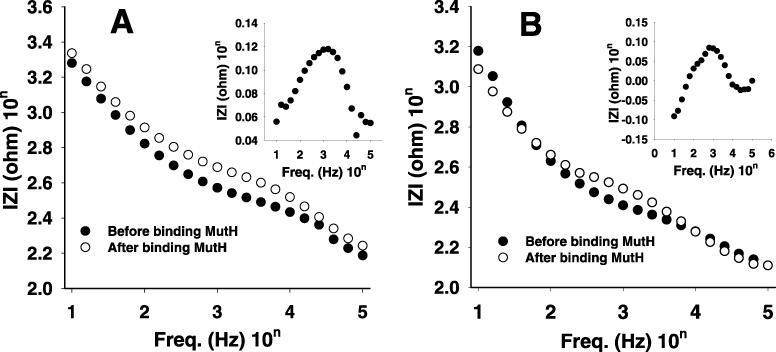
Impedance spectra for (A) 28mer sequence I and (B) sequence II hemimethylated ds-DNAs. The difference impedance for the sequence I [the inset of (A)] and sequence II [the inset of (B)], obtained before and after binding to MutH.
Figure 5B shows the impedance spectra and their difference (inset) recorded for the 28mer-me2 hemimethylated ds-ODN-modified probe before and after binding with MutH. The interaction between the hemimethylated ds-DNA and MutH also causes the decrease of the impedance values. The largest reproducible difference of logarithmic impedance values before and after binding with MutH was ∼0.08 Ω under the previous experimental conditions, which is smaller than that of sequence I by ∼33%. This and the above results likely indicate that the ds-DNA affinity of MutH significantly depends on the DNA sequence itself rather than on methylation even though the hemimethylated ds-d(GATC) DNA sequence is essential for the full endonuclease activity of MutH at the initiation of methyl directed DNA mismatch repair. The co-crystal structures of MutH with hemimethylated and unmethylated DNAs should answer this assumption.
The concentration dependence of proteins on their interactions with DNA
The responses of the ss-DNA probe according to the concentration of mPurβ were compared to the impedance difference, ΔIZI, before and after the reaction as shown in the inset of Figure 6(A). The probe response to mPurβ was examined in the concentration range from 5 to 465 μg/ml. The difference became larger with increasing protein concentration. It was seen that the ss-DNA/mPurβ complex formed through the interaction was more conductive than that of ss-DNA and produced decreases in impedance values. The maximum of ΔlZls (lZlmax) shifted as the concentration changed. ΔlZlmax at low concentrations of mPurβ was ∼104.5 Hz. When the concentration of mPurβ increased to 465 μg/ml, the ΔlZlmax shifted to 103 Hz. These are due to the change of the protein amount bound to the probe surface, but do not depend on the structure of the protein–DNA complex. When the concentration of protein increased, the more protein–DNA complex formed on the surface. Based on these data, a calibration plot of ΔlZl at ΔlZlmax according to the mPurβ concentration is shown in Figure 6A. The linear response was observed in the mPurβ concentration range from 5 to 465 μg/ml. In this case, the slope was ∼1.41 × 10−3 Ω/cm (r2 = 0.986) with the detection limit of 3 μg/ml (88 nM). It has been known that a typical fluorescence method for the protein/ds-DNA interaction shows the detection limit in the range of 100 pM to 10 nM (27). Thus, the detection limit of mPurβ/ss-DNA interaction is comparable with a fluorescence method.
Figure 6.
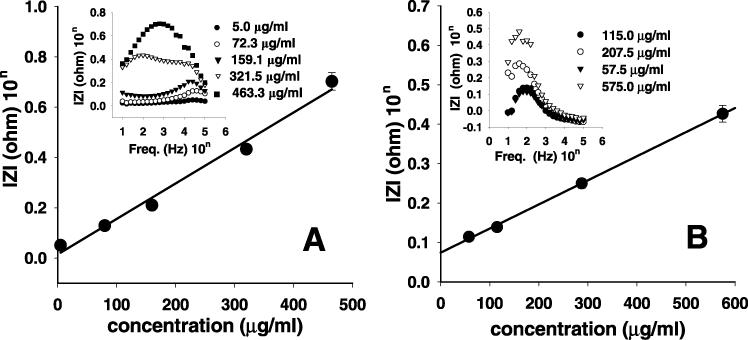
(A) The calibration plots for the determination of mPurβ with ss-DNA-modified electrodes (inset: the difference of impedance spectra for ss-DNA before and after binding with mPurβ in difference concentrations). (B) The calibration plots for the determination of MutH with ds-DNA-modified electrodes (inset: the difference in the impedance spectra for ds-DNA before and after binding with MutH at different concentrations). The highest difference represents the higher concentration of protein.
A ds-DNA-modified probe was applied to determine the concentration of MutH in a buffer solution. Here, the concentration of MutH was changed in the range from 5 to 575 μg/ml. The impedance difference before and after the reaction with MutH became larger with increasing protein concentration, as shown in the inset of Figure 6B. The maximum ΔlZls were observed to be ∼102 Hz. Based on these data, a calibration plot of ΔlZl at 102 Hz versus the different concentrations of MutH is shown in Figure 6B. The linear response was observed in the MutH concentration range from 57.5 to 575 μg/ml. The slope was ∼6.11 × 10−4 Ω/cm (r2 = 0.999) with the detection limit of 25 μg/ml (890 nM).
DISCUSSION
A simple and sensitive electrochemical method to detect the DNA–protein interaction has been developed. A DNA probe having a specific sequence showed a selective response due to the interaction with a protein. The difference in the impedance values obtained before and after interaction between a protein and a specific sequence DNA reflected the biological binding event between the DNA and proteins. This successfully proved that MutH binds only with ds-DNA, while mPurβ binds with ss-DNA. Moreover, DPVs recorded for the DNA probes before and after the reaction with proteins certified that the electrochemical response due to the oxidation of bases of the DNA probes was changed after binding to proteins through the interaction between them. The results showed that the developed interaction detection method might be promising for usage in related research fields.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by the Center for Integrated Molecular Systems through KOSEF and the POSRIP research grant (1RC0402301) to C.B.
REFERENCES
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis, ASM Press, Washington, DC. [Google Scholar]
- 2.Lewin B. (2000) Genes VII. Oxford University Press, New York, NY. [Google Scholar]
- 3.Fried M.G. and Crothers,D.M. (1981) Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res., 9, 6505–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galas D.J. and Schmitz,A. (1978) A DNA footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res., 5, 3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palecek E. and Foita,M. (2001) DNA hybridization and damage. Anal. Chem., 75A–83A. [DOI] [PubMed] [Google Scholar]
- 6.Drummond T.G., Hill,M.G. and Barton,J.K. (2003) Electrochemical DNA sensors. Nat. Biotechnol., 21, 1192–1199. [DOI] [PubMed] [Google Scholar]
- 7.Souteyrand E., Cloarec,J.P., Martin,J.R., Wilson,C., Lawrence,I., Mikkelsen,S. and Lawrence,M.F. (1997) Direct detection of the hybridization of synthetic homo-oligomer DNA sequences by field effect. J. Phys. Chem. B, 101, 2980–2985. [Google Scholar]
- 8.Lee T.Y. and Shim,Y.B. (2001) Direct DNA hybridization detection based on the oligonucleotide-functionalized conductive polymer. Anal. Chem., 73, 5629–5632. [DOI] [PubMed] [Google Scholar]
- 9.Wang J. (1985) Stripping analysis: Instrumentation and Application. Verlag Chemie, Deerfield Beach, FL. [Google Scholar]
- 10.Bard A.J. and Faulkner,A.R. (2001) Electrochemical Methods. John Wiley & Sons, Inc., New York, NY. [Google Scholar]
- 11.Lee T.Y., Shim,Y.B. and Shin,S.C. (2002) Simple preparation of terthiophene-3′-carboxylic acid characterization of its polymer. Synth. Metals, 126, 105–110. [Google Scholar]
- 12.Kissenger P.T. and Heineman,W.R. (1984) Laboratory Techniques in Electroanalytical Chemistry, Marcel Dekker, Inc., New York, NY. [Google Scholar]
- 13.Kelm R.J. Jr, Wang,S.-X., Polikandriotis,J.A. and Strauch,A.R. (2003) Structure/function analysis of mouse Purβ, a single-stranded DNA-binding repressor of vascular smooth muscle α-actin gene transcription. J. Biol. Chem., 278, 38749–38757. [DOI] [PubMed] [Google Scholar]
- 14.Kapust R.B. and Waugh,D.S. (2000) Controlled intracellular processing of fusion proteins by TEV protease. Proc. Exp. Purif., 19, 312–318. [DOI] [PubMed] [Google Scholar]
- 15.Feng G. and Winkler,E. (1995) Single-step purifications of his6-MutH, his6-MutL and His6-MutS repair proteins of Escherichia coli K-12. BioTechniques, 19, 956–965. [PubMed] [Google Scholar]
- 16.Ban C. and Yang,W. (1998) Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J., 17, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J., Rivas,G., Cai,X., Palecek,E., Nielsen,P., Shiraishi,H., Dontha,N., Luo,D., Parrado,C., Chicharro,M. et al. (1997) DNA electrochemical biosensors for environmental monitoring. A review. Anal. Chim. Acta, 347, 1–8. [Google Scholar]
- 18.Kelm R.J. Jr., Elder,P.K. and Getz,M.J. (1999) The single-stranded DNA-binding proteins, Purα, Purβ, and MSY1 specifically interact with an exon 3-derived mouse vascular smooth muscle α-actin messenger RNA sequence. J. Biol. Chem., 274, 38268–38275. [DOI] [PubMed] [Google Scholar]
- 19.Kelm R.J. Jr, Cogan,J.G., Elder,P.K., Strauch,A.R. and Getz,M.J. (1999) Molecular interactions between single-stranded DNA-binding proteins associated with an essential MCAT element in the mouse smooth muscle α-actin promoter. J. Biol. Chem., 274, 14238–14245. [DOI] [PubMed] [Google Scholar]
- 20.Carlini L.E., Getz,M.J., Strauch,A.R. and Kelm,R.J.,Jr (2002) Cryptic MCAT enhancer regulation in fibroblasts and smooth muscle cells. Suppression of TEF-1 mediated activation by the single-stranded DNA-binding proteins, Pur alpha, Pur beta, and MSY1. J. Biol. Chem., 277, 8682–8692. [DOI] [PubMed] [Google Scholar]
- 21.Welsh K.M., Lu,A.-L., Clark,S. and Modrich,P. (1987) Isolation and characterization of the Escherichia coli MutH gene product. J. Biol. Chem., 262, 15624–15629. [PubMed] [Google Scholar]
- 22.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 23.Obmolova G., Ban,C., Heish,P. and Yang,W. (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature, 407, 703–710. [DOI] [PubMed] [Google Scholar]
- 24.Wu T., Loh,T. and Marinus,M.G. (2002) The function of Asp70, Glu77 and Lys79 in the Escherichia coli MutH protein. Nucleic Acids Res., 30, 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedhoff P., Thomas,E. and Pingoud,A. (2003) Tyr212: a key residue involved in strand discrimination by the DNA mismatch repair endonuclease MutH. J. Mol. Biol., 325, 285–297. [DOI] [PubMed] [Google Scholar]
- 26.Kramer B., Kramer,W. and Fritz,H.J. (1984) Different base/base mismatches are corrected with different efficiencies by themethyl-directed DNA mismatch-repair system of E. coli. Cell, 95, 541–552. [DOI] [PubMed] [Google Scholar]
- 27.Rippe K. (1997) Analysis of protein-DNA binding at equilibrium. B. I. F. Futura, 12, 20–26. [Google Scholar]