

Abstract
Proper control and maintenance of gene expression is critical for cellular identity and maintenance. Transcription of RNA from the genome is intimately controlled by post-translational chemical modification of histone tails and DNA. Recent studies have demonstrated that chromatin-remodeling complexes seek out their target genomic loci through the help of noncoding RNA molecules. Within this Review, we will outline how the use of biochemical techniques has shed light on the mechanisms employed by RNA to guide these complexes and therefore control gene expression.
Graphical abstract
Proper control of gene expression is essential for the cellular response to extracellular cues or stressors and ultimately organismal survival. While DNA content from cell to cell is largely the same, distinct gene sets are turned on or off in concert to orchestrate a systems-level reaction and ensure proper cellular function. To control such gene sets, DNA is organized into a proteinaceous unit termed chromatin. Chromatin structures, which are modulated through DNA looping, nuclear bodies, promoter-enhancer elements, and transcription factories, must be tightly controlled to ensure proper gene expression.1,2 Recent methods have been developed to understand the structure and functional mechanisms that regulate the chromatin state on a global level.3,4
Chemical modification of histone proteins is a key mechanism for controlling gene expression. Many histone modifications have been reported to date.5 The two most abundant modifications—methylation and acetylation—have been widely studied and are well characterized. Their dynamic regulation has been demonstrated to be critical to chromatin maintenance across many species.6 Acetylation and deacetylation occur on lysine residues near the N-terminal tail of histone proteins.7 Histone methylation and demethylation have been implicated in both transcriptional activation (H3K4, K36, K79) and silencing (H3K9, H3K27, H4K20).8 Chromatin-modification enzymes control the deposition and removal of chromatin modifications for modulating RNA expression. Histone acetyl transferases (HATs) and histone deacetylases (HDACs) are responsible for lysine acetylation (Figure 1A). Histone methyltransferases and demethylases control histone methylation status (Figure 1B).8 Histone acetylation eliminates the positively charged lysines on histone tails, the result of which is decreased interaction with DNA. The DNA is subsequently more loosely wrapped around histones and is available for the binding of transcription factors and proteins that initiate transcription.1 The opposite is true for histone deacetylation, wherein the restored positive charge on lysine causes greater interaction with DNA and thus condenses chromatin to prevent transcription.7 Chromatin condensation can also be brought about by methylation via indirect mechanisms. Relaxed, transcriptionally active DNA is referred to as euchromatin, and transcriptionally silent DNA (condensed) is heterochromatin. The dynamic nature of chromatin chemical modification has been explored for many decades. However, because the chemical and structural composition of histones is very similar from gene to gene, the mechanism for loci selection was largely unknown.
Figure 1.
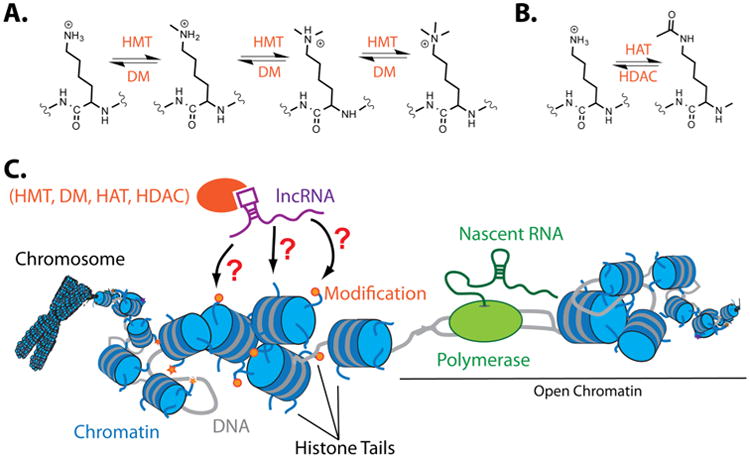
Mechanisms of chromatin modification and RNA-guided modification. (A) Methylation and demethylation of histone tails. (B) Acetylation and deacetylation of histone tails. (C) Depiction of guidance of histone modification enzymes by RNA.
Transcription factors are well-known for their ability to bind to specific sequences or structures in genomic DNA.9 However, many chromatin-modifying complexes do not have proteins that bind to DNA directly.10 In some cases transcription factors can guide chromatin-modifying complexes. For example, recent studies have recently shown that the Jumanji protein Jarid2 is responsible for recruiting the histone demethylase polycomb repressive complex 2 (PRC2) to its target sites in mouse embryonic stem cells.11 However, Jarid2 expression is very low in differentiated cells.12 Therefore, for some time, it was unknown how PRC2 is targeted to chromatin in other cell types. In addition, there are many chromatin-modifying complexes, which target specific loci, without discovered DNA-binding proteins.13 This represents just one example but is a common gap in our understanding of how chromatin modification is controlled and targeted.
The sequencing of higher-order mammalian transcriptomes has revealed that a substantial fraction of DNA is transcribed to yield many short or long noncoding RNAs (lncRNAs) with limited protein-coding capacity. Long noncoding RNAs are a set of noncoding transcripts that are greater than 200 nt in length. Similar to coding mRNAs (mRNAs), lncRNAs are transcribed by RNA Polymerase II, capped, spliced and polyadenylated.14 Profiling unique chromatin marks (K4–K36 bivalency) and reconstructing RNA-Seq maps have definitively demonstrated that lncRNAs are unique transcripts.15 Additional sequencing technologies capturing both the 5′-7-methyguano-sine cap and polyA tail have further enhanced our understanding of the complexity of lncRNA expression.16,17 RNA sequencing coupled with protein-coding potential prediction programs have added additional levels of stringency for predicting lncRNAs and separating them from protein-coding mRNAs.18 Each of these methods have developed robust and complementary maps of lncRNA expression. Comprehensive lncRNA expression analysis in several mouse and human cell types has revealed that lncRNA expression is very unique to cell-type and may even be more specific than the mRNA expression profile.19 Molecular profiling and genetic screens have identified lncRNAs that play a role in dosage compensation,20 imprinting,21 developmental gene expression, and reprogramming of human induced pluripotent stem cells.22
Perhaps the first well-characterized lncRNA was X-inactive-specific transcript, or XIST.23 XIST is a 17 kb participant in X-chromosome inactivation during early development in females.24 XIST RNA spreads along the X-chromosome in cis and recruits different chromatin remodeling complexes, most notably polycomb repressive complex 2 (PRC2), to enforce gene silencing. PRC2 is a chromatin modifying complex consisting of H3K27 methyltransferase subunits EZH2 (Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit), SUZ12 (Suppressor of Zeste 12), and EED (Embryonic Ectoderm Development).25 The observation that XIST interacts directly with PRC2 has been challenged, as PRC2 has been demonstrated to have a high level of nonspecific promiscuity, and super-resolution microscopy has revealed a spatial separation of XIST and PRC2 on the X-chromosome.26–28 Recent findings have also demonstrated that XIST may instead bind the protein SHARP to recruit HDAC3 (histone deacetylase 3) and initiate histone deacetylation and PRC2 recruitment to exclude Pol II across the X chromosome.29 The molecular mechanism of XIST targeting is an evolving story; nevertheless, XIST has served as a paradigm for understanding the mechanisms by whicn lncRNAs can control chromatin state.
Another of the first functionally characterized lncRNAs, HOTAIR, was shown to associate with and target PRC2 to distantly located genes.30 Unlike XIST, which targets gene repression in cis, HOTAIR works by silencing distant loci in trans. This work therefore provided evidence that lncRNAs may globally affect gene expression by bringing complexes to repress gene transcription. Another RNA, HOTTIP, was also shown to bind to the mixed-lineage leukemia (MLL) histone H3K4 methyltransferase complex, bringing MLL to specific sites on the genome to activate transcription.31 These observations have been extended by others to show that many well-characterized intergenic RNAs are indeed binding to these complexes,32,33 thereby suggesting that lncRNAs may serve as guides to bring chromatin remodeling complexes to specific loci (outlined in Figure 1C).
lncRNAs have been implicated to control many diverse biological functions, beyond chromatin modification. For example, the lncRNA NRON complexes with an importin protein to regulate the subcellular trafficking of NFAT.34 A UCHL1 antisense lncRNA complexes with the UCHL1 mRNA to regulate protein synthesis of the UCHL1 protein.35 Alu elements within cytoplasmic lncRNAs can form complementary duplexes with Alu elements in the 3′-UTRs of mRNAs to recruit the Staufen1 complex and enhance mRNA decay.36 The many functions of lncRNAs, from chromatin remodeling to translation control, have prompted the need to develop biochemical tools to study lncRNA function within living cells.
There are many questions that remain to be answered in order to fully characterize the content of chromatin-associated RNAs. Where and when do lncRNAs bind to the genome? What are the proteins that interact with lncRNAs, and what is the RNA–protein interface? How does the structure of lncRNAs contribute to their biological function? The recent developments in methods to address these questions have foundations in chemical biology, with chemical reagent design being a critical component to the progress of the field. Within this Review, we discuss the recent utilization of chemical biology and chemical techniques to increase our understanding of how lncRNAs work and target specific chromatin modifying complexes to regulate gene expression.
Chemical Cross-Linking to Decipher Where and When RNAs Bind to the Genome
The observation that lncRNAs can bind to specific loci on the genome has prompted the development of biochemical methods that can assay their specific locations and the macromolecules they associate with. Many of these methods are rooted in the same principals that were first applied to assay chromatin marks and the localization of DNA-binding proteins and chromatin-modification enzymes. Chromatin immunopurification followed by deep sequencing, or ChIP-Seq,37 is a common method used to assay the genomic localization of chromatin chemical modifications and proteins. In a ChIP experiment, a protein is cross-linked to DNA using formaldehyde. An antibody is then used to enrich the protein of interest. The associated DNA is then isolated and transformed into a sequencing library. ChIP has since been used as an experimental paradigm for the design of protocols used to isolate regions of the genome associated with an RNA of interest.
A few different protocols have been developed to assay the chromosomal location of noncoding RNAs associated with the regulation of epigenetic processes. Each of these methods begins with a critical chemical cross-linking step that “freezes” the localization of an RNA on the genome (Figure 2, A). The first of these, chromatin isolation by RNA purification or ChIRP,38 first relies on in-cell glutaraldehyde treatment (Figure 2B; compound 2). Glutaraldehyde forms a variety of adducts upon reacting with biomolecules, particularly with primary amines, such as lysine.39 Glutaraldehyde undergoes a Mannich-type reaction to form imines, which tautomerize to enamines rendering the products irreversible. The products of these reactions can be both direct and indirect targets of an RNA.
Figure 2.
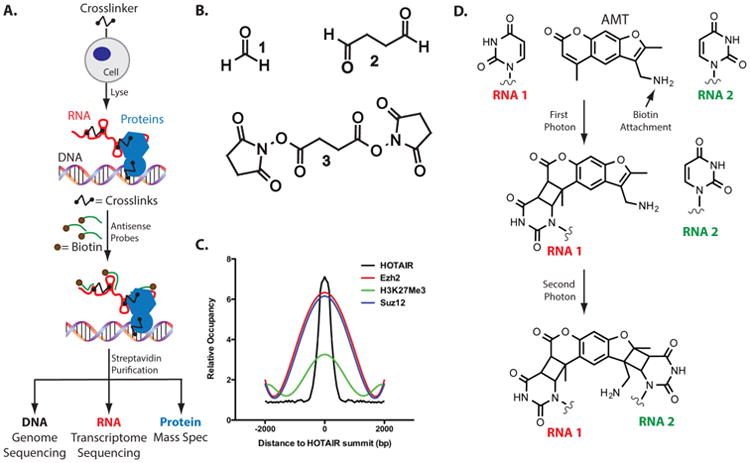
Methods for assaying RNA localization on chromatin. (A) Schematic of methods to isolate RNAs, based on hybridization techniques. (B) Chemical structure of cross-linkers used to capture in vivo RNA interactions. (C) Localization profile demonstrating HOTAIR genomic occupancy in comparison to proteins in the PRC2 complex. (Figure adapted from ref 38. Copyright Elsevier 2011). (D) Chemical scheme depicting RNA–RNA cross-linking using AMT cross-linker.
Capture hybridization analysis of RNA targets (CHART) was an analogous technique developed in parallel to ChIRP.40 In CHART, formaldehyde is used as the in vivo cross-linking chemical reagent (Figure 2B; compound 1). Extensive formaldehyde treatment (3% for 30 min) is expected to yield both direct and indirect cross-linking products containing RNA. Unlike glutaraldehyde cross-linking, formaldehyde forms an intermediary Schiff-base adduct, which is reversible.41 Such reversibility may be advantageous, as the cross-linked nucleic acid and proteins can be easily retrieved in their near-native form by hydrolysis.
The most stringent chemical cross-linking protocol developed is associated with the methodology termed RNA antisense purification, or RAP.42 RAP utilizes disuccinimidyl glutarate, or DSG (Figure 2B; compound 3), along with formaldehyde to cross-link RNA-containing complexes in vivo. DSG has two N-hydroxysuccinimide esters that are activated for nucleophilic attack with stronger nucleophiles, such as primary amines. The cross-linked products are formed via highly stable amides, and they are thought to be a mixture of RNA–protein, RNA–DNA, and DNA–protein. Each of these chemical cross-linking compounds has unique characteristics for their reactivity and stability, and thus they have all found utility in methods that are focused on describing the lncRNA–genome interface.
In all of the methods mentioned above, in vivo cross-linking promotes the formation of stable complexes that can preserve both protein and DNA interactions with RNAs. In each protocol (as summarized in Figure 2A), the cells are subsequently lysed. Following lysis, biotin-appended antisense oligos can be used to enrich for an RNA of interest. Once an RNA is enriched, the cross-linked complex can be digested to obtain cross-linked proteins or genomic coordinates. Below, we summarize how such powerful biochemical techniques have increased our understanding of the ways lncRNAs bind to the genome.
The utilization of ChIRP to study RNA genomic binding sites was employed on the long noncoding RNA HOTAIR.38 HOTAIR associates with the chromatin modifying complex PRC2 to deposit methylation marks through its lysine methyltransferase activity, which leads to chromatin silencing. Incredibly, an unbiased comparison between the sites of HOTAIR binding demonstrated enrichment for genes involved in pattern specification processes, consistent with the model that HOTAIR enforces the epigenomic state of distal and posterior positional identity. Such a strong comparison is consistent with the suggestion that the HOTAIR–chromatin interaction is associated with PRC2 relocalization and gene silencing. Overlap of the HOTAIR ChIRP signal with the genomic occupancy of the PRC2 components (Figure 2C) clearly shows very focal organization of HOTAIR genomic binding. The overlap between PRC2 target genes and HOTAIR occupancy suggested two models: HOTAIR may actively recruit PRC2 to it targets genes or HOTAIR is a scaffold that is transported with PRC2. The power of CHIRP was further demonstrated upon EZH2 (a core component of PRC2 that binds to histones) knockdown. The results of this test showed that HOTAIR is able to find its target loci, independent of PRC2. These ChIRP results were the first to show how focal lncRNA binding is, which is highly suggestive of lncRNAs' ability to seek out their target loci. Overall, such hybridization-based techniques demonstrated their power in revealing the intricacies of RNA targeting to chromatin. The additional utility of cross-linker selection has been extended to further increase our mechanistic understanding of how RNAs choose their respective chromatin targets.
The mechanisms for how RNAs choose to target specific chromatin loci is still largely unresolved. Choosing an appropriate cross-linker to trap unique complexes inside the cell can aid in attaining a deeper understanding. As one example, for some time it was hypothesized that intermolecular RNA–RNA interactions can be used by many ncRNAs to achieve their diverse functions (for example, snoRNAs43). However, this mechanism had not been proven in the context of genomic targeting. Using this approach was critical for unraveling the RNA–RNA interactions which provide evidence for RNA-mediated targeting to the genome. Psoralen, and derivatives such as 4′aminomethyltrioxsalen or AMT, can crosslink two opposing uridine nucleobases in double-stranded RNA regions (Figure 2D). Upon light-exposure, AMT generates interstrand cross-links between uridine bases in RNA without reacting with proteins.44 In a modified RAP protocol (see above), AMT was used to cross-link RNA–RNA interactions inside cells. To showcase the utility of RNA–RNA cross-linking by AMT, two ncRNAs implicated in RNA processing were investigated: U1 small nuclear RNA, a component of the spliceosome, and Malat1, a large ncRNA that localizes to nuclear speckles. U1 snRNA forms base-pair contacts with pre-mRNAs at 5′ splice sites. RAP confirmed that U1 directly hybridizes to 5′ splice sites and 5′ splice site motifs throughout introns. It also revealed that Malat1 interacts with pre-mRNAs indirectly through protein intermediates. Interactions with nascent pre-mRNAs cause U1 and Malat1 to localize proximally to chromatin at active genes, demonstrating that ncRNAs can use RNA–RNA interactions, which come from other RNAs that are being actively transcribed during their activation, to target specific pre-mRNAs and genomic sites. Thus, RAP-RNA (using AMT cross-linking) can accurately and specifically identify RNA–RNA interactions mediated by direct hybridization. As illustrated by this section, there are now several effective methods and techniques that can be used to identify where, and to some extent how, ncRNAs can localize to specific regions of the genome. The next logical steps following such characterization are focused on elucidating the structure of ncRNAs and the RNA–protein interface that controls ncRNA function.
Using Chemical Probing to Understand the Structure of Lncrnas
Each individual RNA can have unique primary sequences that can give rise to different structural components and mofits. Each of these motifs can perform different biological functions. For example, single-stranded regions often serve as landing pads for proteins.45 Extended RNA structures have been implicated in RNA-based diseases,46 and disease-associated mutations have been demonstrated to alter the structural landscape of RNA.47 Therefore, discerning the function of noncoding RNAs and their control over epigenetic regulation critically relies on a more comprehensive understanding of their structures.
RNA structure mapping has been employed for decades to understand the physical properties of several RNAs. Several reagents have been developed for obtaining high confidence structure maps of RNAs. RNases that are specific for an RNA structural motif are used to obtain low-resolution measurements of RNA secondary structure.48,49
As an alternative, structure probing using small molecules has a rich history of providing RNA structure measurements at high resolution. Chemical probing of RNA structure first appeared in the 1980s.50,51 In these early experiments, dimethylsulfate was used to interrogate the solvent accessibility of guanosine and interrogate the base-pairing characteristics of adenosine and cytosine (Figure 3A). N1-methylation of adenosine and N3-methylation of cytosine can be can be read out through reverse transcription, from which a corresponding cDNA sequence is created that is truncated at the modification site (explained below).52,53 These early investigations established the paradigm of using chemical methods to probe RNA structure.
Figure 3.
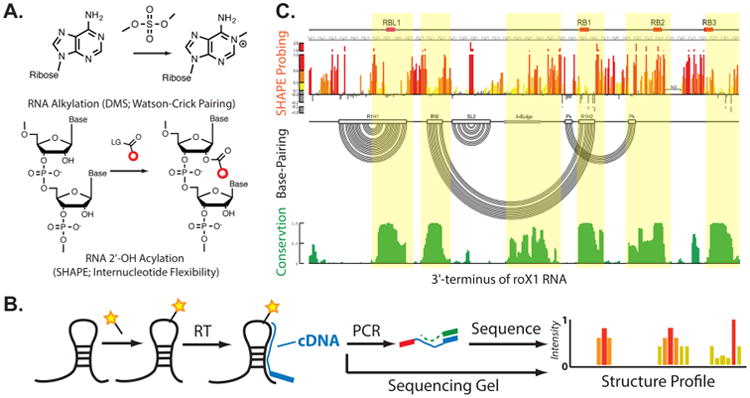
Chemical and genomic methods for analyzing RNA structure. (A) Chemical modification by RNA structure readers DMS and SHAPE. (B) Schematic for analysis of adduct formation through RNA structure measurements. The experimenter can either analyze adducts through denaturing gel electrophoresis, or more recently, RNA sequencing. (C) SHAPE analysis of RoX RNA. A comparison between RNA structure probing and conservation suggests the lower SHAPE reactive sites (double stranded) are highly conserved and thus are important for function (Figure adapted from ref 77. Copyright Elsevier 2013).
Additional chemical methods have more recently become available. Hydroxyl radical cleavage of solvent exposed residues, or metal catalyzed cleavage, are currently the most thorough methods for determining the secondary and tertiary structure of RNAs. Cleavage results in the formation of hydroxyl radicals that abstract the hydrogen from the C5′-position of the backbone, one product of which is a 3′-phosphate and 5′-aldehyde.49,54–56
Selective Hydroxyl Acylation analyzed by Primer Extension, or SHAPE, is the most widely used method of chemical modification to probe RNA structure (Figure 3A). SHAPE takes advantage of two key observations: first, that electrophilic reagents can acylate the 2′-hydroxyl group of RNA nucleotides and, second, that the reactivity of these 2′-hydroxyl groups differs depending on local RNA structure. Nucleotides at flexible positions sample conformations that transiently enhance their nucleophilicity and thus are more highly modified.57,58 All together, these chemical methods provide an increasingly holistic approach to probing RNA secondary structure.
RNA chemical modifications that present as adduct formation or strand cleavage products are traditionally analyzed by denaturing gel electrophoresis.59 For adduct formation, reverse transcription is performed to synthesize cDNA molecules. Each cDNA can be identified on a gel and mapped back to the primary sequence, where the intensity of cDNA stops is related to the number of modifications on the RNA at a given position. For RNA cleavage, the RNA is traditionally radiolabeled at the 5′-end. It is then directly loaded into a denaturing gel, and the cut sites can be identified and mapped back to the sequence (Figure 3B). More comprehensive reviews on methods to analyze RNA structure have recently been published.59
Interpretation of RNA structure data (from denaturing gels or sequencing) has its roots in the merger of RNA structure predictions and chemical probing experiments. RNA structure predictions originate in careful stability measurements on model RNAs; such measurements have given rise to the prevailing nearest-neighbor model developed by Turner and colleagues.60–62 To expand this model, free energy calculations for base pairs, helix stacking, mismatches, and even modified nucleosides have been empirically determined through optical melting experiments. A more comprehensive review on this important subject was very recently published.63 The merger of RNA structure measurements and structure prediction using nearest neighbor parameters can dramatically increase the true positive rates of base pairs and accuracy of secondary structure predictions.64,65 RNA structure prediction algorithms, such as RNAstructure,66 UNAfold,67 or RNAfold,68 can now be routinely used with chemical probing experiments to create reasonably accurate RNA structure models.
RNA structure probing by use of chemical probes has been utilized to understand the structure of entire viral RNA genomes,69 to characterize RNA–protein interactions,70 and to characterize RNA folding and structural transitions.57 Further, the information gained from studying RNA folding transitions with chemical probing71 and RNA–protein interactions inside cells for well-characterized RNAs72 set a strong experimental outline for investigations into the structures of lncRNAs and their complexes.
The sites of RNA–protein interaction, which are critical for epigenetic regulation, have recently been elucidated through RNA structure probing techniques. Chemical probing was used to evaluate the complete secondary structure73 of all 2148 nucleotides of HOTAIR, a long-noncoding RNA involved in the targeted recruitment of the polycomb repressive complex (PRC2) to silence gene expression.30 Refolded HOTAIR RNA was subjected to RNA structure probing using SHAPE, DMS, and terbium-catalyzed strand cleavage.74 These results revealed that HOTAIR has several isolated regions that form into modular structured domains. Structure probing comparisons of individual domains to the full-length RNA revealed similar profiles, further demonstrating the stability of modular domains in RNA structure. Overall, it was demonstrated that HOTAIR is highly structured, as there are 56 helical segments, 38 terminal loops, 34 internal loops, and 19 junction regions. Furthermore, greater than 50% of the nucleotides in HOTAIR are involved in base pairing.
The molecular interface between HOTAIR and PRC2 has yet to be fully determined. Previous deletion experiments on HOTAIR narrowed its protein interaction sites down to two modular regions: (1) a 300-nt-long region at the 5′ end of HOTAIR (nucleotides 1–300, HOTAIR300), which binds PRC2, and (2) a 646-nt-long region at the 3′ end of HOTAIR, which binds LSD1/CoREST/REST.75 Analysis of primary sequence conservation and covariation analyses revealed that the regions associated with putative PRC2 binding are highly structured and also have highly conserved predicted secondary structures. A parallel analysis of the HOTAIR–PRC2 interface further demonstrated that the PRC2 interaction site is composed of structured motifs that are critical for PRC2 recognition.76 These results demonstrate the overall utility of chemical probing to interrogate the structure and function of ncRNAs and proteins critical for epigenetic regulation.
Chemical probing of RNA structure has also been used to reveal the mechanism by which RNAs control the epigenetic state via recruitment of chromatin-modifying complexes. As one example, structure probing and RNA structure conservation analyses revealed the critical nature of RNA structure elements responsible for dosage compensation in Drosophila.77,78 Dosage compensation is an epigenetic phenomenon wherein proteins and lncRNAs associate for the purpose of transcriptional upregulation of genes on the male X chromosome. In order for this to occur, two RNAs called roX1 and roX2 control X chromosome-wide histone acetylation at H4 lysine 16. Dosage compensation complex (DCC) contains the male-specific lethal proteins MSL1–MSL3, a helicase MLE, and the histone acetyltransferase.79,80 A key missing piece in this epigenetic puzzle was how the structures of roX1 and roX2 RNAs were precisely formed and remodeled to control the nucleation of the dosage compensation complex.
Chromatography and immunopurification analysis demonstrated that MLE and the MSL proteins form direct interactions with roX RNAs. As such, MLE and MSL RNA binding sites were then characterized using individual-nucleotide resolution cross-linking and immunoprecipitation (iCLIP, discussed above). This analysis revealed that the MLE RNA helicase and MSL2 ubiquitin ligase bind to evolutionarily conserved domains containing tandem stem-loops in roX1 and roX2 RNAs in vivo. These sites are consistent with conserved elements known as roX boxes, which could play an important role in dosage compensation.81 Concurrent structural analysis by SHAPE probing demonstrated that MLE binding sites are embedded into long stretches of sequence that are less structured and might be functionally irrelevant. Overall structure models revealed a similar architecture between both roX RNAs: stable helices formed by paired roX box motifs that are strung together by flexible single-stranded linkers. Additionally, analysis between structure probing and structure conservation revealed that structural flexibility is inversely correlated with conservation. This highly suggests that the double stranded regions of the RNA are structurally important (see example in Figure 3C). The structural conservation of roX box motifs was further underscored by demonstrating that different roX RNA domains have overlapping function; only combinatorial mutations in the tandem stem-loops result in a severe loss of dosage compensation and consequently male-specific lethality. This analysis serves as the basis for a model where repetitive structural motifs in lncRNAs could provide plasticity during multiprotein complex assemblies to ensure efficient targeting in cis or in trans along chromosomes. This analysis revealed that a combination of chemical probing and biochemical methods to interrogate the RNA–protein interface could reveal the mechanism of epigenetic regulation by noncoding RNAs.
The methods for measuring RNA structure with small molecules have also been extended to incorporate transcriptome-spanning technologies, such as RNA sequencing. DMS and SHAPE have been used in this capacity to provide transcriptome-wide measurements of RNA structure.82–84 These efforts have revealed the architecture of RNAs within cells and how RNA structure elements can influence post-transcriptional interactions, such as protein binding and methylation. Furthermore, more focused sequencing-based methods have been already used to reconstruct the entire secondary structure of the XIST noncoding RNA inside cells.85 As such, the marrying of chemical and transcriptomic approaches will undoubtedly reveal the even more fascinating roles that RNA structure plays to regulate the chromatin state.
Using Chemical Methods to Characterize the RNA–Protein Interface
Within cells, RNA molecules are rarely without a protein counterpart. RNA–protein interactions are critical for all aspects of RNA biology, from transcription to export, localization, and decay.86 The prevailing model in the field is that chromatin-associated RNAs are intimately tied to proteinacious chromatin remodeling complexes. Therefore, gaining molecular insights of the RNA–protein interface is critical for a holistic understanding of RNA-based mechanisms for controlling gene expression.
Perhaps the most widely used method for analyzing RNA–protein interactions is RNA immunopurification, or RIP.87 In RIP, formaldehyde cross-linking is used to freeze RNA–protein complexes. The complex is then purified by utilizing an antibody either against the native protein sequence or a protein containing tag. qRT-PCR or RNA sequencing then identifies the associated RNAs. Formaldehyde cross-linking can capture RNA–protein or protein–protein interactions that are not at the RNA–protein interface. As such, the associated RNAs may not be in direct contact with a protein of interest enriched through purification. This limitation can result in false-positives or an unnatural RNA-protein interaction that forms post lysis.88,89 To overcome these methods, more precise assays have been developed to study the RNA–protein interface with high resolution.
Cross-linking and immunoprecipitation or CLIP of in vivo RNA targets of RNA-binding proteins is a recently developed method that takes advantage of chemical cross-linking to identify the sites of protein binding on RNA.90 CLIP uses 254 nm ultraviolet light to induce excitation of the RNA nucleobases. This causes the formation of covalent bonds between proteins and nucleic acids that are in close proximity. The RNA–protein interface is then amenable to high stringency washes and denaturing gel electrophoresis. Isolated cross-link sites can then be cloned and identified by deep sequencing, where the actual binding site can be identified (outlined in Figure 4A). Although powerful, a major setback in CLIP experiments is the reliance upon the low-efficiency cross-linking between RNA and proteins, which is estimated to be <1%.
Figure 4.
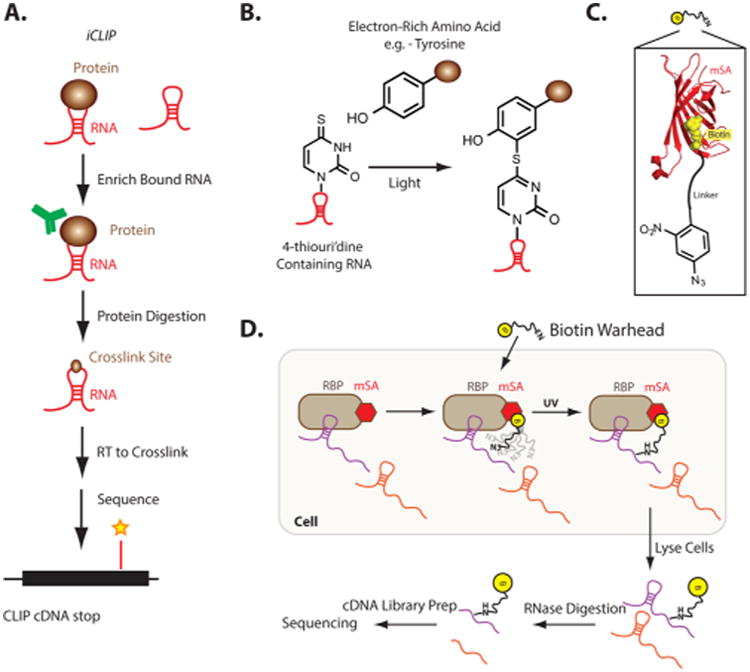
Methods to interrogate the RNA–protein interface. (A) Schematic of CLIP. (B) Chemical cross-linking of 4-thiouridine and tyrosine in PAR-CLIP. (C) Schematic of aryl zide warhead in IPL. (D) Schematic of IPL.
To overcome the limitations of CLIP, an alternative method termed Photoactivatable-Ribonucleoside-Enhanced Cross-linking and Immunoprecipitation or PAR-CLIP was developed. PAR-CLIP leverages the ability of thiol-containing nucleosides to selectively excite with 365 nm light.91 Nascent RNA transcripts can be labeled with modified nucleosides by treating cells with photoreactive ribonucleoside analogs, such as 4-thiouridine (4-SU; Figure 4B) and 6-thioguanosine (6-SG), which will nascently incorporate. Irradiation of the cells by 365 nm light induces efficient cross-linking of RNAs containing photoreactive nucleoside RNAs with electron-rich amino acids that are closely associated (Figure 4B). For some RNA-binding proteins compared head-to-head, UV cross-linking efficiency has been demonstrated to increase from <1% (with uridine) to in some cases near quantitative.91 One potential drawback to PAR-CLIP is the introduction of 4-SU into cellular RNA; however, 4-SU has been demonstrated to be robustly incorporated into many different cell types in vitro.91–93
PAR-CLIP was recently used to characterize the lncRNA-PRC2 interface.94 PRC2 has histone methyltransferase activity and primarily trimethylates lysine 27 of histone H3 (i.e., H3K27me3), a mark of transcriptionally silent chromatin.95 The biochemical function of PRC2 had been known for some time; however, how PRC2 is coordinated to the genome to locate methylation target sites was still not fully resolved. Using PAR-CLIP, it was revealed that PRC2 binds with low occupancy to a majority of promoters. PRC2 was demonstrated to bind to the 5′-end of nascent RNA transcribed at or near the promoter sites of PRC2 enrichment. As such, PAR-CLIP was instrumental for establishing the model in which PRC2 senses nascent RNA transcription and anchors itself at promoters, prompting methylation at those genetic loci. The numerous studies utilizing the methods of CLIP and PAR-CLIP have paved the way to an increased understanding of how RNAs interact with protein complexes and guide them to particular loci on the genome.
Additional tools continue to be developed with the hopes of expanding the methodologies used to probe and identify RNA-binding proteins. This is especially important in the context of RNA–protein interactions that are highly transient. Recent studies have demonstrated that many of the chromatin-associated RNAs are in complexes that act in transit and thus have very short residence time on the genome while complexed. As such, for furthered understanding of chromatin-associated RNAs and the proteins they associate with in vivo, there is a need to develop novel cross-linking methods that have higher efficiencies than CLIP or PAR-CLIP.
One such method was recently developed, and it relies on ligand-induced chemical cross-linking. In vivo proximity labeling (IPL) technology employs an affinity tag, combined with a photoactivatable probe to label RNAs nearby in space (length limited by the linker distance from BSA) to a protein of interest.96 One side of the ligand probe is appended with a biotin, the other with an aryl azide (Figure 4C). Aryl azides are especially powerful for cross-linking with RNA due to their high efficiency. When an aryl azide is exposed to UV light (250 to 350 nm), it forms a nitrene group that can initiate addition reactions with double bonds, insertion into C–H and N–H sites, or subsequent ring expansion to react with a nucleophile (e.g., primary amines). IPL cross-linking relies on tagging of nearby RNAs to the BSA-tagged protein (Figure 4D). The power of IPL was showcased when it was used to identify RNAs associated with the SNRNP70 protein. RNA sequencing of tagged RNAs was used to identify the spliceosomal U1 small nuclear RNA (snRNA) but was unable to detect other highly abundant 5S rRNA and U2 small nuclear RNA (snRNA), both of which are known not to be associated with the SNRNP70 protein. As such, IPL is a novel alternative for utilizing chemical probes to identify protein–RNA interactions inside living cells.
In summation, the number of methods that can be utilized to study RNA–protein interactions continues to grow. This underscores the expanse of their utility in characterizing the RNA–protein interface inside living cells.
Concluding Remarks
Studies focused on RNA biology are undergoing a renaissance. Characterizing their structure and the proteins they interact with was key to further understanding their function. We suspect that it is due in large part to the plethora of chemical tools used to functionally characterize lncRNAs, and their interaction with chromatin will move at a much faster pace. In addition, many other types of RNA are associated with chromatin. For example, antisense RNAs have been demonstrated to interact with chromatin complexes and control gene expression and even RNA decay.97,98 Furthermore, small RNAs have been shown to form duplex structures with nascent RNA to guide protein complexes for chromatin modification.99 The tools and techniques described herein have demonstrated great utility in furthering our understanding of the many types of RNAs controlling chromatin and the mechanisms used. Moreover, further development and optimization of biological protocols based on the principals of chemistry will immensely aid in this effort.
Acknowledgments
We thank members of the Spitale lab for critical reading of the manuscript. S.N. is supported as a Vertex Fellow. RNA research in the Spitale lab is supported by startup funds through the University of California, Irvine and the National Institutes of Health Director's New Innovator Award (1DP2GM119164).
Footnotes
Notes: The authors declare no competing financial interest.
References
- 1.Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 3.Ho JW, Bishop E, Karchenko PV, Negre N, White KP, Park PJ. ChIP-chip versus ChIP-seq: lessons for experimental design and data analysis. BMC Genomics. 2011;12:134. doi: 10.1186/1471-2164-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 2006;34:2653–2662. doi: 10.1093/nar/gkl338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 8.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 10.Khalil AM, Rinn JL. RNA-protein interactions in human health and disease. Semin Cell Dev Biol. 2011;22:359–365. doi: 10.1016/j.semcdb.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009;139:1290–1302. doi: 10.1016/j.cell.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pethe P, Nagvenkar P, Bhartiya D. Polycomb group protein expression during differentiation of human embryonic stem cells into pancreatic lineage in vitro. BMC Cell Biol. 2014;15:18. doi: 10.1186/1471-2121-15-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassan AH, Neely KE, Vignali M, Reese JC, Workman JL. Promoter targeting of chromatin-modifying complexes. Front Biosci, Landmark Ed. 2001;6:D1054–1064. doi: 10.2741/hassan. [DOI] [PubMed] [Google Scholar]
- 14.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawaji H, Lizio M, Itoh M, Kanamori-Katayama M, Kaiho A, Nishiyori-Sueki H, Shin JW, Kojima-Ishiyama M, Kawano M, Murata M, Ninomiya-Fukuda N, Ishikawa-Kato S, Nagao-Sato S, Noma S, Hayashizaki Y, Forrest AR, Carninci P Consortium F. Comparison of CAGE and RNA-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Res. 2014;24:708–717. doi: 10.1101/gr.156232.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beck AH, Weng Z, Witten DM, Zhu S, Foley JW, Lacroute P, Smith CL, Tibshirani R, van de Rijn M, Sidow A, West RB. 3′-end sequencing for expression quantification (3SEQ) from archival tumor samples. PLoS One. 2010;5:e8768. doi: 10.1371/journal.pone.0008768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, Wu YM, Robinson DR, Beer DG, Feng FY, Iyer HK, Chinnaiyan AM. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinn JJ, Zhang QC, Georgiev P, Ilik IA, Akhtar A, Chang HY. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes Dev. 2016;30:191–207. doi: 10.1101/gad.272187.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanduri C. Long noncoding RNAs: Lessons from genomic imprinting. Biochim Biophys Acta, Gene Regul Mech. 2016;1859:102–111. doi: 10.1016/j.bbagrm.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Flynn RA, Chang HY. Long noncoding RNAs in cell-fate programming and reprogramming. Cell stem cell. 2014;14:752–761. doi: 10.1016/j.stem.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992;71:515–526. doi: 10.1016/0092-8674(92)90519-i. [DOI] [PubMed] [Google Scholar]
- 24.Clemson CM, McNeil JA, Willard HF, Lawrence JB. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol. 1996;132:259–275. doi: 10.1083/jcb.132.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidovich C, Cech TR. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA. 2015;21:2007–2022. doi: 10.1261/rna.053918.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davidovich C, Wang X, Cifuentes-Rojas C, Goodrich KJ, Gooding AR, Lee JT, Cech TR. Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol Cell. 2015;57:552–558. doi: 10.1016/j.molcel.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerase A, Smeets D, Tang YA, Gdula M, Kraus F, Spivakov M, Moindrot B, Leleu M, Tattermusch A, Demmerle J, Nesterova TB, Green C, Otte AP, Schermelleh L, Brockdorff N. Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. Proc Natl Acad Sci U S A. 2014;111:2235–2240. doi: 10.1073/pnas.1312951111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, Lander ES, Hess S, Plath K, Guttman M. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–236. doi: 10.1038/nature14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, Schultz PG. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 35.Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, Forrest AR, Carninci P, Biffo S, Stupka E, Gustincich S. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491:454–457. doi: 10.1038/nature11508. [DOI] [PubMed] [Google Scholar]
- 36.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migneault I, Dartiguenave C, Bertrand MJ, Waldron KC. Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques. 2004;37(790-796):798–802. doi: 10.2144/04375RV01. [DOI] [PubMed] [Google Scholar]
- 40.Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. 2011;108:20497–20502. doi: 10.1073/pnas.1113536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thavarajah R, Mudimbaimannar VK, Elizabeth J, Rao UK, Ranganathan K. Chemical and physical basics of routine formaldehyde fixation. J Oral Maxillofac Pathol. 2012;16:400–405. doi: 10.4103/0973-029X.102496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, Plath K, Guttman M. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dieci G, Preti M, Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 2009;94:83–88. doi: 10.1016/j.ygeno.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 44.Lustig Y, Wachtel C, Safro M, Liu L, Michaeli S. ‘RNA walk’ a novel approach to study RNA-RNA interactions between a small RNA and its target. Nucleic Acids Res. 2010;38:e5. doi: 10.1093/nar/gkp872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, Na H, Irimia M, Matzat LH, Dale RK, Smith SA, Yarosh CA, Kelly SM, Nabet B, Mecenas D, Li W, Laishram RS, Qiao M, Lipshitz HD, Piano F, Corbett AH, Carstens RP, Frey BJ, Anderson RA, Lynch KW, Penalva LO, Lei EP, Fraser AG, Blencowe BJ, Morris QD, Hughes TR. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernat V, Disney MD. RNA Structures as Mediators of Neurological Diseases and as Drug Targets. Neuron. 2015;87:28–46. doi: 10.1016/j.neuron.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. 2010;6:e1001074. doi: 10.1371/journal.pgen.1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trang P, Hsu AW, Liu F. Nuclease footprint analyses of the interactions between RNase P ribozyme and a model mRNA substrate. Nucleic Acids Res. 1999;27:4590–4597. doi: 10.1093/nar/27.23.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tijerina P, Mohr S, Russell R. DMS footprinting of structured RNAs and RNA-protein complexes. Nat Protoc. 2007;2:2608–2623. doi: 10.1038/nprot.2007.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peattie DA, Gilbert W. Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci U S A. 1980;77:4679–4682. doi: 10.1073/pnas.77.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stern S, Moazed D, Noller HF. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 1988;164:481–489. doi: 10.1016/s0076-6879(88)64064-x. [DOI] [PubMed] [Google Scholar]
- 53.Been MD, Barfod ET, Burke JM, Price JV, Tanner NK, Zaug AJ, Cech TR. Structures involved in Tetrahymena rRNA self-splicing and RNA enzyme activity. Cold Spring Harbor Symp Quant Biol. 1987;52:147–157. doi: 10.1101/sqb.1987.052.01.019. [DOI] [PubMed] [Google Scholar]
- 54.Latham JA, Cech TR. Defining the inside and outside of a catalytic RNA molecule. Science. 1989;245:276–282. doi: 10.1126/science.2501870. [DOI] [PubMed] [Google Scholar]
- 55.Pan T. Probing RNA structure by lead cleavage. Current Protocols in Nucleic Acid Chemistry. 2000:6.3.1. doi: 10.1002/0471142700.nc0603s00. [DOI] [PubMed] [Google Scholar]
- 56.Ingle S, Azad RN, Jain SS, Tullius TD. Chemical probing of RNA with the hydroxyl radical at single-atom resolution. Nucleic Acids Res. 2014;42:12758–12767. doi: 10.1093/nar/gku934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilkinson KA, Merino EJ, Weeks KM. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J Am Chem Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
- 58.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 59.Kubota M, Tran C, Spitale RC. Progress and challenges for chemical probing of RNA structure inside living cells. Nat Chem Biol. 2015;11:933–941. doi: 10.1038/nchembio.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turner DH, Sugimoto N, Jaeger JA, Longfellow CE, Freier SM, Kierzek R. Improved parameters for prediction of RNA structure. Cold Spring Harbor Symp Quant Biol. 1987;52:123–133. doi: 10.1101/sqb.1987.052.01.017. [DOI] [PubMed] [Google Scholar]
- 61.Jaeger JA, Turner DH, Zuker M. Predicting optimal and suboptimal secondary structure for RNA. Methods Enzymol. 1990;183:281–306. doi: 10.1016/0076-6879(90)83019-6. [DOI] [PubMed] [Google Scholar]
- 62.Serra MJ, Turner DH. Predicting thermodynamic properties of RNA. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 63.Andronescu M, Condon A, Turner DH, Mathews DH. The determination of RNA folding nearest neighbor parameters. Methods Mol Biol. 2014;1097:45–70. doi: 10.1007/978-1-62703-709-9_3. [DOI] [PubMed] [Google Scholar]
- 64.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathews DH. RNA Secondary Structure Analysis Using RNAstructure. Current Protocols in Bioinformatics. 2014;46:12 16 11–12 16 25. doi: 10.1002/0471250953.bi1206s46. [DOI] [PubMed] [Google Scholar]
- 67.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods in molecular biology. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 68.Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGinnis JL, Duncan CD, Weeks KM. High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol. 2009;468:67–89. doi: 10.1016/S0076-6879(09)68004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Banerjee AR, Jaeger JA, Turner DH. Thermal unfolding of a group I ribozyme: the low-temperature transition is primarily disruption of tertiary structure. Biochemistry. 1993;32:153–163. doi: 10.1021/bi00052a021. [DOI] [PubMed] [Google Scholar]
- 72.Clatterbuck Soper SF, Dator RP, Limbach PA, Woodson SA. In vivo X-ray footprinting of pre-30S ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol Cell. 2013;52:506–516. doi: 10.1016/j.molcel.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, Pyle AM. HOTAIR forms an intricate and modular secondary structure. Mol Cell. 2015;58:353–361. doi: 10.1016/j.molcel.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, Pyle AM. HOTAIR forms an intricate and modular secondary structure. Mol Cell. 2015;58:353–361. doi: 10.1016/j.molcel.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu L, Murat P, Matak-Vinkovic D, Murrell A, Balasubramanian S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry. 2013;52:9519–9527. doi: 10.1021/bi401085h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, Chang HY, Akhtar A. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell. 2013;51:156–173. doi: 10.1016/j.molcel.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maenner S, Muller M, Frohlich J, Langer D, Becker PB. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol Cell. 2013;51:174–184. doi: 10.1016/j.molcel.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 79.Amrein H, Axel R. Genes expressed in neurons of adult male Drosophila. Cell. 1997;88:459–469. doi: 10.1016/s0092-8674(00)81886-3. [DOI] [PubMed] [Google Scholar]
- 80.Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL. roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell. 1997;88:445–457. doi: 10.1016/s0092-8674(00)81885-1. [DOI] [PubMed] [Google Scholar]
- 81.Hallacli E, Lipp M, Georgiev P, Spielman C, Cusack S, Akhtar A, Kadlec J. Msl1-mediated dimerization of the dosage compensation complex is essential for male X-chromosome regulation in Drosophila. Mol Cell. 2012;48:587–600. doi: 10.1016/j.molcel.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 82.Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–490. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 84.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fang R, Moss WN, Rutenberg-Schoenberg M, Simon MD. Probing Xist RNA Structure in Cells Using Targeted Structure-Seq. PLoS Genet. 2015;11:e1005668. doi: 10.1371/journal.pgen.1005668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Galgano A, Gerber AP. RNA-binding protein immunopurification-microarray (RIP-Chip) analysis to profile localized RNAs. Methods Mol Biol. 2011;714:369–385. doi: 10.1007/978-1-61779-005-8_23. [DOI] [PubMed] [Google Scholar]
- 88.Riley KJ, Yario TA, Steitz JA. Association of Argonaute proteins and microRNAs can occur after cell lysis. RNA. 2012;18:1581–1585. doi: 10.1261/rna.034934.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jensen KB, Darnell RB. CLIP: crosslinking and immunoprecipitation of in vivo RNA targets of RNA-binding proteins. Methods Mol Biol. 2008;488:85–98. doi: 10.1007/978-1-60327-475-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, Amit I, Regev A. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miller C, Schwalb B, Maier K, Schulz D, Dumcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dolken L, Martin DE, Tresch A, Cramer P. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol. 2011;7:458. doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol. 2013;20:1258–1264. doi: 10.1038/nsmb.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015;350:aac4383. doi: 10.1126/science.aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beck DB, Narendra V, Drury WJ, 3rd, Casey R, Jansen PW, Yuan ZF, Garcia BA, Vermeulen M, Bonasio R. In vivo proximity labeling for the detection of protein-protein and protein-RNA interactions. J Proteome Res. 2014;13:6135–6143. doi: 10.1021/pr500196b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Beltran M, Garcia de Herreros A. Antisense non-coding RNAs and regulation of gene transcription. Transcription. 2016;7:39–43. doi: 10.1080/21541264.2016.1148804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14:880–893. doi: 10.1038/nrg3594. [DOI] [PubMed] [Google Scholar]
- 99.Moazed D. Small RNAs in transcriptional gene silencing and genome defence. Nature. 2009;457:413–420. doi: 10.1038/nature07756. [DOI] [PMC free article] [PubMed] [Google Scholar]