

ABSTRACT
Saffold virus (SAFV), a human cardiovirus, is occasionally detected in infants with neurological disorders, including meningitis and cerebellitis. We recently reported that SAFV type 3 isolates infect cerebellar glial cells, but not large neurons, in mice. However, the impact of this infection remained unclear. Here, we determined the neuropathogenesis of SAFV type 3 in the cerebella of neonatal ddY mice by using SAFV passaged in the cerebella of neonatal BALB/c mice. The virus titer in the cerebellum increased following the inoculation of each of five passaged strains. The fifth passaged strain harbored amino acid substitutions in the VP2 (H160R and Q239R) and VP3 (K62M) capsid proteins. Molecular modeling of the capsid proteins suggested that the VP2-H160R and VP3-K62M mutations alter the structural dynamics of the receptor binding surface via the formation of a novel hydrophobic interaction between the VP2 puff B and VP3 knob regions. Compared with the original strain, the passaged strain showed altered growth characteristics in human-derived astroglial cell lines and greater replication in the brains of neonatal mice. In addition, the passaged strain was more neurovirulent than the original strain, while both strains infected astroglial and neural progenitor cells in the mouse brain. Intracerebral inoculation of either the original or the passaged strain affected brain Purkinje cell dendrites, and a high titer of the passaged strain induced cerebellar hypoplasia in neonatal mice. Thus, infection by mouse-passaged SAFV affected cerebellar development in neonatal mice. This animal model contributes to the understanding of the neuropathogenicity of SAFV infections in infants.
IMPORTANCE Saffold virus (SAFV) is a candidate neuropathogenic agent in infants and children, but the neuropathogenicity of the virus has not been fully elucidated. Recently, we evaluated the pathogenicity of two clinical SAFV isolates in mice. Similar to other neurotropic picornaviruses, these isolates showed mild infectivity of glial and neural progenitor cells, but not of large neurons, in the cerebellum. However, the outcome of this viral infection in the cerebellum has not been clarified. Here, we examined the tropism of SAFV in the cerebellum. We obtained an in vivo-passaged strain from the cerebella of neonatal mice and examined its genome and its neurovirulence in the neonatal mouse brain. The passaged virus showed high infectivity and neurovirulence in the brain, especially the cerebellum, and affected cerebellar development. This unique neonatal mouse model will be helpful for elucidating the neuropathogenesis of SAFV infections occurring early in life.
INTRODUCTION
Saffold virus (SAFV) belongs to the species Cardiovirus B, the genus Cardiovirus, and the family Picornaviridae (1–3). After its initial identification, SAFV was extensively detected or isolated from fecal or throat swab samples from infants with acute gastroenteritis or upper respiratory symptoms (4–16). SAFV is classified into at least 11 genotypes based on the capsid protein VP1 genome sequence, and seroepidemiological studies suggest that SAFV type 2 (SAFV-2) and SAFV-3 circulate in the human population early in life (5, 9, 10, 13, 15, 17, 18). SAFV is occasionally detected in fecal or cerebrospinal fluid (CSF) specimens collected from children with neurological diseases such as acute flaccid paralysis, aseptic meningitis, and cerebellitis (4, 6, 18–23). However, it is unclear whether SAFV causes neurological diseases in humans, primarily because SAFV is often detected alongside enteric or respiratory viruses such as norovirus, rotavirus, bocavirus, and influenza virus (4, 5, 7, 8, 11, 12, 14, 16, 19, 24, 25) and even in healthy individuals (6).
Recent studies have reported infection of mouse models with SAFV-2 and -3 strains (26–28). These strains show similar tropism for the murine brain, spinal cord, heart, and/or pancreas and are thus likely to be rodent cardioviruses. In our previous study, we performed pathological, virological, and immunological studies of SAFV-3-infected neonatal and young mice by using two SAFV-3 isolates and examined the neuropathogenesis of the virus (29). These two isolates of SAFV-3 were derived from two different clinical cases; the JPN 08-404 strain was isolated from the CSF of an aseptic meningitis patient (21), and the Gunma/176/2008 strain was isolated from a throat swab from a patient with upper respiratory tract inflammation (12). Both strains exhibited mild neurovirulence after intracerebral inoculation into neonatal ddY mice. The two isolates infected glial cells and neural progenitor cells, but not large neurons, in the brains and cerebella of neonatal ddY mice. In addition, the JPN 08-404 strain was more infectious to glial cells in the cerebellum than the Gunma/176/2008 strain was.
Because cerebellar glial cell tropism is not observed with other neurotropic picornaviruses (30), the present study examined the neurotropism of the JPN08-404 strain of SAFV-3 in the cerebella of neonatal mice. We assessed the phenotype of the serially passaged JPN 08-404 strain in the cerebella of neonatal mice. We found that the passaged strain showed three amino acid substitutions in the VP2 and VP3 capsid proteins, replicated more efficiently in the brains and cerebella of neonatal mice, and was more highly neurovirulent than the original strain. We also found that infection with the passaged strain after intracerebral inoculation affected the development of the cerebellar cortex. These results suggest that the SAFV-3 infection of glial cells in the cerebella of neonatal mice may impact cerebellar development.
MATERIALS AND METHODS
Viruses and cells.
The clinical isolate of the SAFV-3 JPN 08-404 strain, referred to here as the AM strain, was a kind gift from Takushi Hosomi (The Public Health Institute of Kochi Prefecture, Kochi, Japan) (21). The AM strain was obtained from the CSF of a patient with aseptic meningitis in 2008. After isolation in rhesus monkey kidney epithelial (LLC-MK2) cells, the viral stock was passaged three times in LLC-MK2 cells that were cultured in minimal essential medium (Gibco, Life Technologies Corporation, Carlsbad, CA) supplemented with 2% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (2MEM). The virus-infected LLC-MK2 cells were disrupted by three freeze-thaw cycles. The supernatant was obtained by centrifugation at 2,000 × g for 15 min and stored at −80°C until it was used. The titers of the stock viruses were expressed in 50% cell culture infective doses (CCID50s) per milliliter in LLC-MK2 cells, which were calculated by the method of Behrens and Kärber (76). All work with infectious SAFV-3 was performed under biosafety level 2 conditions.
Mice.
Pregnant BALB/c mice and ddY mice were purchased from Japan SLC (Shizuoka, Japan) and housed at the animal facility of the National Institute of Infectious Diseases. Pregnant mice were closely monitored for date and approximate time of delivery. Three-week-old ddY mice were purchased from Japan SLC and used as control animals. Animal studies were carried out in strict accordance with the Guidelines for Proper Conduct of Animal Experiments of the Science Council of Japan, and all animal experiments were conducted in strict compliance with animal husbandry and welfare regulations. All animal experiments were approved by the Committee on Experimental Animals at the National Institute of Infectious Diseases in Japan, and all experimental animals were handled in biosafety level 2 animal facilities according to the guidelines recommended by this committee.
Serial in vivo passage of SAFV-3 AM strain in neonatal mice.
Five neonatal BALB/c mice within 24 h of birth were inoculated intracerebrally with 104 CCID50 of the AM strain in a volume of 10 μl. These five mice were sacrificed by excess anesthesia with isoflurane on day 3 postinoculation (p.i.) for collection of their cerebella. Ten percent (wt/vol) tissue homogenates of the collected cerebella were prepared in 2MEM. The cerebellum homogenate was centrifuged at 2,000 × g for 10 min, and 10 μl of supernatant was used as the inoculum in the next passage. During the serial passages, the supernatants of the cerebellum homogenates (first passage, 1Cb; second passage, 2Cb; third passage, 3Cb; fourth passage, 4Cb; fifth passage, 5Cb) were obtained and stored at −80°C until they were used. After five passages, the supernatants from the cerebellum homogenates (5Cb) were used to infect LLC-MK2 cells. The cells were infected with 150 μl of the supernatant in 10 ml of 2MEM. The infected cell cultures were continuously incubated at 37°C with 5% CO2. Once cytopathic effects (CPEs) appeared, the cells were harvested and then frozen and thawed three times. After centrifugation at 2,000 × g for 10 min, the supernatant was used as the passaged virus (referred to as the AM-5Cb strain) (Fig. 1A).
FIG 1.

In vivo passage of SAFV-3 in neonatal mice. (A) Schematic diagram showing the serial in vivo passage of SAFV-3. Within 24 h of birth, neonatal BALB/c mice (n = 5) were inoculated intracerebrally at 104 CCID50s/10 μl with SAFV-3 (AM strain) collected from a patient with aseptic meningitis. On day 3 p.i., five cerebella were collected from neonatal mice and 10% tissue homogenates were prepared. After centrifugation, 10 μl of the supernatant (termed 1Cb) was inoculated intracerebrally into the next group of neonatal mice within 24 h of their birth. This process was conducted a total of five times. After propagation of the virus from the supernatant of 5Cb in LLC-MK2 cells, the culture supernatant, called the AM-5Cb strain, was stored as stock virus. (B) Amino acid substitutions occurring during the in vivo serial passages. These passaged strains acquired three sequential amino acid substitutions in viral structural proteins, VP2-160, VP2-239, and VP3-62. The three amino acid substitutions were VP2-H160R, VP2-Q239R, and VP3-K62M. (C) Viral RNA copy number during the serial passage of SAFV-3 in the mouse cerebellum. The number of viral RNA copies was determined by real-time RT-PCR and is expressed relative to the number of mouse beta-actin RNA copies. (**, P < 0.01; ***, P < 0.001; one-way ANOVA).
Viral genome sequencing.
The viral RNA was extracted from the supernatants of the cerebellar homogenates 1Cb, 2Cb, 3Cb, 4Cb, and 5Cb and the AM-5Cb strain with the High Pure Viral RNA kit (Roche, Basel, Switzerland) or the RNeasy Plus minikit (Qiagen, Hilden, Germany). Reverse transcription (RT)-PCR was performed with the OneStep RT-PCR kit (Qiagen) and specific primers (18, 31). The PCR-amplified DNA products were purified with a MonoFas DNA purification kit I (GL Sciences Inc., Tokyo, Japan) and then sequenced with a BigDye Terminator v3.1 Cycle Sequencing kit (Life Technologies Corporation) and an ABI 3130 genetic analyzer (Applied Biosystems, Life Technologies Corporation). The nucleotide sequences were analyzed with Sequencher software (ver. 4.10.1; Gene Codes Corporation, Ann Arbor, MI).
Molecular modeling of SAFV VP1, VP2, and VP3 capsid proteins.
Molecular models of the VP1, -2, and -3 proteins on the virion surface of the AM and AM-5Cb strains were constructed by homology modeling with the Molecular Operating Environment system (Chemical Computing Group Inc., Montreal, Canada) as described for the human rhinovirus VP1, VP2, and VP3 complex modeling (32). We used the X-ray-derived crystal structure of particles of a prototype (DA) strain of Theiler's murine encephalomyelitis virus (TMEV), which is a Cardiovirus B species, at a resolution of 2.80 Å (Protein Data Bank accession number 1TME) (33) as the template for homology modeling.
Growth of mouse-passaged SAFV-3 in cultured cell lines.
The growth of the AM and AM-5Cb strains in vitro was assessed with a nonhuman primate LLC-MK2 cell line and human medulloblastoma (ONS-76), astrocytoma (U-251 MG), neuroblastoma (SK-N-SH), and rhabdomyosarcoma RD cells. ONS-76 and U-251 MG cells were purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). Each cell line was cultured in six-well culture plates and then inoculated with the AM or AM-5Cb strain in triplicate at a multiplicity of infection of 0.1. On days 0, 1, 2, 3, and 5 p.i., the supernatants from the infected cells were obtained as extracellular virus samples. After the cells had been washed twice in phosphate-buffered saline (PBS), 2MEM was added to collect the cell pellets, which were frozen and thawed three times and collected as intracellular virus samples. All samples were centrifuged at 2,000 × g for 15 min. Virus titers were determined with LLC-MK2 cells and expressed in CCID50s.
Experimental infection of neonatal mice.
The ddY outbred albino strain of mice showed good reproductive performance, growth, and susceptibility to SAFV-3 and was thus used for the neurovirulence study (29). We performed three SAFV-3 infection experiments with neonatal mice. In the first experiment, neonatal ddY mice were inoculated intracerebrally within 24 h of birth with 104 CCID50s of the AM or AM-5Cb strain in 10 μl of 2MEM. These mice were observed for clinical manifestations, and their body weights were measured daily for 21 days (n = 7). On days 3, 5, 7, and 21 p.i., the inoculated animals were sacrificed by excess anesthesia with isoflurane and examined by virological and pathological methods (n = 3, 4, or 7 mice per group). For this experiment, the 12 neonatal mice used as negative controls were inoculated intracerebrally with 2MEM. In the second experiment, an additional 12 neonatal ddY mice were intracerebrally inoculated within 24 h of birth with 106 CCID50s of the AM-5Cb strain in 10 μl of 2MEM. These mice were observed for clinical manifestations, and their body weights were measured daily for 19 days. Additional inoculated animals were sacrificed on days 3, 7, and 19 p.i. by excess anesthesia with isoflurane for histopathologic examination (n = 4 mice per group). In the third experiment, 10 neonatal ddY mice were intracerebrally inoculated within 24 h of birth with 104 CCID50s of the AM or AM-5Cb strain in 10 μl of 2MEM (n = 5 mice per group). On day 22 p.i., the inoculated animals were sacrificed by excess isoflurane anesthesia for high-resolution fluorescent imaging. Three-week-old mice born on the same day as the other mice were used as controls (n = 5).
Histopathology and immunohistochemistry.
Neonatal mice were sacrificed by excess isoflurane anesthesia and perfused with 10% phosphate-buffered formalin injected directly into their hearts. Their harvested tissues were immersed in a formalin solution overnight. The whole head, including the brain, was sliced into sagittal sections, which were then immersed in a formalin solution for 2 or 3 days. After fixation, the sections were dehydrated in ethanol (30 to 50% solutions) and decalcified in a buffered EDTA 2Na solution (Dojindo, Kumamoto, Japan) as necessary. The tissue samples were then embedded in paraffin and stained with hematoxylin and eosin (H&E).
A polymer-based detection system, Nichirei-Histofine Simple Stain Mouse MAX PO (R) (Nichirei Biosciences, Inc., Tokyo, Japan), was used for the immunohistochemical detection of virus antigens on paraffin-embedded tissue. Antigen retrieval was performed with a retrieval solution (pH 6) (Nichirei Biosciences) at 121°C for 10 min in an autoclave. The IgG fraction of the primary antibody, a polyclonal antibody against the anti-AM strain of SAFV-3 (21), was purified with the Melon Gel IgG Spin Purification kit (Thermo Fisher Scientific Inc., Waltham, MA), according to the manufacturer's instructions. Peroxidase activity was detected with 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO), and the sections were counterstained with hematoxylin.
Immunofluorescence staining.
Double immunofluorescence staining of paraffin-embedded tissues was performed with rabbit antiserum against SAFV-3, a rat monoclonal antibody (Musashi-1, clone D270-3; Medical & Biological Laboratories, Nagoya, Japan), a mouse anti-glial fibrillary acidic protein (GFAP) monoclonal antibody (clone GA5; Merck Millipore, Billerica, MA), a mouse anti-glutamate-aspartate transporter (GLAST) monoclonal antibody (clone ACSA-1; Miltenyi Biotec, Auburn, CA), and a rabbit anti-vitamin D-dependent calcium-binding protein (calbindin) polyclonal antibody (ab25085; Abcam, Cambridge, United Kingdom). The antigens were retrieved by autoclaving the sections in retrieval solution (pH 6.0) (Nichirei Biosciences) at 121°C for 10 min. The sections were incubated with the first monoclonal antibody against Musashi-1, GFAP, or GLAST, followed by incubation with the calbindin polyclonal antibody or antiserum against the SAFV-3 antigen. Goat anti-rabbit antibody conjugated with Alexa Fluor 568 (for calbindin and SAFV-3 antigen), goat anti-rat antibody conjugated with Alexa Fluor 488 (for Musashi-1), and goat anti-mouse antibody conjugated with Alexa Fluor 488 (for GFAP and GLAST) were used as secondary antibodies (all secondary antibodies were from Molecular Probes, Life Technologies Corporation). The sections were incubated with the antibodies for 30 min at room temperature before being mounted with SlowFade Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes). Double fluorescence images were captured with a laser scanning confocal microscope (FV1000-D; Olympus, Tokyo, Japan).
Quantitative analysis of Purkinje cell morphology.
Neonatal mice (n = 5) were sacrificed on day 22 p.i. by excess isoflurane anesthesia and perfused via the heart with 4% paraformaldehyde in phosphate-buffered saline (PBS) following perfusion with heparin sodium (50 IU/ml in saline) to help remove blood from the cardiovascular system before the fixative entered. Fixative perfusions were performed with a peristaltic pump (ATTA, Tokyo, Japan) at a setting of 2 ml/min for ≥3 min. Harvested brains were immersed in paraformaldehyde solution overnight at 4°C. Fixed brains were dissected midline sagittally into 1-mm slices with a stainless steel brain matrix (Brain Science Idea Co. Ltd., Osaka, Japan) dissection kit. For high-resolution fluorescence imaging, we prepared cleared tissue by a modification of the optical clearing method of Ke et al. with an optical clearing agent (SeeDB2) (34).
First, fixed-tissue slices were immersed in PBS containing 2% Triton X-100 for 2 h at 4°C, incubated in blocking solution (PBS containing 10% normal goat serum, 2% Triton X-100, and 0.02% sodium azide) for 1 h, and then incubated with anti-calbindin polyclonal antibody (ab25085; Abcam) for 3 days at 4°C. After being washed with PBS, the sections were incubated with anti-rabbit antibody conjugated with Alexa Fluor 568 (Molecular Probes) for 2 days at 4°C. The sections were then treated with a 1:2 mixture of Omnipaque 350 (Daiichi-Sankyo Co. Ltd., Tokyo, Japan) and 1% Triton X-100 in water for 10 h and then with a 1:1 mixture of Omnipaque 350 and 1% Triton X-100 in water for 10 h. Finally, the sections were incubated in Omnipaque 350 with 1% Triton X-100 for 12 h. The SeeDB2-treated sections were stored in Omnipaque 350. After immunofluorescence staining and clearing treatment, the resultant images were captured from four areas in each mouse, the sulcus regions of the cerebellar lobules from III to VI and apex regions of lobules VII and VIII, without artificial damage (35). Serial confocal images along the z axis at an average depth of 104 μm (46 to 175 μm) and with a 0.46-μm step size in the z direction were obtained with a laser scanning confocal microscope (FV1000-D; Olympus) equipped with a silicone oil immersion objective lens (0.8-mm working distance, UPLSAPO30XS; Olympus). The reconstructed Purkinje cell layer in the three-dimensional images was used for assessments of Purkinje cell morphology with the neuron tracing program of Neurolucida (version 11.09 64 bit; MBF Bioscience, Williston, VT). The widths of the Purkinje and molecular layers were measured at four sites within one area (four areas per mouse;16 sites/mouse). Purkinje cell somata were counted in four areas per mouse at several depths (46 to 175 μm). Somata and main dendrites of Purkinje cells were extracted digitally with the tracing application of Neurolucida software. The main dendrites extracted from 4 to 10 Purkinje cells were measured under high-power field magnification, resulting in the coverage of a total area of 2,882,828 μm2 in four areas per mouse. In total, an average of 35 cells (25 to 40 cells) were assessed per mouse. The diameter of the main dendrite was measured by tracing. Main dendrite length was also measured from the surface of the soma to the first major branch point by tracing. The tracing results were analyzed with Neurolucida Explorer (MBF Bioscience), which generated data including the width of the cerebellar cortex, Purkinje cell soma number and area, and the average diameter and length of the main dendrites of the Purkinje cells (36, 37).
Virus titration.
The brains of neonatal mice were separated into two parts, the cerebellum and the remaining brain, including the cerebrum and brainstem. The brain samples (except neonatal mouse cerebella) were homogenized with a Bio Masher II (Nippi, Incorporated, Tokyo, Japan) and diluted in 2MEM to yield 10% homogenates, whereas the samples of the neonatal mouse cerebella were homogenized with 1 ml of 2MEM. After centrifugation at 8,000 × g for 5 min, the supernatants from the homogenates were used for titration in LLC-MK2 cells. The inoculated cells were examined twice within 2 weeks and then assessed for CPEs at 2 weeks p.i. The detection limit was 101.5 CCID50s/g of tissue.
Real-time RT-PCR.
The total RNA of tissue homogenates from the virus-inoculated mice was extracted with an RNeasy Plus minikit (Qiagen). The expression levels of the mRNA from GLAST (accession no. AF330257.1), hairy and enhancer of split 5 (Hes5) (accession no. NM_010419.4), calbindin (accession no. NM_009788.4), and delta/notch epidermal growth factor-related receptor (DNER; accession no. NM_152915.1) were assessed by real-time RT-PCR methods. Primer Express software (Applied Biosystems) was used to design the primers and probes. The TaqMan probe and primer sets used (Sigma-Aldrich) are listed in Table 1. The viral genome and expression of type 1 interferon (IFN) mRNA, including mouse IFN-α4 and IFN-β, were assessed by real-time RT-PCR methods (38, 39). Mouse beta-actin mRNA (a housekeeping gene) was also quantified. The real-time RT-PCR round was conducted for 30 min at 50°C for RT and 15 min at 95°C for Taq polymerase activation, followed by 40 cycles of 94°C for 15 s and 60°C for 60 s. The second round of PCR was conducted for 3 min at 95°C, followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min (40).
TABLE 1.
Primer and probe sequences used for real-time RT-PCR
| Test, primers, and probe | Genome sequence (5′–3′) |
|---|---|
| Real-time RT-PCR for Saffold virus | |
| Forward | AAACCATGCCACAAACACCAT |
| Reverse | GCCYTGACCAACTACCCACAT |
| Probe (FAM-TAMRA) | CTTGCCGAYACACGTGACCCACA |
| Real-time RT-PCR for murine Hes5 | |
| Forward | CCTGAAACACAGCAAAGCCTTC |
| Reverse | GGAGTAGCCCTCGCTGTAGTC |
| Probe (FAM-TAMRA) | CCGGCCCCAAGAGCCTGCACC |
| Real-time RT-PCR for murine DNER | |
| Forward | TGTCCTAGACCCATGCAGAAATG |
| Reverse | CAAGCGGAGCCGAAGTACC |
| Probe (FAM-TAMRA) | TGAACCCGCTGAGGCTGGACACACA |
| Real-time RT-PCR for murine GLAST | |
| Forward | GATGCTGCAGATGCTGGTCTT |
| Reverse | TTACTATCTAGGGCCGCCATTC |
| Probe (FAM-TAMRA) | CCCCTGATCATCTCCAGTCTCGTCACA |
| Real-time RT-PCR for murine Calbindin | |
| Forward | GCTCCGCGCACTCTCAA |
| Reverse | GAGATGACTGCAGGTGGGATTC |
| Probe (FAM-TAMRA) | AGCCGCTGCACCACGATGGC |
Statistical analysis.
Data are expressed as the mean with the standard error of the mean. Statistical analysis was performed with GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA). Intergroup comparisons were performed with unpaired t tests, one-way analysis of variance (ANOVA) followed by Tukey's post hoc test, or two-way ANOVA. A P value of <0.05 was considered statistically significant.
Accession number(s).
The complete nucleotide sequence of the AM-5Cb strain was submitted to the DNA Data Bank of Japan and assigned accession no. AB983595.
RESULTS
Mouse-passaged SAFV-3 acquired three amino acid mutations.
The complete genomes of the original AM and mouse-passaged AM-5Cb strains of SAFV-3 were compared. The nucleotide sequence of the complete genome of the AM-5Cb strain (accession no. AB983595) was 99.9% identical to that of the AM strain (accession no. HQ902242). The genome of the AM-5Cb strain had five nucleotide differences from that of the AM strain, and two of the differences were synonymous changes in 2C at position 4701 (AM, C; AM-5Cb, T) and 3C at position 6453 (AM, G; AM-5Cb, A), whereas three of the differences were nonsynonymous in VP2, at positions 1949 (AM, A; AM-5Cb, G) and 2192 (AM, A; AM-5Cb, G), and in VP3, at position 2468 (AM, A; AM-5Cb, T). These three amino acid substitutions were in structural protein VP2 at positions 160 (AM, H; AM-5Cb, R) and 239 (AM, Q; AM-5Cb, R) and in VP3 at position 62 (AM, K; AM-5Cb, M). We then compared the extracted viral genome RNA at these substituted regions from the supernatants of the cerebellum homogenates during the serial passages (Fig. 1B). The first-passage 1Cb strain attained one amino acid substitution, at position 239 (AM, Q; 1Cb, R) of the VP2 protein. The third-passage 3Cb strain showed one amino acid substitution, at position 160 (AM, H; 3Cb, R) of the VP2 protein. One amino acid mutation at position 62 (AM, K; 5Cb, M) of the VP3 protein was obtained in the fifth-passage 5Cb strain. The number of viral RNA copies in each supernatant of the cerebellum homogenates during the serial passages was quantified by real-time RT-PCR for SAFV-3 RNA (Fig. 1C). The amino acid substitutions of the mouse-passaged SAFV-3 appeared to be correlated with increasing viral RNA levels in the cerebellum during the serial passages.
Mutations in the passaged strain affect interactions between residues at positions 62 and 160.
The VP2 and VP3 proteins of Cardiovirus B species are known to form the pentamer lattice on the virion surface (33) and play critical roles in viral infection (41–44). To address the structural impacts of the three mutations in the capsid proteins of the AM-5Cb strain, we constructed molecular models of the VP2 and VP3 proteins in the virion. The models showed that the three mutations occurred in the exposed loop regions. Specifically, the H160R substitution occurred at the stem of the loop in the VP2 puff B region (Fig. 2A), the Q239R substitution occurred at the tip of a loop of VP2 (Fig. 2A), and the K62M substitution occurred at the stem of the loop in the knob region of VP3 (Fig. 2A) (45). Notably, VP3-62 is located in close proximity to VP2-160 in the reported TMEV receptor binding site (Fig. 2B and C). The three-dimensional locations of VP3-62M and VP2-160R enabled their side chains to form a hydrogen bond only in the AM-5Cb strain (Fig. 2D). These results suggest that the VP3-K62M and VP2-H160R mutations can alter the structural dynamics of the receptor binding surface via the formation of a novel hydrophobic interaction. The structural alterations may, in turn, affect the binding affinity of the receptor for the virus or antibodies.
FIG 2.
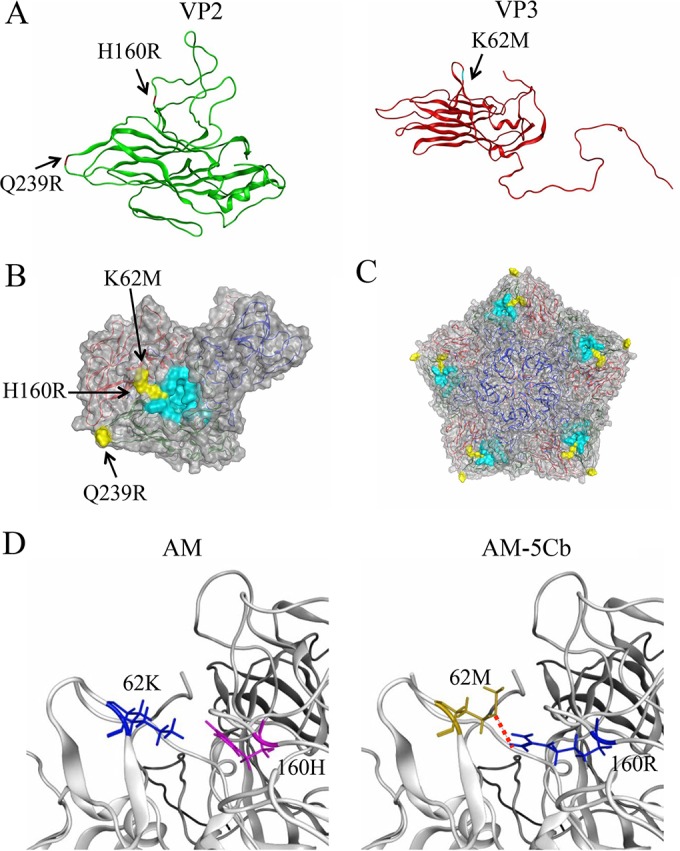
Structural models of SAFV-3 capsid proteins. The models were constructed by homology modeling based on the X-ray-derived crystal structure of the virus particle of the TMEV DA strain. (A) Models of the VP2 (green) and VP3 (red) proteins of the AM-5Cb strain. Substituted residues in the AM-5Cb strain indicated are H160R at the loop of the VP2 alias puff B structure, Q239R at the loop of VP2, and K62M at the loop in the VP3 knob structure. (B, C) Models of the VP1, VP2, and VP3 complex (B) and the pentamer of the complex (C) on the virion surface. Three substituted residues in the AM-5Cb strain (yellow) and residues of the receptor binding region of TMEV (cyan) are highlighted. (D) Closeup view around the putative receptor binding domain of the SAFV-3 VP2 and VP3 proteins: AM (left) and AM-5Cb (right) strains. A red dotted line indicates that a novel hydrogen bond forms between side chains of the mutated residues (VP2-160R and VP3-62M) in the AM-5Cb strain.
Growth properties of mouse-passaged SAFV-3 in human glial cell lines.
To examine changes in the growth phenotype of the AM-5Cb strain, viruses were inoculated into four human cell lines (ONS-76 medulloblastoma, U-251 MG astrocytoma, SK-N-SH neuroblastoma, and rhabdomyosarcoma RD) (Fig. 3). After inoculation with the AM or AM-5Cb strain, all human cells showed a CPE, including cytolysis and degeneration, indicating that they were susceptible to these viruses. Compared with the AM strain, the AM-5Cb strain exhibited earlier and stronger growth in ONS-76 cells, which are derived from a human cerebellar medulloblastoma, and in astrocytoma U-251 MG cells; however, human neuroblastoma SK-N-SH cells were less susceptible. On the other hand, although the AM-5Cb strain showed earlier growth in human rhabdomyosarcoma RD cells, there was no difference in the yield of the titer. These results suggest that the mouse-passaged AM-5Cb strain shows stronger growth in human glial cell lines than the original strain.
FIG 3.

Growth curves of original and mouse-passaged SAFV-3 in various cell lines. Human cell lines (medulloblastoma, ONS-76; astrocytoma, U-251 MG; neuroblastoma, SK-N-SH; rhabdomyosarcoma, RD) were infected with the AM (original) or AM-5Cb (mouse-passaged) SAFV-3 strains at a multiplicity of infection of 0.1. Extracellular virus samples were collected in cell culture medium, and intracellular virus samples were isolated by freezing and thawing the cell pellets. The amount of virus present in each sample is expressed as the CCID50. The dotted line represents the viral titer detection limit. Shown are representative data from two experiments, each with similar results.*, P < 0.05; ***, P < 0.001; two-way ANOVA.
Mouse-passaged SAFV-3 is neurovirulent in neonatal ddY mice.
To evaluate the neurovirulence of mouse-passaged SAFV-3, neonatal ddY mice were inoculated intracerebrally with the AM or AM-5Cb strain of SAFV-3. The rate of weight gain by the AM-5Cb-inoculated neonatal mice was lower than that of the 2MEM-inoculated control mice during the observation period, whereas there was no difference between AM-inoculated and control mice (Fig. 4A). The AM-5Cb-inoculated mice showed neurological symptoms, including ataxia, running, rolling, or staggering, from days 2 to 4 p.i. (Fig. 4A, arrows), and the AM strain-inoculated mice developed mild neurological signs, such as ataxia, from days 7 to 9 p.i. From day 11 p.i., two (29%) out of seven AM-5Cb strain-inoculated mice displayed dome-shaped heads and were moribund within 21 days p.i., whereas none of the AM-inoculated or control mice developed this sign. Histopathologically, the moribund mice with dome-shaped heads had enlarged ventricles with a necrotic cerebra. The brainstems, cerebella, and spinal cords of the moribund mice showed slight inflammatory infiltration (Table 2 and Fig. 4B). Other AM-5Cb-inoculated mice showed perivascular cuffing with mononuclear cells and swelled endothelial cells in the cerebellar medulla, while the AM-inoculated mice did not (Fig. 4C). In addition, inflammatory reactions were found in the spinal cords and skeletal muscles of the AM-5Cb-inoculated mice (Table 2).
FIG 4.
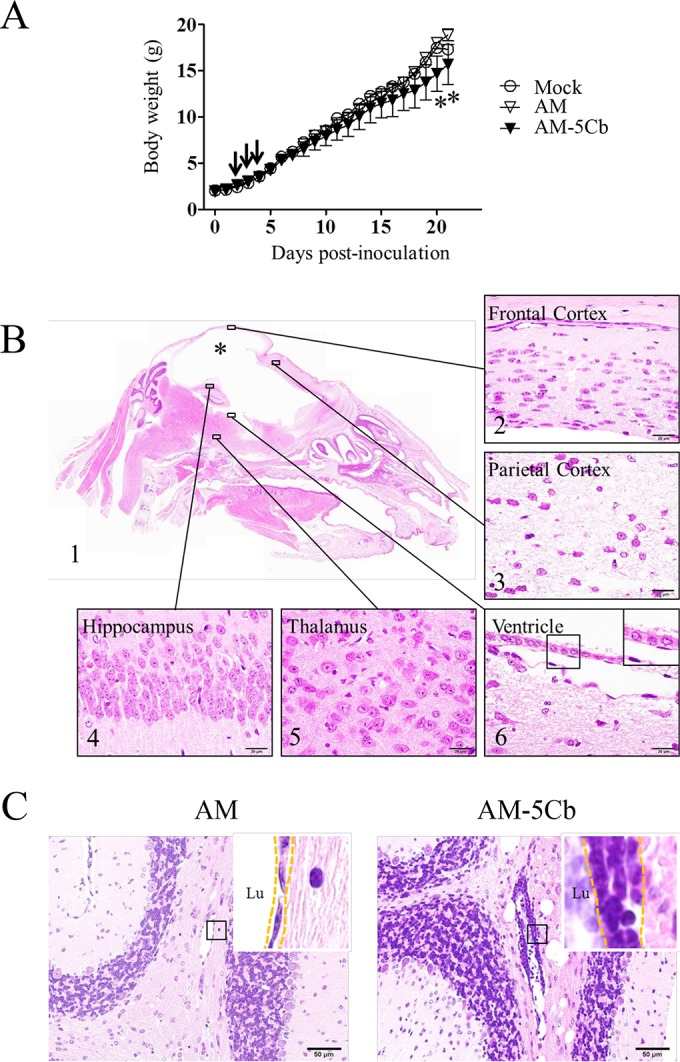
Neurovirulence of mouse-passaged SAFV-3 in neonatal ddY mice. Within 24 h of birth, neonatal ddY mice were inoculated intracerebrally at 104 CCID50s/10 μl with SAFV-3 derived from a case of aseptic meningitis (AM strain) or with mouse-passaged SAFV-3 (AM-5Cb strain). (A) Body weights of neonatal ddY mice after SAFV inoculation. Animals were observed for clinical manifestations, and body weights were measured daily for 21 days (n = 4). One AM-inoculated mouse showed mild neurological signs, including rolling and ataxia, from days 7 to 9 p.i., whereas some AM-5Cb-inoculated mice showed obvious rolling and ataxia from days 2 to 4 p.i. (arrow). On days 20 and 21 p.i., AM-5Cb-inoculated mice showed significantly less weight gain than mock-infected control mice (*, P < 0.05; one-way ANOVA). (B, C) Sagittal brain sections of representative neonatal ddY mice intracerebrally infected with the AM or AM-5Cb strain on day 21 p.i. H&E staining. The cystic lesion (asterisk) is filled with CSF, and the cerebral cortex is nearly absent (B1). The brainstem and cerebellum remain. Thinning of the cerebral cortex (B2) and severe necrosis (B3) are observed in the residual cerebral cortex. The hippocampus (B4) and thalamus (B5) appear normal. The cystic lesion in the cerebrum is lined with ciliated ependymal cells (B6, inset). Perivascular inflammation (C, dotted frame in inset) is observed in the cerebellar medulla of AM-5Cb-inoculated mice (C, right side) but not in that of AM-inoculated mice (C, left side). Lu, lumen. Original magnifications, ×20 (B1), ×400 (C), and ×1,000 (B2, -3, -4, -5, and -6 and all insets). Bars, 20 μm (B) and 50 μm (C).
TABLE 2.
Tissue distribution of viral antigen and inflammatory reactions following the intracerebral inoculation into neonatal ddY mice of the original Saffold virus strain (AM) or the strain passaged in mouse cerebellum (AM-5Cb)
| Virus strain and day of sacrifice p.i. | No. of mice | Central nervous system tissuea |
Peripheral tissuea |
||||
|---|---|---|---|---|---|---|---|
| Cerebrum | Brainstem | Cerebellum | Spinal cord | Muscle | Peripheral nerve | ||
| AM | |||||||
| 3 | 3 | 3/0 | 3/0 | 3/1 | 1/0 | 2/0 | 0/0 |
| 5 | 3 | 3/0 | 0/0 | 3/0 | 1/0 | 3/0 | 3/0 |
| 7 | 3 | 1/0 | 0/0 | 3/0 | 1/0 | 1/0 | 0/0 |
| 21 | 7 | 0/1 | 0/2 | 0/0 | 0/0 | 0/0 | 0/0 |
| AM-5Cb | |||||||
| 3 | 3 | 3/0 | 3/0 | 3/0 | 2/0 | 3/0 | 3/0 |
| 5 | 3 | 3/2 | 2/0 | 3/3 | 2/0 | 2/0 | 3/0 |
| 7 | 3 | 3/1 | 2/1 | 2/0 | 3/0 | 3/0 | 0/0 |
| 21 | 7 | 1/7 | 0/4 | 0/3 | 0/4 | 0/5 | 0/0 |
Values represent the number of animals positive for viral antigen/number of animals positive for degeneration or inflammatory reactions.
Viral proliferation and immune response to mouse-passaged SAFV-3 in neonatal ddY mice.
To examine viral proliferation in neonatal ddY mice, we determined the viral titers and viral RNA copy numbers in the brains of AM- and AM-5Cb-inoculated mice. Viral replication was measured separately in the cerebellum and other brain regions (cerebrum and brainstem, referred to as cerebrum/brainstem). On day 3 p.i., both the cerebrum/brainstem and cerebellum of the AM-5Cb-inoculated neonatal mice had significantly higher viral titers than those of the AM-inoculated mice (Fig. 5A). Similarly, the levels of viral RNA in both the cerebrum/brainstem and cerebellum of AM-5Cb-inoculated mice on day 3 p.i. were higher than in those of AM-inoculated mice (Fig. 5B). We next examined the antiviral responses to the mouse-passaged SAFV-3 infection in the brains of neonatal mice. The expression levels of both IFN-α4 and IFN-β in the brains of AM-5Cb-inoculated mice were significantly higher than those in the brains of AM-inoculated mice on day 3 p.i. (Fig. 5C). Thus, the proliferative capacity of the AM-5Cb strain in the neonatal mouse brain was increased by in vivo passage, and infection by this strain induced a stronger type 1 IFN response and longer-term inflammatory infiltration than infection with the AM strain.
FIG 5.

Viral replication and type 1 IFN expression in neonatal ddY mice after intracerebral inoculation of the original (AM) or mouse-passaged (AM-5Cb) SAFV-3 strain. Virus titers (A) and the numbers of viral RNA copies per mouse (B) were measured in the cerebrum/brainstem and cerebellum on days 3, 7, and 21 p.i. with the SAFV-3 strains (104 CCID50s/10 μl; n = 4 per day). The dotted line in panel A represents the viral titer detection limit. The expression levels of mouse type 1 IFN-α4 and -β (C) were determined on days 3, 7, and 21 p.i. (n = 4 per day). The brains of mock-infected mice were used as a negative control. The numbers of viral RNA copies and type 1 IFN mRNA copies are expressed relative to the number of mouse beta-actin RNA copies. **, P < 0.01; ***, P < 0.001; one-way ANOVA.
Mouse-passaged SAFV-3 infects glial and neural progenitor cells, but not large neurons, in neonatal ddY mice.
We also evaluated the histopathological changes in the brains of AM-5Cb strain-inoculated mice by histopathological and immunohistochemical approaches (Table 2 and Fig. 6). On day 3 p.i., many degenerated cells with eccentric nuclei and moderate inflammation were seen in the cerebral ventricles and cerebella of both the AM- and AM-5Cb-inoculated mice. The AM-5Cb-inoculated mice showed more severe lesions than the AM-inoculated mice (Fig. 6A). The number of viral antigen-positive cells in the cerebrum/brainstem of AM-5Cb-inoculated mice was significantly higher than that in AM-inoculated mice on day 3 p.i.; in addition, a greater number of antigen-positive cells was observed in the cerebellum of AM-5Cb-inoculated mice than in that of AM-inoculated mice, although the difference was not significant (Fig. 6B and C).
FIG 6.
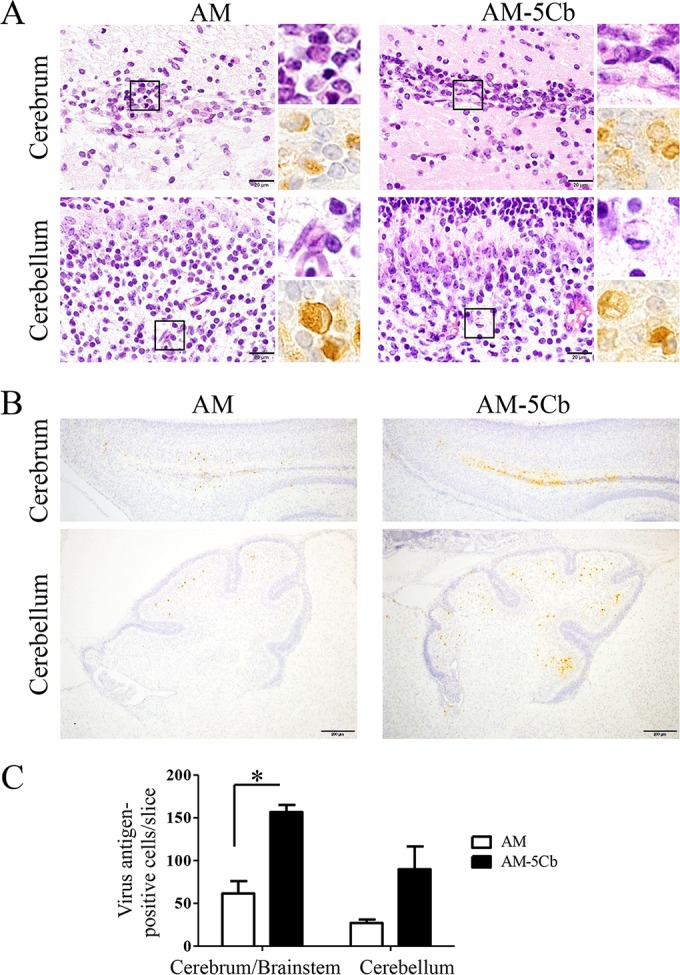
Histopathological results and viral antigens in the brains of neonatal mice after intracerebral inoculation with mouse-passaged SAFV-3. Neonatal ddY mice were intracerebrally inoculated with the AM or AM-5Cb strain at 104 CCID50s/10 μl. (A) The areas of the cerebral ventricles and cerebellum of a neonatal mouse after infection. Top left and right, H&E staining; bottom right, immunohistochemical staining with an anti-SAFV antibody. Degenerated neural cells are present in the cerebral ventricles and cerebella of AM- and AM-5Cb-inoculated mice. The degenerated neural cells with eccentric nuclei are positive for viral antigens (insets). (B) Viral antigen-positive cells are located mainly in the areas of the cerebral ventricles and cerebella of AM- and AM-5Cb-inoculated mouse strains. Original magnifications, ×1,000 (panel A and all insets) and ×100 (B). Bar, 20 μm (left side of panel A) and 200 μm (B). (C) Viral antigen-positive cells were counted in two slices per mouse (n = 3 each group). Numbers of viral antigen-positive cells in the cerebrum/brainstem and cerebellum were compared. *, P < 0.05; unpaired t tests.
Double immunofluorescence staining indicated that on day 3 p.i., viral antigens were present in the GLAST+ glial cells of the cerebellum and in the GFAP+ glial cells and Musashi-1+ neural progenitor cells around the lateral cerebral ventricle in the brains of AM-5Cb-inoculated neonatal mice (Fig. 7). This result is consistent with that of our previous study showing that the viral antigen-positive cells in AM-inoculated mice were glial and neural progenitor cells but not large neurons (29). Thus, in vivo passage did not alter the neurotropism of the AM strain in neonatal mice. Also, because the intensity of the staining for viral antigens was strong in neural progenitor cells around the lateral cerebral ventricle, we speculated that the hydrocephalus observed after AM-5Cb infection might be an obstructive hydrocephalus (46, 47) caused by restricted flow of CSF past the damaged area. However, the outcome of SAFV-3 infection of the cerebellum remains unclear.
FIG 7.
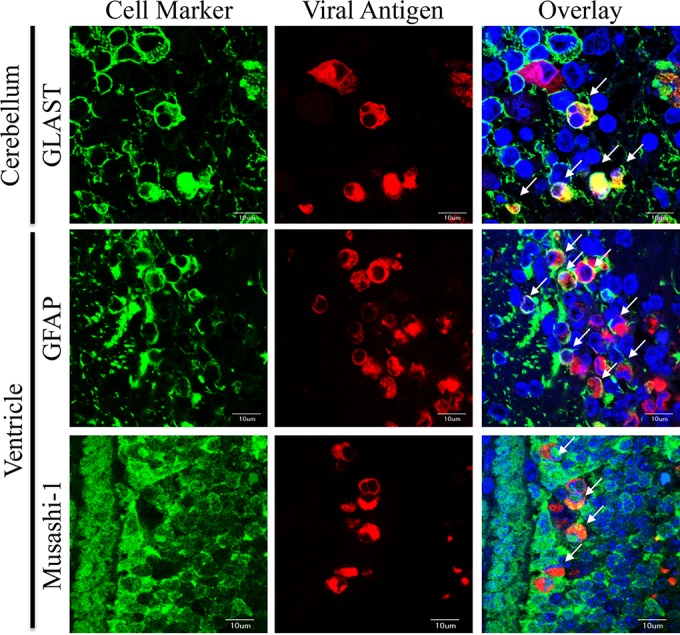
Identification of SAFV-3 AM-5Cb-infected cells in the brains of ddY neonatal mice. Shown are representative double immunofluorescence images of cells positive for the cell markers (green) GLAST (radial astrocytes), GFAP (astrocytes), and Musashi-1 (neural progenitor cells) and for the viral antigen (red) in the cerebral ventricles and cerebella of AM-5Cb-inoculated (104 CCID50s/10 μl) neonatal ddY mice on day 3 p.i. Viral antigen-positive cells are GLAST+ radial astrocytes (Bergmann glial cells) in the cerebellum, as well as GFAP+ radial astrocytes and Musashi-1+ neural progenitor cells (neuroepithelial cells) in the cerebral ventricle. Arrows, viral antigen-positive and neural marker-positive cells. Original magnification, ×600. Bars, 10 μm.
SAFV-3 infection of Bergmann glial cells affects cerebellar development.
We next assessed the pathogenesis of the SAFV-3 infection in the cerebellum of neonatal ddY mice. The viral antigen-positive GLAST+ glial cells in the cerebellum, namely, Bergmann glial cells, are associated with the elongation of Purkinje cell dendrites during cerebellar cortical development (48, 49). We found that both the AM and AM-5Cb strains infected Bergmann glial cells but not Purkinje cells (Fig. 6 and 7). The interaction between Bergmann glial cells and Purkinje cells activates the Notch signaling pathway. The results of our real-time RT-PCR assay revealed that the expression level of the gene for Hes5, which reflects Notch signaling activity in Bergmann glial cells, in the cerebella of AM- and AM-5Cb-inoculated mice was significantly higher than that in 2MEM-inoculated mice on day 3 p.i. However, the expression level of GLAST, which is expressed in Bergmann glial cells, was not different among AM-, AM-5Cb-, and 2MEM-inoculated mice (Fig. 8A). In contrast, the mRNA expression levels of DNER and calbindin in the cerebella of the AM-5Cb-inoculated mice were slightly higher than those of the AM-inoculated mice (Fig. 8B). DNER protein is expressed in Purkinje cell dendrites and is related to Notch signaling activity through direct cell-to-cell contact between Purkinje cells and Bergmann glial cells.
FIG 8.

Gene expression associated with cerebellar cortical development in neonatal mice after SAFV-3 infection. The original AM or mouse-passaged AM-5Cb SAFV-3 strains (104 CCID50s/10 μl) were intracerebrally inoculated into neonatal ddY mice. The expression levels of genes associated with Bergmann glial cells (A) and Purkinje cells (B) were then determined on days 3, 7, and 21 p.i. (n = 4 per day). *, P < 0.05; ***, P < 0.001; one-way ANOVA.
Hence, we evaluated the morphology of cerebellar Purkinje cells in AM- and AM-5Cb-inoculated mice in three-dimensional images of sections of cleared tissue that had been immunofluorescence stained with anti-calbindin antibody (Fig. 9A to E). Analyses of the widths of the Purkinje layer and the molecular layer (Fig. 9B), the number and area of Purkinje cell soma (Fig. 9D), and the average diameter and length of the Purkinje cells (Fig. 9E) showed that the lengths of the main dendrites of Purkinje cells of AM-5Cb-inoculated mice were significantly shorter than those of the control mice. There were no differences in the number or area of soma or the average diameter of main dendrites among control or AM- or AM-5Cb-inoculated mice. However, small round soma with narrow main dendrites were observed in the AM- and AM-5Cb-inoculated mice whereas large round soma with thick main dendrites were observed in control mice. These results suggest that SAFV-3 infection affects the expression of genes associated with Bergmann glial cells, which may result in impaired differentiation and growth of Purkinje cells in the absence of direct Purkinje cell infection.
FIG 9.

Saffold virus infection affects the development of the Purkinje cell. On day 22 p.i., the original AM or mouse-passaged AM-5Cb strain intracerebrally inoculated (104 CCID50s/10 μl) neonatal ddY mice were used for high-resolution fluorescence imaging. (A) Three-dimensional images of sections of the cerebellar layer at a depth of 50 μm that had been immunofluorescence stained with anti-calbindin antibody. The stack images were acquired with a 0.46-μm step size in the z direction. y-z and x-z images are shown in the sagittal slice of a stack image. (B) The width of the Purkinje cell layer and the molecular layer of the stack images at a 50-μm depth were determined with Neurolucida software. Data are from four areas per mouse. (C) A representative tracing image of the soma and main dendrites of a Purkinje cell. (D) Soma number and area in Purkinje cells were measured by using Neurolucida software. Data are from four areas per mouse. (E) Average diameter and length of the main dendrites of Purkinje cells. An average of 35 cells were measured in four areas of the cerebellum per mouse. The main dendrites of AM-5Cb-inoculated mice are significantly shorter than those of control mice. *, P < 0.05; one-way ANOVA. Original magnification, ×300 (A, C).
To clarify the outcome of mouse cerebellar development after severe SAFV infection, we next investigated the impact of inoculating the mouse cerebellum with a higher titer of the AM-5Cb strain. Within 24 h of birth, neonatal ddY mice were intracerebrally inoculated with the AM-5Cb strain at 106 CCID50s/10 μl. On day 3 p.i., the AM-5Cb-inoculated mice appeared supersensitive to touch and sound stimulation and then gradually became less active. After 19 days p.i., all four infected mice developed dome-shaped heads and were moribund. As determined through histopathological analysis, AM-5Cb-inoculated mice showed marked morphological changes in the cerebral cortex and cerebellum compared with control mice (Fig. 10A, top). All four AM-5Cb-inoculated mice showed hydrocephalus and small cerebella. In addition, the arbor vitae of the cerebellum were not apparent in the any of the four AM-5Cb-inoculated neonatal mice (Fig. 10A, bottom). Inflammatory cell infiltration and mild necrosis were also observed in the cerebellum medulla. Double immunofluorescence staining with anti-GLAST and anti-calbindin antibodies revealed that the three layers consisting of Bergmann glial cells and Purkinje cells in the cerebellum were not apparent in the AM-5Cb-inoculated mice, while control mice had apparently normal cerebellar cortices (Fig. 10B). In addition, moderate inflammation was observed in the cerebra, brainstems, and spinal cords of AM-5Cb-inoculated mice (Table 3).
FIG 10.
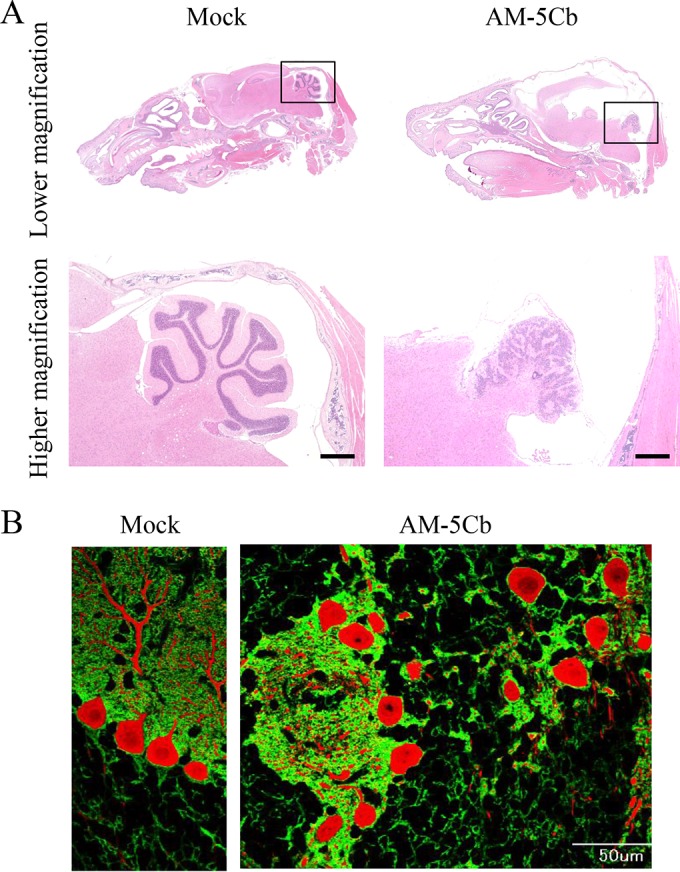
High titers of the mouse-passaged SAFV-3 strain induce malformation of the cerebellar cortex in neonatal ddY mice. Within 24 h of birth, neonatal ddY mice were intracerebrally inoculated with the mouse-passaged AM-5Cb strain of SAFV-3 at 106 CCID50s/10 μl. (A) H&E staining. (B) Double immunofluorescence staining with anti-GLAST (radial astrocytes; green) and anti-calbindin (Purkinje cells; red) antibodies. (Left side of panels A and B) Representative sagittal brain sections from a mock-infected neonatal mouse at day 19 postinjection with 2MEM. (Right side of panels A and B) Representative sagittal brain sections from an AM-5Cb-inoculated neonatal mouse at day 19 p.i. The arbor vitae of the cerebellum appear normal in the mock neonatal mouse (left side of panel A). In contrast, hydrocephalus of the cerebral cortex and small cerebella are evident in all four AM-5Cb-inoculated neonatal mice (upper right side of panel A). The arbor vitae of the cerebellum are unclear in the AM-5Cb-inoculated mouse (lower right side of panel A). The cerebellar cortex of the mock-inoculated mouse can be divided into three layers: the molecular layer, the Purkinje cell layer, and the granular layer (left side of panel B). In contrast, the three layers are not apparent in the cerebellar cortex of the AM-5Cb-inoculated mouse (right side of panel B). Original magnifications: top of panel A, ×20; bottom of panel A, ×100; panel B, ×400. Bars, 500 μm (bottom of panel A) and 50 μm (right side of panel B).
TABLE 3.
Histopathological analysis results following an intracerebral inoculation of a high viral titer of mouse-passaged Saffold virus AM-5Cb strain into neonatal ddY mice
| Day of sacrifice p.i.a | No. of mice | Central nervous system tissueb |
Peripheral tissueb |
||||
|---|---|---|---|---|---|---|---|
| Cerebrum | Brainstem | Cerebellum | Spinal cord | Muscle | Peripheral nerve | ||
| 3 | 4 | 4/3 | 4/2 | 4/4 | 4/0 | 4/0 | 4/0 |
| 7 | 4 | 4/4 | 4/4 | 4/4 | 4/0 | 4/0 | 2/0 |
| 19 | 4 | 1/4 | 1/2 | 1/4 | 3/4 | 0/0 | 0/0 |
The virus strain used was AM-5Cb.
Values represent the number of animals positive for viral antigen/number of animals positive for degeneration or inflammatory reactions.
DISCUSSION
The prototype strain of the Cardiovirus B species, TMEV, is widely used in mouse models used to study neuroviral infections (50–52). The natural host of TMEV is the house mouse (Mus musculus), and the infection is spread and maintained in mouse colonies via the fecal-oral route (53, 54). TMEV isolates show various degrees of neurovirulence in mice; the highly neurovirulent strain causes lethal encephalomyelitis upon acute infection of neurons, whereas the low-virulence strain persistently infects oligodendrocytes, leading to multiple sclerosis (44, 55, 56). Exchanging the viral capsid coding region, which is associated with the receptor binding site, between highly virulent and less virulent strains influences the outcome of the infection by directing the tropism of the virus to the central nervous system (57–59). Here, we found that the AM strain of SAFV acquired three amino acid substitutions within virion surface proteins VP2 and VP3 after five passages in the mouse cerebellum. These substitutions accumulated in a stepwise manner and might be associated with the consecutive increases in viral RNA levels detected in cerebellum homogenates tested at each passage. Specifically, the level of viral RNA markedly increased after amino acid substitutions at H160R and K62M in the VP2 puff B and VP3 knob regions, suggesting that these two substitutions play central roles in altering viral replication efficiency in the neonatal mouse brain.
The constructed molecular models of the capsid proteins revealed that these two substitutions occurred at adjacent loops on the virion surface. Moreover, the models suggest a noticeable difference in the capsid surface between the AM and AM-5Cb strains, namely, the absence and presence, respectively, of a hydrophobic interaction between loops of the VP2 puff B and VP3 knob regions. The structural dynamics of protein surfaces play a key role in protein interactions. Therefore, on the basis of these structural data, it is reasonable to postulate that these two substitutions induce alterations in the interactions of the virus with the host.
Previous studies examining members of the family Picornaviridae consistently suggested the importance of the puff B and knob regions in regulating viral infectivity and neutralization sensitivity. The Cardiovirus and Enterovirus genera have five distinctive structures on the capsid protein: loop 1 and loop 2 of VP1, the puff A and puff B regions of VP2, and the knob region of VP3. These regions are putative phenotypic determinants, such as sialic acid binding and neutralization antibody binding sites, in TMEV (60, 61). Additionally, in coxsackieviruses belonging to the genus Enterovirus, neutralization escape is reportedly related to structural alterations in the puff B region of VP2 and the knob of VP3 (62). Our in vitro experiments examining viral growth kinetics revealed that the mouse-passaged AM-5Cb strain showed earlier and stronger growth in the human cell lines ONS-76 and U-251 MG (which are derived from human neural progenitor cell and human astroglial cell tumors, respectively) than the original strain. In addition, in neonatal ddY mice, the passaged AM-5Cb strain also showed greater infectivity of neuroepithelial cells in the cerebral ventricle and astroglial cells in the cerebellum than the AM strain. Thus, it is likely that the increased proliferative capacity of the AM strain used in the present study resulted from the two substitutions that altered the affinity of the viral capsid for the virus receptor. However, because no cellular receptors for SAFV have been identified, further studies are necessary to address this issue.
The AM-5Cb-inoculated neonatal mice showed cerebellar ataxia at an earlier phase of viral infection than the AM-inoculated neonatal mice. Bergmann glial cells play an important role in the glutamic acid intake of the cerebellum, and impaired glutamate transport in Bergmann glial cells secondarily leads to Purkinje cell degeneration, which causes cerebellar ataxia (63–65). On the basis of our results, we speculated that the AM-5Cb infection-induced degeneration and necrosis of Bergmann glial cells impaired their glutamic acid intake function, generating excess excitatory postsynaptic currents in Purkinje cells, which led to the abnormal neurological signs seen. Because interactions between neurites of Bergmann glial cells and Purkinje cells activate the Notch signaling pathway during cerebellar cortical development, both Bergmann glial cells and Purkinje cells play roles in neuronal differentiation and neurite elongation (48, 66–69). The activation of the Notch signaling pathway begins with direct binding between the Notch of Bergmann glial cells and the DNER of Purkinje cells and depends on the expression of the gene for Hes5. The loss of Bergmann glial cells after AM-5Cb infection induced repair reactions in the undifferentiated neural progenitor cells, causing overexpression of the gene for Hes5. This overexpression in the Notch signaling pathway might enhance the differentiation of Bergmann glial cells and provoke the abnormal elongation of Purkinje cell neurites and may also explain the immature extension of the main dendrites of Purkinje cells in AM-5Cb-inoculated mice.
Inoculation of neonatal ddY mice with a high titer of the AM-5Cb strain clearly induced dysplasia of the cerebellar cortex. We speculated that a large number of Bergmann glial cells were infected with the AM-5Cb strain and died because of disruption of the balance in the interactions between Bergmann glial cells and Purkinje cells. Although there are no reports of hypoplasia after picornavirus infection, parvovirus, lymphocytic choriomeningitis virus, and border disease virus infections induce hypoplasia of the cerebellar cortex (70–73). Borna disease virus also induces cerebellar disorders in neonatal rats (74, 75). These viruses infect Purkinje cells, neural progenitor cells, or astroglia in the cerebellum to repress translocation of immature nerve cells, which leads to cerebellar hypoplasia. In contrast, the SAFV-3 strains in the present study infected Bergmann glial cells but not Purkinje cells in the cerebellum, indirectly affecting the development of the cerebellar cortex. Thus, the neurovirulence of SAFV-3 in mice is unique compared with that of other picornaviruses and other neurovirulent viruses that infect the cerebellum.
In conclusion, the SAFV-3 isolate acquired high infectivity and neurovirulence in the brains of neonatal mice after in vivo passage. Infection by the mouse-passaged SAFV-3 strain induced obstructive hydrocephalus and affected cerebellar development in neonatal mice. Because SAFV-3 has the potential for pathogenicity in the cerebella of human infants (22), this neonatal mouse model will be useful for increasing our understanding of the neurovirulence of SAFV. In addition, this animal model will further our understanding of the neuropathogenicity of viral infections early in life.
ACKNOWLEDGMENTS
We thank Takushi Hosomi (The Meat Inspection Center of Kochi Prefecture, Kochi, Japan) for providing the JPN08-404 isolate of Saffold virus. We also thank our colleagues at the National Institute of Infectious Diseases in Tokyo, Japan, particularly Ayako Harashima, Mihoko Fujino, and Moeko Aida, for their technical assistance and Shin-ichi Tamura, Hideki Asanuma, Akira Ainai, and Minoru Tobiume for valuable discussions. We are also grateful to Chikako Kataoka and Tomofumi Nakamura (Department of Virology II at the National Institute of Infectious Diseases, Japan) for their technical expertise with respect to sequencing of the passaged virus genome.
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grants-in-Aid for Scientific Research no. 23590554 and 15K08511) and the Ministry of Health, Labor, and Welfare of Japan (Grants-in-Aid for Research on Emerging and Reemerging Infectious Diseases no. H23-Shinko-Ippan-007 and H25-Shinko-Ippan-012).
REFERENCES
- 1.Jones MS, Lukashov VV, Ganac RD, Schnurr DP. 2007. Discovery of a novel human picornavirus in a stool sample from a pediatric patient presenting with fever of unknown origin. J Clin Microbiol 45:2144–2150. doi: 10.1128/JCM.00174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang Z, Manoj Kumar AS, Jones MS, Knowles NJ, Lipton HL. 2008. Phylogenetic analysis of the species Theilovirus: emerging murine and human pathogens. J Virol 82:11545–11554. doi: 10.1128/JVI.01160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed). 2012. Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Academic Press, Ltd., London; United Kingdom. [Google Scholar]
- 4.Chiu CY, Greninger AL, Kanada K, Kwok T, Fischer KF, Runckel C, Louie JK, Glaser CA, Yagi S, Schnurr DP, Haggerty TD, Parsonnet J, Ganem D, DeRisi JL. 2008. Identification of cardioviruses related to Theiler's murine encephalomyelitis virus in human infections. Proc Natl Acad Sci U S A 105:14124–14129. doi: 10.1073/pnas.0805968105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drexler JF, Luna LK, Stocker A, Almeida PS, Ribeiro TC, Petersen N, Herzog P, Pedroso C, Huppertz HI, Ribeiro Hda C Jr, Baumgarte S, Drosten C. 2008. Circulation of 3 lineages of a novel Saffold cardiovirus in humans. Emerg Infect Dis 14:1398–1405. doi: 10.3201/eid1409.080570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blinkova O, Kapoor A, Victoria J, Jones M, Wolfe N, Naeem A, Shaukat S, Sharif S, Alam MM, Angez M, Zaidi S, Delwart EL. 2009. Cardioviruses are genetically diverse and cause common enteric infections in South Asian children. J Virol 83:4631–4641. doi: 10.1128/JVI.02085-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren L, Gonzalez R, Xiao Y, Xu X, Chen L, Vernet G, Paranhos-Baccala G, Jin Q, Wang J. 2009. Saffold cardiovirus in children with acute gastroenteritis, Beijing, China. Emerg Infect Dis 15:1509–1511. doi: 10.3201/eid1509.081531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu ZQ, Cheng WX, Qi HM, Cui SX, Jin Y, Duan ZJ. 2009. New Saffold cardiovirus in children, China. Emerg Infect Dis 15:993–994. doi: 10.3201/eid1506.090109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zoll J, Erkens Hulshof S, Lanke K, Verduyn Lunel F, Melchers WJ, Schoondermark-van de Ven E, Roivainen M, Galama JM, van Kuppeveld FJ. 2009. Saffold virus, a human Theiler's-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog 5:e1000416. doi: 10.1371/journal.ppat.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu CY, Greninger AL, Chen EC, Haggerty TD, Parsonnet J, Delwart E, Derisi JL, Ganem D. 2010. Cultivation and serological characterization of a human Theiler's-like cardiovirus associated with diarrheal disease. J Virol 84:4407–4414. doi: 10.1128/JVI.02536-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren L, Gonzalez R, Xie Z, Xiao Y, Li Y, Liu C, Chen L, Yang Q, Vernet G, Paranhos-Baccala G, Jin Q, Shen K, Wang J. 2010. Saffold cardioviruses of 3 lineages in children with respiratory tract infections, Beijing, China. Emerg Infect Dis 16:1158–1161. doi: 10.3201/eid1607.091682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsukagoshi H, Masuda Y, Mizutani T, Mizuta K, Saitoh M, Morita Y, Nishina A, Kozawa K, Noda M, Ryo A, Kimura H. 2010. Sequencing and phylogenetic analyses of Saffold cardiovirus (SAFV) genotype 3 isolates from children with upper respiratory infection in Gunma, Japan. Jpn J Infect Dis 63:378–380. [PubMed] [Google Scholar]
- 13.Chua KB, Voon K, Yu M, Ali WN, Kasri AR, Wang LF. 2011. Saffold virus infection in children, Malaysia, 2009. Emerg Infect Dis 17:1562–1564. doi: 10.3201/eid1708.101380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai XQ, Yuan CL, Yu Y, Zhao W, Yang ZB, Cui L, Hua XG. 2011. Molecular detection of Saffold virus in children in Shanghai, China. J Clin Virol 50:186–187. doi: 10.1016/j.jcv.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Galama J, Lanke K, Zoll J, Roivainen M, van Kuppeveld F. 2011. Seroepidemiology of Saffold cardiovirus type 2. Emerg Infect Dis 17:1572–1573. doi: 10.3201/eid1708.101953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itagaki T, Abiko C, Aoki Y, Ikeda T, Mizuta K, Noda M, Kimura H, Matsuzaki Y. 2011. Saffold cardiovirus infection in children associated with respiratory disease and its similarity to coxsackievirus infection. Pediatr Infect Dis J 30:680–683. doi: 10.1097/INF.0b013e31821608a8. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi M, Tsukagoshi H, Ishioka T, Mizuta K, Noda M, Morita Y, Ryo A, Kozawa K, Kimura H. 2013. Seroepidemiology of Saffold cardiovirus (SAFV) genotype 3 in Japan. J Infect 66:191–193. doi: 10.1016/j.jinf.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 18.Naeem A, Hosomi T, Nishimura Y, Alam MM, Oka T, Zaidi SS, Shimizu H. 2014. Genetic diversity of circulating Saffold viruses in Pakistan and Afghanistan. J Gen Virol 95:1945–1957. doi: 10.1099/vir.0.066498-0. [DOI] [PubMed] [Google Scholar]
- 19.Zhang XA, Lu QB, Wo Y, Zhao J, Huang DD, Guo CT, Xu HM, Liu EM, Liu W, Cao WC. 2015. Prevalence and genetic characteristics of Saffold cardiovirus in China from 2009 to 2012. Sci Rep 5:7704. doi: 10.1038/srep07704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. 2009. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol 83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Himeda T, Hosomi T, Asif N, Shimizu H, Okuwa T, Muraki Y, Ohara Y. 2011. The preparation of an infectious full-length cDNA clone of Saffold virus. Virol J 8:110. doi: 10.1186/1743-422X-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen AC, Bottiger B, Banner J, Hoffmann T, Nielsen LP. 2012. Serious invasive Saffold virus infections in children, 2009. Emerg Infect Dis 18:7–12. doi: 10.3201/eid1801.110725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nix WA, Khetsuriani N, Penaranda S, Maher K, Venczel L, Cselko Z, Freire MC, Cisterna D, Lema CL, Rosales P, Rodriguez JR, Rodriguez W, Halkyer P, Ronveaux O, Pallansch MA, Oberste MS. 2013. Diversity of picornaviruses in rural Bolivia. J Gen Virol 94:2017–2028. doi: 10.1099/vir.0.053827-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khamrin P, Thongprachum A, Kikuta H, Yamamoto A, Nishimura S, Sugita K, Baba T, Kobayashi M, Okitsu S, Hayakawa S, Shimizu H, Maneekarn N, Ushijima H. 2013. Three clusters of Saffold viruses circulating in children with diarrhea in Japan. Infect Genet Evol 13:339–343. doi: 10.1016/j.meegid.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Brañas P, Garcia M, Prieto C, Folgueira L. 2015. Saffold virus respiratory infection in children and immunocompromised patients in Spain. J Infect 70:679–680. doi: 10.1016/j.jinf.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Hertzler S, Liang Z, Treso B, Lipton HL. 2011. Adaptation of Saffold virus 2 for high-titer growth in mammalian cells. J Virol 85:7411–7418. doi: 10.1128/JVI.00265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan SZ, Chua KB, Xu Y, Prabakaran M. 2016. The pathogenesis of Saffold virus in AG129 mice and the effects of its truncated L protein in the central nervous system. Viruses 8:E24. doi: 10.3390/v8020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorgeloos F, Lardinois C, Jacobs S, van Kuppeveld FJ, Kaspers B, Michiels T. 2016. Neurotropism of Saffold virus in a mouse model. J Gen Virol 97:1350–1355. doi: 10.1099/jgv.0.000452. [DOI] [PubMed] [Google Scholar]
- 29.Kotani O, Naeem A, Suzuki T, Iwata-Yoshikawa N, Sato Y, Nakajima N, Hosomi T, Tsukagoshi H, Kozawa K, Hasegawa H, Taguchi F, Shimizu H, Nagata N. 2016. Neuropathogenicity of two Saffold virus type 3 isolates in mouse models. PLoS One 11:e0148184. doi: 10.1371/journal.pone.0148184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotani O, Iwata-Yoshikawa N, Suzuki T, Sato Y, Nakajima N, Koike S, Iwasaki T, Sata T, Yamashita T, Minagawa H, Taguchi F, Hasegawa H, Shimizu H, Nagata N. 2015. Establishment of a panel of in-house polyclonal antibodies for the diagnosis of enterovirus infections. Neuropathology 35:107–121. doi: 10.1111/neup.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li TC, Ami Y, Suzaki Y, Yasuda SP, Yoshimatsu K, Arikawa J, Takeda N, Takaji W. 2013. Characterization of full genome of rat hepatitis E virus strain from Vietnam. Emerg Infect Dis 19:115–118. doi: 10.3201/eid1901.121007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuroda M, Niwa S, Sekizuka T, Tsukagoshi H, Yokoyama M, Ryo A, Sato H, Kiyota N, Noda M, Kozawa K, Shirabe K, Kusaka T, Shimojo N, Hasegawa S, Sugai K, Obuchi M, Tashiro M, Oishi K, Ishii H, Kimura H. 2015. Molecular evolution of the VP1, VP2, and VP3 genes in human rhinovirus species C. Sci Rep 5:8185. doi: 10.1038/srep08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant RA, Filman DJ, Fujinami RS, Icenogle JP, Hogle JM. 1992. Three-dimensional structure of Theiler virus. Proc Natl Acad Sci U S A 89:2061–2065. doi: 10.1073/pnas.89.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ke MT, Nakai Y, Fujimoto S, Takayama R, Yoshida S, Kitajima TS, Sato M, Imai T. 2016. Super-resolution mapping of neuronal circuitry with an index-optimized clearing agent. Cell Rep 14:2718–2732. doi: 10.1016/j.celrep.2016.02.057. [DOI] [PubMed] [Google Scholar]
- 35.Nedelescu H, Abdelhack M. 2013. Comparative morphology of dendritic arbors in populations of Purkinje cells in mouse sulcus and apex. Neural Plast 2013:948587. doi: 10.1155/2013/948587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donald S, Humby T, Fyfe I, Segonds-Pichon A, Walker SA, Andrews SR, Coadwell WJ, Emson P, Wilkinson LS, Welch HC. 2008. P-Rex2 regulates Purkinje cell dendrite morphology and motor coordination. Proc Natl Acad Sci U S A 105:4483–4488. doi: 10.1073/pnas.0712324105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamodeh S, Eicke D, Napper RM, Harvey RJ, Sultan F. 2010. Population based quantification of dendrites: evidence for the lack of microtubule-associate protein 2a,b in Purkinje cell spiny dendrites. Neuroscience 170:1004–1014. doi: 10.1016/j.neuroscience.2010.08.021. [DOI] [PubMed] [Google Scholar]
- 38.Ichinohe T, Watanabe I, Ito S, Fujii H, Moriyama M, Tamura S, Takahashi H, Sawa H, Chiba J, Kurata T, Sata T, Hasegawa H. 2005. Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J Virol 79:2910–2919. doi: 10.1128/JVI.79.5.2910-2919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwata-Yoshikawa N, Uda A, Suzuki T, Tsunetsugu-Yokota Y, Sato Y, Morikawa S, Tashiro M, Sata T, Hasegawa H, Nagata N. 2014. Effects of Toll-like receptor stimulation on eosinophilic infiltration in lungs of BALB/c mice immunized with UV-inactivated severe acute respiratory syndrome-related coronavirus vaccine. J Virol 88:8597–8614. doi: 10.1128/JVI.00983-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katano H, Kano M, Nakamura T, Kanno T, Asanuma H, Sata T. 2011. A novel real-time PCR system for simultaneous detection of human viruses in clinical samples from patients with uncertain diagnoses. J Med Virol 83:322–330. doi: 10.1002/jmv.21962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarousse N, Fiette L, Grant RA, Hogle JM, McAllister A, Michiels T, Aubert C, Tangy F, Brahic M, Pena Rossi C. 1994. Chimeric Theiler's virus with altered tropism for the central nervous system. J Virol 68:2781–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarousse N, Grant RA, Hogle JM, Zhang L, Senkowski A, Roos RP, Michiels T, Brahic M, McAllister A. 1994. A single amino acid change determines persistence of a chimeric Theiler's virus. J Virol 68:3364–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michiels T, Jarousse N, Brahic M. 1995. Analysis of the leader and capsid coding regions of persistent and neurovirulent strains of Theiler's virus. Virology 214:550–558. doi: 10.1006/viro.1995.0066. [DOI] [PubMed] [Google Scholar]
- 44.Lipton HL. 1980. Persistent Theiler's murine encephalomyelitis virus infection in mice depends on plaque size. J Gen Virol 46:169–177. doi: 10.1099/0022-1317-46-1-169. [DOI] [PubMed] [Google Scholar]
- 45.Pevear DC, Luo M, Lipton HL. 1988. Three-dimensional model of the capsid proteins of two biologically different Theiler virus strains: clustering of amino acid difference identifies possible locations of immunogenic sites on the virion. Proc Natl Acad Sci U S A 85:4496–4500. doi: 10.1073/pnas.85.12.4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayashi K, Iwasaki Y, Yanagi K. 1986. Herpes simplex virus type 1-induced hydrocephalus in mice. J Virol 57:942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson RT, Johnson KP. 1969. Hydrocephalus as a sequela of experimental myxovirus infections. Exp Mol Pathol 10:68–80. doi: 10.1016/0014-4800(69)90049-5. [DOI] [PubMed] [Google Scholar]
- 48.Komine O, Nagaoka M, Watase K, Gutmann DH, Tanigaki K, Honjo T, Radtke F, Saito T, Chiba S, Tanaka K. 2007. The monolayer formation of Bergmann glial cells is regulated by Notch/RBP-J signaling. Dev Biol 311:238–250. doi: 10.1016/j.ydbio.2007.08.042. [DOI] [PubMed] [Google Scholar]
- 49.Yamada K, Fukaya M, Shibata T, Kurihara H, Tanaka K, Inoue Y, Watanabe M. 2000. Dynamic transformation of Bergmann glial fibers proceeds in correlation with dendritic outgrowth and synapse formation of cerebellar Purkinje cells. J Comp Neurol 418:106–120. [PubMed] [Google Scholar]
- 50.Theiler M. 1934. Spontaneous encephalomyelitis of mice—a new virus disease. Science 80:122. [DOI] [PubMed] [Google Scholar]
- 51.Dal Canto MC, Lipton HL. 1977. Multiple sclerosis. Animal model: Theiler's virus infection in mice. Am J Pathol 88:497–500. [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez M, Oleszak E, Leibowitz J. 1987. Theiler's murine encephalomyelitis: a model of demyelination and persistence of virus. Crit Rev Immunol 7:325–365. [PubMed] [Google Scholar]
- 53.Lipton HL, Kim BS, Yahikozawa H, Nadler CF. 2001. Serological evidence that Mus musculus is the natural host of Theiler's murine encephalomyelitis virus. Virus Res 76:79–86. doi: 10.1016/S0168-1702(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 54.Descoteaux JP, Mihok S. 1986. Serologic study on the prevalence of murine viruses in a population of wild meadow voles (Microtus pennsylvanicus). J Wildl Dis 22:314–319. [DOI] [PubMed] [Google Scholar]
- 55.Aubert C, Chamorro M, Brahic M. 1987. Identification of Theiler's virus infected cells in the central nervous system of the mouse during demyelinating disease. Microb Pathog 3:319–326. [DOI] [PubMed] [Google Scholar]
- 56.Dal Canto MC, Lipton HL. 1982. Ultrastructural immunohistochemical localization of virus in acute and chronic demyelinating Theiler's virus infection. Am J Pathol 106:20–29. [PMC free article] [PubMed] [Google Scholar]
- 57.Calenoff MA, Faaberg KS, Lipton HL. 1990. Genomic regions of neurovirulence and attenuation in Theiler murine encephalomyelitis virus. Proc Natl Acad Sci U S A 87:978–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu JL, Stein S, Rosenstein L, Bodwell T, Routbort M, Semler BL, Roos RP. 1990. Neurovirulence determinants of genetically engineered Theiler viruses. Proc Natl Acad Sci U S A 87:4125–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McAllister A, Tangy F, Aubert C, Brahic M. 1990. Genetic mapping of the ability of Theiler's virus to persist and demyelinate. J Virol 64:4252–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo M, Toth KS, Zhou L, Pritchard A, Lipton HL. 1996. The structure of a highly virulent Theiler's murine encephalomyelitis virus (GDVII) and implications for determinants of viral persistence. Virology 220:246–250. [DOI] [PubMed] [Google Scholar]
- 61.Zhou L, Luo Y, Wu Y, Tsao J, Luo M. 2000. Sialylation of the host receptor may modulate entry of demyelinating persistent Theiler's virus. J Virol 74:1477–1485. doi: 10.1128/JVI.74.3.1477-1485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stadnick E, Dan M, Sadeghi A, Chantler JK. 2004. Attenuating mutations in coxsackievirus B3 map to a conformational epitope that comprises the puff region of VP2 and the knob of VP3. J Virol 78:13987–14002. doi: 10.1128/JVI.78.24.13987-14002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. 2006. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci 9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 64.Takatsuru Y, Takayasu Y, Iino M, Nikkuni O, Ueda Y, Tanaka K, Ozawa S. 2006. Roles of glial glutamate transporters in shaping EPSCs at the climbing fiber-Purkinje cell synapses. Neurosci Res 54:140–148. doi: 10.1016/j.neures.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 65.Takayasu Y, Iino M, Shimamoto K, Tanaka K, Ozawa S. 2006. Glial glutamate transporters maintain one-to-one relationship at the climbing fiber-Purkinje cell synapse by preventing glutamate spillover. J Neurosci 26:6563–6572. doi: 10.1523/JNEUROSCI.5342-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stump G, Durrer A, Klein AL, Lutolf S, Suter U, Taylor V. 2002. Notch1 and its ligands Delta-like and Jagged are expressed and active in distinct cell populations in the postnatal mouse brain. Mech Dev 114:153–159. doi: 10.1016/S0925-4773(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 67.Eiraku M, Tohgo A, Ono K, Kaneko M, Fujishima K, Hirano T, Kengaku M. 2005. DNER acts as a neuron-specific Notch ligand during Bergmann glial development. Nat Neurosci 8:873–880. doi: 10.1038/nn1492. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka M, Maeda N, Noda M, Marunouchi T. 2003. A chondroitin sulfate proteoglycan PTPzeta/RPTPbeta regulates the morphogenesis of Purkinje cell dendrites in the developing cerebellum. J Neurosci 23:2804–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fukazawa N, Yokoyama S, Eiraku M, Kengaku M, Maeda N. 2008. Receptor type protein tyrosine phosphatase zeta-pleiotrophin signaling controls endocytic trafficking of DNER that regulates neuritogenesis. Mol Cell Biol 28:4494–4506. doi: 10.1128/MCB.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Url A, Truyen U, Rebel-Bauder B, Weissenbock H, Schmidt P. 2003. Evidence of parvovirus replication in cerebral neurons of cats. J Clin Microbiol 41:3801–3805. doi: 10.1128/JCM.41.8.3801-3805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonthius DJ, Perlman S. 2007. Congenital viral infections of the brain: lessons learned from lymphocytic choriomeningitis virus in the neonatal rat. PLoS Pathog 3:e149. doi: 10.1371/journal.ppat.0030149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Résibois A, Coppens A, Poncelet L. 2007. Naturally occurring parvovirus-associated feline hypogranular cerebellar hypoplasia—a comparison to experimentally induced lesions using immunohistology. Vet Pathol 44:831–841. doi: 10.1354/vp.44-6-831. [DOI] [PubMed] [Google Scholar]
- 73.Toplu N, Oguzoglu TC, Epikmen ET, Aydogan A. 2011. Neuropathologic study of border disease virus in naturally infected fetal and neonatal small ruminants and its association with apoptosis. Vet Pathol 48:576–583. doi: 10.1177/0300985810371309. [DOI] [PubMed] [Google Scholar]
- 74.Eisenman LM, Brothers R, Tran MH, Kean RB, Dickson GM, Dietzschold B, Hooper DC. 1999. Neonatal Borna disease virus infection in the rat causes a loss of Purkinje cells in the cerebellum. J Neurovirol 5:181–189. [DOI] [PubMed] [Google Scholar]
- 75.Pletnikov MV, Moran TH, Carbone KM. 2002. Borna disease virus infection of the neonatal rat: developmental brain injury model of autism spectrum disorders. Front Biosci 7:d593–d607. doi: 10.2741/pletnik. [DOI] [PubMed] [Google Scholar]
- 76.World Health Organization. 1997. Manual for the virological investigation of poliomyelitis. Publication WHO/EPI/GEN/97.01. World Health Organization, Geneva, Switzerland. [Google Scholar]