

ABSTRACT
Respiratory syncytial virus (RSV) is the leading cause of lower respiratory tract infections in infant and elderly populations worldwide. Currently, there is no efficacious vaccine or therapy available for RSV infection. The molecular mechanisms underlying RSV-induced acute airway disease and associated long-term consequences remain largely unknown; however, experimental evidence suggests that the lung inflammatory response plays a fundamental role in the outcome of RSV infection. High-mobility group box 1 (HMGB1) is a nuclear protein that triggers inflammation when released from activated immune or necrotic cells and drives the pathogenesis of various infectious agents. Although HMGB1 has been implicated in many inflammatory diseases, its role in RSV-induced airway inflammation has not been investigated. This study investigates the molecular mechanism of action of extracellularly released HMGB1 in airway epithelial cells (A549 and small airway epithelial cells) to establish its role in RSV infection. Immunofluorescence microscopy and Western blotting results showed that RSV infection of human airway epithelial cells induced a significant release of HMGB1 as a result of translocation of HMGB1 from the cell nuclei to the cytoplasm and subsequent release into the extracellular space. Treating RSV-infected A549 cells with antioxidants significantly inhibited RSV-induced HMGB1 extracellular release. Studies using recombinant HMGB1 triggered immune responses by activating primary human monocytes. Finally, HMGB1 released by airway epithelial cells due to RSV infection appears to function as a paracrine factor priming epithelial cells and monocytes to inflammatory stimuli in the airways.
IMPORTANCE RSV is a major cause of serious lower respiratory tract infections in young children and causes severe respiratory morbidity and mortality in the elderly. In addition, to date there is no effective treatment or vaccine available for RSV infection. The mechanisms responsible for RSV-induced acute airway disease and associated long-term consequences remain largely unknown. The oxidative stress response in the airways plays a major role in the pathogenesis of RSV. HMGB1 is a ubiquitous redox-sensitive multifunctional protein that serves as both a DNA regulatory protein and an extracellular cytokine signaling molecule that promotes airway inflammation as a damage-associated molecular pattern. This study investigated the mechanism of action of HMGB1 in RSV infection with the aim of identifying new inflammatory pathways at the molecular level that may be amenable to therapeutic interventions.
INTRODUCTION
Respiratory syncytial virus (RSV) is a ubiquitous, negative-sense, enveloped, single-stranded RNA virus that frequently causes upper and lower respiratory tract infections in infants, young children, the elderly, and immunocompromised individuals. Epidemiological evidence indicates that severe pulmonary disease caused by RSV infection in infancy is associated with recurrent wheezing and the development of asthma later in childhood. No safe and efficacious therapies for RSV infection exist and natural immunity is incomplete, resulting in repeated attacks of acute respiratory tract infections throughout life (1, 2). The molecular mechanisms underlying RSV-induced acute airway disease and associated long-term consequences remain largely unknown; however, experimental evidence suggests that the lung inflammatory response plays a fundamental role in the outcome of RSV infection. Major targets of RSV infection are airway epithelial cells, which respond to infection by producing a variety of proinflammatory mediators, such as cytokines and chemokines involved in lung immune/inflammatory responses. The mechanisms by which pattern recognition epithelial cells trigger inflammatory responses have been extensively investigated (3–5). More recently, oxidative stress was shown to play an important role in the pathogenesis of many lung inflammatory diseases, such as asthma and chronic obstructive pulmonary disease (COPD) (6, 7). RSV infection induces reactive oxygen species (ROS) production in vitro and oxidative lung injury in vivo (8, 9), suggesting that oxidative stress plays a role in its pathogenesis; however, the mechanism of RSV-induced cellular oxidative stress has not been extensively investigated.
Extensive research has shed light on the role of high-mobility group box 1 protein (HMGB1) in the pathogenesis of many infectious and noninfectious inflammatory diseases. While studies on HMGB1 have extensively focused on its involvement in many pathological states, there has been no report of its involvement in RSV-induced human lung pathogenesis, with the exception of one article showing that the HMGB1 protein levels were induced in mouse lung homogenates (10). HMGB1 is a ubiquitous redox-sensitive, highly conserved nuclear protein that functions as a structural protein of chromatin and also as a transcription factor (reviewed in references 11 and 12). HMGB1 belongs to the Alarmins family, members of which alert the immune system to tissue damage and trigger immediate response (13). Recently, extracellular HMGB1 has been identified as a key signaling molecule involved in many pathological conditions, such as cancer (14), cardiovascular disease, ischemia/reperfusion (I/R) injury (15), and lung inflammatory diseases (16, 17, 17–20). HMGB1 can be released passively by necrotic or damaged cells (21) or can be actively secreted by various cell types, including monocytes, macrophages, natural killer cells, dendritic cells, and hepatocytes, in response to exogenous and endogenous stimuli, such as cytokines, lipopolysaccharide (LPS), hypoxia, and infection (13, 22–26). Upon release, HMGB1 mediates innate and adaptive immune responses to infection and injury through the receptor for advanced glycation end products (RAGE) and some Toll-like receptors (TLRs) (27–30). HMGB1 signaling through RAGE leads to activation of the NF-κB pathway, as well as signal transduction through extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein (MAP) kinase, while HMGB1 interactions with TLR2 and TLR4 mediate immune activation, thereby leading to cytokine production and cell survival (31). The functionality of actively secreted HMGB1 was shown to be modulated by posttranslational modifications, such as acetylation and phosphorylation (32, 33).
This study focused on the molecular mechanism of the active release of HMGB1 as a result of RSV infection of A549 cells, a human alveolar type II-like epithelial cell line, and small alveolar epithelial (SAE) cells which are primary human airway epithelial cells derived from terminal bronchioles of a normal individual. We have used an in vitro model of RSV infection of A549 and SAE cells to demonstrate secretion of HMGB1 from these cells and also to study its role in RSV pathogenesis. We found that RSV infection led to the translocation of HMGB1 from the nucleus to the cytoplasm and its subsequent significant release into the extracellular medium after stimulation. In addition, we observed a dose-dependent inhibitory effect from antioxidant treatment on the extracellular HMGB1 release in RSV-infected A549 cells. We also investigated the possible role of HMGB1 in RSV infection using recombinant human HMGB1 (rHMGB1) protein and found that HMGB1 activated primary human monocytes and induced an inflammatory response. Thus, our results provide evidence for the molecular basis of HMGB1 release from airway epithelial cells and suggest that HMGB1 plays an important role in triggering inflammatory responses via activating neighboring immune cells in the airways.
MATERIALS AND METHODS
Materials.
F12K medium, EDTA, and Hanks' balanced salt solution (HBSS) without Mg2+ or Ca2+ were purchased from Gibco-BRL (Grand Island, NY). Novex 10%, 12%, and 4 to 12% mini gels were obtained from Invitrogen (Carlsbad, CA). Eukarion salen-manganese complex (EUK-8) and the ProteoExtract subcellular proteome extraction kit were purchased from Calbiochem (San Diego, CA). HEPES and RPMI 1640 were from Cellgro (Manassas, VA). Small airway epithelial cell (SAEC) growth medium was from Lonza (Houston, TX). tert-Butylhydroquinone (tBHQ), Histopaque-1077, and dextran were obtained from Sigma-Aldrich (St. Louis, MO). The 10× Tris-glycine buffer, 10× Tris-glycine-SDS electrophoresis buffer, RC DC protein assay kit, human 27-Plex Bio-Plex kit, and Bio-Rad protein assay reagent were from Bio-Rad (Hercules, CA). Amersham full-range rainbow molecular weight markers were from GE Healthcare (Piscataway, NJ). Polyvinylidene difluoride membranes and Immobilon Western horseradish peroxidase (HRP) substrate were obtained from Millipore (Billerica, MA). Recombinant human HMGB1 (rHMGB1) was purchased from R&D Systems (Minneapolis, MN). Rabbit polyclonal anti-human HMGB1 antibody was from Abcam (Cambridge, MA), and the fluorescein isothiocyanate (FITC)-conjugated secondary antibody was from Southern Biotech (Birmingham, AL).
RSV preparation.
The RSV Long strain was grown in HEp-2 cells and purified by centrifugation on discontinuous sucrose gradients as described previously (34, 35). The virus titer of the purified RSV pools was 8 to 9 log10 PFU/ml using a methylcellulose plaque assay (36). No contaminating cytokines were found in these sucrose-purified viral preparations. Lipopolysaccharide (LPS) was not detected using the Limulus hemocyanin agglutination assay. Virus pools were aliquoted, quick-frozen on dry ice-alcohol, and stored at −80°C until used.
Cell culture and RSV infection of airway epithelial cells.
We used A549 cells, a human alveolar type II-like epithelial cell line (American Type Culture Collection, Manassas, VA), and small alveolar epithelial (SAE) cells, which are primary human airway epithelial cells derived from terminal bronchioli of a normal individual (Clonetics, San Diego, CA), were grown according to the manufacturer's instructions. A549 cells were maintained in F12K medium containing 10% fetal bovine serum (FBS), 10 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. SAE cells were maintained in SAEC growth medium containing 7.5 mg/ml bovine pituitary extract (BPE), 0.5 mg/ml hydrocortisone, 0.5 μg/ml human epidermal growth factor (hEGF), 0.5 mg/ml epinephrine, 10 mg/ml transferrin, 5 mg/ml insulin, 0.1 μg/ml retinoic acid, 0.5 μg/ml triiodothyronine, 50 mg/ml gentamicin, and 50 mg/ml bovine serum albumin (BSA). Monolayers of undifferentiated SAE cells were cultured in 25-cm2 flasks at 37°C and 5% CO2 with an SAEC basal medium supplied with growth factors. Cells were used in the experiments at passage three (34). When SAE cells were used for RSV infection, the cells were changed to basal medium and not supplemented with growth factors 6 h prior to and throughout the length of the experiment. At 80 to 90% confluence, cell monolayers were infected with RSV at a multiplicity of infection (MOI) of 1 (unless otherwise stated), as previously described (37). An equivalent amount of a 30% sucrose solution was added to uninfected A549 and SAE cells as a control. In some experiments, cells were pretreated with antioxidants such as EUK-8 and tBHQ or rHMGB1 for 1 h and then infected with RSV in the presence of the selected compound. Since EUK-8 and tBHQ were diluted in ethanol, an equal amount of ethanol was added to untreated cells as a control. The total number of cells, cell viability, and viral replication were measured by trypan blue exclusion and plaque assay, respectively. There was no significant change in either cell viability or viral replication when cells were incubated in the presence of the antioxidants or rHMGB1. To inactivate RSV, aliquots of the virus were placed in the center wells of a 24-well plate, and a transilluminator (UVP model UVL-56; Entela, Upland, CA) was kept on top of the uncovered plate. Exposure to UV radiation was done for 15 min (365 nm; dose of 1.5 × 10−2 μW/mm2) by placing the plate containing the virus on ice to eliminate the viral infectivity without altering the conformation of viral proteins and mediators.
Isolation of human monocytes from peripheral blood mononuclear cells and viral infection.
Monocytes were isolated from human peripheral blood mononuclear cells using whole blood from healthy, nonsmoking individuals (18 to 50 years old) with donor consent under a human subject study protocol approved by the institutional review board at the University of Texas Medical Branch at Galveston (IRB no. 04371). Blood samples (60 ml) were mixed with 1.5 ml of dextran (15%) and 1.5 ml of EDTA (0.25 M) and allowed to sediment for 30 to 40 min at room temperature. After sedimentation, the leukocyte-containing layer was overlaid onto Histopaque-1077 and centrifuged (720 × g) at room temperature for 40 min. The layer of mononuclear cells was collected and washed several times with RPMI 1640 medium supplemented with 2 mM l-glutamine, 2% FBS, 50 μM 2-mercaptoethanol, and 1,000 U/liter penicillin-streptomycin medium. Cells were allowed to adhere for 4 h, and nonadherent cells were removed by several washes with RPMI 1640 medium. Adherent monocytes (5 × 105) were pretreated with 100 μl of rHMGB1 for 1 h followed by infection with RSV at an MOI of 5 for 1 to 2 h at 37°C (viral adsorption phase), and then the cells were washed twice with RPMI. For the remaining time of infection the cells were placed in a 24-well plate in a total volume of 1 ml of RPMI.
Measurement of cytokines.
After 24 h, cell-free supernatants were collected and tested for multiple cytokines using the Bio-Plex human cytokine 27-plex panel (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. The panel included the following cytokines: interleukin-1β (IL-1β), IL-1RA, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 p70, IL-13, IL-15, IL-17, eotaxin, fibroblast growth factor (FGF)-basic, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage CSF (GM-CSF), gamma interferon (IFN-γ), IP-10, monocyte chemoattractant protein 1 (MCP-1) (MCAF), MIP-1α, MIP-1β, platelet-derived growth factor (PDGF)-bb, RANTES, tumor necrosis factor alpha (TNF-α), and vascular endothelial growth factor (VEGF).
Western blotting and densitometric analyses.
Cytoplasmic and nuclear extracts were prepared from A549 and SAE cells that were left uninfected or infected with RSV for various lengths of time using the hypotonic/nonionic detergent lysis method as previously described (37). Total cell lysates were prepared from uninfected and infected A549 and SAE cells by adding ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 1 mM EGTA, 0.25% sodium deoxycholate, 1 mM Na3VO4, 1 mM NaF, 1% Triton X-100, and 1 μg/ml of aprotinin, leupeptin, and pepstatin). After incubation on ice for 10 min, the lysates were centrifuged at 4°C at 14,000 × g to remove the detergent-insoluble cell debris. Proteins (10 to 20 μg per sample) were boiled in 2× Laemmli buffer and fractionated by SDS-PAGE. Proteins were then transferred onto a Hybond-polyvinylidene difluoride membrane (Amersham, Piscataway, NJ), and the membranes were incubated for 30 min in 10 mM Tris-buffered saline-Tween (TBST; Tris-HCl, pH 7.6, containing 150 mM NaCl and 0.05% Tween 20) and 5% bovine serum albumin. After blocking the nonspecific binding sites, membranes were washed with TBST, incubated sequentially with the primary antibody overnight at 4°C, and then incubated with anti-rabbit peroxidase-conjugated secondary antibody (1:10,000 in TBST) at room temperature for 30 min. Membranes were washed and signal was detected using enhanced chemiluminescence (ECL) substrate (Amersham) according to the manufacturer's protocol. The primary antibody for Western blotting was anti-HMGB1 rabbit polyclonal antibody from Abcam, anti-rabbit peroxidase-conjugated secondary antibody from Cell Signaling Technology (Danvers, MA), and anti-β-actin monoclonal antibody from Sigma-Aldrich (St. Louis, MO). Densitometric analysis of Western blot band intensities was performed using Alpha Ease software, version 2200 (2.2d) (Alpha Innotech Co., San Leandro, CA). Bands in RSV-infected samples were normalized to uninfected sample background for each time point.
ELISA.
HMGB1 was quantitated in cell-free supernatants using a DuoSet sandwich enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) by following the manufacturer's protocol. The sensitivity of the assay was 7.8 to 2,000 pg/ml.
Cell viability.
Total number of cells and cell viability, following RSV infection at an MOI of 1.0, was measured by trypan blue exclusion. There was no significant change in cell viability with RSV infection both at 15 h and 24 h postinfection (p.i.).
Immunocytochemistry.
Cells were cultured in LabTek II chambers (Nalge Nunc, Penfield, NY) and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) at room temperature for 30 min. The cells were then washed with PBS and incubated at 4°C for 10 min with permeabilization buffer (PBS containing 0.1% Triton X-100). After blocking with 5% BSA in PBS for 1 h, cells were incubated with anti-HMGB1 antibody (Abcam), followed by incubation for 1 h with Alexa Flour 488-conjugated or FITC-conjugated secondary antibody (Invitrogen). The cells were coverslip mounted using mounting medium containing the fluorescent nuclear stain 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen), and signals were analyzed using a fluorescence microscope (Nikon, Japan).
qRT-PCR.
Total RNA was extracted from uninfected and RSV-infected A549 and SAE cells with an RNeasy minikit (Qiagen, Hilden, Germany). RNA samples were quantified using a NanoDrop spectrophotometer (NanoDrop Technologies), and the quality was analyzed on RNA nano or pico chips using an Agilent 2100 Bioanalyzer (Agilent Technologies). Synthesis of cDNA was performed with 1 μg of total RNA in a 20-μl reaction mix using the reagents in the TaqMan reverse transcription reagent kit from ABI (no. N8080234; Applied Biosystems). The forward and reverse primers for HMGB1 were 5′-CCATTGCAGTACATTGAGCTCC-3′ and 5′-TGCCTCTCGGCTTAGGAT-3′, respectively. The reaction conditions used were 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min. Quantitative real-time PCR (qRT-PCR) amplifications (performed in triplicate) were done with 1 μl of cDNA in a total volume of 25 μl using the FastStart Universal SYBR green master mix (no. 04913850001; Roche Applied Science). The final concentration of the primers was 300 nM. 18S RNA was used as the housekeeping gene for normalization. PCR assays were run using the Applied Biosystems Prism 7500 sequence detection system under the following conditions: 50°C for 2 min, 95°C for 10 min, 95°C for 15 s, and 60°C for 1 min for 40 cycles. Duplicate cycle threshold (CT) values were analyzed in Microsoft Excel by the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems). The amount of target (2−ΔΔCT) was obtained by normalizing to the endogenous reference (18S) sample.
Statistics.
A two-tailed Student's t test using a 95% confidence level was performed in all experiments. Significance is indicated as a P value of <0.05 (*), <0.005 (**), and <0.0005 (***).
RESULTS
RSV infection induced the secretion of HMGB1 in human airway epithelial cells.
A549 cells, a human alveolar type II-like epithelial cell line, were mock infected or infected with RSV at an MOI of 1 for 6, 15, 24, and 48 h, and the cell-free supernatants were harvested to measure HMGB1 levels by Western blotting and ELISA. We found that RSV induced a substantial increase in HMGB1 release from these cells in a time-dependent manner compared with uninfected cells (Fig. 1A). Densitometric analysis of the Western blot showed that RSV infection for 24 and 48 h increased the HMGB1 release after infection by 28-fold and 23-fold, respectively (Fig. 1B). An ELISA confirmed the Western blot results and showed 2 - and 3-fold increase in HMGB1 released from the cells infected with RSV for 24 and 48 h, respectively (Fig. 1C). To confirm these results, we also examined the effect of RSV infection on HMGB1 release from small alveolar epithelial (SAE) cells, which are primary human airway epithelial cells derived from terminal bronchioles of a normal individual. SAE cells behave very similarly to A549 cells in terms of chemokine/cytokine gene expression and transcription factor and signaling pathway activation after RSV infection (37–39). Our results showed a significant increase in HMGB1 levels in RSV-infected SAE cells compared with uninfected cells (Fig. 1D). The effect of RSV infection on cell viability was determined using trypan blue exclusion. We found that at the 15- and 24-h p.i. time points, at an MOI of 1.0, where most of the experiments were done, RSV infection was not toxic to the cells (Fig. 2A). Together, the results shown in Fig. 1 and 2A suggested that RSV infection triggers an active release of HMGB1 from airway epithelial cells. The molecular mass of nuclear HMGB1 is ∼25 kDa, whereas the extracellular HMGB1 runs at ∼30 kDa, largely due to acetylation. Upon RSV infection, HMGB1 secreted by airway epithelial cells showed multiple protein bands on Western blots; one at ∼30 kDa, another at ∼60 kDa, and another at ∼25-kDa (Fig. 1A). The higher-molecular-mass band could be a complex of HMGB1 with another protein(s) yet to be determined (30). Posttranslational acetylation of key lysine residues within nuclear localization signals of HMGB1 shifts its equilibrium from a predominant nuclear location toward cytoplasm and subsequent extracellular release. Multiple posttranslational modifications (acetylation, phosphorylation, and methylation) of secreted HMGB1 have been reported that give rise to a complex protein pattern on gel electrophoresis (27, 32, 40). The characterization of the secreted multiple molecular forms of HMGB1 resulting from RSV infection is a separate ongoing study in our laboratory.
FIG 1.

RSV infection induces HMGB1 secretion in human lung epithelial cells. (A) A549 cells were infected with RSV at an MOI of 1, and cell-free supernatants were harvested at 6, 15, 24, and 48 h postinfection (p.i.) to measure HMGB1 release by Western blotting using anti-HMGB1 antibody. (B) Densitometric analysis of HMGB1 Western blot band intensity was performed using Alpha Ease software, version 2200 (2.2d) (Alpha Innotech Co., San Leandro, CA). Bands in RSV-infected samples were normalized to uninfected sample background for each time point. Open bars, control; solid bars, RSV. The figure is representative of three independent experiments. A549 (C) and SAE (D) cells were mock infected or infected with RSV, and cell-free supernatants were harvested over time to measure HMGB1 release by ELISA. P values were <0.05 (*), <0.005 (**), and <0.0005 (***) compared with uninfected cells.
FIG 2.

Effects of RSV infection on HMGB1 protein levels in A549 cells. (A) A549 cells were mock infected or infected with RSV at an MOI of 1.0, and the viability of the cells was evaluated by Trypan blue exclusion. (B) Total cell lysates and cytoplasmic and nuclear extracts prepared from uninfected or RSV-infected A549 cells for 6, 15, 24, and 48 h p.i. were resolved on 12% SDS-PAGE, and Western blotting was performed using antibody against HMGB1 protein. Membranes were stripped and reprobed for β-actin as an internal control for protein integrity and loading for total lysates and cytoplasmic extracts, and lamin B was used for nuclear extracts. (C to E) Densitometric analyses of HMGB1 Western blot band intensity for total cell lysates (C), cytoplasmic extracts (D), and nuclear extracts (E). Bands in RSV-infected samples were normalized to the uninfected sample background for each time point. Open bars, control; solid bars, RSV. The figure is representative of three independent experiments. P values were <0.05 (*), <0.005 (**), and <0.0005 (***) compared with uninfected cells.
RSV infection induced translocation of HMGB1 in airway epithelial cells.
To understand the biological relevance of the translocation of HMGB1 in RSV-infected cells, we monitored the subcellular localization of HMGB1 in the airway epithelial cells. A549 and SAE cells were mock infected or infected with RSV for 6, 15, 24, and 48 h and harvested for the preparation of total cell lysates, as well as nuclear and cytoplasmic fractions, to measure HMGB1 levels by Western blotting. We found that HMGB1 protein levels decreased in the total cell lysates and in nuclear and cytoplasmic fractions by RSV infection after 24 and 48 h p.i. (Fig. 2B). Comparative densitometric analyses with uninfected cells of Western blot results are presented in Fig. 2C to E. In this analysis, the comparisons were made for RSV-infected versus uninfected cells at each time point to control for variations in HMGB1 expression by serum and cell density. In nuclear fractions, the HMGB1 protein level increased by 73% (0.6-fold) at 6 h p.i., and thereafter it gradually decreased by 37% (1.6-fold), 41% (1.7-fold), and 72% (3.6-fold), respectively, at 15, 24, and 48 h after RSV infection (Fig. 2E). The HMGB1 protein level in the cytoplasmic fraction gradually decreased throughout the course of RSV infection compared with uninfected cells by 51% (2.0-fold), 80% (5.0-fold), and 94% (17-fold) at 15, 24, and 48 h after infection, respectively (Fig. 2D). Likewise, the HMGB1 level in total cell lysates of RSV-infected A549 cells decreased significantly throughout the course of infection, with 49.5% (∼2-fold), 95.5% (22-fold), and 96.2% (26.3-fold) at 15, 24, and 48 h after infection, respectively (Fig. 2C), compared with control cells. Decreased HMGB1 protein levels were also observed in the nucleus, cytoplasm, and total cell lysates prepared from RSV-infected SAE cells compared with uninfected cells (Fig. 3A, B, C, D, and E). Confocal immunofluorescence microscopy analyses of A549 cells infected with RSV and immunostained with an HMGB1-specific antibody are shown in Fig. 4. HMGB1 protein was detected predominantly in the nucleus of uninfected cells. In comparison, in the cells infected with RSV for 24 h, a significant amount of HMGB1 was observed in the cytoplasm (Fig. 4). Taken together, the results presented in Fig. 2 to 4 suggested that (i) RSV infection resulted in an overall decline in cellular HMGB1 protein levels, (ii) the translocation of HMGB1 from the nucleus was enhanced immediately after RSV infection, and (iii) by 24 h p.i., HMGB1 was largely translocated from cell nuclei to the cytoplasm and released into the extracellular space.
FIG 3.

RSV infection modifies HMGB1 protein expression in SAE cells. (A and B) Total cell lysates and cytoplasmic and nuclear extracts prepared from uninfected or RSV-infected SAE cells for 6, 15, 24, and 48 h p.i. were resolved on 12% SDS-PAGE, and Western blots were performed using antibody against HMGB1 protein. Membranes were stripped and reprobed for β-actin as an internal control for protein integrity and loading for total lysates and cytoplasmic extracts, and lamin B was used for nuclear extracts. (C to E) Densitometric analyses of HMGB1 Western blot band intensity for total cell lysates (C), cytoplasmic extracts (D), and nuclear extracts (E). Bands in RSV-infected samples were normalized to uninfected sample background for each time point. Open bars, control; solid bars, RSV. The figure is representative of three independent experiments. P values were <0.05 (*), <0.005 (**), and <0.0005 (***) compared with uninfected cells.
FIG 4.
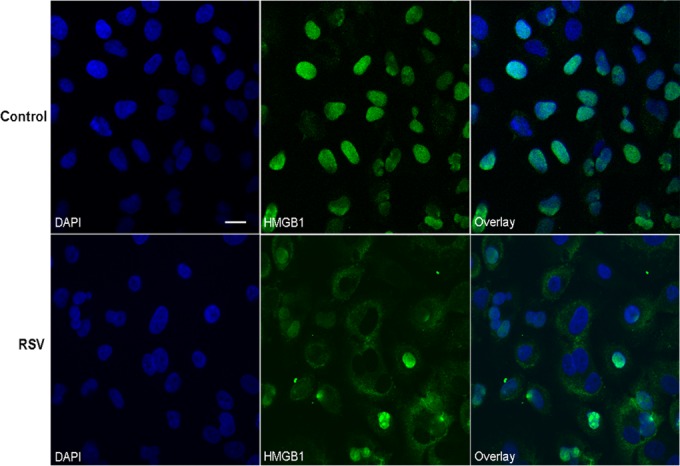
RSV infection induces translocation of HMGB1 in A549 cells. A549 cells were mock infected or infected with RSV, and the cells were incubated after 24 h p.i. with rabbit anti-HMGB1 polyclonal antibody followed by Alexa Flour 488-conjugated secondary antibody. Nuclei were labeled with DAPI and analyzed by confocal immunofluorescence microscopy (original magnification, ×400). Representative fluorescence microscopic images (HMGB1 and DAPI) and merged images (overlay) are shown. Scale bar, 20 μm.
RSV infection downregulated HMGB1 gene expression in airway epithelial cells.
To investigate the mechanism responsible for the observed changes in HMGB1 protein levels in response to RSV infection, we monitored HMGB1 mRNA levels in airway epithelial cells. Total RNA was prepared from the uninfected and RSV-infected A549 cells at 6, 15, 24, and 48 h p.i., and the HMGB1 gene was amplified by quantitative real-time PCR (qRT-PCR). Similar to what we observed for the HMGB1 protein levels, HMGB1 mRNA decreased during the course of RSV infection, particularly at the later time points (15, 24, and 48 h after infection). HMGB1 expression was decreased by ∼30% (1.4-fold), 49% (2.0-fold), and 63% (2.7-fold) at 15, 24, and 48 h p.i., respectively, compared with uninfected cells (Fig. 5A). To confirm our findings in A549 cells, a similar experiment was performed using normal SAE cells. The pattern of HMGB1 mRNA expression in SAE cells infected with RSV was similar to the one found in infected A549 cells (Fig. 5B). HMGB1 mRNA expression in SAE cells infected with RSV decreased by 30% (1.9-fold), 46% (2.7-fold), and 92% (13.7-fold) at 15, 24, and 48 h p.i., respectively, compared with uninfected cells.
FIG 5.
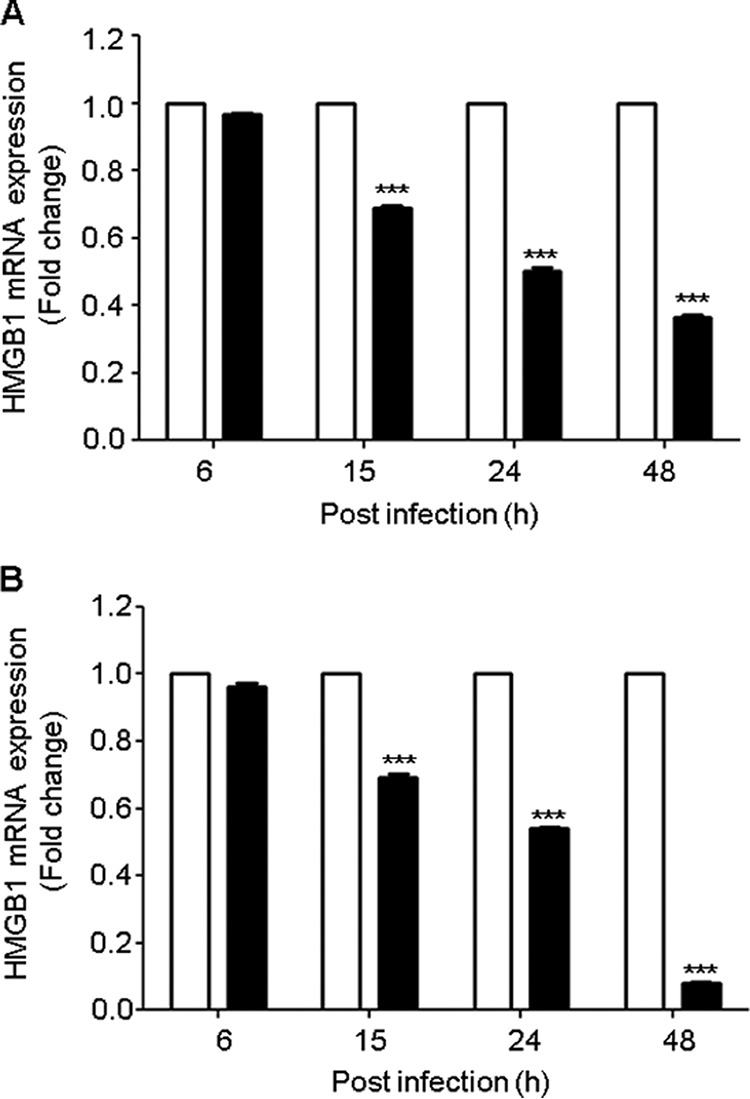
RSV infection inhibits HMGB1 gene expression in A549 and SAE cells. A549 (A) and SAE (B) cells were mock infected (open bars) or infected with RSV (solid bars) at an MOI of 1 and harvested to prepare total RNA at various lengths of time after infection. HMGB1 mRNA expression was measured by quantitative reverse transcriptase PCR (qRT-PCR). Data are representative of three independent experiments run in triplicate. ***, P value of <0.0005 relative to uninfected cells.
Effect of UV inactivation of RSV on HMGB1 release in airway epithelial cells.
To examine whether viral infectivity is required for HMGB1 release in the A549 cells, we monitored the HMGB1 protein levels in the cells incubated with UV-inactivated virus. Aliquots of RSV A2 viral stocks were exposed to UV radiation for 15 min using a transilluminator (365 nm; dose of 1.5 × 10−2 μW/mm2) to eliminate the viral infectivity without altering the conformation of viral proteins and mediators. A549 cells were mock infected or infected with RSV or UV-inactivated virus, and cells harvested at 24 h p.i. were used to prepare cell-free supernatants and total cell lysates to measure HMGB1 levels by Western blotting. We found that UV inactivation of RSV did not induce the release of HMGB1 from A549 cells compared with the cells infected with live RSV (Fig. 6). These results suggested that the active replication by virus was required to trigger HMGB1 release from A549 cells and that secretion of HMGB1 by these cells was viral replication dependent.
FIG 6.
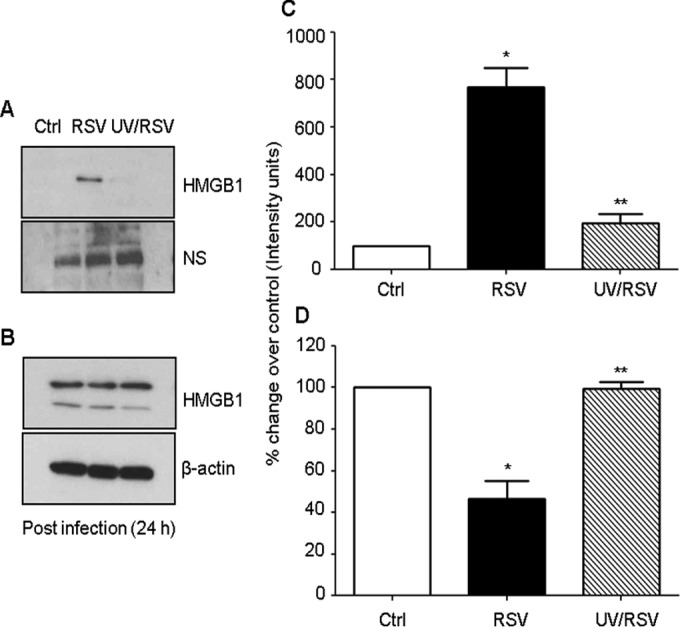
Effect of UV-inactivated RSV on HMGB1 release. A549 cells were mock infected or infected either with RSV or UV-inactivated RSV. Cell-free supernatants (A) and total cell lysates (B) were harvested 24 h p.i. to measure HMGB1 release by Western blotting. Membrane was stripped and reprobed for β-actin as an internal control for protein integrity and loading. NS, nonspecific band from the same blot; Ctrl, control. (C and D) Densitometric analyses of HMGB1 Western blot band intensity for supernatants (C) and total cell lysates (D). The figure is representative of three independent experiments. *, P value of <0.05 compared with uninfected cells; **, P value of <0.005 compared with RSV-infected cells.
Effect of proinflammatory cytokines and chemokines on HMGB1 release.
RSV induces the expression of proinflammatory mediators in airway epithelial cells (41–43). We therefore determined the effect of proinflammatory cytokines and chemokines on HMGB1 release from airway epithelial cells. A549 cells were pretreated with various cytokines and chemokines (IL-6, TNF-α, GM-CSF, IFN-γ, IL-13, and RANTES) and mock infected or infected with RSV and cell-free supernatants, and total cell lysates were prepared at 24 h after treatment to measure HMGB1 protein levels by Western blotting. We found that proinflammatory mediators did not induce the release of HMGB1 from A549 cells as was noted in RSV-infected cells (Fig. 7). These results suggested that RSV infection-induced cytokines and chemokines do not promote HMGB1 release from lung epithelial cells.
FIG 7.
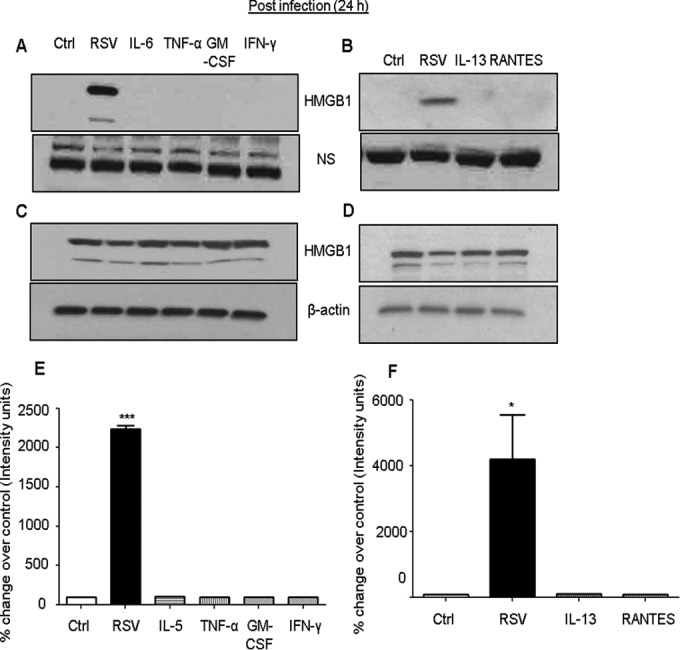
Effect of proinflammatory cytokines and chemokines on HMGB1 release. A549 cells were either infected with RSV or pretreated with 100 ng/ml of various cytokines and chemokines, and HMGB1 levels in cell-free supernatants (A and B) as well as in total cell lysates (C and D) were measured by Western blotting using anti-HMGB1 antibody. Membranes were stripped and reprobed for β-actin as an internal control for protein integrity and loading. NS, nonspecific band from the same blot. (E and F) Densitometric analyses of HMGB1 Western blot band intensity for supernatants. The figure is representative of three independent experiments. P values were <0.05 (*) and <0.0005 (***) compared with uninfected, RSV-infected, and cytokine-treated cells.
Effect of antioxidants on RSV-induced HMGB1 release.
We have previously reported that RSV induces cellular oxidative stress by generating ROS and downregulating antioxidant enzymes, and that treatment with antioxidants decreases virus-induced ROS production (44, 45). Studies have shown that oxidative stress triggers the translocation of HMGB1 from the nucleus to the cytoplasm (46–48). In this study, we investigated the possible mechanism for HMGB1 release in RSV infection and indicate that RSV-induced ROS generation triggers the release of HMGB1. Thus, treatment of RSV-infected cells with ROS scavengers, such as EUK-8, which possesses significant catalase and peroxidase activities in addition to superoxide dismutase (SOD) activity, as well as tBHQ, would inhibit HMGB1 release from the cells and thereby exert a protective effect against RSV-induced cellular stress. A549 cells were pretreated for 1 h before infection and throughout the course of infection with increasing concentrations of EUK-8 or tBHQ. Cells were harvested after 24 h of treatment, and the HMGB1 protein level in cell-free supernatants was measured by Western blotting or ELISA and its mRNA expression by qRT-PCR. EUK-8 as well as tBHQ treatment significantly inhibited the RSV-induced HMGB1 release in A549 cells in a dose-dependent manner (Fig. 8A, C, and D). HMGB1 mRNA expression was also significantly increased by antioxidant treatment in RSV-infected cells (Fig. 8B). The confocal immunofluorescence microscopic results revealed HMGB1 export from the nucleus to the cytoplasm in RSV-infected cells. Antioxidant treatment led to significant retention of HMGB1 in the cytoplasm of RSV-infected cells compared to untreated control cells (Fig. 9). These results suggested that RSV-induced ROS generation promotes the release of HMGB1 from the cells, and that treatment with ROS scavengers significantly inhibited its extracellular release.
FIG 8.

Effect of antioxidant treatment on RSV-induced HMGB1 release and its mRNA expression in A549 cells. A549 cells were pretreated for 1 h with antioxidants like EUK-8 or tert-butylhydroqinone (tBHQ) in the presence or absence of RSV. Cell-free supernatants and total cell lysates were prepared after 24 h p.i. to measure HMGB1 protein levels by ELISA (A) and Western blotting (C and D). Membranes were stripped and reprobed for β-actin as an internal control for protein integrity and loading. NS, nonspecific band from the same blot. (B) Total RNA prepared from RSV-infected A549 cells pretreated with an antioxidant (EUK-8) at various concentrations and from uninfected and RSV-infected cells at 24 h p.i., and HMGB1 mRNA expression was measured by qRT-PCR. P values were <0.05 (*), <0.005 (**), and <0.0005 (***) compared with uninfected, RSV-infected, and EUK-8-treated cells.
FIG 9.
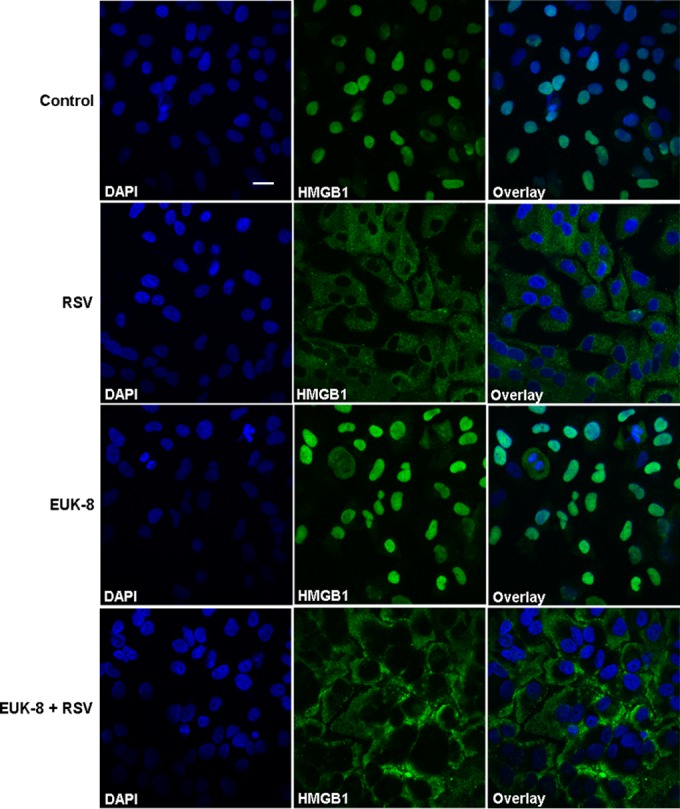
Nuclear translocation of HMGB1 in response to RSV infection and inhibition of its release by antioxidant treatment. A549 cells were pretreated with the antioxidant mimetic EUK-8 at 100 μM for 1 h and then mock infected or infected with RSV at an MOI of 3. Cells were analyzed after 24 h p.i. by confocal immunofluorescence microscopy (original magnification, ×400) using anti-HMGB1 antibody (green). Representative fluorescence microscopic images (HMGB1 and DAPI) and merged images (overlay) are shown. Scale bar, 20 μm.
Effect of HMGB1 on secretion of proinflammatory mediators in airway epithelial cells.
To investigate whether HMGB1 enhances RSV-induced proinflammatory gene expression, RSV-infected A549 cells were incubated in the presence or absence of rHMGB1 (100 ng/ml) for 24 h, and cell-free supernatants were utilized to measure proinflammatory mediators by Bio-Plex assay. Our results showed that rHMGB1 did not itself induce the release of cytokines/chemokines in A549 cells. However, rHMGB1 treatment of the RSV-infected cells resulted in a significant increase in the release of proinflammatory mediators, such as IL-8, RANTES, and G-CSF, compared with that in RSV-infected/untreated cells (Fig. 10). These results suggested that HMGB1 provides a synergistic signal in eliciting proinflammatory gene expression in RSV-infected A549 cells.
FIG 10.
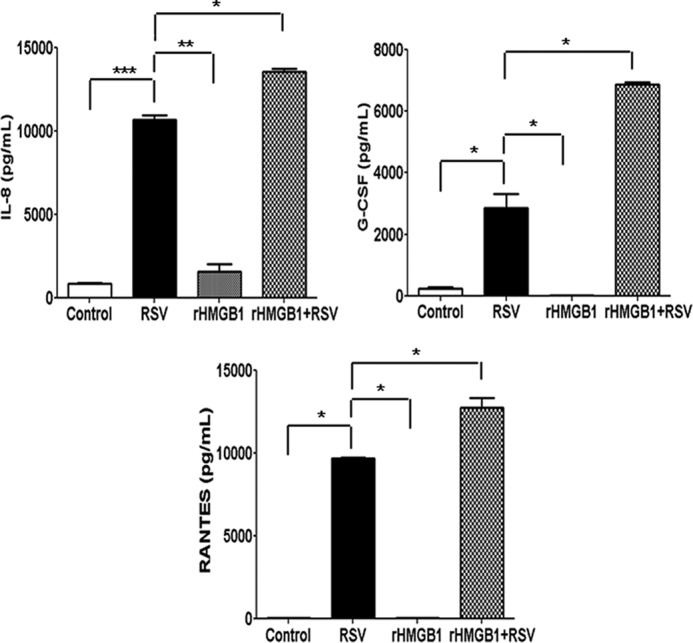
Effect of rHMGB1 on proinflammatory mediator release in A549 cells. Cell-free supernatants from uninfected and RSV-infected A549 cells in the presence or absence of recombinant HMGB1 (100 ng/ml) were harvested after 24 h p.i. to measure the concentrations of different proinflammatory mediators by Bio-Plex. n = 2 independent experiments run in triplicate. P values were <0.05 (*), <0.005 (**), and <0.0005 (***) compared with uninfected, RSV-infected, and rHMGB1-treated cells.
HMGB1 triggered proinflammatory cytokine/chemokine release in human primary monocytes.
We also determined whether RSV infection induces HMGB1 release in immune cells. We infected human primary monocytes with RSV for 24 h, and cell-free supernatants and total cell lysates were utilized for measuring HMGB1 levels by Western blotting. We did not detect HMGB1 release from monocytes in response to RSV infection; however, LPS treatment significantly induced HMGB1 release from the primary monocytes (Fig. 11). Since rHMGB1 is a poor activator of proinflammatory mediators in A549 cells, we wanted to test its effect in monocytes. Primary human monocytes were pretreated with rHMGB1 at 100 ng/ml, and cell-free supernatants were used to measure cytokine/chemokine release by a Bio-Plex assay. Our results showed that the addition of rHMGB1 to cultured human monocytes significantly induced the release of proinflammatory mediators, including IL-1β, IL-1RA, IL-6, IL-8, IL-10, TNF-α, G-CSF, IFN-γ, IP-10, MCP-1, MIP-1α, MIP-1β, RANTES, PDGF, and VEGF (Fig. 12). We also observed a synergistic increase in the levels of many of these cytokines when the cells were treated in combination with rHMGB1 and RSV. These results showed that HMGB1 released from airway epithelial cells can provide danger signals to neighboring immune cells in the airways and promote inflammation and also provides a synergistic signal in eliciting proinflammatory gene expression in RSV-infected human primary monocytes.
FIG 11.
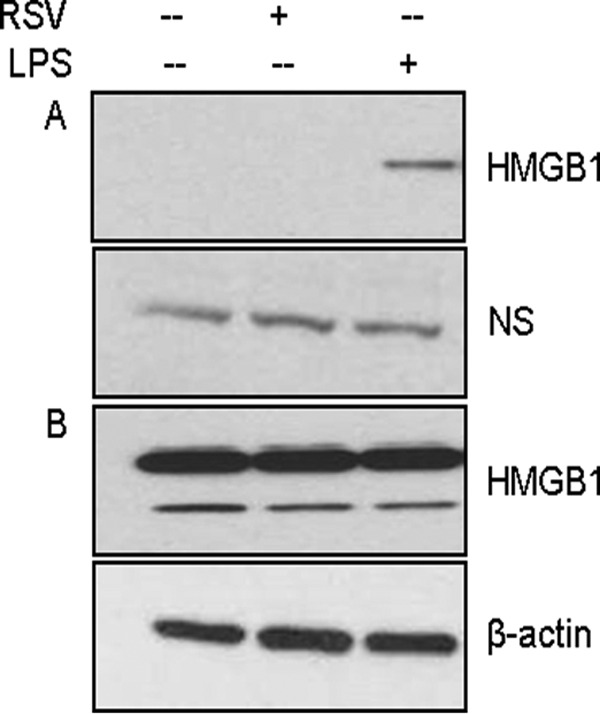
Effect of RSV infection on HMGB1 protein levels in human monocytes. Human primary monocytes were treated with LPS (1 μg/ml) or infected with RSV at an MOI of 5, and cell-free supernatants (A) as well as total cell lysates (B) were prepared after 24 h p.i. to measure HMGB1 protein levels by Western blotting using anti-HMGB1 antibody. Membrane was stripped and reprobed for β-actin as an internal control for protein integrity and loading. NS, nonspecific band from the same blot.
FIG 12.

HMGB1 stimulates the release of proinflammatory mediators in human primary monocytes. Human monocytes cultured with recombinant HMGB1 (100 ng/ml) in the presence or absence of RSV. Cell-free supernatants were harvested after 24 h p.i. to measure the concentrations of different proinflammatory mediators by Bio-Plex (n = 2 independent experiments run in triplicate). (A) Proinflammatory mediators stimulated by RSV. (B) Proinflammatory mediators stimulated by RSV as well as rHMGB1. (C) Proinflammatory mediators stimulated in combination by rHMGB1 and RSV infection. P values were <0.05 (*), <0.005 (**), and <0.0005 (***) compared with uninfected, RSV-infected, and rHMGB1-treated cells.
DISCUSSION
HMGB1 is a 25-kDa DNA-binding protein typically located in the nucleus of the cells, but after significant posttranslational modifications, such as acetylation, phosphorylation, and methylation, it can translocate to the cytoplasm and subsequently be released into the extracellular space during cell activation and/or cell death (32, 33). HMGB1 was initially characterized as a transcription factor and growth factor but later was identified as a cytokine mediator of many inflammatory diseases (13, 24–26). The present work establishes that RSV infection induces the release of HMGB1 from the lung epithelial cells, and our results demonstrated that upon RSV infection HMGB1 secreted by airway epithelial cells showed multiple protein bands on Western blots (Fig. 1A). The extracellular HMGB1 was shown to run at 30-kDa due to its significant posttranslational modifications (13). This study demonstrated that RSV infection triggers the translocation of HMGB1 protein from the cell nucleus to the cytoplasm and induces its secretion into the extracellular milieu (Fig. 1 to 4). Although HMGB1 can be passively released after necrosis, our results demonstrated that active RSV infection in airway epithelial cells leads to translocation of HMGB1 from the nucleus to the cytoplasm and subsequently to the extracellular space, and this phenomenon occurred independent of cell death; we did not see any significant increase in cell apoptosis, necrosis, or cytopathy during the 24 h p.i. (Fig. 2A and 4). To our knowledge, our studies represent the first report of elevated HMGB1 secretion in the RSV-infected airway epithelial cells. HMGB1 release has been shown after infection with various viruses, including RNA viruses (e.g., influenza virus H5N1, hepatitis C virus, West Nile virus, dengue virus, and HIV-1) (22, 49–51) and DNA viruses (e.g., herpes simplex virus type 2) (52), and HMGB1 has been implicated as an important mediator in the pathogenesis of these viral infections (53). Recent reports on asthma and COPD provide strong correlative evidence for HMGB1's involvement in lung inflammation (19, 54–58).
Although our findings indicated that RSV induced HMGB1 secretion, the molecular basis of its nuclear translocation and the mechanism of its release to the extracellular milieu are not fully yet understood. However, a possible mechanism involves ROS generated in RSV-infected airway epithelial cells. We have previously shown that RSV induces severe cellular oxidative stress in vitro and oxidative injury to the lungs in vivo, and that treatment with antioxidants decreases virus-induced ROS production, viral replication, and proinflammatory gene expression (44, 45, 59). Hydrogen peroxide-induced oxidative stress has been shown to stimulate macrophages and monocytes to actively or passively release HMGB1 through MAP kinase- and chromosome region maintenance (CRM1)-dependent mechanisms (48). HMGB1 levels were induced during mouse liver ischemia reperfusion (I/R) injury, and inhibition of its activity with neutralizing antibody significantly decreased liver damage after I/R injury (25). Hyperglycemia-induced ROS increases expression of HMGB1 and RAGE in human endothelial cells (60). HMGB1 has been shown to act as an early mediator of injury and inflammation mediated through TLR-4, TLR-9, and RAGE receptors (61–63). Several studies have demonstrated in various disease states, including liver I/R injury and atherosclerosis, as well as in aging, that oxidative stress triggers the translocation of HMGB1 from the nucleus to the cytoplasm, indicating that under cellular stress HMGB1 release facilitates cellular defenses and alerts the immune system to the damage (46–48, 64, 65).
Using several proinflammatory cytokines and chemokines to treat A549 cells, we found that proinflammatory mediators had no direct role in signaling HMGB1 release from A549 cells (Fig. 7). Likewise, rHMGB1 alone had no effect on inducing proinflammatory gene expression in A549 cells, indicating that cellular stimulation by HMGB1 was specific to monocytes. However, rHMGB1 significantly enhanced the RSV-induced proinflammatory response in A549 cells. These results provide evidence that HMGB1 protein is bioactive only in the presence of external stimulus, such as RSV, and can provide a synergistic functionality (Fig. 10). Synergy among cytokines has been reported (66).
In this study, we monitored the effect of antioxidant treatment (with EUK-8 and tBHQ) (Fig. 8A to D and 9) and UV inactivation of the virus (Fig. 6A to D) on RSV-induced HMGB1 release in A549 cells. We showed that HMGB1 release from the cells is virus replication dependent, and inhibition of HMGB1 release from the cells by antioxidants may suppress the HMGB1-induced inflammatory responses. Moreover, we showed that HMGB1 mRNA expression was significantly increased after antioxidant treatment, which was progressively decreased after RSV infection. We have shown that blocking HMGB1 release in A549 cells through antioxidant treatment is the result of reduced viral replication by antioxidant treatment (44). In recent years, several classes of synthetic antioxidant mimetics have been tested as a potential therapeutic approach to oxidant-related lung damage. The salen class of antioxidant enzyme mimetics includes compounds that mainly have SOD activity, as well as compounds that also exhibit catalase and peroxidase activity. These compounds have shown protective effects in various experimental disease models, including injury models of brain, lung, and systemic shock (reviewed in reference 67). Our previous studies have shown that treating A549 cells with EUK-134 significantly inhibited RSV-induced IL-8 and RANTES secretion (45). Several antioxidants, such as ethyl pyruvate, green tea, quercetin, N-acetyl cysteine, and curcumin, play a significant role in the setting of experimental inflammation, partly by attenuating systemic HMGB1 accumulation (reviewed in reference 47). These observations suggested that the elevated levels of ROS in RSV-infected cells contribute, at least in part, to the translocation of HMGB1.
In the current study, we focused on the function of HMGB1 protein in RSV infection. Our results with rHMGB1 protein and human primary monocytes (Fig. 11) suggested that HMGB1 secreted by airway epithelial cells in response to RSV infection signal the monocytes/macrophages to release proinflammatory cytokines and chemokines (Fig. 12). Other investigators have reported similar observations in human monocytes treated with purified HMGB1 (32, 68). Monocytes and macrophages are the key players in orchestrating the immune response to infection and injury. Our observation that HMGB1 can activate additional downstream cytokine/chemokine cascades has widespread implications in RSV infection, as HMGB1 failed to stimulate A549 cells by itself but showed some effect only in the presence of RSV. Our findings also suggested that monocytes are the cellular targets for the cytokine-inducing activity of HMGB1, as it is not released by monocytes. Moreover, secreted HMGB1 likely triggers inflammatory responses by activating neighboring immune cells in the airways to release more proinflammatory cytokines (69). Therefore, HMGB1 by itself or in combination with other proinflammatory cytokines may also contribute to chronic inflammation of airway epithelial cells. These observations support the view that secreted HMGB1 contributed to the triggering and maintaining of a pathological state. Confirming the roles of HMGB1 in RSV pathology and the physiological activities of HMGB1 during RSV infection will require further investigations using appropriate animal model systems. These results led us to conclude that RSV infection induced the secretion of HMGB1 protein into the extracellular milieu and that secreted HMGB1 triggered the activation of immune cells to release proinflammatory mediators, thereby alerting the immune system to the potential damage (Fig. 13).
FIG 13.
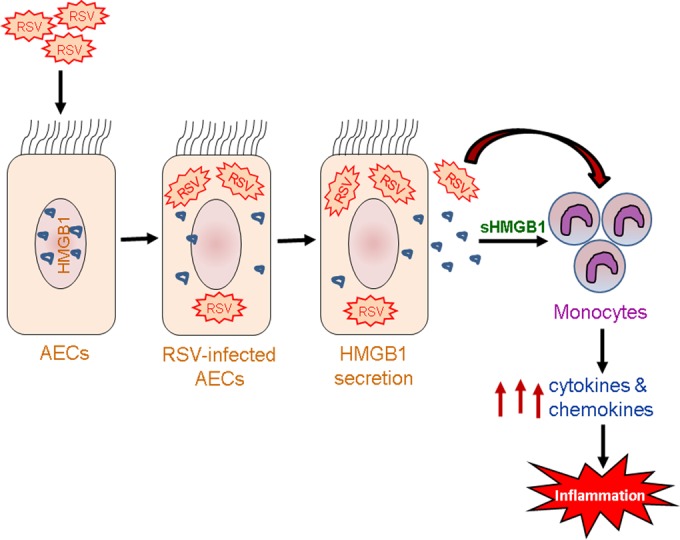
Schematic representation of HMGB1's role in RSV infection. RSV-induced HMGB1 secretion from airway epithelial cells (AECs) activates primary human monocytes in the airways to release proinflammatory mediators and promote inflammation.
It is worth emphasizing the potential significance of the translocation of HMGB1 from the nucleus to the cytoplasm and to understand its role in autophagy, apoptosis, necrosis, and other cellular processes that play a major role in host immunity against respiratory viral infections. Recent studies on autophagy in RSV infection suggest that autophagy facilitates intracellular pathogen recognition, dendritic cell (DC) maturation, and proinflammatory cytokine production (70–72). Because RSV enters the host cell cytosol directly through membrane fusion (73), DC activation relies on the autophagic machinery to mediate endosomal TLR-dependent cytokine production and proper immune responses. Recent studies have demonstrated the relationship between HMGB1 and autophagy, where it promotes starvation- and oxidative stress-induced autophagy in immortalized mouse embryonic fibroblasts and cancer cells, through direct interaction with Beclin-1, a critical regulator of autophagy and apoptosis (33, 40). HMGB1-mediated autophagy also regulated acute promyelocytic leukemia cell differentiation via controlling the degradation of protein of promyelocytic leukemia and retinoic acid receptor-α (74). Further investigations on HMGB1's role in the airway inflammation caused by RSV infection will provide new insights on the mechanisms involved that facilitate the development of novel therapeutic strategies to treat and prevent virus-induced lung inflammatory diseases in pediatric and elderly populations.
ACKNOWLEDGMENTS
We thank Nisha Garg, David Konkel, and Animesh Chandra for manuscript editing. We also thank Konrad Pazdrak and John E. Wiktorowicz for helpful discussions and Susan Stafford for technical assistance.
A.K. is a recipient of a grant from MedImmune, LLC, Gaithersburg, Maryland, USA.
Funding Statement
This study was supported by a Young Clinical Scientist Award from the Flight Attendant Medical Research Institute (FAMRI) to Y.M.H. (grant 123385). Funding was also provided by NIH/NHLBI contract HHSN268201000037-C-0-01 (N01-HV-00245) to A.K., MedImmune, LLC, grant MA-409325 to A.K., and a contract from the University of Texas System Proteomics Network to A.K. Additional funding was provided by grants from NIAID, Signaling in Airway Inflammation, PO1 AI068865 (A.R.B., R.P.G., and A.C.), University of Texas Medical Branch (UTMB) Clinical Translational Sciences Award (CTSA), UL1TR001439 (A.R.B. and Y.M.H.), and NIEHS P30 ES006676 (A.R.B., R.P.G., and A.K.).
REFERENCES
- 1.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 2.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N Engl J Med 360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brasier AR, Tian B, Jamaluddin M, Kalita MK, Garofalo RP, Lu M. 2011. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J Virol 85:11752–11769. doi: 10.1128/JVI.05360-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian B, Zhang Y, Luxon BA, Garofalo RP, Casola A, Sinha M, Brasier AR. 2002. Identification of NF-kappaB-dependent gene networks in respiratory syncytial virus-infected cells. J Virol 76:6800–6814. doi: 10.1128/JVI.76.13.6800-6814.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian B, Zhao Y, Kalita M, Edeh CB, Paessler S, Casola A, Teng MN, Garofalo RP, Brasier AR. 2013. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J Virol 87:7075–7092. doi: 10.1128/JVI.03399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macnee W. 2001. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol 429:195–207. doi: 10.1016/S0014-2999(01)01320-6. [DOI] [PubMed] [Google Scholar]
- 7.Rahman I, Morrison D, Donaldson K, Macnee W. 1996. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 154:1055–1060. doi: 10.1164/ajrccm.154.4.8887607. [DOI] [PubMed] [Google Scholar]
- 8.Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, Garofalo RP, Casola A. 2006. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med 174:1361–1369. doi: 10.1164/rccm.200603-319OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu T, Castro S, Brasier AR, Jamaluddin M, Garofalo RP, Casola A. 2004. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J Biol Chem 279:2461–2469. doi: 10.1074/jbc.M307251200. [DOI] [PubMed] [Google Scholar]
- 10.Hou CC, Zhao HJ, Cai SX, Li WJ, Tong WC, Liu LY. 2010. Respiratory syncytial virus increases the expression and release of high mobility group Box-1 protein in the lung tissue of mice. Nan Fang Yi Ke Da Xue Xue Bao 30:700–703. [PubMed] [Google Scholar]
- 11.Stros M. 2010. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Ulloa L, Batliwalla FM, Andersson U, Gregersen PK, Tracey KJ. 2003. High mobility group box chromosomal protein 1 as a nuclear protein, cytokine, and potential therapeutic target in arthritis. Arthritis Rheum 48:876–881. doi: 10.1002/art.10854. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 14.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. 2010. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 15.Oozawa S, Mori S, Kanke T, Takahashi H, Liu K, Tomono Y, Asanuma M, Miyazaki I, Nishibori M, Sano S. 2008. Effects of HMGB1 on ischemia-reperfusion injury in the rat heart. Circ J 72:1178–1184. doi: 10.1253/circj.72.1178. [DOI] [PubMed] [Google Scholar]
- 16.Ferhani N, Letuve S, Kozhich A, Thibaudeau O, Grandsaigne M, Maret M, Dombret MC, Sims GP, Kolbeck R, Coyle AJ, Aubier M, Pretolani M. 2010. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 181:917–927. doi: 10.1164/rccm.200903-0340OC. [DOI] [PubMed] [Google Scholar]
- 17.Hamada N, Maeyama T, Kawaguchi T, Yoshimi M, Fukumoto J, Yamada M, Yamada S, Kuwano K, Nakanishi Y. 2008. The role of high mobility group box1 in pulmonary fibrosis. Am J Respir Cell Mol Biol 39:440–447. doi: 10.1165/rcmb.2007-0330OC. [DOI] [PubMed] [Google Scholar]
- 18.Lee CC, Lai YT, Chang HT, Liao JW, Shyu WC, Li CY, Wang CN. 2013. Inhibition of high-mobility group box 1 in lung reduced airway inflammation and remodeling in a mouse model of chronic asthma. Biochem Pharmacol 86:940–949. doi: 10.1016/j.bcp.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Ma L, Zeng J, Mo B, Wang C, Huang J, Sun Y, Yu Y, Liu S. 2015. High mobility group box 1: a novel mediator of Th2-type response-induced airway inflammation of acute allergic asthma. J Thorac Dis 7:1732–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang F, Huang G, Hu B, Fang LP, Cao EH, Xin XF, Song Y, Shi Y. 2014. Anti-HMGB1 neutralizing antibody ameliorates neutrophilic airway inflammation by suppressing dendritic cell-mediated Th17 polarization. Mediators Inflamm 2014:257930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scaffidi P, Misteli T, Bianchi ME. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 22.Jung JH, Park JH, Jee MH, Keum SJ, Cho MS, Yoon SK, Jang SK. 2011. Hepatitis C virus infection is blocked by HMGB1 released from virus-infected cells. J Virol 85:9359–9368. doi: 10.1128/JVI.00682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, Susarla S, Czura C, Mitchell RA, Chen G, Sama AE, Tracey KJ, Wang H. 2003. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol 170:3890–3897. doi: 10.4049/jimmunol.170.7.3890. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, Inoue K, Yamada S, Ijiri K, Matsunaga S, Nakajima T, Komiya S, Maruyama I. 2003. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum 48:971–981. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 25.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. 2005. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. 2004. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 27.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. 2003. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lotze MT, Tracey KJ. 2005. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 29.Tang D, Shi Y, Jang L, Wang K, Xiao W, Xiao X. 2005. Heat shock response inhibits release of high mobility group box 1 protein induced by endotoxin in murine macrophages. Shock 23:434–440. doi: 10.1097/01.shk.0000159556.95285.df. [DOI] [PubMed] [Google Scholar]
- 30.Vande WL, Kanneganti TD, Lamkanfi M. 2011. HMGB1 release by inflammasomes. Virulence 2:162–165. doi: 10.4161/viru.2.2.15480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. 2004. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 32.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. 2000. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT. 2010. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olszewska-Pazdrak B, Casola A, Saito T, Alam R, Crowe SE, Mei F, Ogra PL, Garofalo RP. 1998. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol 72:4756–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueba O. 1978. Respiratory syncytial virus. I. Concentration and purification of the infectious virus. Acta Med Okayama 32:265–272. [PubMed] [Google Scholar]
- 36.Kisch AL, Johnson KM. 1963. A plaque assay for respiratory syncytial virus. Proc Soc Exp Biol Med 112:583–589. doi: 10.3181/00379727-112-28111. [DOI] [PubMed] [Google Scholar]
- 37.Garofalo R, Sabry M, Jamaluddin M, Yu RK, Casola A, Ogra PL, Brasier AR. 1996. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol 70:8773–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casola A, Burger N, Liu T, Jamaluddin M, Brasier AR, Garofalo RP. 2001. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J Biol Chem 276:19715–19722. [DOI] [PubMed] [Google Scholar]
- 39.Pazdrak K, Olszewska-Pazdrak B, Liu T, Takizawa R, Brasier AR, Garofalo RP, Casola A. 2002. MAPK activation is involved in posttranscriptional regulation of RSV-induced RANTES gene expression. Am J Physiol Lung Cell Mol Physiol 283:L364–L372. doi: 10.1152/ajplung.00331.2001. [DOI] [PubMed] [Google Scholar]
- 40.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III, Lotze MT. 2010. Endogenous HMGB1 regulates autophagy. J Cell Biol 190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garofalo RP, Goldman AS. 1999. Expression of functional immunomodulatory and anti-inflammatory factors in human milk. Clin Perinatol 26:361–377. [PubMed] [Google Scholar]
- 42.Graham BS, Johnson TR, Peebles RS. 2000. Immune-mediated disease pathogenesis in respiratory syncytial virus infection. Immunopharmacology 48:237–247. doi: 10.1016/S0162-3109(00)00233-2. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Luxon BA, Casola A, Garofalo RP, Jamaluddin M, Brasier AR. 2001. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J Virol 75:9044–9058. doi: 10.1128/JVI.75.19.9044-9058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hosakote YM, Komaravelli N, Mautemps N, Liu T, Garofalo RP, Casola A. 2012. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am J Physiol Lung Cell Mol Physiol 303:L991–L1000. doi: 10.1152/ajplung.00192.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hosakote YM, Liu T, Castro SM, Garofalo RP, Casola A. 2009. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am J Respir Cell Mol Biol 41:348–357. doi: 10.1165/rcmb.2008-0330OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. 2006. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res 312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 47.Tang D, Kang R, Zeh HJ III, Lotze MT. 2011. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal 14:1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang D, Shi Y, Kang R, Li T, Xiao W, Wang H, Xiao X. 2007. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 81:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barqasho B, Nowak P, Abdurahman S, Walther-Jallow L, Sonnerborg A. 2010. Implications of the release of high-mobility group box 1 protein from dying cells during human immunodeficiency virus type 1 infection in vitro. J Gen Virol 91:1800–1809. doi: 10.1099/vir.0.016915-0. [DOI] [PubMed] [Google Scholar]
- 50.Hou XQ, Qin JL, Zheng XX, Wang L, Yang ST, Gao YW, Xia XZ. 2014. Potential role of high-mobility group box 1 protein in the pathogenesis of influenza H5N1 virus infection. Acta Virol 58:69–75. doi: 10.4149/av_2014_01_69. [DOI] [PubMed] [Google Scholar]
- 51.Ong SP, Lee LM, Leong YF, Ng ML, Chu JJ. 2012. Dengue virus infection mediates HMGB1 release from monocytes involving PCAF acetylase complex and induces vascular leakage in endothelial cells. PLoS One 7:e41932. doi: 10.1371/journal.pone.0041932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borde C, Barnay-Verdier S, Gaillard C, Hocini H, Marechal V, Gozlan J. 2011. Stepwise release of biologically active HMGB1 during HSV-2 infection. PLoS One 6:e16145. doi: 10.1371/journal.pone.0016145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H, Ward MF, Fan XG, Sama AE, Li W. 2006. Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol 19:3–9. doi: 10.1089/vim.2006.19.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cuppari C, Manti S, Chirico V, Caruso R, Salpietro V, Giacchi V, Lagana F, Arrigo T, Salpietro C, Leonardi S. 2015. Sputum high mobility group box-1 in asthmatic children: a noninvasive sensitive biomarker reflecting disease status. Ann Allergy Asthma Immunol 115:103–107. doi: 10.1016/j.anai.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 55.Hou C, Kong J, Liang Y, Huang H, Wen H, Zheng X, Wu L, Chen Y. 2015. HMGB1 contributes to allergen-induced airway remodeling in a murine model of chronic asthma by modulating airway inflammation and activating lung fibroblasts. Cell Mol Immunol 12:409–423. doi: 10.1038/cmi.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hou C, Zhao H, Liu L, Li W, Zhou X, Lv Y, Shen X, Liang Z, Cai S, Zou F. 2011. High mobility group protein B1 (HMGB1) in asthma: comparison of patients with chronic obstructive pulmonary disease and healthy controls. Mol Med 17:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanazawa H, Tochino Y, Asai K, Ichimaru Y, Watanabe T, Hirata K. 2012. Validity of HMGB1 measurement in epithelial lining fluid in patients with COPD. Eur J Clin Investig 42:419–426. doi: 10.1111/j.1365-2362.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- 58.Watanabe T, Asai K, Fujimoto H, Tanaka H, Kanazawa H, Hirata K. 2011. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir Med 105:519–525. doi: 10.1016/j.rmed.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 59.Hosakote YM, Jantzi PD, Esham DL, Spratt H, Kurosky A, Casola A, Garofalo RP. 2011. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med 183:1550–1560. doi: 10.1164/rccm.201010-1755OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yao D, Brownlee M. 2010. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 59:249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. 2010. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 51:621–632. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kruger B, Krick S, Dhillon N, Lerner SM, Ames S, Bromberg JS, Lin M, Walsh L, Vella J, Fischereder M, Kramer BK, Colvin RB, Heeger PS, Murphy BT, Schroppel B. 2009. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A 106:3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng S, Dun H, Ippagunta N, Rosario R, Zhang QY, Lefkowitch J, Yan SF, Schmidt AM, Emond JC. 2009. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. J Hepatol 50:929–936. doi: 10.1016/j.jhep.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 64.Kregel KC, Zhang HJ. 2007. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol 292:R18–R36. [DOI] [PubMed] [Google Scholar]
- 65.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, Wang H, Warren HS, Moldawer LL, Fink MP, Andersson U, Tracey KJ, Yang H. 2003. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med 9:37–45. [PMC free article] [PubMed] [Google Scholar]
- 66.Bianchi ME. 2009. HMGB1 loves company. J Leukoc Biol 86:573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 67.Kinnula VL, Crapo JD. 2003. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med 167:1600–1619. doi: 10.1164/rccm.200212-1479SO. [DOI] [PubMed] [Google Scholar]
- 68.El-Saghire H, Michaux A, Thierens H, Baatout S. 2013. Low doses of ionizing radiation induce immune-stimulatory responses in isolated human primary monocytes. Int J Mol Med 32:1407–1414. [DOI] [PubMed] [Google Scholar]
- 69.Gremion C, Cerny A. 2005. Hepatitis C virus and the immune system: a concise review. Rev Med Virol 15:235–268. doi: 10.1002/rmv.466. [DOI] [PubMed] [Google Scholar]
- 70.Lee FE, Walsh EE, Falsey AR, Lumb ME, Okam NV, Liu N, Divekar AA, Hall CB, Mosmann TR. 2007. Human infant respiratory syncytial virus (RSV)-specific type 1 and 2 cytokine responses ex vivo during primary RSV infection. J Infect Dis 195:1779–1788. doi: 10.1086/518249. [DOI] [PubMed] [Google Scholar]
- 71.Morris S, Swanson MS, Lieberman A, Reed M, Yue Z, Lindell DM, Lukacs NW. 2011. Autophagy-mediated dendritic cell activation is essential for innate cytokine production and APC function with respiratory syncytial virus responses. J Immunol 187:3953–3961. doi: 10.4049/jimmunol.1100524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reed M, Morris SH, Jang S, Mukherjee S, Yue Z, Lukacs NW. 2013. Autophagy-inducing protein beclin-1 in dendritic cells regulates CD4 T cell responses and disease severity during respiratory syncytial virus infection. J Immunol 191:2526–2537. doi: 10.4049/jimmunol.1300477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srinivasakumar N, Ogra PL, Flanagan TD. 1991. Characteristics of fusion of respiratory syncytial virus with HEp-2 cells as measured by R18 fluorescence dequenching assay. J Virol 65:4063–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang L, Chai W, Wang Y, Cao L, Xie M, Yang M, Kang R, Yu Y. 2015. Reactive oxygen species regulate the differentiation of acute promyelocytic leukemia cells through HMGB1-mediated autophagy. Am J Cancer Res 5:714–725. [PMC free article] [PubMed] [Google Scholar]