

ABSTRACT
The recent discovery of multiple giant double-stranded DNA (dsDNA) viruses blurred the consensual distinction between viruses and cells due to their size, as well as to their structural and genetic complexity. A dramatic feature revealed by these viruses as well as by many positive-strand RNA viruses is their ability to rapidly form elaborate intracellular organelles, termed “viral factories,” where viral progeny are continuously generated. Here we report the first isolation of viral factories at progressive postinfection time points. The isolated factories were subjected to mass spectrometry-based proteomics, bioinformatics, and imaging analyses. These analyses revealed that numerous viral proteins are present in the factories but not in mature virions, thus implying that multiple and diverse proteins are required to promote the efficiency of viral factories as “production lines” of viral progeny. Moreover, our results highlight the dynamic and highly complex nature of viral factories, provide new and general insights into viral infection, and substantiate the intriguing notion that viral factories may represent the living state of viruses.
IMPORTANCE Large dsDNA viruses such as vaccinia virus and the giant mimivirus, as well as many positive-strand RNA viruses, generate elaborate cytoplasmic organelles in which the multiple and diverse transactions required for viral replication and assembly occur. These organelles, which were termed “viral factories,” are attracting much interest due to the increasing realization that the rapid and continuous production of viral progeny is a direct outcome of the elaborate structure and composition of the factories, which act as efficient production lines. To get new insights into the nature and function of viral factories, we devised a method that allows, for the first time, the isolation of these organelles. Analyses of the isolated factories generated at different times postinfection by mass spectrometry-based proteomics provide new perceptions of their role and reveal the highly dynamic nature of these organelles.
INTRODUCTION
An exciting recent development in cellular biology is the realization that intracellular organelles previously considered to be randomly organized are in fact exquisitely ordered and that this order crucially affects their function. A prominent example is provided by replication cycles of positive-strand RNA [(+)RNA] viruses, which were shown to involve massive reorganization of the host cytoskeleton and membrane networks into well-defined cytoplasmic platforms, termed “viral factories” (VFs), within which genome replication and virion assembly are effectively coordinated (1–4). An additional example of highly ordered virus-generated intracellular organelles is the replication cycle of nucleocytoplasmic large DNA viruses (NCLDVs), which include Poxviridae, Phycodnaviridae, Iridoviridae, Asfarviridae, Mimiviridae, and Marseilleviridae (5–7). Specifically, these viruses were shown to partially or exclusively replicate within large and elaborate cytoplasmic VFs. These VFs mediate spatial and temporal coordination of viral assembly and promote effective recruitment of host factors, thus enabling rapid, continuous, and efficient generation of multiple viral progeny (4, 8–12).
Studies of the infection cycles of large DNA viruses such as orthopoxviruses led to the eukaryogenesis hypothesis, according to which eukaryotic nuclei have evolved from virus-generated intracellular organelles, specifically, the complex VFs (13–15). The exquisite spatial order that characterizes mimivirus VFs (10) further supports the hypothesis that these organelles can be considered “mini-nuclei” (11) that eventually might have evolved to eukaryotic nuclei (13, 15–17). This conjecture is intriguing in light of the realization that eukaryotic nuclei are highly ordered, as are VFs (18), and that this order plays a central role in their function (19, 20). It was further suggested that VFs should be considered the actual living stage of viruses, whereas mature virions might be viewed as mere seeds that mediate intercellular transfer of genetic material (21, 22).
The studies reported here were prompted by these findings and conjectures, as well as by the realization that VFs provide an exciting example of efficient intracellular self-assembly processes, in addition to the fact that the giant viruses that generate VFs are abundant and diverse (23, 24). Accordingly, we investigated the structure and dynamic composition of the factories produced by mimivirus in the cytoplasm of its host, Acanthamoeba polyphaga. With a diameter of 750 nm and harboring about 1,000 genes (25), the mimivirus, which belongs to the Mimiviridae family, represents one of the largest viruses known to date. Notably, the mimivirus infection cycle occurs entirely within the host cytoplasm (12, 26), in contrast to other double-stranded DNA (dsDNA) viruses, with the exception of vaccinia virus (8, 27) and pithovirus (23). Thus, host nuclei that generally provide optimal platforms for replication of dsDNA viruses (18) do not function as such with these viruses. In and of itself, this observation underscores the stringent requirements imposed upon cytoplasmic VFs that enable them to act as efficient sites for autonomous generation of viral progeny. The finding that only a relatively small fraction of the ∼1,000 proteins encoded by the mimivirus genome are incorporated into the mature virion (28, 29 [and see below]) further highlights the central role displayed by VFs by implying that many proteins are specifically required to produce and propagate these organelles.
MATERIALS AND METHODS
A. polyphaga growth and mimivirus infection.
A. polyphaga cells were grown in fresh PYG medium (https://www.dsmz.de/microorganisms/medium/pdf/DSMZ_Medium104.pdf) (including ampicillin [100 μg/ml] and kanamycin [50 μg/ml]) to near-confluence and infected with mimivirus at a multiplicity of infection (MOI) of ∼5 as previously described (30). The infection was synchronized by washing away virions with PYG 1 h after infection.
Viral factory isolation and purification.
At the relevant postinfection (p.i.) time points, A. polyphaga cells were shaken vigorously until detachment occurred. Cells were centrifuged at 1,000 × g for 10 min at 4°C. Pellets were resuspended in ice-cold 20 mM Tris-HCl (pH 7.4)–5 mM MgOAc–0.5 mM EDTA (pH 8.0)–5% sucrose and centrifuged at 1,000 × g at 4°C for 10 min. Pellets were resuspended in 80 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] (pH 6.8)–1 mM MgCl2–1 mM EGTA–0.5% Triton X-100–10% glycerol and centrifuged at 1,000 × g for 10 min at 4°C. The pellet was resuspended in 20 mM Tris-HCl (pH 7.4)–1% Triton X-100–10 mM CaCl2–150 mM NaCl and centrifuged at 1,000 × g for 10 min at 4°C. This step was repeated twice, and the final suspension was subjected to OptiPrep (Sigma) density gradient centrifugation (10% to 50%) and mixed with 20 mM Tris-HCl (pH 7.4)–5 mM MgOAc–0.5 mM EDTA (pH 8.0)–5% sucrose in an ultracentrifuge at 30,000 rpm (Sorvall; SW41 rotor) at 4°C for 2 h. The clearly distinct band containing the viral factories was extracted by pipetting and was dialyzed against 20 mM Tris-HCl (pH 8.0)–1 mM EDTA–0.5% NP-40–150 mM NaCl–10% glycerol–1 mM dithiothreitol (DTT). The content of the dialysis bag was centrifuged and the pellet resuspended in 50 mM Tris-HCl (pH 8.5)–5 mM DTT–0.5 M NaCl. The suspension was sonicated using VCX-750 with a tapered tip (Sonics, USA) at 40% amplitude, for 10 cycles (10 s on and 40 s off), on ice.
Mimivirus isolation and purification.
A. polyphaga cells were infected with mimivirus at an MOI of ∼5. Cells were grown in PYG medium (including ampicillin [100 μg/ml] and kanamycin [50 μg/ml] at 30°C until they were lysed. Lysed cells were centrifuged at 100 × g for 20 min at 4°C. The supernatant was centrifuged at 10,000 × g for 30 min at 4°C. The pellet was resuspended in 10 ml phosphate-buffered saline (PBS) and centrifuged at 10,000 × g for 30 min at 4°C. The resulting pellet was resuspended in 10 ml PBS (including ampicillin [100 μg/ml] and kanamycin [50 μg/ml]) and filtered through a 1.2-μm-pore-size filter. Viruses were further purified by shaking in PBS supplemented with 1% NP-40 for 2 h at room temperature. After 3 washes with PBS, 45 mM CaCl2 and 50 μg/ml proteinase K were added and the suspension was shaken for 4 h at room temperature and then overnight at 37°C. Viral particles were further purified by the use of an OptiPrep (Sigma) gradient and lysed by sonication under conditions identical to those used to lyse the viral factories.
Immunofluorescence.
Cells were seeded on glass coverslips in PYG medium, infected with mimivirus particles at an MOI of 5, and incubated for the indicated times before being fixed with 4% paraformaldehyde for 15 min. Cells were permeabilized in PBS supplemented with 0.1% Triton X-100 and incubated with anti-mimivirus antibodies raised against virion particles and then counterstained with DAPI (4′,6-diamidino-2-phenylindole). Fluorescence images were obtained with a DeltaVision system and deconvoluted with a conservative SoftWorx package (Applied Precision).
Scanning electron microscopy (SEM).
Viral factories isolated and purified as described above were fixed with 2% glutaraldehyde–cacodylate buffer for 1 h. Factories were then deposited on 1 mg/ml poly-l-lysine-coated silicon chips. Dehydration in increasing ethanol concentrations was followed by critical point drying (CPD; Bal-Tec, Lichtenstein). Samples were sputter coated with 2 nm Cr and visualized by SEM using a FEG Ultra55 scanning electron microscope (Zeiss).
Sample preparation for mass spectrometry (MS).
Samples from three biological replicates of isolated viral factories from three different time points (4, 5.5, and 7 h postinfection) as well as three biological replicates of purified mature virus particles were prepared as described above. Protein concentrations were determined using the bicinchoninic acid (BCA) assay. Proteins were initially reduced through incubation with 5 mM DTT for 30 min at 60°C and alkylated with 10 mM iodoacetamide (Sigma) in the dark for 30 min at 21°C. Proteins were then subjected to trypsin digestion (Promega, Fitchburg, WI) at a 1:50 trypsin/protein ratio for 16 h at 37°C. Digestion was stopped with trifluoroacetic acid (1%). Triton X-100 and NP-40 were removed using detergent removal columns (Pierce, Rockford, IL, USA), and samples were desalted using solid-phase extraction columns (Oasis HLB; Waters, Milford, MA, USA).
Liquid chromatography high-resolution mass spectrometry (LC/MS).
LC/MS-grade solvents were used for all chromatographic steps. Each sample was loaded using splitless nano-ultraperformance liquid chromatography (nanoUPLC) (NanoAcquity; Waters, Milford, MA, USA). The mobile phase consisted of solution A (H2O–0.1% formic acid) and solution B (acetonitrile–0.1% formic acid). Sample desalting was performed using a reversed-phase C18 trapping column (Waters) (180 μm internal diameter, 20 mm length, 5 μm particle size). Peptides were separated using a HSS T3 nanocolumn (Waters) (75 μm internal diameter, 250 mm length, 1.8 μm particle size) at 0.3 μl/min. Peptides were injected into the mass spectrometer using the following gradient: 4% to 35% solution B for 150 min, 35% to 90% solution B for 5 min, maintained at 95% B for 5 min, and back to the initial conditions. The nanoUPLC was coupled through a nano-electrospray ionization (nanoESI) emitter (New Objective; Woburn, MA, USA) (10 μm tip) to a quadrupole Orbitrap mass spectrometer (Q Exactive; Thermo Scientific) using a FlexIon nanospray.
Data were acquired in DDA mode, using a Top12 method (31). Raw data were imported into Expressionist software (version 9.2.4; Genedata, Switzerland) and processed as described previously (32). The software was used for retention time alignment and peak detection of precursor peptides. A master peak list was generated from all tandem MS (MS/MS) events and sent for database searching using Mascot v2.5.1 (Matrix Sciences). Data were searched against a database containing mimivirus and Acanthamoeba castellanii forward and reversed protein sequences (http://www.uniprot.org/), as well as 125 common laboratory contaminants, for a total of 31,293 searches. Fixed modification was set to carbamidomethylation of cysteines, and variable modifications were set to oxidation of methionines and deamidation of asparagines or glutamines. Search results were then filtered using the PeptideProphet (33) algorithm to achieve a maximum false-discovery rate of 1% at the protein level. Peptide identifications were imported back to Expressionist to annotate identified peaks. Quantification of proteins from the peptide data was performed using an in-house script (32). A Student's t test was used after logarithmic transformation to identify significant differences across the biological replicates. Fold changes were calculated based on the ratio of the geometric means of the different sample groups. Data were normalized on the basis of total ion current. Protein abundance was derived by summing up the three most intense peptides per protein, except for the instances in which the protein was detected with two peptides, in which case only the two were used. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via PRIDE (http://www.proteomexchange.org/) (see below).
Statistics.
Purified viral factories and mature mimivirus particles as well as noninfected A. polyphaga cells were subjected to mass spectrometry analyses in a random sequence and with at least three biological replicates for each p.i. time point. Only proteins identified by at least two unique peptides whose intensities were at least twice those seen with the uninfected cells were included in our analyses. Data in Table S1 in the supplemental material were determined by averaging the intensities determined for the MS results from three biological repeats. Standard deviation was calculated according to the following equation:
| (1) |
where μ is the mean and N is the number of samples. The standard error is the standard deviation divided by the root square of the number of independent biological repeats. Venn diagrams were constructed with Venny 2.0.2 software (http://bioinfogp.cnb.csic.es/tools/venny/index2.0.2).
Accession number(s).
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via PRIDE (http://www.proteomexchange.org/) (partner repository with data set identifier PXD004203).
RESULTS AND DISCUSSION
Previous studies revealed that, shortly after the release of the mimivirus genome into the host cytoplasm by the opening of a modified icosahedral capsid vertex, termed the “stargate” (34), initial replication centers are generated (26). Each replication center originates from one infecting virus, as was also demonstrated for the vaccinia virus (27). These centers, already detected at 4 h postinfection (p.i.), progressively increase in size and eventually coalesce into a single factory that, at 7 h p.i., contains viruses at various assembly and DNA-packaging stages, as well as apparent mature virions (Fig. 1A to C).
FIG 1.

Mimivirus factories within host cells and following isolation. (A to C) Amoeba cells infected with mimivirus at 3 successive postinfection (p.i.) time points (4 h [A], 5.5 h [B], and 7 h [C]) were stained with antibodies against mature virions (red) and counterstained with DAPI (blue). Cell and nucleus contours were derived from differential interference contrast (DIC) micrographs. (D to F) SEM images of isolated viral factories at 4 h (D), 5.5 h (E), and 7 h (F) p.i. Both SEM and fluorescence studies revealed the coalescence of initial viral replication centers into a single viral factory. Insets in panels E and F show stargate structures. (G) Low-magnification SEM micrograph depicting a large field of isolated factories at 7 h p.i. The micrograph reveals that the factories are essentially free from large host components. (H) Purified virions appear pure when surveyed by low-magnification TEM. Scale bars: panels A to C, 5 μm; panel D, 200 nm; panels E to F, 1 μm; panel G, 10 μm; panel H, 500 nm.
Isolation of mimivirus viral factories from different infection stages.
To unravel the composition and dynamic development of mimivirus replication centers and VFs, we devised a novel methodology of isolating and purifying the organelles generated at 4, 5.5, and 7 h p.i. Isolated VFs were subjected to scanning electron microscopy (SEM) and immunofluorescence microscopy, as well as mass spectrometry-based proteomics and bioinformatics analyses, in order to identify the factors responsible for the efficiency of these platforms in rapidly producing multiple complex viruses. SEM analyses revealed that the virus-generated organelles were essentially pure, containing no visible host-derived contaminants (Fig. 1D to G). Replication centers isolated at 4 h p.i. revealed multiple compact particles that did not yet exhibit obvious viral characteristics (Fig. 1D). Replication centers isolated at 5.5 h p.i. appeared as larger, partially fused particles in which stargate structures were already evident (Fig. 1E). Mimivirus factories isolated at 7 h p.i. were larger and studded with fibril-coated mature and fibril-free immature viral particles and exhibited conspicuous stargates (Fig. 1F). Significantly, the morphologies of the purified viral replication centers and factories were consistent with their in situ structures within infected amoeba cells (Fig. 1A to C), implying that the isolated virus-generated organelles retained their integrity.
Protein content in mimivirus factories is highly dynamic.
Purified viral replication centers and factories at each p.i. time point and mature mimivirus particles, as well as mock-infected A. polyphaga amoeba cells, were subjected to mass spectrometry (MS) analyses in a random sequence with at least three repeats. Only proteins that were identified by at least two unique peptides and whose intensities were at least twice those seen in mock-infected A. polyphaga cells were included in our analyses. Since the A. polyphaga genome has been sequenced only partially, the data were searched against Acanthamoeba castellanii strain Neff as well as mimivirus sequences, and we focused only on proteins encoded by mimivirus in the present study.
Our proteomic assays were supported by examination of two viral proteins with known localizations. Specifically, our MS analyses (see Table S1 in the supplemental material) revealed that the L410 core protein was included in the VFs. This observation was confirmed by Western blot analyses (Fig. 2A and B) as well as by immuno-transmission electron microscopy (immuno-TEM) (Fig. 2C). In contrast, eukaryotic translation initiation factor 4E-like protein L496 is present only in the cytoplasm of infected cells and not at any stage in the VFs (Fig. 3A). This finding is corroborated by in situ TEM studies that showed ribosomes surrounding the VFs but not incorporated into these organelles (Fig. 3B). These observations, along with the absence of conserved ribosomal proteins in the VFs (http://www.proteomexchange.org/ [identifier PXD004203]), imply that translation of viral proteins is carried out in the host cytoplasm and not in the VFs. Notably, this finding significantly reinforces the notion of a parallel between viral factories and eukaryotic nuclei.
FIG 2.

Isolated viral factories contain the core protein L410. (A) Western blots of total cell lysates at different postinfection time points were incubated with anti-core protein (L410) antibodies. L410 was detected only at later stages of infection, matching our mass spectroscopy findings. (B) Western blots of purified viral factories were incubated with anti-L410 antibodies. L410 was detected in viral factories, as well as in the virion, only at 7 h p.i., thus further substantiating our MS results. (C) Immunolabeling of cryopreserved cells at 8 h p.i. with rabbit anti-L410 antibodies, followed by exposure to gold-conjugated secondary antibody, revealed that the core protein is present in the viral factory as well as in the assembling virions. Scale bar: 1 μm.
FIG 3.
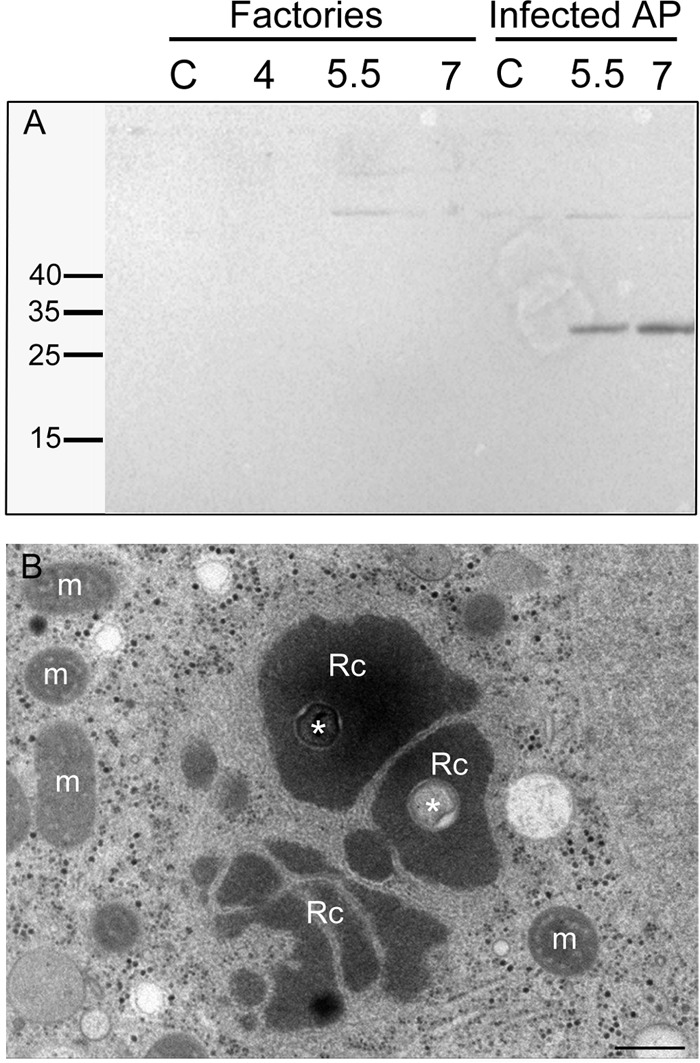
Eukaryotic translation initiation factor 4E-like protein L496 and ribosomes are not incorporated into the viral factories. (A) Western blots of total cell lysates at different postinfection time points were incubated with anti-4E-like protein (L496) antibodies. L496 was detected only in the total cell lysates and not in the factories, as indeed indicated by our MS findings. (B) TEM of intracellular replication centers at 4 h p.i. in the process of coalescing into a single viral factory. The micrograph reveals ribosomes (dark dots with diameters of ∼25 to ∼30 nm) that surround the replication centers but are not incorporated into these organelles. Rc and m, replication centers and mitochondria, respectively. Asterisks represent the cores of the original infecting virus (26). Scale bar: 500 nm.
Venn diagrams were used to visualize the dynamic nature of the viral factories and to provide insights into the mimivirus replication cycle (Fig. 4; see also Tables S1 and S2 in the supplemental material). A total of 303 mimivirus proteins were detected in viral factories. Specifically, 201, 255, and 287 proteins were present in viral factories purified at 4, 5.5, and 7 h p.i., respectively (Fig. 4A). Only one protein was exclusive to the 4-h-p.i. replication centers (L538; RNA helicase). Three unique proteins (R721 [chemotaxis protein CheD], R548 [thioredoxin-like protein], and R841 [an ankyrin repeat-containing protein]) were found in 5.5-h-p.i. viral factories, whereas 36 unique proteins were found in 7-h-p.i. viral factories. These included L540 and R563 (RNA helicases), L425 (capsid), R362 and R443 (thioredoxin domain-containing proteins), L288 (lectin), L484 (ankyrin repeat-containing protein), L293 (hydrolase), L593 (prolyl 4-hydroxylase), and 20 uncharacterized proteins. The presence of five annotated structural proteins (L410 [core], L71 and L668 [collagen proteins], R440 [capsid], and L725 [fibrils]) (35) at both 5.5 and 7 h p.i. is consistent with previous observations according to which extensive assembly processes of viral components such as membranes and capsid generation take place at these p.i. stages (1, 18). It was previously demonstrated by diverse imaging techniques that membranes are present in mature mimivirus virions (36) as well as in VFs (10, 37). Here, 30 membrane proteins were detected in the purified viral factories at the three p.i. time points (see Table S1). This observation suggests that the membrane network remains associated with the VFs following their isolation and purification.
FIG 4.
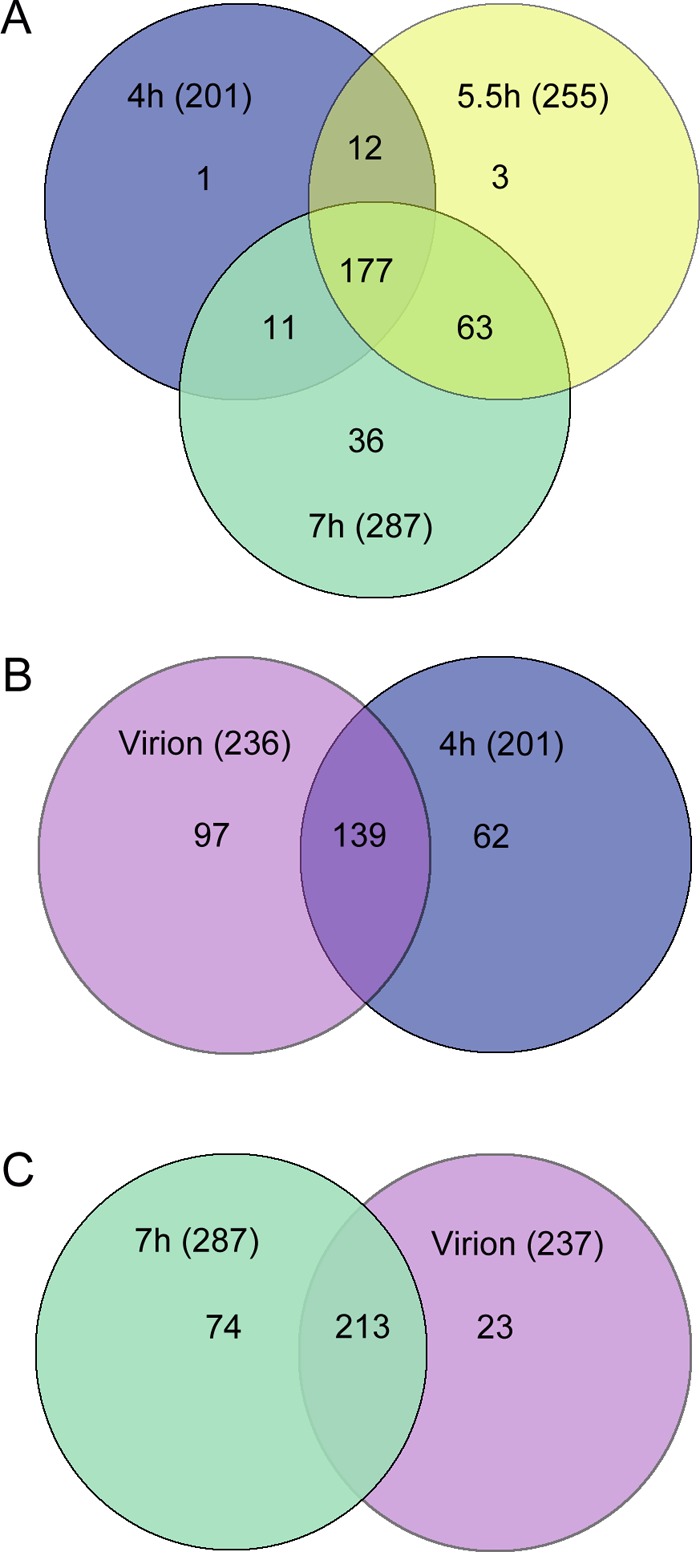
Venn diagrams depicting protein content in mimivirus factories and in mature virions. (A) Proteins detected in isolated viral factories at 4, 5.5, and 7 h p.i. (blue, yellow, and green circles, respectively). The diagram demonstrates that the compositions of viral factories along the infection cycle differ substantially, thus underscoring the dynamic nature of these virus-generated organelles. (B) Relations between proteins detected in mature virions and proteins present in viral factories at 4 h (magenta and blue circles, respectively). (C) Relations between proteins detected in viral factories at 7 h and proteins found in mature virions (green and magenta circles, respectively). This diagram reveals that many proteins were present in the factories but not included in mature virions, implying that these proteins act as part of a production line, on par with the notion that viral factories represent intracellular organelles where vigorous viral assembly occurs.
We found that 177 proteins were common to viral factories at all three time points (Fig. 4A). This result implies that multiple infection processes occur continuously during the infection cycle. Proteins detected throughout the infection cycle include enzymes involved in DNA replication and transcription, DNA repair, and protein degradation (Fig. 5), as well as in redox processes and ankyrin repeat-containing proteins. The presence of multiple viral DNA replication and transcription enzymes in the cytoplasmic factories is consistent with continuous generation of new viral genomes and proteins and corroborates the finding that mimivirus infection occurs exclusively in the host cytoplasm (5, 21, 26, 34). This notion is further supported by the observation that as many as 14 different putative helicases (of 15 encoded helicases) are found in the viral factories. Notably, whereas some helicases were present in factories generated at all of the time points probed, others were detected only in earlier or later infection stages (Fig. 5A; see also Tables S1 and S2 in the supplemental material), implying that the large number of helicases does not indicate redundancy but instead indicates specific roles required at different infection stages.
FIG 5.

Dynamic composition of the mimivirus cytoplasmic factories. (A1 to A3) Distribution of three helicases in viral factories and mature viral particles. (B1 to B3) Distribution of DNA repair proteins in viral factories and in mature viral particles. (C1 to C3) Distribution of DNA-replication-related proteins. (D1 to D3) Distribution of protein-degradation-related proteins. Intensities represent relative protein amounts. The figure highlights the fact that the relative amounts of proteins belonging to similar functional groups in viral factories substantially differ at progressive postinfection time points, as well as in mature virions.
Although the mimivirus virion proteome has already been reported (28, 29), we were interested to reexamine the composition of mature virions by applying mass spectrometry procedures identical to those used for examination of the factories. A total of 236 mimivirus proteins were detected in mature viral particles (see Tables S1 and S2 in the supplemental material) in contrast to the 114 reported in reference 29, a discrepancy likely due to the higher sensitivity of the instrumentation used in this study. Comparison of the protein content of mature virions to the protein content of 4-h VFs (Fig. 4B) revealed that 139 proteins are shared, including six helicases and seven transcription factors (see Tables S1 and S2), supporting the notion that massive replication and transcription processes occur already at this stage (26). In comparisons of the protein content of 7-h viral factories to that of mature virions, 213 proteins were found to be common (Fig. 4C). There were 13 proteins in the virion that were not detected in the factories at all stages investigated here. Three were oxidation-related proteins (R135, R419, and L894), one was an ankyrin repeat-containing protein (L371), and eight were uncharacterized (see Table S2). The comparison also demonstrated that the number of proteins present in the factory is larger than the number detected in mature virions. This finding supports the conjecture that VFs should be considered “production lines” of viruses (10, 18) in which numerous proteins are required for virus production but not for the onset of the infection process.
Notably, 23 proteins, including R135, a putative glucose-methanol-choline (GMC)-type oxidoreductase shown to be a component of mimivirus fibrils (35), were detected in virion particles but not in our MS studies of the relatively late 7-h VFs (Fig. 4C; see also Tables S1 and S2 in the supplemental material). In contrast, antibodies raised against fibril-containing virions did interact with 7-h factories (Fig. 1C), implying the presence of R135 in these factories. A possible interpretation is that R135, which was shown to be glycosylated (35), may undergo glycosylation on host membranes outside the VFs and may only then be supplemented to the assembling virions. Another interpretation could be that some proteins are present in the VFs in small amounts and are therefore undetectable.
In addition to depicting the highly dynamic distribution of DNA processing enzymes such as representative helicases, DNA repair proteins, and DNA-replication-related proteins in factories from various p.i. times (Fig. 5A, B, and C, respectively), the results shown in Fig. 5D imply that significant protein regulation occurs at the protein level. Specifically, 11 proteins predicted to be involved in protein degradation were detected in the viral factories as well as in mature virions, indicating that protein degradation processes are taking place throughout mimivirus infection cycle. The levels of these proteins from 4-h to 7-h factories differ, however. Thus, nine degradation-related proteins were found in 4-h factories, eight were detected in 5.5-h factories, and all 11 enzymes were present at 7 h. Six degradation-related proteins were present in mature virions, hinting that degradation of host and viral proteins is already required at the onset of the infection, as well as throughout the process. The possibility that protein degradation also occurs in mature virions is intriguing and will be examined.
Genes encoding ankyrin repeat-containing proteins were previously shown to be the largest group in the mimivirus genome, with 66 members (38). The ankyrin repeat is 33 amino acids long, appearing in proteins involved in multiple cellular tasks such as protein-protein interactions, cytoskeleton generation, and cell signaling. We manually reannotated the list of ankyrin repeat-containing proteins in the genome of mimivirus (NC_014649.1) and found 98 proteins, of which only 18 were found in the mimivirus factories; 13 were present from 4 h to 7 h in factories but only 1 in mature virions (see Tables S1 and S2 in the supplemental material). This observation implies functions carried out mainly outside the factories. The latter conjecture is supported by the previous findings indicating that ankyrin repeat-containing proteins in vaccinia virus (39) and in Paramecium bursaria chlorella virus 1 (PBCV-1) virus (40) are involved in ubiquitination of host proteins in the host cytoplasm. This finding substantiates the notion suggested above that protein regulation through protein degradation is a crucial process throughout viral infection both inside and outside the viral factories.
The majority of proteins in the viral factories are encoded by the central part of the mimivirus genome.
We have mapped the distribution of proteins detected in mimivirus factories at three successive time points, as well as in mature virions, on the mimivirus genome (Fig. 6). Notably, we found that the majority of these proteins were encoded by the central part of the genome, whereas the extremities of the mimivirus genome genes were detected to a markedly lesser degree. This observation corroborates previous studies where mimivirus virions propagated under axenic conditions revealed that a substantial reduction of their genome specifically occurred at the edges of the genome (35). It was proposed that these regions encode proteins involved in control of competitor and hence are redundant under axenic conditions (35). In addition, it has been demonstrated that the central region of poxvirus genomes is highly conserved whereas genes at the terminal regions are more divergent (41, 42). The termini of the genome of African swine fever virus (ASFV), which also belongs to the NCLDV family, are similarly variable in different isolates (43).
FIG 6.

Genomic organization of proteins detected in viral factories and mature virions. Our proteomic analyses indicate that the majority of proteins found in the viral factories of mimivirus are encoded by genes located in the center of the mimivirus chromosome. This observation is consistent with previous findings according to which mimivirus virions propagated under axenic conditions undergo substantial reduction of their genome that specifically occurs at the extremities of the genome and yet is not detrimental to mimivirus infectivity (35). CDSs (coding DNA sequences) are represented as blue arrowheads. Proteins detected in VFs at 4, 5.5, and 7 h p.i. are labeled in blue, purple, and maroon rectangles, respectively. Proteins found in mature mimivirus particles are labeled in green. The linear mimivirus genome is depicted as a circle starting from the site indicated by an arrow. The figure was constructed with the CGView Comparison Tool (CCT) (46).
Proteome studies of large viruses, including Cafeteria roenbergensis virus (CroV) (44) and PBCV-1 virions (45), were conducted. The large number of proteins in PBCV-1 raised the issue of why giant viruses contain so many genes (45). We propose that a partial answer is provided by the current report, which highlights the elaborate and highly dynamic protein composition of VFs. In addition, the issue of duplicated genes and genomic regions in the mimivirus was raised (38). Our results imply that proteins that seem to have similar functions actually have different expression levels at various stages of infection, implying specificity rather than redundancy. Still, only about 300 proteins are found in the factories, and 200 of those proteins are uncharacterized; the roles of these proteins as well as of the remaining ∼700 mimivirus proteins need to be elucidated. Finally, we claim that the procedure reported here for the isolation of mimivirus factories and their proteomic analyses, along with the fact that this methodology can be extended to other large dsDNA and RNA viruses, may provide new insights into the pathway of viral infection as well as into intracellular self-assembly processes in general.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eyal Shimoni and Tamar Unger for helpful discussions.
Y.F.-S., E.M., and A.M. designed the research; Y.F.-S., E.M., Y.M., S.B.-D., Y.L., A.S., and E.K. and conducted the experiments; Y.F.-S., E.M., Y.M., S.B.-D., Y.L., A.S., and A.M. analyzed the data; Y.F.-S. and A.M. wrote the paper.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01319-16.
REFERENCES
- 1.de Castro IF, Volonte L, Risco C. 2013. Virus factories: biogenesis and structural design. Cell Microbiol 15:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol 64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 3.Netherton C, Wileman T. 2011. Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr Opin Virol 1:381–387. doi: 10.1016/j.coviro.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abergel C, Legendre M, Claverie JM. 2015. The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiol Rev 39:779–796. doi: 10.1093/femsre/fuv037. [DOI] [PubMed] [Google Scholar]
- 6.Iyer LA, Balaji S, Koonin EV, Aravind L. 2006. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res 117:156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 7.Iyer LM, Aravind L, Koonin EV. 2001. Common origin of four diverse families of large eukaryotic DNA viruses. J Virol 75:11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milrot E, Mutsafi Y, Fridmann-Sirkis Y, Shimoni E, Rechav K, Gurnon J, Van Etten JL, Minsky A. 2016. Virus-host interactions: insights from the replication cycle of the large Paramecium bursaria chlorella virus. Cell Microbiol 18:3–16. doi: 10.1111/cmi.12486. [DOI] [PubMed] [Google Scholar]
- 10.Mutsafi Y, Shimoni E, Shimon A, Minsky A. 2013. Membrane assembly during the infection cycle of the giant mimivirus. PLoS Pathog 9:e1003367. doi: 10.1371/journal.ppat.1003367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tolonen N, Doglio L, Schleich S, Krijnse Locker J. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell 12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Etten JL, Lane LC, Dunigan DD. 2010. DNA viruses: The really big ones (giruses). Annu Rev Microbiol 64:83–99. doi: 10.1146/annurev.micro.112408.134338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell PJL. 2001. Viral eukaryogenesis: was the ancestor of the nucleus a complex DNA virus? J Mol Evol 53:251–256. doi: 10.1007/s002390010215. [DOI] [PubMed] [Google Scholar]
- 14.Bell PJL. 2006. Sex and the eukaryotic cell cycle is consistent with a viral ancestry for the eukaryotic nucleus. J Theor Biol 243:54–63. doi: 10.1016/j.jtbi.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 15.Takemura M. 2001. Poxviruses and the origin of the eukaryotic nucleus. J Mol Evol 52:419–425. [DOI] [PubMed] [Google Scholar]
- 16.Forterre P. 2006. The origin of viruses and their possible roles in major evolutionary transitions. Virus Res 117:5–16. doi: 10.1016/j.virusres.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Forterre P, Prangishvili D. 2013. The major role of viruses in cellular evolution: facts and hypotheses. Curr Opin Virol 3:558–565. doi: 10.1016/j.coviro.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Mutsafi Y, Fridmann-Sirkis Y, Milrot E, Hevroni L, Minsky A. 2014. Infection cycles of large DNA viruses: emerging themes and underlying questions. Virology 466:3–14. [DOI] [PubMed] [Google Scholar]
- 19.Meaburn KJ, Misteli T. 2007. Chromosome territories. Nature 445:379–381. doi: 10.1038/445379a. [DOI] [PubMed] [Google Scholar]
- 20.Misteli T. 2004. Spatial positioning: a new dimension in genome function. Cell 119:153–156. doi: 10.1016/j.cell.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 21.Claverie JM, Abergel C. 2009. Mimivirus and its virophage. Annu Rev Genet 43:49–66. doi: 10.1146/annurev-genet-102108-134255. [DOI] [PubMed] [Google Scholar]
- 22.Claverie JM, Abergel C. 2010. Mimivirus: the emerging paradox of quasi-autonomous viruses. Trends Genet 26:431–437. doi: 10.1016/j.tig.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Legendre M, Bartoli J, Shmakova L, Jeudy S, Labadie K, Adrait A, Lescot M, Poirot O, Bertaux L, Bruley C, Coute Y, Rivkina E, Abergel C, Claverie JM. 2014. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc Natl Acad Sci U S A 111:4274–4279. doi: 10.1073/pnas.1320670111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Philippe N, Legendre M, Doutre G, Coute Y, Poirot O, Lescot M, Arslan D, Seltzer V, Bertaux L, Bruley C, Garin J, Claverie JM, Abergel C. 2013. Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341:281–286. doi: 10.1126/science.1239181. [DOI] [PubMed] [Google Scholar]
- 25.Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, La Scola B, Suzan M, Claverie JM. 2004. The 1.2-megabase genome sequence of mimivirus. Science 306:1344–1350. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- 26.Mutsafi Y, Zauberman N, Sabanay I, Minsky A. 2010. Vaccinia-like cytoplasmic replication of the giant mimivirus. Proc Natl Acad Sci U S A 107:5978–5982. doi: 10.1073/pnas.0912737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallardo M, Leithe E, Schleich S, Roos N, Doglio L, Krijnse Locker J. 2002. Relationship between vaccinia virus intracellular cores, early mRNAs, and DNA replication sites. J Virol 76:5167–5183. doi: 10.1128/JVI.76.10.5167-5183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claverie JM, Abergel C, Ogata H. 2009. Mimivirus, p 89–121. In Van Etten JL. (ed), Lesser known large dsDNA viruses, vol 328 Springer-Verlag, Berlin, Germany. [Google Scholar]
- 29.Renesto P, Abergel C, Decloquement P, Moinier D, Azza S, Ogata H, Fourquet P, Gorvel JP, Claverie JM. 2006. Mimivirus giant particles incorporate a large fraction of anonymous and unique gene products. J Virol 80:11678–11685. doi: 10.1128/JVI.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.La Scola B, Audic S, Robert C, Jungang L, de Lamballerie X, Drancourt M, Birtles R, Claverie JM, Raoult D. 2003. A giant virus in amoebae. Science 299:2033. doi: 10.1126/science.1081867. [DOI] [PubMed] [Google Scholar]
- 31.Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. 2012. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res 11:3487–3497. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- 32.Shalit T, Elinger D, Savidor A, Gabashvili A, Levin Y. 2015. MS1-Based label-free proteomics using a quadrupole orbitrap mass spectrometer. J Proteome Res 14:1979–1986. doi: 10.1021/pr501045t. [DOI] [PubMed] [Google Scholar]
- 33.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 34.Zauberman N, Mutsafi Y, Ben Halevy D, Shimoni E, Klein E, Xiao C, Sun S, Minsky A. 2008. Distinct DNA exit and packaging portals in the virus Acanthamoeba polyphaga mimivirus. PLoS Biol 6:e114. doi: 10.1371/journal.pbio.0060114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyer M, Azza S, Barrassi L, Klose T, Campocasso A, Pagnier I, Fournous G, Borg A, Robert C, Zhang XZ, Desnues C, Henrissat B, Rossmann MG, La Scola B, Raoult D. 2011. Mimivirus shows dramatic genome reduction after intraamoebal culture. Proc Natl Acad Sci U S A 108:10296–10301. doi: 10.1073/pnas.1101118108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao CA, Chipman PR, Battisti AJ, Bowman VD, Renesto P, Raoult D, Rossmann MG. 2005. Cryo-electron microscopy of the giant mimivirus. J Mol Biol 353:493–496. doi: 10.1016/j.jmb.2005.08.060. [DOI] [PubMed] [Google Scholar]
- 37.Suárez C, Welsch S, Chlanda P, Hagen W, Hoppe S, Kolovou A, Pagnier I, Raoult D, Krijnse Locker J. 2013. Open membranes are the precursors for assembly of large DNA viruses. Cell Microbiol 15:1883–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suhre K. 2005. Gene and genome duplication in Acanthamoeba polyphaga mimivirus. J Virol 79:14095–14101. doi: 10.1128/JVI.79.22.14095-14101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonnberg S, Seet BT, Pawson T, Fleming SB, Mercer AA. 2008. Poxvirus ankyrin repeat proteins are a unique class of F-box proteins that associate with cellular SCF1 ubiquitin ligase complexes. Proc Natl Acad Sci U S A 105:10955–10960. doi: 10.1073/pnas.0802042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noel EA, Kang M, Adamec J, Van Etten JL, Oyler GA. 2014. Chlorovirus Skp1-binding ankyrin repeat protein interplay and mimicry of cellular ubiquitin ligase machinery. J Virol 88:13798–13810. doi: 10.1128/JVI.02109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gubser C, Hué S, Kellam P, Smith GL. 2004. Poxvirus genome: a phylogenetic analysis. J Gen Virol 85:105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- 42.Moss B. 2006. Poxviridae: the viruses and their replication, p 2905–2945. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 43.Dixon LK, Chapman DAG, Netherton CL, Upton C. 2013. African swine fever virus replication and genomics. Virus Res 173:3–14. doi: 10.1016/j.virusres.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 44.Fischer MG, Kelly I, Foster LJ, Suttle CA. 2014. The virion of Cafeteria roenbergensis virus (CroV) contains a complex suite of proteins for transcription and DNA repair. Virology 466:82–94. [DOI] [PubMed] [Google Scholar]
- 45.Dunigan DD, Cerny RL, Bauman AT, Roach JC, Lane LC, Agarkova IV, Wulser K, Yanai-Balser GM, Gurnon JR, Vitek JC, Kronschnabel BJ, Jeanniard A, Blanc G, Upton C, Duncan GA, McClung OW, Ma F, Van Etten JL. 2012. Paramecium bursaria chlorella virus 1 proteome reveals novel architectural and regulatory features of a giant virus. J Virol 86:8821–8834. doi: 10.1128/JVI.00907-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grant JR, Arantes AS, Stothard P. 2012. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 13:202. doi: 10.1186/1471-2164-13-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.