

Abstract
Pyrococcus horikoshii Dph2 (PhDph2) is an unusual radical SAM enzyme involved in the first step of diphthamide biosynthesis. It catalyzes the reaction by cleaving S-adenosylmethionine (SAM) to generate a 3-amino-3-carboxy propyl (ACP) radical. To probe the reaction mechanism, we synthesized a SAM analog (SAMCA), in which the ACP group of SAM is replaced with a 3-carboxyallyl group. SAMCA is cleaved by PhDph2, yielding a paramagnetic (S = 1/2) species, which is assigned to a complex formed between the reaction product, α-sulfinyl-3-butenoic acid, and the [4Fe-4S] cluster. Electron-nuclear double resonance (ENDOR) measurements with 13C and 2H isotopically labeled SAMCA support a π-complex between the C=C double bond of α-sulfinyl-3-butenoic acid, and the unique iron of the [4Fe-4S] cluster. This is the first example of a radical SAM related [4Fe-4S]+ cluster forming an organometallic complex with an alkene, shedding additional light into the mechanism of PhDph2 and expanding our current notions for the reactivity of [4Fe-4S] clusters in radical SAM enzymes.
Graphical Abstract
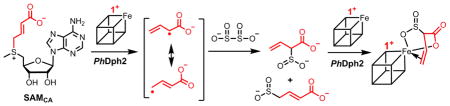
Diphthamide is a post-translationally modified histidine residue on archaeal and eukaryotic translation elongation factor 2 (EF2), a protein essential for ribosomal protein synthesis. Its biosynthesis involves at least seven proteins.1,2 The first step involves the transfer of 3-amino-3-carboxypropyl (ACP) group derived from S-adenosyl-L-methionine (SAM) to the histidine residue of EF2. In vitro, the [4Fe-4S]-containing radical SAM (RS) enzyme PhDph23 or the eukaryotic Dph1-Dph2 homodimer4 is sufficient for this step, with dithionite as the reducing agent. In contrast to classical RS enzymes that cleave the C5′,Ade–S bond of SAM to form 5′-deoxyadenosyl radical (5′-dA•) (Scheme 1A)5, PhDph2 cleaves the Cγ,Met–S bond to form an ACP radical (Scheme 1B).3
Scheme 1.

PhDph2 is different from classical RS enzymes.
The formation of the ACP radical is supported by the PhDph2-catalyzed generation of 2-aminobutyric acid and homocysteine sulfinic acid in the absence of the substrate protein, PhEF2.3 However, the ACP radical has not been directly observed even when we carried out freeze quench-electron paramagnetic resonance (EPR) experiments (Figure S1). This is not surprising because the ACP radical is expected to be short-lived. Following the strategy developed by Frey and coworkers to generate a more stable allylic analog of the 5′-dA•6.7, we synthesized a SAM analog, SAMCA, in which the ACP group of SAM was replaced with a 3-carboxyallyl group (Scheme 2). If PhDph2 could accept SAMCA as a substrate, a 3-carboxyallyl radical would be generated, which should be more stable and allow direct observation by EPR. As shown below, even with SAMCA, we could not detect the radical intermediate. However, surprisingly, we found that a dithionite quenched radical product formed an organometallic complex with the Fe-S cluster, revealing an interesting reactivity of the [4Fe-4S] cluster in RS enzymes.
Scheme 2.
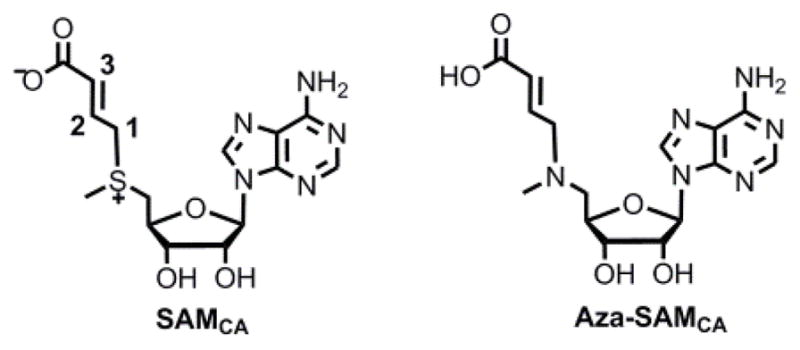
Structures of SAMCA and aza-SAMCA.
We initially examined whether SAMCA could serve as a substrate of PhDph2. High-performance liquid chromatography (HPLC) showed that all the SAMCA was converted to 5′-deoxy-5′-methylthioadenosine (MTA) within 20 min, whereas in the absence of dithionite or PhDph2, no SAMCA was cleaved (Figure 1A). These results demonstrate that PhDph2 is able to catalyze the cleavage of the ‘correct’ C-S bond of SAMCA. In the presence of the substrate protein PhEF2, the same reaction product was detected (Figure S2). However, PhEF2 was not modified by SAMCA. This was likely because SAMCA generated a more stable radical (see more experimental evidence for the stability below), which was not active enough to react with PhEF2.
Figure 1.

A). HPLC traces of the SAMCA cleavage reaction (reaction time 20 min) catalyzed by PhDph2 at room temperature. B). X-band CW EPR spectra of PhDph2 in the absence and presence of SAMCA. A spectral simulation for the S = 1/2 species is shown as green dotted line. T = 12 K.
We then prepared freeze quench samples and used EPR to detect radical signals. The dithionite-reduced PhDph2 in the absence of SAMCA showed a typical [4Fe-4S]1+ EPR signal at 12 K (Figure 1B),3 which was temperature-dependent and broadened beyond detection at 30 K (Figure S1). When PhDph2 in the presence of dithionite was allowed to react with SAMCA for 30 s, the [4Fe-4S]1+ signal was replaced by a nearly axial signal, with a split feature at g1 (Figure 1B). The ratio of this g1 splitting at X- and Q-band scaled with the ratio of microwave frequencies (Figure S3), showing that the splitting represents two different conformations and not a hyperfine interaction with a paramagnetic nucleus. Simulation of the spectra gave for the one conformer principal g-values g = [2.092, 2.010, 1.990], and for the second one g = [2.074, 2.010, 1.990]. Both signals exhibited similar relaxation properties and were hardly detectable above 70 K (Figure S1). The large g-anisotropy and the absence of proton hyperfine splittings suggest that it is not a free allyl radical.6,8 In contrast, the signals of the new species formed in the reaction of the reduced PhDph2 with SAMCA exhibited an “isotropic” g-value, giso = (g11+g22+g33)/3 > ge = 2.0023,9 which is reminiscent of that observed in high potential iron-sulfur proteins (HiPIP) and [4Fe-4S]3+ intermediates trapped with the [4Fe-4S] proteins IspG10 and IspH11. With 57Fe-enriched PhDph2, the EPR signal became broadened (Figure 1B), further supporting that the new species (termed CA) is associated with the [4Fe-4S] cluster.
When we monitored the reaction at different time points, the EPR signal of CA persisted for >30 min, even after all SAMCA was consumed (Figure S4). This suggested that CA was a complex of the reaction product with the [4Fe-4S]1+ cluster, the latter regenerated by dithionite reduction of the [4Fe-4S]2+ produced in the reaction. To test this hypothesis, protein-free aliquots of the reaction products were added to ‘fresh’ dithionite-reduced PhDph2. The same anisotropic EPR signal as that of CA was obtained, supporting that CA is a product complex (Figure S5).
To understand the structure of CA, we analyzed the reaction products using 1H-NMR. Several new peaks were observed from the reaction containing PhDph2, SAMCA, and dithionite (Figure S6), which were absent in control reactions without dithionite. Some of the new peaks were assigned to MTA. Crotonic acid and its isomer, 2-butenoic acid, which are the hydrogen abstraction products of the carboxyallyl radical, were not formed by comparison to the spectra of their standards. Three new peaks (a–c) were assigned to γ-sulfinylcrotonic acid (Figure S6) and the remaining three peaks (d, e and f) around 5.1 and 5.8 ppm were assigned to α-sulfinyl-3-butenoic acid (Figure S6); the remaining proton on α-sulfinyl-3-butenoic acid is localized at 3.4 ppm based on 1H-1H COSY (correlation spectroscopy) (Figure S14, S15). Using 1,1-2H2-SAMCA as the substrate, peaks c, e and f (Figure S7) disappeared, which supported the assignments. The assignments were confirmed by 1H/13C HMBC (Heteronuclear Multiple Bond Correlation), HSQC (Heteronuclear Single Quantum Correlation) and 1H-1H COSY (Figure S8 – S15). The reaction products were also chemically modified to allow detection by LC-MS (Figure S16). Our data thus demonstrated the formation of γ-sulfinylcrotonic and α-sulfinyl-3-butenoic acid, which likely resulted from the recombination of the carboxyallyl radical with the dithionite-derived SO2−• radical, similar to previously reported reactions of radicals with dithionite 3,12,13. The NMR signal intensities suggested roughly equal formation of the two products.
We then performed 35 GHz CW/pulse 13C, 1H and 2H ENDOR with 1-13C, 2-13C, 3-13C, 1,1-2H2 and 1,1,2,3-2H4 labeled SAMCA (see Scheme 2). Field-modulated 35 GHz CW ENDOR spectra obtained from reactions with 1-13C, 2-13C labeled SAMCA both exhibit 13C doublets with broad component lines, split by the 13C hyperfine coupling, A ~ 5–8 MHz (Figure 2). An estimate of the magnitude of the hyperfine coupling tensors for these two 13C sites of SAMCA (Table S1) was obtained through analysis of 2D field-frequency patterns of ENDOR spectra collected across the EPR envelopes of the SAMCA isotopologs (Figures S17, S18). The analysis showed comparable isotropic hyperfine tensors, Aiso ~ 7 MHz for both 1-13C and 2-13C. For 3-13C labeled SAMCA, a considerably smaller isotropic coupling, Aiso ~ 0.6 MHz, was estimated using Mims pulsed ENDOR (Figure 2A, Inset).
Figure 2.

(A) 35 GHz CW 13C ENDOR spectra for CA with 1-13C and 2-13C-labeled SAMCA collected at g2; the doublets are split by the hyperfine coupling, and centered at the Larmor frequency as described in SI. Inset: Mims ENDOR spectra of 3-13C SAMCA. (B) 1H Davies ENDOR difference spectra of CA generated by subtracting from the spectrum of 1H-SAMCA (natural abundance) the spectra for 1,1,2,3-2H-labeled SAMCA (upper) and 1,1-2H-labeled SAMCA (lower). Spectra were collected at g2 and normalized by EPR echo height.
To further probe the structure of CA, 1H and 2H pulse ENDOR measurements were performed with 1,1-2H2, and 1,1,2,3-2H4 labeled SAMCA. Only the pulsed ENDOR techniques resolved individual features, but the lower sensitivity of pulsed vs CW ENDOR allowed spectra to be collected only at the intensity maximum of the EPR envelope, near g2. The 1H signals from the labeled sites were obtained by subtracting the spectra obtained using the natural-abundance SAMCA (Figure 2B). After subtraction of the 1,1-2H2 spectrum, a doublet of 1H peaks assignable to the 1,1-1H nuclei was obtained with coupling constant of A ~ 7 MHz; subtraction with the 1,1,2,3-2H4 labeled SAMCA showed an enhanced intensity for this doublet (Figure 2B), indicating that the 1,1-1H and 2-1H nuclei had comparable couplings (assigned based on 2-13C data). In addition, weak broad features corresponding to a doublet with A ~ 2 MHz were assigned to 3-1H. Mims 2H ENDOR spectra (Figure S19 and S20) provided support for this assignment. The substantial isotropic couplings to 1-13C and 2-13C (Aiso ~ 7 MHz), the deviations of the g-values from g~2, and the 57Fe broadening imply the spin is primarily localized on the cluster, but with close C-Fe interaction.
The similarity of 1-13C and 2-13C hyperfine couplings, as well as the similarity of the 1,1-2H and the 2-2H couplings, suggests that both 1-C and 2-C have similar direct interactions with the unique Fe of the [4Fe-4S] cluster. In contrast, the weaker 3-13C and 3-2H hyperfine couplings show that 3-C does not experience a significant direct interaction with the Fe. These results suggest that CA only consists of one of the two products, α-sulfinyl-3-butenoic acid, coordinated to the unique Fe site of the [4Fe-4S] cluster. If CA also involved a complex with γ-sulfinylcrotonic acid, 2-C and 3-C should have similar strong hyperfine couplings, and 1-C should have weaker coupling, contrary to our observations (Figure 2A). We further confirmed this by utilizing the instability of α-sulfinyl-3-butenoic acid. After incubating the protein-free aliquots of the reaction products at room temperature overnight, the peaks corresponding to α-sulfinyl-3-butenoic acid disappeared in the 1H-NMR spectrum (Figure S21), while the signals from γ-sulfinylcrotonic acid remained. The instability of α-sulfinyl-3-butenoic acid is likely due to the acidity of the α-proton, which is adjacent to two strong electron-withdrawing groups.14 When the mixture containing only the γ-sulfinylcrotonic acid product was added to ‘fresh’ dithionite-reduced PhDph2, the signal associated with CA was not be detected by EPR (Figure S22). This result supports the notion that γ-sulfinylcrotonic acid does not form an EPR-active complex with the [4Fe-4S] cluster and that CA is best described as a complex formed between α-sulfinyl-3-butenoic acid and the [4Fe-4S] cluster.
We then probed whether the sulfinyl and carboxyl of α-sulfinyl-3-butenoic acid groups are required for CA formation. We synthesized a non-cleavable analog of SAMCA, aza-SAMCA (Scheme 2)15. The freeze quench-EPR experiment with aza-SAMCA showed only a [4Fe-4S]+ signal (Figure S23) that was similar to that of the reduced cluster in PhDph2 in the absence of SAMCA. Therefore, CA is not formed by binding of uncleaved SAMCA to the [4Fe-4S] cluster. Addition of crotonic acid to PhDph2 also failed to generate the CA species. Thus, the sulfinic group in α-sulfinyl-3-butenoic acid is likely involved in the formation of CA. Similarly, adding allylsulfinic acid to PhDph2 did not generate the CA signal either. Thus, the carboxyl group in α-sulfinyl-3-butenoic acid is also necessary. The results suggest that both the sulfinic group and the carboxylate of α-sulfinyl-3-butenoic acid bind to the Fe-S cluster, facilitating the formation of a π-complex between that Fe and the C=C double bond of the product (Figure 3).
Figure 3.

Plausible structures of CA and formation mechanism.
CA can have two formal resonance structures (Figure 3, structures a and b). To further probe the oxidation state of CA, we attempted to reduce CA by low-temperature γ-irradiation (cryoreduction)16 (Figure S24). The EPR signal did not decrease upon γ-irradiation, as would be expected for a [4Fe-4S]3+ cluster. Similarly, extra dithionite could not reduce CA, which is inconsistent with the expectation for a [4Fe-4S]3+ cluster (Figure S25)17. In contrast, when we tried to oxidize CA with K3[Fe(CN)6], CA was oxidatively degraded to a [3Fe-4S]+ cluster (Figure S25), an outcome commonly observed in similar treatments of [4Fe-4S]+ clusters of RS enzymes with chemical oxidants. Thus, the Fe-S core in CA behaves like a [4Fe-4S]+ cluster. We also sought to use 57Fe Mössbauer spectroscopy. However, owing to the rather modest yield of CA (50% of total Fe) and the presence of at least one additional EPR-silent Fe-containing product, the analysis of the spectra does not permit definitive conclusions about the electronic structure of CA (Figures S26, 27 and 28).
With the characterization of reaction products and the structure of CA, we proposed a plausible mechanism for the formation of CA (Figure 3). The [4Fe-4S]+ cluster in PhDph2 provides one electron to SAMCA, cleaving the CCA-S bond and generating a 3-carboxyallyl radical, MTA, and a [4Fe-4S]2+ cluster. The 3-carboxyallyl radical reacts with a dithionite-derived SO2−• radical, forming γ-sulfinylcrotonic acid and α-sulfinyl-3-butenoic acid. The sulfinic group, the carboxyl group, and the C=C double bond of α-sulfinyl-3-butenoic acid, coordinate the [4Fe-4S]+ cluster (reduced by extra dithionite) to generate the π-complex. The coordination of the sulfinic and carboxyl group helps to place the double bond in a favorable position for π interactions with the unique Fe. The two conformers revealed by EPR (Figure 1B) could be attributed to different binding orientations of the α-sulfinyl-3-butenoic acid to [4Fe-4S]+ cluster (the three chelating functional groups may exchange positions).
In summary, using SAMCA, we have detected an EPR-active species, CA, formed in the enzymatic reaction of PhDph2. Various experiments demonstrate that CA is a complex formed between the [4Fe-4S]1+ cluster and one of the reaction products, α-sulfinyl-3-butenoic acid, coordinated to the unique cluster Fe through the sulfinyl and carboxylate oxygens, with the C=C double bond forming an organometallic π-complex with the Fe. Although we could not accumulate the radical intermediate that we set out to detect with SAMCA, formation of both α-sulfinyl-3-butenoic acid and γ-sulfinylcrotonic acids provides additional support for the radical mechanism of PhDph2 reaction, as a nucleophilic mechanism cannot account for their formation. Interestingly, although the EPR g tensors of CA shows similarity to those of [4Fe-4S]3+-like clusters observed in two enzymes involved in the non-mevalonate pathway, IspG and IspH, the cluster in CA behaves like a [4Fe-4S]+ cluster.
The present case can be described as the reaction of the unique Fe of the [4Fe-4S]+ cluster with the alkene product to form a stable organometallic complex. Such an alkene complex has reported for the nitrogenase FeMo-cofactor and the ethylenic product of alkyne reduction18, and for mutants of IspH and its substrate HMBPP.11,19 However, similar organometallic complex had not been demonstrated for RS enzymes until very recently, when the [4Fe-4S]2+ cluster of the RS enzyme, pyruvate formatelyase activating enzyme is shown to react with the 5′-dA• to form a highly reactive organometallic intermediate.20 This study and our current result suggest that the unique iron of [4Fe-4S] clusters in RS enzymes can readily form a C-Fe complex. Such a property not only may help to explain the unusual chemistries catalyzed by the [4Fe-4S]-containing RS enzymes, but also may open up new avenues to tune the activity or develop new chemistry for this enzyme class or for other [4Fe-4S]-containing enzymes.
Supplementary Material
Acknowledgments
This work is supported by NIH (GM088276, P41-GM103521, and GM111097). We thank Dr. Ivan Keresztes and Mr. Anthony Condo for assistance with NMR; Prof. H. Halpern at University of Chicago for access to the 60Co Gammacell irradiator.
Footnotes
The authors declare no competing financial interests.
Experimental materials and methods, supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Su X, Lin Z, Lin H. Crit Rev Biochem Mol Biol. 2013;48:515. doi: 10.3109/10409238.2013.831023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaffrath R, Abdel-Fattah W, Klassen R, Stark MJ. Mol Microbiol. 2014;94:1213. doi: 10.1111/mmi.12845. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, Lin H. Nature. 2010;465:891. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong M, Su X, Dzikovski B, Dando EE, Zhu X, Du J, Freed JH, Lin H. J Am Chem Soc. 2014;136:1754. doi: 10.1021/ja4118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magnusson OT, Reed GH, Frey PA. J Am Chem Soc. 1999;121:9764. [Google Scholar]
- 7.Horitani M, Byer AS, Shisler KA, Chandra T, Broderick JB, Hoffman BM. J Am Chem Soc. 2015;137:7111. doi: 10.1021/jacs.5b00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mcmanus HJ, Fessenden RW, Chipman DM. J Phys Chem. 1988;92:3778. [Google Scholar]
- 9.Belinskii M. Chem Phys. 1993;172:189. [Google Scholar]
- 10.Wang W, Li J, Wang K, Huang C, Zhang Y, Oldfield E. Proc Natl Acad Sci USA. 2010;107:11189. doi: 10.1073/pnas.1000264107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Wang K, Span I, Jauch J, Bacher A, Groll M, Oldfield E. J Am Chem Soc. 2012;134:11225. doi: 10.1021/ja303445z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ko Y, Ruszczycky MW, Choi SH, Liu HW. Angew Chem Int Ed Engl. 2015;54:860. doi: 10.1002/anie.201409540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-de-Choudens S, Fontecave M, Atta M. J Biol Chem. 2008;283:36361. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khatik GL, Kumar R, Chakraborti AK. Org Lett. 2006;8:2433. doi: 10.1021/ol060846t. [DOI] [PubMed] [Google Scholar]
- 15.Joce C, Caryl J, Stockley PG, Warriner S, Nelson A. Org Biomol Chem. 2009;7:635. doi: 10.1039/b816495a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davydov R, Hoffman BM. Arch Biochem Biophys. 2011;507:36. doi: 10.1016/j.abb.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Torre A, Lara C, Yee BC, Malkin R, Buchanan BB. Arch Biochem Biophys. 1982;213:545. doi: 10.1016/0003-9861(82)90582-3. [DOI] [PubMed] [Google Scholar]
- 18.Lee HI, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2004;126:9563. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 19.Wang W, Wang K, Liu YL, No JH, Li J, Nilges MJ, Oldfield E. Proc Natl Acad Sci U S A. 2010;107:4522. doi: 10.1073/pnas.0911087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horitani M, Shisler K, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM, Broderick JB. Science. 2016;352:822. doi: 10.1126/science.aaf5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.