

Abstract
BACKGROUND
The type 3 Dearing reovirus (Reolysin) is a naturally occurring virus that preferentially infects and causes oncolysis in tumor cells with a Ras-activated pathway. It induces host immunity and cell cycle arrest and acts synergistically with cytotoxic agents.
METHODS
This study evaluated Reolysin combined with paclitaxel and carboplatin in patients with metastatic/recurrent KRAS-mutated or epidermal growth factor receptor (EGFR)–mutated/amplified non–small cell lung cancer.
RESULTS
Thirty-seven patients were treated. Molecular alterations included 20 KRAS mutations, 10 EGFR amplifications, 3 EGFR mutations, and 4 BRAF-V600E mutations. In total, 242 cycles (median, 4; range, 1-47) were completed. The initial doses were area under the curve (AUC) 6 mg/mL/min for carboplatin, 200 mg/m2 for paclitaxel on day 1, and 3×1010 50% tissue culture infective dose for Reolysin on days 1 to 5 of each 21-day cycle. Because of diarrhea and febrile neutropenia (in the first 2 patients), subsequent doses were reduced to 175 mg/m2 for paclitaxel and AUC 5 mg/mL/min for carboplatin. Toxicities included fatigue, diarrhea, nausea/vomiting, neutropenia, arthralgia/myalgia, anorexia, and electrolyte abnormalities. Response Evaluation Criteria in Solid Tumors 1.0 responses included the following: partial response for 11 patients, stable disease (SD) for 20 patients, progressive disease for 4 patients, and not evaluable for 2 patients (objective response rate, 31%; 90% 1-sided lower confidence interval, 21%). Four SD patients had >40% positron emission tomography standardized uptake value reductions. The median progression-free survival, median overall survival, and 12-month overall survival rate were 4 months, 13.1 months, and 57%, respectively. Seven patients were alive after a median follow-up of 34.2 months; they included 2 patients without disease progression at 37 and 50 months.
CONCLUSIONS
Reolysin in combination with paclitaxel and carboplatin was well tolerated. The observed response rate suggests a benefit of the reovirus for chemotherapy. A follow-up randomized study is recommended. The proportion of patients surviving longer than 2 years (30%) suggests a second/third-line treatment effect or possibly the triggering of an immune response after tumor reovirus infiltration.
Keywords: BRAF, epidermal growth factor receptor (EGFR), lung cancer, KRAS, oncolytic virus
INTRODUCTION
Epidermal growth factor receptor (EGFR) dysregulation and KRAS mutations occur commonly in non–small cell lung cancer (NSCLC), and both lead to downstream activation of Ras-dependent pathways. Patients with non–EGFR-mutated/EGFR-amplified tumors derive little benefit from EGFR tyrosine kinase inhibitors (TKIs), whereas no effective KRAS-targeted therapy is currently available. Targeting Ras-dependent pathways is, therefore, a major area of unmet therapeutic need in NSCLC.
The type 3 Dearing strain reovirus (Reolysin) is a naturally occurring, ubiquitous, nonenveloped human reovirus with a genome that consists of 10 segments of double-stranded RNA. In preclinical studies, it has been shown to induce host immunity and cell cycle arrest and to act synergistically with chemotherapy.1 A reovirus infection begins with the internalization of the virus via the attachment of the reovirus sigma 1 protein to the cell surface sialic acid residues.2 Enhanced infection efficiency has been observed with either functional EGFR or the v-erb B oncogene.3,4 In unsusceptible cells, a reovirus infection results in the autophosphorylation of double-stranded RNA–activated protein kinase R (PKR). The phosphorylation event activates PKR, which in turn phosphorylates the α subunit of eukaryotic initiation factor 2 and subsequently inhibits viral protein synthesis.5,6 In reovirus-susceptible cells, the active Ras-signaling pathway inhibits the autophosphorylation of PKR and thereby permits the synthesis of viral proteins and results in the lysis of the host cell (Fig. 1). In EGFR-, Sos-, or ras-transformed cells, PKR is held in a nonphosphorylated state, and the replication of the reovirus proceeds uninhibited.6 The dependence of reovirus activity on activation or mutation of the Ras pathway may be indication-specific.7,8
Figure 1.
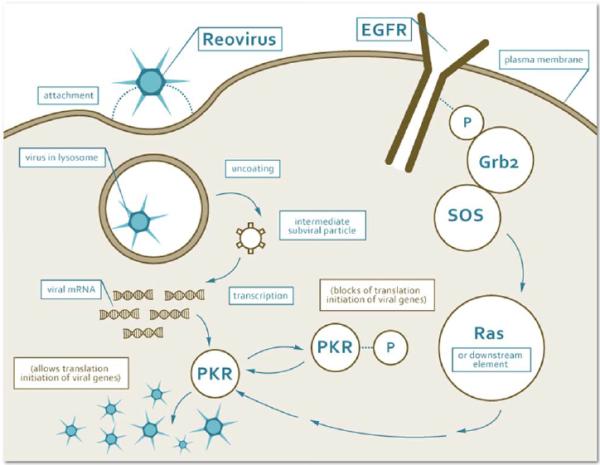
Stepwise and schematic representation of the reovirus type 3 Dearing mechanism of action in the cytoplasm of cancer cells with an activated ras signaling pathway. The phosphorylated proteins (EGFR, and PKR) are tagged with P. EGFR indicates epidermal growth factor receptor; mRNA, messenger RNA; PKR, protein kinase R.
A reovirus administered with paclitaxel was synergistic in all NSCLC cell lines examined, including those with high-level resistance to paclitaxel or the reovirus.9 Phase 1 clinical trials involving Reolysin dispensed as a monotherapy or in combination with gemcitabine, cyclophosphamide, docetaxel, or paclitaxel and carboplatin demonstrated its good tolerability as monotherapy (mild to moderate flulike symptoms and gastrointestinal symptoms were the major side effects) and a lack of exacerbation of the toxicities of the chemotherapeutic agents when it was given in combination.10-13
Because of the preferential activity for Reolysin observed in Ras-activated cells, we set out to screen NSCLC patients to select those with a KRAS-activated pathway through KRAS mutations or EGFR mutations or amplification and to establish the safety and efficacy of Reolysin in combination with paclitaxel and carboplatin in those patients.
MATERIALS AND METHODS
Patients
The institutional review boards of Ohio State University and Georgetown University approved this study (NCT 00861627 at ClinicalTrials.gov). Patients who were 18 years old or older and had recurrent or metastatic NSCLC with EGFR activation (EGFR-activating mutations in exons 18-21 or EGFR fluorescence in situ hybridization [FISH] amplification) or KRAS mutations (exon 2, codons 12, 13, and 61) and no previous cytotoxic chemotherapy for metastatic disease (except previous therapy for localized disease or TKIs for EGFR-mutant patients) were eligible. Other eligibility requirements included the following: the signing of written informed consent, a lapse of at least 6 months from prior adjuvant chemotherapy or chemoradiation therapy, an Eastern Cooperative Oncology Group performance status≤2, and normal organ and marrow function (absolute neutrophil count≥1.5×109, platelets≥100×109, hemoglobin ≥9g/dL, serum creatinine and bilirubin ≤1.5 times the upper limit of normal, and aspartate aminotransferase/alanine aminotransferase ≤2.5 times the upper limit of normal). TKI treatment and palliative radiation must have been discontinued for at least 4 weeks, and the toxicities must have been reduced to grade 1 or lower. Exclusion criteria included pregnancy, brain metastases (resected oligometastases were allowed), grade 2 or higher peripheral neuropathy, uncontrolled concurrent illness (including a known cardiac ejection fraction < 40%), known human immunodeficiency virus infection, and active hepatitis B or C.
Molecular Characterization
All molecular studies were performed centrally at the Molecular Pathology Core Facility at Ohio State University (OSU-PCF). FISH for EGFR amplification was performed with a commercially available Vysis dual-color, dual-probe kit to determine the ratio of the EGFR gene (orange) to CEP7 (green) in the tumor cells under a fluorescence microscope, and it was interpreted by a board-certified pathologist. Positive amplification was scored if any of the following were observed: 1) EGFR/CEP7 ratio≥2, 2)≥15 copies of EGFR per cell in 10% or more of analyzed cells, and 3) EGFR copy numbers per nucleus≥4 in 20% or more of analyzed cells.
For the identification of EGFR and KRAS mutations, genomic DNA was extracted from formalin-fixed, paraffin-embedded tissue with the Qiagen DNA preparation kit, and the target sequences containing the targeted mutations were amplified by polymerase chain reaction with specific primers for the genes of interest. After purification, the polymerase chain reaction products were sequenced with an ABI 3130xl DNA analyzer and were interpreted by a pathologist at OSU-PCF.
Study Design and Treatment Plan
The study was a single-arm, Fleming-A'Hern, single-stage, open-label, phase 2 study14,15 with a 6-patient run in a mini–phase 1 portion to ensure the appropriateness of chemotherapy doses for combination with Reolysin. Paclitaxel was administered as a 3-hour intravenous infusion, and carboplatin was administered as a 30-minute intravenous infusion. The chemotherapy was followed by Reolysin, which was administered as a 60-minute intravenous infusion at a dose of 3×1010 50% tissue culture infective dose (TCID50) on day 1. On days 2 through 5, Reolysin alone was administered at the same dose and with the same method used on day 1.
The study was designed to reject the combination therapy with a 90% chance (10% 1-sided type I error) if the objective response rate (ORR) was 20% and to consider the combination worthy of further study with a high chance (90%; 10% type II error) if the true ORR was 40% or more. Thirty-six response-evaluable patients were planned for recruitment, and an improvement was determined if more than 10 of the 36 patients were observed to have objective response. A partial response (PR) required a Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 computed tomography (CT) response or at least a 40% uptake reduction in positron emission tomography (PET)–avid lesions.
For the phase 1 portion, dose-limiting toxicity was defined as grade 4 neutropenia for > 7 days or with sepsis or fever, grade 4 thrombocytopenia, or grade 3/4 nonhematologic toxicity. Toxicities were graded according to the National Cancer Institute's Common Terminology Criteria for Adverse Events (version 3.0). The starting doses were area under the curve (AUC) 6 mg/mL/min (Calvert formula) for carboplatin and 200 mg/m2 for paclitaxel with a planned dose reduction for subsequent patients in the phase 2 portion to AUC 5 mg/mL/min for carboplatin and 175 mg/m2 for paclitaxel if the first dose was intolerable in at least 2 of 6 patients. Cycles were repeated every 3 weeks.
Dose Modifications
For subsequent treatment courses in patients with toxicities meeting the aforementioned criteria for dose-limiting toxicity, dose reductions (25% of the dose of paclitaxel and decreases of 1 mg/mL/min in the AUC dose of carboplatin) were planned as long as a clinical benefit could be documented. Paclitaxel was discontinued for grade 3 or worse peripheral neuropathy. Dose reductions of Reolysin were to be undertaken only for moderate to severe symptoms not attributable to chemotherapy, such as flulike symptoms, rhinorrhea, and diarrhea. For these events, the Reolysin dose was decreased to 1×1010 TCID50. In the absence of intolerable toxicity or tumor progression, the combination regimen was given for 4 to 6 cycles (according to physician preference), and this was followed by Reolysin as single-agent maintenance with the same schedule of 5 days every 3 weeks.
Study Endpoints
The primary endpoint was the ORR (complete response + PR) as per a RECIST 1.0 CT response16 or at least a 40% reduction of uptake in PET-avid lesions.17 CT was performed at the baseline and after every 2 cycles for all patients, whereas PET was introduced in this fashion, after an amendment, for the last 26 patients. Secondary endpoints included progression-free survival (PFS), overall survival (OS), safety, and tolerability.
Statistical Methods
Exact binomial confidence intervals (CIs) for the ORR were calculated to summarize the primary endpoint for all response-evaluable patients and by subgroup.18 Survival endpoints were summarized with the methods of Kaplan and Meier,19 with Brookmeyer-Crowley CIs used for the median survival time and with Greenwood's method used for 6- and 12-month survival estimates. PFS was defined from the date of study entry to the date of disease progression. Patients who withdrew from the study, were lost to follow-up, died without progressive disease, or began alternative treatments before progression was noted were censored on the date of withdrawal, last follow-up, or new treatment initiation. OS was determined as the time from study entry to the date of death or was censored on the date on which a patient was last known to be alive. The final review was completed on March 15, 2015. All analyses were unadjusted for multiple comparisons and were performed with STATA (version 13.1; StataCorp, College Station, Texas).
RESULTS
Screening
Patients with metastatic or recurrent NSCLC who were found with a routine EGFR amplification/KRAS mutation assessment performed at OSU-PCF were referred for participation in the study. From March 2009 to February 2013, 37 patients were enrolled at the Ohio State University Medical Center (n = 33) and Georgetown University Medical Center (n = 4). The characteristics of these patients are depicted in Table 1. The majority of tumors had an adenocarcinoma histology (n = 27), which reflected the association of testing for KRAS and EGFR mutations with adenocarcinomas; 2 patients with a mixed adenosquamous histology, 1 patient with squamous carcinomas, and 1 patient with a bronchioloalveolar histology were also enrolled. Six patients had NSCLC that was not otherwise specified. KRAS mutations were present in 20 patients, whereas EGFR amplification alone was present in 10 patients. Three patients’ tumors exhibited EGFR-activating mutations, and 4 patients’ tumors (all EGFR-amplified) also had concurrent BRAF-V600E mutations.
TABLE 1.
Patient Characteristics
| Enrolled patients, No. | 37 |
|---|---|
| Age, y | |
| Median | 65 |
| Range | 47-82 |
| Sex | |
| Men | 15 |
| Women | 22 |
| Race/ethnicity | |
| White | 32 |
| African American | 5 |
| Hispanic/Latino | 0 |
| ECOG performance status | |
| 0 | 23 |
| 1 | 13 |
| 2 | 1 |
| Stage | |
| IVA | 14 |
| IVB | 23 |
| Recurrent | 8 |
| Histology | |
| Adenocarcinoma | 26 |
| Adenosquamous | 2 |
| Squamous | 2 |
| Bronchioloalveolar | 1 |
| NSC NOS | 6 |
| Molecular Abnormality | |
| KRAS mutation only | 14 |
| EGFR-amplified only | 10 |
| EGFR mutant only | 1 |
| KRAS mutation + EGFR amplification | 6 |
| EGFR mutation + EGFR amplification | 2 |
| BRAF mutation + EGFR amplification | 4 |
| Prior anticancer therapies | |
| Targeted agents | 3 |
| Chemotherapy (adjuvant) | 4 |
| Surgery | 13 |
| Thoracic surgery for NSCLC | 9 |
| Resection of brain metastasis | 4 |
| Radiation | 15 |
| Adjuvant (mediastinum) | 5 |
| Palliative | 12 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; NOS, not otherwise specified; NSC, non–small cell; NSCLC, non–small cell lung cancer.
Dose Evaluation and Toxicities
Two patients were treated at the starting doses of AUC 6 for carboplatin, 200 mg/m2 for paclitaxel, and 3×1010 TCID50 for Reolysin, and both experienced unacceptable toxicities (febrile neutropenia in one and grade 3 diarrhea in the other). These patients were dose-reduced for subsequent cycles to AUC 5 for carboplatin and 175 mg/m2 for paclitaxel; Reolysin was kept at 3×1010 TCID50 as previously planned, and these starting doses were used for all subsequently enrolled patients. Overall, 242 cycles (including 105 for Reolysin-only maintenance) were administered to the 37 patients (median, 4; range, 1-47). Chemotherapy was further reduced because of adverse events for only 6 additional patients (17%; 18 of 240 cycles or 8%). Only 1 patient had to reduce the Reolysin dose; more than 95% of planned doses were administered at the full dose over the course of the study. The type, grade, and frequency of the toxicities observed with the combination are depicted in Table 2. The combination at the reduced doses was generally well tolerated. The most frequent moderate to severe toxicities included fatigue (8 patients with grade 3, 1 patient with grade 4), neutropenia (7 patients with grade 3, 1 patient with grade 4), diarrhea (5 patients with grade 3), and nausea/vomiting (3 patients with grade 3). Four patients developed hypotension, and 2 patients developed confusion. Fever was noted only during Reolysin-only maintenance.
TABLE 2.
Toxicities for Reolysin in Combination With Paclitaxel and Carboplatin
| Toxicity | Reolysin Plus Paclitaxel With Carboplatin at 200mg/m2/AUC 6 (n = 2), % | Reolysin Plus Paclitaxel With Carboplatin at 175 mg/m2/AUC 5 (n = 36), % | ||||||
|---|---|---|---|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Hematologic | ||||||||
| Neutropenia | 0 | 0 | 0 | 50 | 0 | 14 | 19 | 0 |
| Anemia | 0 | 0 | 50 | 0 | 6 | 14 | 3 | 0 |
| Thrombocytopenia | 0 | 50 | 0 | 0 | 14 | 3 | 3 | 0 |
| Lymphocytopenia | 0 | 0 | 0 | 0 | 8 | 11 | 6 | 0 |
| Febrile neutropenia | 0 | 0 | 50 | 0 | 0 | 0 | 0 | 0 |
| Non hematologic | ||||||||
| Nausea | 50 | 50 | 0 | 0 | 64 | 8 | 3 | 0 |
| Vomiting | 0 | 0 | 0 | 0 | 22 | 8 | 8 | 0 |
| Anorexia | 0 | 50 | 0 | 0 | 17 | 11 | 0 | 0 |
| Diarrhea | 0 | 0 | 100 | 0 | 72 | 17 | 8 | 0 |
| Confusion | 0 | 0 | 0 | 0 | 6 | 3 | 3 | 0 |
| Headaches | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| Transaminitis | 0 | 0 | 0 | 0 | 14 | 6 | 3 | 0 |
| Fatigue/asthenia | 0 | 50 | 50 | 0 | 83 | 33 | 19 | 3 |
| Arthralgia | 0 | 0 | 0 | 0 | 11 | 17 | 0 | 0 |
| Myalgia | 0 | 0 | 0 | 0 | 17 | 28 | 0 | 0 |
| Peripheral neuropathy | 50 | 0 | 0 | 0 | 14 | 3 | 0 | 0 |
| Hypotension | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Peripheral edema | 0 | 0 | 50 | 0 | 3 | 0 | 0 | 0 |
| Fever | 0 | 0 | 0 | 0 | 36 | 8 | 3 | 0 |
Abbreviation: AUC, area under the curve.
Antitumor Activity
Table 3 depicts the best response according to RECIST tumor assessment results, the percentage change in the standardized uptake value (SUV) maximum sum of tumor lesions on PET scans performed after 2 cycles versus the baseline, and PFS and OS according to molecular and histologic characteristics. Two patients were not evaluable for a response because of consent withdrawal shortly after they had received 1 cycle. Eleven of 35 evaluable patients had a RECIST response; all were partial (ORR, 31%; 90% 1-sided lower CI, 21%) as assessed by investigators and were confirmed at a 4-week scan. Twenty patients had stable disease (SD) as their best response, whereas 4 patients experienced disease progression on their first postbaseline assessment. The percentage of patients who had a RECIST PR with a KRAS tumor genotype (5 PRs among 18 evaluable patients) and the percentage of patients who had a PR with an EGFR amplification–alone tumor genotype (3 PRs among 10 evaluable patients) were similar (odds ratio of EGFR amplification alone vs KRAS, 1.1; 95% CI, 0.20-6.11; P = .90). Two PRs occurred among 4 patients with BRAF-mutated tumors (50%; 95% CI, 7%-93%). Twenty-four patients had PET scans performed simultaneously with the CT assessments, before treatment, and after every 2 cycles. Ten of these patients (6 with PRs and 4 with SD according to RECIST) had a > 40% decrease in the SUV sum of the lesions after 2 cycles. No significant increases in SUV were observed after 2 cycles in any of the patients treated. Thus, the RECIST + PET ORR was 43% (90% 1-sided lower CI, 31%).
TABLE 3.
Clinical Benefit Assessment of Paclitaxel, Carboplatin, and Reolysin in Non-Small Cell Lung Cancer
| Patient No. | Abnormality | Amino Acid | Nucleotide | Histology | Cycle No. | Best Response | PET SUV Change, % | Progression-Free Survival, d | Overall Survival, d |
|---|---|---|---|---|---|---|---|---|---|
| 0023 | KRAS, EGFR Amp | G12C | c.34G>T | Adenocarcinoma | 2 | PD | IC | 31 | 193 |
| 0004 | KRAS, EGFR Amp | G12C | c.34G>T | NSC NOS | 6 | SD | −49.8 | 120 | 1195 |
| 0006 | KRAS, EGFR Amp | G12D | c.34G>A | Adenocarcinoma | 6 | SD | ND | 119 | 690 |
| 0020 | KRAS, EGFR Amp | G12C | c.34G>T | Adenocarcinoma | 4 | SD | 1.4 | 78 | 343 |
| 0026 | KRAS, EGFR Amp | G12C | c.34G>T | NSC NOS | 8 | SD | −51.8 | 196 | 279 |
| 0027 | KRAS, EGFR Amp | G12V | c.34G>T | Adenocarcinoma | 4 | SD | −22.3 | 80 | 508 |
| 0001 | KRAS | G12R | c.34G>C | Adenocarcinoma | 3 | PR | −80.6 | 170 | 227 |
| 0002 | KRAS | G12C | c.34G>T | Adenocarcinoma | 4 | PR | ND | 624 | >2164 |
| 0007 | KRAS | G13R | c.37G>C | Adenocarcinoma | 1 | NE | ND | N/A | Unknown |
| 0013 | KRAS | G12C | c.34G>T | Adenocarcinoma | 4 | SD | ND | 80 | 96 |
| 0014 | KRAS | G13C | c.37G>T | Adenocarcinoma | 4 | SD | ND | 78 | 182 |
| 0017 | KRAS | G12C; G12V | c.34G>T; c.35G>T | Adenocarcinoma | C4D1 | SD | −64.0 | 235 | 290 |
| 0025 | KRAS | G12C | c.34G>T | Adenocarcinoma | 1 | NE | ND | 90 | 228 |
| 0028 | KRAS | G12S | c.34G>A | NSC NOS | 4 | SD | 0.0 | 88 | 172 |
| 0032 | KRAS | G12V | c.35G>T | Adenocarcinoma | 14 | PR | −48.5 | 302 | >999 |
| 0034 | KRAS | G12V | c.35G>T | Adenocarcinoma | 4 | SD | −36.3 | 80 | 175 |
| 0035 | KRAS | G12A | c.35G>C | Adenosquamous | 6 | SD | −34.5 | 120 | >828 |
| 0037 | KRAS | G12C | c.34G>T | Adenocarcinoma | 6 | PR | −0.1 | Died without PD | 216 |
| 0031 | KRAS | G12A | c.35G>C | Adenocarcinoma | 47 | PR | −100.0 | >1105 | >1105 |
| 0038 | KRAS | G12C | c.34G>T | Adenocarcinoma | 2 | PD | ND | 38 | 118 |
| 0005 | EGFRm, EGFR Amp | p.E746_T51 del | c.2237_2251del15 | Bronchioloalveolar | 6 | PR | ND | 139 | 498 |
| 0011 | EGFRm, EGFR Amp | p.E746_T51 del | c.2237_2251del15 | NSC NOS | 1 | PD | ND | 4 | 105 |
| 0018 | EGFRm | p.E746_T51 del | c.2237_2251del15 | Adenocarcinoma | C6D1 | SD | −13.9 | >1486 | >1486 |
| 0003 | EGFR Amp | Adenosquamous | 3 | SD | −37.6 | 88 | 359 | ||
| 0008 | EGFR Amp | NSC NOS | 8 | SD | ND | 170 | 589 | ||
| 0015 | EGFR Amp | Adenocarcinoma | 4 | SD | ND | 78 | 129 | ||
| 0019 | EGFR Amp | Adenocarcinoma | 10 | PR | −34.3 | 210 | 399 | ||
| 0022 | EGFR Amp | Adenocarcinoma | 6 | SD | 0.0 | 120 | 249 | ||
| 0024 | EGFR Amp | Adenocarcinoma | 6 | SD | −50.2 | 121 | 1067 | ||
| 0029 | EGFR Amp | Adenocarcinoma | 4 | PR | −63.1 | 93 | 369 | ||
| 0030 | EGFR Amp | Adenocarcinoma | 14 | SD | −18.9 | 316 | 658 | ||
| 0033 | EGFR Amp | Adenocarcinoma | 14 | SD | −30.7 | 248 | >837 | ||
| 0021 | EGFR Amp | Squamous | 8 | PR | −80.8 | 185 | 468 | ||
| 0009 | BRAF, EGFR Amp | V600E | c.1799T>A | Adenocarcinoma | 6 | SD | −34.0 | 575 | 854 |
| 0010 | BRAF, EGFR Amp | V600E | c.1799T>A | Adenocarcinoma | 2 | PD | 0.0 | 36 | 382 |
| 0012 | BRAF, EGFR Amp | V600E | c.1799T>A | Adenocarcinoma | 4 | PR | ND | 1460 | >1850 |
| 0016 | BRAF, EGFR Amp | V600E | c.1799T>A | NSC NOS | 8 | PR | −91.3 | 168 | 803 |
Abbreviations: Amp, amplified; C4D1, cycle 4 day 1; C6D1, cycle 6 day 1; EGFR, epidermal growth factor receptor; EGFRm, EGFR mutated; IC, incomplete; N/A, not available; ND, not determined; NE, not evaluable; NOS, not otherwise specified; NSC, non-small cell; PD, progressive disease; PET, positron emission tomography; PR, partial response; SD, stable disease; SUV, standardized uptake value.
The median PFS, OS, and 12-month OS rate were 4 months (95% CI, 2.9-6.1 months), 13.1 months (95% CI, 9.2-21.6 months), and 57% (95% CI, 39%-72%), respectively (Table 3). Ten patients went on to receive Reolysin-only maintenance after 4 to 6 cycles of the combination, whereas 3 patients opted to switch to pemetrexed maintenance at the completion of paclitaxel, carboplatin, and Reolysin without progression or significant toxicity, and 1 patient (carrying an EGFR-activating mutation) was switched to erlotinib after 6 cycles. These patients were censored at the time of the switch to the alternative therapy for the PFS calculations. Kaplan-Meier PFS and OS plots according to the molecular phenotype are depicted in Figures 2 and 3. Seven patients were alive after a median follow-up of 34.2 months (range, 26.9-71.5 months); they included 2 patients (with an EGFR mutation and a KRAS G12 mutation) with no evidence of disease progression to date (37 and 50 months, respectively; Table 3).
Figure 2.
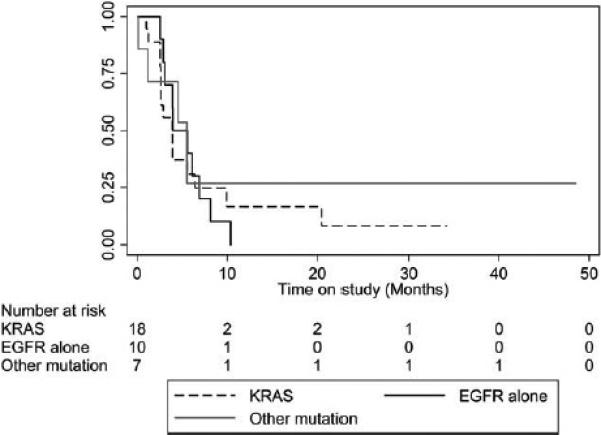
Progression-free survival rates versus time for patients receiving Reolysin in combination with paclitaxel and carboplatin according to molecular abnormalities. The data are presented as months. EGFR indicates epidermal growth factor receptor.
Figure 3.
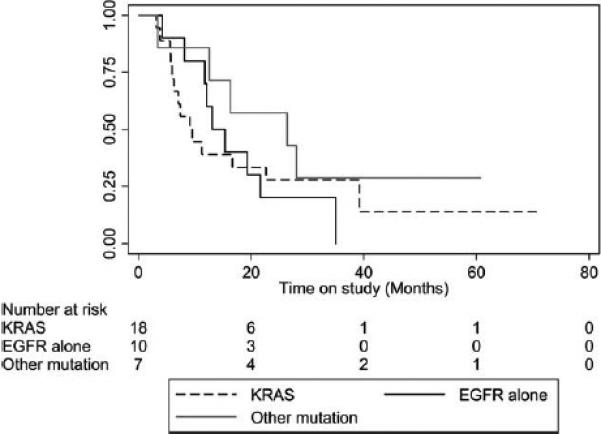
Overall survival rates versus time for patients receiving Reolysin in combination with paclitaxel and carboplatin according to molecular abnormalities. The data are presented as months. EGFR indicates epidermal growth factor receptor.
DISCUSSION
A systematic evaluation of tumor genetics in NSCLC has led to the identification of tumor drivers and, in some cases, to effective targeted therapies. Notable examples are EGFR mutations and anaplastic lymphoma kinase trans-locations, for which several targeted agents are available for clinical use.20,21 KRAS constitutive activation has been more challenging; this has led some to conclude that it is not a druggable target. The preclinical data for the Reolysin virus showing selectivity for Ras have, therefore, high appeal for an evaluation in Ras-activated NSCLC patients. The Ras pathway can be activated through an upstream event, such as EGFR mutations and amplification, or through a downstream event, such as BRAF mutations. This study included a group of patients with diverse molecular aberrations that had the common denominator of Ras activation, and it showed good feasibility for the population-enriched approach undertaken as well as a good safety profile for the combination of Reolysin with chemotherapy in this population.
In this study, the RECIST response rate for paclitaxel, carboplatin, and Reolysin according to the investi gators’ assessment was significantly increased (31%; 90% 1-sided lower CI, 21%) in comparison with the assumed historical response rate for paclitaxel and carboplatin alone (20%). However, the firmness of historical data is limited, and the comparison being used here is hypothesis-generating because the prognostic or predictive value of KRAS mutations is still a matter of debate. Studies in early-stage disease offer contradictory information.22-24 The First-Line Erbitux in Lung Cancer (FLEX) study, performed with patients with metastatic NSCLC, randomized EGFR-expressing patients by immunohisto-chemistry to receive either cisplatin and vinorelbine or this chemotherapy in combination with cetuximab.25 OS, but not PFS, was better for the cetuximab-containing arm. In a retrospective molecular characterization study that used tumors from patients in the FLEX trial,26 the response rate for patients with KRAS mutations was 36.8% for patients receiving cetuximab and chemotherapy and 21.6% for those receiving chemotherapy alone. PFS was 5.5 months (95% CI, 3.1-6.9 months) for cetuximab and chemotherapy and 2.9 months (95% CI, 1.8-5.8 months) for patients with KRAS mutations receiving chemotherapy alone. However, OS was only 8.9 months for the combination versus 11.1 months for chemotherapy alone. In the group of patients enrolled with evidence of EGFR amplification by FISH, the response rates were 36.7% and 26.4% for the cetuximab/chemotherapy and chemotherapy arms, respectively, whereas PFS was 4.2 and 4.4 months and OS was 11.6 and 9.9 months for the cetuximab/chemotherapy and chemotherapy arms, respectively.
Although the response rate (31% according to RECIST) and the median survival rate (13.1 months) for our trial compare favorably with those for the chemotherapy-alone arm in the FLEX trial, the lack of a randomized control group in our trial does not allow us to make firm interpretations of superior antitumor efficacy for the approach tested in this study. Furthermore, we did not observe significant differences in clinical outcomes for patients enrolled according to the genotype leading to Ras activation (Figs. 2 and 3), and because of the lack of a non–Ras-activated control cohort, we could not confirm or reject the utility of Ras activation as a predictor of Reolysin/chemotherapy activity.
Significant increases in neutralizing antireoviral antibodies (median, 250-fold) have been demonstrated in humans with advanced cancers after exposure to Reolysin,27 and coadministration of cyclophosphamide was shown not to be able to attenuate the host antiviral response to a reovirus.28 However, despite the presence of neutralizing antibodies, it has been demonstrated that a reovirus can evade neutralization by associating with peripheral blood mononuclear cells for up to 10 days after treatment in patient samples.28,29 Further demonstration of the ability of a reovirus to escape neutralization can be found in the persistence of an infectious, replicating virus in patient tumor samples at various periods up to weeks after reovirus administration despite the presence of neutralization antibodies. Infectious reoviruses have been isolated from various solid tumor types after systemic administration, including ovarian cancer, melanoma, pleural mesothelioma, breast cancer, pancreatic cancer, colon cancer, and head and neck cancer,10,12,29-33 and from hematopoietic malignancies such as multiple myeloma.34
Thus, it is very tempting to speculate on the potential for Reolysin to create immunogenicity against the tumor cells infiltrated by the virus. This response could theoretically be capable of bypassing operating mechanisms of immune tolerance and result in longer survival. The current study was designed for patients to receive maintenance reovirus alone after initial induction with Reolysin and chemotherapy; this was done except for the 4 patients who switched to pemetrexed or erlotinib for maintenance. An intriguing and potentially better approach could be the use of a checkpoint inhibitor in that setting, which could benefit from the immunogenicity created by the virus during the induction phase. In support of this hypothesis, CD8+-enriched splenocytes have been shown to secrete augmented levels of interferon-γ when they are cocultured with Reolysin ver sus an ultraviolet-inactivated reovirus; this indicates reovirus recognition by CD8+ cells and proinflammatory response stimulation through interferon-γ secretion.35 Furthermore, therapy combining a reovirus with programmed death 1 blockade has been shown to produce a significant survival benefit by augmenting tumor-specific natural killer responses and specifically attenuating tumor-specific immunosuppression.36
In summary, Reolysin in combination with paclitaxel and carboplatin was well tolerated, and the observed response rate met study specifications. A randomized trial that could elucidate both the contribution of Reolysin to the effect of chemotherapy and the utility of a predictive biomarker is planned. It would be interesting to explore serum immune response markers as well as tumor biopsy before and after Reolysin therapy to evaluate the extent of tumor infiltration with markers of an immune response such as Fox-P3+ regulatory T cells. Because of the benefits shown with immune checkpoint inhibitor therapy in lung cancer and the intriguing data of immune stimulation after viral exposure, a study combining the 2 approaches would be an exciting follow-up study.
Acknowledgments
FUNDING SUPPORT
An institutional grant to The Ohio State University was received from Oncolytics Biotech, Incorporated, to conduct the clinical trial.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
Gregory Otterson reports grants from Pfizer, Genentech, Boeh-ringer Ingelheim, Clovis, NewLink Genetics, Celgene, and Bristol-Myers Squibb outside the submitted work. George M. Gill is employed by Oncolytics Biotech, Incorporated, as chief safety officer. Matthew Coffey is a cofounder of Oncolytics Biotech, Incorporated (US Patent 6,110,461). Giovanni Selvaggi is Vice President of Clinical Development for Oncolytics Biotech, Incorporated. Bo Chao is currently an employee of Eli Lilly and Company.
REFERENCES
- 1.Chakrabarty R, Tran H, Selvaggi G, Hagerman A, Thompson B, Coffey M. The oncolytic virus, pelareorep, as a novel anticancer agent: a review. Invest New Drugs. 2015;33:761–764. doi: 10.1007/s10637-015-0216-8. [DOI] [PubMed] [Google Scholar]
- 2.Danthi P, Holm GH, Stehle T, Dermody TS. Reovirus receptors, cell entry, and proapoptotic signaling. Adv Exp Med Biol. 2013;790:42–71. doi: 10.1007/978-1-4614-7651-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strong JE, Tang D, Lee PW. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology. 1993;197:405–411. doi: 10.1006/viro.1993.1602. [DOI] [PubMed] [Google Scholar]
- 4.Strong JE, Lee PW. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J Virol. 1996;70:612–616. doi: 10.1128/jvi.70.1.612-616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hershey JW. Translational control in mammalian cells. Annu Rev Biochem. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- 6.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roulstone V, Pedersen M, Kyula J, et al. BRAF- and MEK-targeted small molecule inhibitors exert enhanced antimelanoma effects in combination with oncolytic reovirus through ER stress. Mol Ther. 2015;23:931–942. doi: 10.1038/mt.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maitra R, Seetharam R, Tesfa L, et al. Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget. 2014;5:2807–2819. doi: 10.18632/oncotarget.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sei S, Mussio JK, Yang Q, et al. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents against non–small cell lung cancer (NSCLC). Mol Cancer. 2009;8:47. doi: 10.1186/1476-4598-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vidal L, Pandha HS, Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14:7127–7137. doi: 10.1158/1078-0432.CCR-08-0524. [DOI] [PubMed] [Google Scholar]
- 11.Lolkema MP, Arkenau HT, Harrington K, et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011;17:581–588. doi: 10.1158/1078-0432.CCR-10-2159. [DOI] [PubMed] [Google Scholar]
- 12.Comins C, Spicer J, Protheroe A, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16:5564–5572. doi: 10.1158/1078-0432.CCR-10-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karapanagiotou EM, Roulstone V, Twigger K, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18:2080–2089. doi: 10.1158/1078-0432.CCR-11-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming TR. One-sample multiple testing procedure for phase II clinical trials. Biometrics. 1982;38:143–151. [PubMed] [Google Scholar]
- 15.A'Hern RP. Sample size tables for exact single stage phase II designs. Stat Med. 2001;20:859–866. doi: 10.1002/sim.721. [DOI] [PubMed] [Google Scholar]
- 16.Eisenhauer E, Therasse P, Bogaerts J, et al. New response criteria in solid tumors: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Shankar LK, Hoffman JM, Bacharach S, et al. Consensus recommendations for the use of 18F-FDG PET as an indicator of therapeutic response in patients in National Cancer Institute trials. J Nucl Med. 2006;47:1059–1066. [PubMed] [Google Scholar]
- 18.Brown LD, Cai T, DasGupta A. Interval estimation for a binomial proportion. Stat Sci. 2001;16:101–117. [Google Scholar]
- 19.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 20.Steuer CE, Khuri FR, Ramalingam SS. The next generation of epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of lung cancer. Cancer. 2015;121:E1–E6. doi: 10.1002/cncr.29139. [DOI] [PubMed] [Google Scholar]
- 21.Gainor JF, Sherman CA, Willoughby K, et al. Alectinib salvages CNS relapses in ALK-positive lung cancer patients previously treated with crizotinib and ceritinib. J Thorac Oncol. 2015;10:232–236. doi: 10.1097/JTO.0000000000000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metro G, Chiari R, Bennati C, et al. Clinical outcome with platinum-based chemotherapy in patients with advanced nonsquamous EGFR wild-type non–small-cell lung cancer segregated according to KRAS mutation status. Clin Lung Cancer. 2014;15:86–92. doi: 10.1016/j.cllc.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Martin P, Leighl N, Tsao M, Shepherd L. KRAS mutations as prognostic and predictive markers in non–small cell lung cancer. J Thorac Oncol. 2013;8:530–542. doi: 10.1097/JTO.0b013e318283d958. [DOI] [PubMed] [Google Scholar]
- 24.Mascaux C, Iannino N, Martin B, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92:131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pirker R, Pereira JR, Szczesna A, et al. Cetuximab plus chemotherapy in patients with advanced non–small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525–1531. doi: 10.1016/S0140-6736(09)60569-9. [DOI] [PubMed] [Google Scholar]
- 26.O'Byrne KJ, Gatzemeier U, Bondarenko I, et al. Molecular bio-markers in non–small-cell lung cancer: a retrospective analysis of data from the phase 3 FLEX study. Lancet Oncol. 2011;12:795–805. doi: 10.1016/S1470-2045(11)70189-9. [DOI] [PubMed] [Google Scholar]
- 27.White CL, Twigger KR, Vidal L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008;15:911–920. doi: 10.1038/gt.2008.21. [DOI] [PubMed] [Google Scholar]
- 28.Roulstone V, Khan K, Pandha HS, et al. Phase I trial of cyclophosphamide as an immune modulator for optimizing oncolytic reovirus delivery to solid tumors. Clin Cancer Res. 2015;21:1305–1312. doi: 10.1158/1078-0432.CCR-14-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4:138ra77. doi: 10.1126/scitranslmed.3003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galanis E, Markovic SN, Suman VJ, et al. Phase II trial of intravenous administration of Reolysin(®) (reovirus serotype-3-Dearing strain) in patients with metastatic melanoma. Mol Ther. 2012;20:1998–2003. doi: 10.1038/mt.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gollamudi R, Ghalib MH, Desai KK, et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs. 2010;28:641–649. doi: 10.1007/s10637-009-9279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahalingam D, Patel S, Nuovo G, et al. The combination of intravenous Reolysin and gemcitabine induces reovirus replication and endoplasmic reticular stress in a patient with KRAS-activated pancreatic cancer. BMC Cancer. 2015;15:513. doi: 10.1186/s12885-015-1518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nuovo GJ, Garofalo M, Valeri N, et al. Reovirus-associated reduction of microRNA-let-7d is related to the increased apoptotic death of cancer cells in clinical samples. Mod Pathol. 2012;25:1333–1344. doi: 10.1038/modpathol.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sborov D, Nuovo G, Stiff A, et al. A phase I trial of single agent Reolysin in patients with relapsed multiple myeloma. Clin Cancer Res. 2014;20:5946–5955. doi: 10.1158/1078-0432.CCR-14-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Spurrel J, Shi ZQ, Chen WQ, Morris DG. Oncolytic viral therapy with immune modulation is an effective novel treatment strategy for non–small cell lung cancer [abstract]. Cancer Res. 2015;75:5355. [Google Scholar]
- 36.Rajani K, Parrish C, Kottke T, et al. Combination therapy with reo-virus and anti–PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther. doi: 10.1038/mt.2015.156. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]