

ABSTRACT
Canine parvovirus (CPV) is a highly contagious pathogen that causes severe disease in dogs and wildlife. Previously, a panel of neutralizing monoclonal antibodies (MAb) raised against CPV was characterized. An antibody fragment (Fab) of MAb E was found to neutralize the virus at low molar ratios. Using recent advances in cryo-electron microscopy (cryo-EM), we determined the structure of CPV in complex with Fab E to 4.1 Å resolution, which allowed de novo building of the Fab structure. The footprint identified was significantly different from the footprint obtained previously from models fitted into lower-resolution maps. Using single-chain variable fragments, we tested antibody residues that control capsid binding. The near-atomic structure also revealed that Fab binding had caused capsid destabilization in regions containing key residues conferring receptor binding and tropism, which suggests a mechanism for efficient virus neutralization by antibody. Furthermore, a general technical approach to solving the structures of small molecules is demonstrated, as binding the Fab to the capsid allowed us to determine the 50-kDa Fab structure by cryo-EM.
IMPORTANCE Using cryo-electron microscopy and new direct electron detector technology, we have solved the 4 Å resolution structure of a Fab molecule bound to a picornavirus capsid. The Fab induced conformational changes in regions of the virus capsid that control receptor binding. The antibody footprint is markedly different from the previous one identified by using a 12 Å structure. This work emphasizes the need for a high-resolution structure to guide mutational analysis and cautions against relying on older low-resolution structures even though they were interpreted with the best methodology available at the time.
INTRODUCTION
The characteristics of a successful host antibody response and the resulting neutralization mechanisms are poorly understood despite the key role played by antibodies in protecting animals against virus infection. Virus neutralization may be achieved through one or more mechanisms, which can differ widely, depending on the specific antibody. Neutralization mechanisms that have been defined include cross-linking or aggregation of capsids (1, 2), prevention of uncoating (3, 4), interference with receptor binding by sterically blocking the virus-receptor interaction (5–7), and induction of a capsid conformational change (8, 9). A better understanding of these and other specific antibody-virus interactions will be useful for improving vaccines that elicit protective antibodies.
Canine parvovirus (CPV) is a highly contagious virus that causes severe enteritis in dogs and wild carnivores, and antibodies are a key component of protective immunity to this virus (10, 11). The single-stranded DNA genome is packaged into a nonenveloped icosahedral capsid 26 nm in diameter. The virus is assembled with a major capsid protein, VP2, and a few copies of a minor structural protein, VP1, which contains a unique N-terminal peptide that is packaged within the capsid interior. All of the capsid subunits contain an eight-stranded antiparallel β-barrel motif commonly found in many virus structural proteins. Loops connecting the β strands make up most of the CPV surface. Distinctive “spikes” surrounding each icosahedral 3-fold axis mediate host receptor recognition and antigenicity (12–15). The CPV capsid binds to transferrin receptor type 1 on its host cells, relying on highly specific interactions with residues of the 3-fold spike (12, 13, 16, 17).
A panel of eight antibodies to CPV was previously characterized by using a structural analysis to complement binding and neutralization studies. Some of the Fabs, including Fab E, were shown to neutralize the virus at nonsaturating concentrations of <20 Fab molecules per capsid, whereas others were poorly neutralizing even at high concentrations of Fab molecules per binding site (18). The cryo-electron microscopy (cryo-EM) reconstructions previously described provided virus-Fab complex maps ranging from 9 to 18 Å in resolution (19). Web-based antibody models (WAM) were used to interpret these maps and to identify the antibody-binding surfaces or “footprints” on the capsid exterior (20). However, the 12 Å resolution cryo-EM map of the CPV capsid-Fab E complex did not show the interaction in detail or reveal a specific mechanism of neutralization (19).
Improvements in microscopy, image detection, and software have made it possible to obtain cryo-EM structures of atomic and near-atomic resolution, allowing the de novo building of atomic models (21–23). Here we redefine the CPV capsid-Fab E complex by solving the structure at near-atomic resolution. Virus building was initiated by first fitting the crystal structure, and the Fab was built de novo into density. The residues in the interaction interface were unambiguously identified even though steric collisions limit the binding of Fab to one Fab molecule per icosahedral 2-fold axis. A local resolution map revealed that Fab binding induced significant changes in the nearby 3-fold spikes, which became poorly resolved likely because of flexibility that does not exist in this region in wild-type virus structures. Thus, the high-resolution map reported here has provided additional information that reveals a likely mechanism of MAb and Fab neutralization.
MATERIALS AND METHODS
Cryo-EM data collection.
Capsids were purified by sucrose gradient centrifugation as previously described (12). The IgGs were purified from hybridoma supernatants by protein G chromatography, and the Fab was isolated after digestion with pepsin as described previously (18). Purified Fab E was incubated with CPV capsids at a ratio of four Fab molecules to each of the 60 binding sites on the capsid. The virus-Fab complex was incubated at room temperature for 1 h, and 3 μl of the complex was applied to a Quantifoil grid (Quantifoil Micro Tools GmbH, Jena, Germany), blotted to remove the excess, and plunge-frozen in a liquid ethane-propane mixture with a Mk III Vitrobot (FEI, Hillsboro, OR). Low-dose micrographs were recorded with an FEI Polara G2 microscope operating at 300 kV and a nominal magnification of 78,000× with defocus values ranging from −1.5 to −4.0 μm. Images were collected under the control of the EPU software program with an FEI Falcon 2 direct electron detector with a postcolumn magnification of 1.4×, yielding a calibrated pixel size at the sample of 1.37 Å.
Data processing.
Defocus values for the micrographs were estimated with the program ctffind4 (24). Particle selection was automated with the autopicking algorithm of the program Relion (25). According to the current single-particle reconstruction gold standard, two independent groups of particles were separately reconstructed by first sorting particles into two-dimensional (2D) class averages to exclude bad particles. This step resulted in a total of 47,562 particles that were sorted for final 3D refinement. A random model was generated with the program AUTO3DEM (26) and low-pass filtered at 60 Å to initiate the reconstruction. The contrast transfer function was fully corrected. Resolution was determined with the gold standard resolution determination in Relion. The map was sharpened with a B factor of 260 U to reach a final resolution of 4.1 Å (Table 1). Resolution for the virus and Fab alone in the CPV-FabE complex was determined by masking out each density separately with a mask with a soft edge of 10 pixels and measuring resolution with Bsoft (27). An absolute pixel size of 1.37 Å was assessed in Chimera (28) by scaling the crystal structure of CPV (PDB ID 1C8D) (14) to the density map. The CPV control map was processed by using the same approaches.
TABLE 1.
Cryo-EM statistics
| Measurement | CPV-Fab | CPV control |
|---|---|---|
| No. of micrographs | 1,424 | 828 |
| Defocus range (μm) | 1.5–4.0 | 1.1–5.6 |
| No. of particles selected | 86,950 | 94,432 |
| No. of particles used | 47,563 | 82,448 |
| Resolution range (Å) | 3.9–4.7 | 4.1 |
Virus and Fab building and interface determination.
The heavy chain of Fab E was built de novo into the density with the program Coot (29) by using the fitted WAM E positioned as a guide. When the initial model (WAM light chain plus de novo-built heavy chain) was completed, it was placed into the corresponding Fab density in conjunction with placing the virus structure into the virus density and both were refined simultaneously in Phenix. MolProbity was used to validate the structures and this procedure was iterated through refinement.
For the cross-validation test of overfitting, Phenix was used to randomly displace the atomic coordinates of the CPV and Fab models with a mean error value equal to 2. The models were refined into half of the data by the procedure described above. Fourier shell correlation (FSC) curves were generated with Bsoft (27) for each model against the map or half map used for refinement (Fig. 1D). Contacts between virus and Fab molecules were identified as residues that had the correct geometry and were separated by a distance of <5 Å in the previous low-resolution (12 Å) map or <4 Å in the experimental map. Contacts were identified by the CCP4 programs contact, NCONT, and Chimera (28, 30). The types of bonds between residues in the footprint were determined with the programs Chimera (28), PyMOL (The PyMOL Molecular Graphics System, version 1.7.4; Schrödinger LLC), and PISA (31). Rigid-body fitting was performed in independent experiments as described previously (32) with Fab E WAM model (Protein Data Bank [PDB] ID 3IY6), the de novo Fab model, and the CPV crystal structure (PDB ID 1C8D) with the program Situs (19, 33, 34). Footprints were generated with the program RIVEM (35).
FIG 1.

CPV in complex with neutralizing Fab E (B5A8). (A) Surface-rendered cryo-EM map of virus (black arrow, blue tints)-and-Fab (white arrow, green) complex with icosahedral symmetry axes labeled (2, 3, and 5) colored according to radius (color key at the bottom) to illustrate topology. (B) Central section through the complex map indicating the quality of the reconstruction and showing that the magnitude of the map density is different between the capsid and bound Fab. (C) Cryo-EM map density (gray mesh) has sufficiently high resolution to allow building of the alpha carbon backbone (green ball and stick) and placement of bulkier side chains. (D) As a cross-validation test against overfitting, two FSC curves are plotted. In blue is the final CPV-Fab model versus the final density map. In red is the model refined into half of the data versus the density map corresponding to half of the data.
Cloning and scFv production.
The heavy and light variable domains of MAb E were synthesized by joining a linker sequence of three repeats of Gly-Gly-Gly-Ser to form a single-chain variable fragment (scFv). This sequence was fused to the baculovirus gp68 signal sequence at the N terminus and also linked through a flexible linker to the Fc portion of human IgG1 with a 6His tag at the C terminus. The complete clones were prepared in pFastbac plasmids (Invitrogen, Carlsbad, CA). Bacmids were produced through recombination of the DH10Bac (Invitrogen) vector and the pFastbac donor plasmids, and baculoviruses were produced. After growth in Hi5 cells, the scFv-Fc was isolated with nitrilotriacetic acid-Ni resin and then purified with a Sephacryl S-200 chromatography column in phosphate-buffered saline (PBS).
Measurement of binding affinity.
Binding was measured with a BioLayer Interferometry (BLItz) system (ForteBio, Menlo Park, CA) (36) using biosensors that were protein A coated. Biosensors were first blocked and rehydrated with kinetics buffer (PBS with 0.01% bovine serum albumin, 0.02% Tween 20, and 0.005% NaN3) to minimize nonspecific binding. The timing series used was 300 s at the initial baseline in kinetics buffer, 300 s of scFv-Fc or IgG loading, a 60-s wash step in kinetics buffer, 300 s of viral capsid association, and 300 s of dissociation. Antibodies or scFv-Fcs were incubated to give consistent amounts of bound protein, with 0.4-nm loading shown. Viral capsids were incubated at 20 μg/ml with kinetics buffer.
Accession number(s).
The cryo-EM map of the CPV-Fab complex has been deposited in the EMDataBank (www.emdatabank.org/) under accession number EMD-6629. Models built for the virus and Fab have been deposited in the PDB under ID 3JCX.
RESULTS AND DISCUSSION
Purified virus was incubated with excess Fab molecules (240 Fab molecules per capsid—a virus binding site to Fab ratio of 1:4) to make complexes for the cryo-EM reconstruction (Fig. 1 and Table 1). In the 3D map, obvious densities of pairs of Fab molecules are located at each icosahedral 2-fold symmetry axis of the capsid and the high quality of the map is particularly evident in the central section. A global resolution of 4.1 Å was assessed for the unmasked virus-Fab cryo-EM map, which was sufficient to trace the alpha carbon backbone and locate bulkier side chains. Local calculations showed that the resolution of the core portions of the capsid shell were nearer a 3 Å resolution, whereas residues at the inner- and outermost radii had lower resolutions approaching 6 Å (Fig. 2). Masking routines were used to assess the resolution of the Fab and virus independently to confirm these variations. On the virus capsid, the 3-fold spikes had remarkably lower resolution than the other regions, suggesting flexibility of the loops making up the spikes (Fig. 2A). Resolution of the Fab density ranged from 4 Å in the heavy chain that is bound to the capsid to 6 Å in the light chain that is positioned away from the capsid surface (Fig. 2C). The magnitude of the Fab density was weaker than that of the density of the capsid, indicating that Fab did not occupy all of the 60 potentially available binding sites, and quantification of density suggests about 25 Fab molecules per capsid, on average.
FIG 2.

Local resolution estimations. (A) Surface-rendered cryo-EM map of the virus-Fab complex colored according to local resolution (color key at the far right). The map has regions that vary in resolution from 3 to 6 Å. The 3-fold spike region of the capsid is shown in the inset and illustrates these isolated islands of lowest resolution of the capsid shell. The identity of residue 426 (yellow sphere) is different in different strains of CPV. The loops composed of residues 88 to 97 and 219 to 227 (yellow and white arrows, respectively) likely have enhanced flexibility, which is induced by the bound Fab. (B) Surface-rendered cryo-EM map of the CPV capsid alone colored according to local resolution by the same color key as the complex map. The control map of the capsid shows consistently high resolution of ∼3 to 3.5, with no flexibility or low resolution at the 3-fold spike (black dashed line). (C) Density at the virus-Fab interface reveals that the Fab heavy chain (light blue arrow) is bound to the capsid surface (traced by a thin black line) and has better resolution than the light chain (dark blue arrow) that is oriented away from the capsid surface. (D) Central section of the local resolution map reveals regions of high resolution making up the capsid shell. (E) FSC curves showing resolution of Fab (blue), virus (red), and complex (green) maps that were calculated individually. The dotted line was drawn at 0.143 for gold standard resolution measurement.
An X-ray crystal structure of CPV (PDB ID 1C8D) was fitted into the cryo-EM density to begin building the virus structure (34). Adjustments of the atomic model were made manually where it protruded out of the cryo-EM density map, followed by real space refinement and validation with PHENIX and MolProbity, respectively (37, 38). This iterative approach continued throughout refinement, resulting in good structural statistics for the virus capsid as assessed by rotamer and Ramachandran outliers. For the variable domain of the Fab structure, the alpha carbon atoms of the heavy-chain domain were built de novo with local refinement algorithms in the program Coot. Amino acid side chains were added, and the heavy chain was fitted into the cryo-EM map density as a rigid body. By the same approach as with the virus capsid, the newly built Fab heavy chain was refined in real space by using PHENIX with applied secondary-structure restraints to optimize Ramachandran angles and rotamers. During the refinement process, the light chain of the WAM E model was used to initiate light-chain building and was refined as described above for the virus.
Because of the interlocking structure of CPV coat proteins, three different molecules of the major capsid protein were found to contribute to each Fab-binding interface. Fab heavy- and light-chain structures were therefore combined with the appropriate three copies of the virus coat protein, and the complex was refined together to minimize clashes between molecules while improving the atom positions within the density map. Refinement was done with the flexible fitting routine of Phenix. Finally, icosahedral symmetry was applied to the model for a round of refinement of the complex in the context of the intact icosahedral capsid. The final model resulted in 0% rotamer outliers and 1.6% Ramachandran outliers (Fig. 3). A cross-validation test was performed to assess for possible overfitting. The atomic coordinates of the output CPV-Fab structure were randomly displaced and refined into a density map reconstructed from half of the data. The FSC curve was calculated for each model against the map into which it was refined. The alignment of the two FSC curves attests to lack of overfitting for the final model (Fig. 1D).
FIG 3.

Final models of Fab and virus. (A) Superimposition of Fab (yellow) on the WAM model (blue) shows minor differences that map mostly to complementarity-determining region loop H1. (B) Superimposition of virus (yellow) on the crystal structure of CPV (blue) (PDB ID 1C8D) shows nearly identical structures (C) with a zoomed view of the region of major difference, which maps to the HI loop composed of residues 359 to 375. (D) There was no significant difference found at the capsid VP2 loop containing residue 300.
The refined capsid model showed only small differences from the crystal structure (root mean square deviation [RMSD] = 1.9 Å) (Fig. 3). A flexible loop of VP2, corresponding to residues 359 to 375, exhibited the greatest conformational change. This loop has previously been shown to change conformation because of changes in pH and the presence or absence of a divalent ion, most likely calcium, which binds close to the capsid surface (34). Fab E interacts directly with this calcium-binding site, making contact with one of the residues that coordinate the ion, VP2 Asp 237. No significant change was detected in the loop that contains VP2 residues 299 to 301, which are key players in the control of parvovirus tropism and confer differences in binding to the entry receptor. However, this loop maps to the 3-fold spike where the capsid resolution is poorest in the complex map. Such a local drop in resolution could be due to loss of density or to flexibility/movement in the loops. The regions of 3 Å resolution throughout the capsid core support good alignments during the reconstruction process and since none of the crystal structures (PDB IDs 1IJS, 2CAS, 4QYK, 4DPV, and 1C8D) (14, 34, 39–41) indicate flexibility of the 3-fold spike, we conclude that the conformational changes resulting in enhanced flexibility and movement of the 3-fold loops have likely been initiated by Fab binding (Fig. 2A).
A combined approach was used to identify interactions within the interface between the Fab and the virus in our 4.1 Å map (see Materials and Methods). All interactions but one were between the Fab heavy chain and the virus surface near the 3-fold spike (Table 2). Each Fab covered a surface area on the capsid of 45,080 Å2 and made contact with multiple residues within each of three symmetrically arrayed capsid proteins. Each of the three copies of the structural protein was predicted to participate in the total of six hydrogen bonds (Table 2), indicating that the Fab bound through multiple contacts with three coat proteins simultaneously. The angle of Fab binding across the icosahedral 2-fold axis shows that there are structural collisions between Fab constant domains, although the exact clashes were difficult to assess because of the weak density in this region of the Fab. Consequently, only 30 Fab molecules are capable of binding to a capsid, 1 at each of the 2-fold axis-related binding sites. However, as few as 10 Fab E molecules are sufficient for neutralization, which is likely accomplished by inducing the conformational changes that destabilize or change the receptor binding site at the 3-fold spike (18).
TABLE 2.
Virus-Fab contacts identified from the final modelsa
| VP2 protein and CPV residue | B5A8 residue | van der Waals | Hydrogen | Polar | Hydrophobic | Proximity |
|---|---|---|---|---|---|---|
| 1 | ||||||
| Arg 81 | Asp 155 | x | x | |||
| Asn 85 | Leu 204 | x | ||||
| Met 87 | Leu 204 | x | ||||
| Tyr 206 | x | x | x | |||
| Asp 88 | Tyr 206 | x | ||||
| Ala 91 | Tyr 206 | x | ||||
| Ile101 | Leu 204 | x | ||||
| Asn 231 | Leu 204 | x | ||||
| Ile 232 | Gly 203 | x | ||||
| Tyr 233 | Leu 204 | x | ||||
| Gly 203 | x | |||||
| His 234 | Pro 202 | x | ||||
| Gly 235 | Pro 202 | x | ||||
| Leu 131 | x | |||||
| Val 151 | x | |||||
| Thr 236 | Leu 131 | x | ||||
| Asp 237 | Leu 131 | x | ||||
| 2 | ||||||
| Pro 123 | Trp 153 | x | ||||
| Lys 387 | Trp 153 | x | x | |||
| Asn 154 | x | |||||
| Thr 390 | Asn 161 | x | ||||
| Thr 391 | Asn 161 | x | ||||
| Asn 554 | Ile 170 | x | ||||
| Val 555 | Leu 168 | x | ||||
| Ile 170 | x | |||||
| Asp 556 | Met 152 | x | ||||
| Arg 167 | x | |||||
| Leu 168 | x | |||||
| Asn 557 | Met 152 | x | ||||
| Gln 558 | Ser 131 | x | x | |||
| Val 151 | x | |||||
| Met 152 | x | |||||
| Phe 559 | Met 152 | x | ||||
| Trp 153 | x | |||||
| Asn 154 | x | |||||
| Asp 155 | x | |||||
| Asn 560 | Met 152 | x | ||||
| Trp 153 | x | x | ||||
| Tyr 561 | Trp 153 | x | x | |||
| Asn 154 | x | |||||
| Val 562 | Trp 153 | x | ||||
| Asn 154 | x | |||||
| Pro 563 | Asn 154 | x | ||||
| Ser 564 | Asn 154 | x | x | x | ||
| Asn 565 | Asp 155 | x | ||||
| Gly 568 | Asn 154 | x | ||||
| Lys 570 | Met 152 | x | ||||
| Trp 153 | x | x | ||||
| 3 | ||||||
| Gly 299 | Tyr 92* | x | ||||
| Asp 155 | x | |||||
| Gly 156 | x | |||||
| Asp 157 | x | |||||
| Ala 300 | Tyr 92* | x | ||||
| Asp 155 | x | |||||
| Gly 156 | x | x | ||||
| Asp 157 | x | |||||
| Glu 200 | x | |||||
| Leu 201 | x | |||||
| Thr 301 | Tyr 92* | x | ||||
| Glu 200 | x | |||||
| Asn 302 | Tyr 92* | x | ||||
| Trp 545 | Trp 153 | x | x | |||
| Asn 546 | Trp 153 | x |
Three copies of the VP2 capsid protein are listed with the Fab contacts that were identified by a distance of 4 Å. *, location on light chain of the Fab.
The residues identified in the virus-Fab interface included VP2 residue 300, which is the site of a previously identified virus escape mutation where the change from Ala to Asp abolishes Fab E binding (10). Introducing mutations into Fab E allowed us to test the capsid-binding capabilities of the altered antibody variable domains we constructed. The antibody E heavy and light chains of the variable region were expressed as a scFv linked to the Fc sequence of human IgG1. The scFv-Fc was then used to test the contribution to binding conferred by specific residues Asp 155, Leu 204, and Thr 205, which were predicted from the previous low-resolution structure to contribute to capsid binding (19). Asp 155→Ala-substituted scFv-Fc bound to capsids similarly to the wild type, but the Asp 155→Lys change reduced binding (Fig. 4A), suggesting that whereas the loss of a negative charge at this position in the antibody-binding site can be tolerated, the addition of a positive charge cannot. A contribution to binding by charged residues was also supported on the basis of the electrostatic potential found in the interface (Fig. 4B). There was a charged patch on the capsid composed mostly of residues 231 to 236 and 554 to 556, whereas the charge on the binding surface of the Fab included residue 155.
FIG 4.
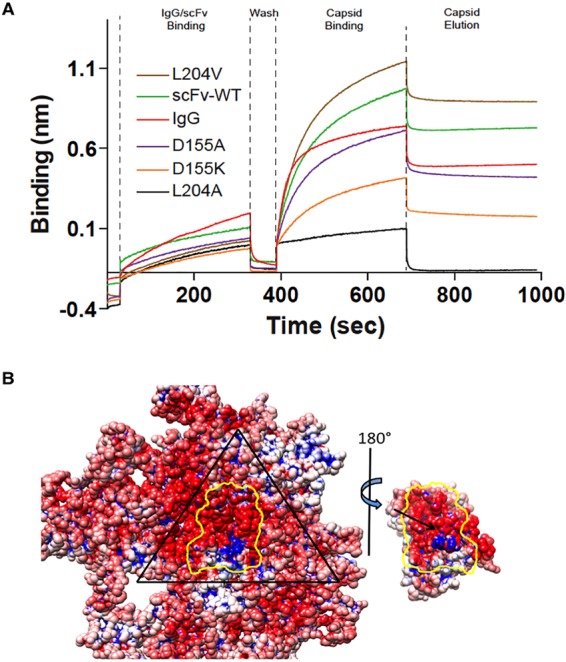
Binding between virus and Fab. (A) BLItz (36) profiles showing the binding of the IgG of MAb E, wild-type (WT) scFv-Fc, and the Leu 204→Ala, Leu 204→Val, Asp 155→Lys and Asp 155→Ala scFv-Fc mutant forms. Protein associated with the probe over time, and the interference with the reflective wavelength was measured in relative intensity units (nanometers) (see Materials and Methods). The time course procedure included association with the probe, washing, and capsid-binding and elution steps, as indicated at the top. (B) The three copies of the coat protein and the Fab variable domain contribute to the binding interface (outlined in yellow). One asymmetric unit of the virus capsid is outlined in black, and the electrostatic potential is shown in blue (+) and red (−) (42). Fab residue Asp 155 (black arrow) is located in the center of the footprint.
Fab residue Leu 204→Ala, within the antibody combining site, also prevented binding, but the Leu 204→Val mutation had no effect, suggesting that sufficient virus-Fab interactions can still take place with a Val occupying this site. Both the Fab Thr 205→Leu and Thr 205→Tyr mutations had no effect on capsid binding. The results of the mutational analysis complemented those of the structural analysis, as two of the antibody residues, Asp 155 and Leu 204, mapped to the combining site identified here. Although Thr 205 had been predicted to be in the Fab footprint by the previous structural study, it was not found to be in the binding interface in this higher-resolution analysis, explaining the lack of contribution to binding. Previously, we established a direct effect of cryo-EM map resolution on the correct identification of residues at the virus-protein interface (32). We showed that despite the application of rigorous fitting algorithms and proven methodologies, accurate positioning of the bound protein relative to the capsid depended on map resolution.
Previously, in the absence of a high-resolution Fab E crystal structure, a WAM was used for interpretation of Fab E binding (19, 20); however, the predicted footprint obtained by that approach was different from the experimental one identified here. To determine whether this discrepancy was due to the poorer resolution of the previous map or to the use of the homology model of Fab E (WAM E), WAM E was fitted into the new 4.1 Å cryo-EM map of the complex along with our atomic model of the virus. The Fab and virus were separately positioned and fitted together by using an iterative rigid-body fitting process to minimize clashes between the two molecules, maximize structures within densities, and minimize structures outside densities (33). The refined fit of the WAM into the high-resolution map superimposed on the previous WAM (fitted into the low-resolution map) with an RMSD of 14 Å (Fig. 5A), indicating significant differences in the position of the WAM due to the resolution of the maps. The newly fitted WAM was then used to define an antibody footprint on the capsid surface, which identified five of the same residues as in the previously described footprint (Fig. 5 and 6); however, there was disagreement among an additional four residues identified.
FIG 5.

Effect of resolution on virus-Fab interface identification based on the final model compared to previous models. (A) WAM E fitted into the previous low-resolution map (blue) (19) superimposed on WAM E fitted into the new high-resolution map (red). (B) The de novo Fab structure fitted into the previous low-resolution map (green) superimposed on the de novo Fab structure built into the new high-resolution map (yellow). (C, D) Contacts on the surface of the virus and Fab, respectively, are identified from the fitted structures, with colors corresponding to the ribbon color of each fitted Fab. The list of contacts on the virus surface also indicates which antibody complementarity-determining region (CDR) loop is the binding partner: L3, H1, H2, H3, or none (---). The CDR loops are also labeled on the Fab footprint.
FIG 6.

Virus-Fab complex footprints shown as roadmaps. The asymmetric unit of the viral capsid is composed of interlocking viral proteins (pastel colors) and shown as a stereographic projection of the surface amino acids. ϕ and θ angles represent the longitude and latitude of the viral surface, respectively (35). (A, B) Footprints of WAM E fitted into the previous low-resolution map (blue) and the new high-resolution map (red). (C, D) Footprints of the de novo Fab structure fitted into the previous low-resolution map (green) and the new high-resolution map (yellow).
The de novo Fab model built into our 4.1 Å map superimposed with the WAM with an RMSD of 9.1 Å, with the major difference occurring at the H1 and H2 loops of the heavy chain (Fig. 5). To test how much of the disagreement in antibody footprints might have been due to the use of the WAM, the de novo Fab structure was fitted into the low-resolution map (19). In this fit, 10 of the 38 capsid contacts were correctly identified and 3 capsid contacts were incorrectly identified (Fig. 5). The fit of WAM and the fit of de novo Fab to interpret the low-resolution maps produced similar footprints (Fig. 5 and 6). When all of the footprints were plotted as “roadmaps,” projections of the virus surface, it was obvious that both the low resolution of the maps and the use of a model affected the identification of contacts (Fig. 6). This aspect of our study highlights the importance of resolution for correctly determining protein-protein interactions of cryo-EM complex maps and presents a caution for reliance on previous low-resolution structural solutions to drive ongoing research.
We note that although recent advances in cryo-EM have propelled the field into atomic- and near-atomic-resolution structures, there remains a dependence on nuclear magnetic resonance analysis or crystallography for the structural determination of small molecules of <100 kDa. However, here we have successfully built the model of a 50-kDa Fab molecule, which is too small by itself to be solved by cryo-EM single-particle methods. By complexing Fab with CPV, the virus provided a multivalent macromolecular protein platform to present the 50-kDa Fab protein for structural solution by cryo-EM. This approach suggests a general approach to solving small protein structures by using larger symmetrical binding partners to hold the protein in complexes.
ACKNOWLEDGMENTS
This work was funded in part by Pennsylvania Department of Health CURE funds and by the Office of The Director, National Institutes of Health, under awards S10OD019995 and S10OD011986, as well as NIH grants R01AI107121 (S.H.), R01AI092571 (C.R.P.), and T32CA060395 (L.J.O.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We have no potential conflicts of interest to declare.
L.J.O. prepared all of the samples for cryo-EM and data collection. K.H. and S.I. prepared the antibody expression clones for analysis and analyzed their binding to the capsid. R.E.A. prepared samples, analyzed initial data, and collected data. A.M.M. and J.F.C. interpreted and collected cryo-EM data. L.J.O. performed image processing and model building. S.H. and L.J.O. analyzed the data and interpreted the structures. H.L. performed image processing and model building for the native virus map. L.J.O., C.R.P., J.F.C., and S.H. oversaw the projects, designed the experiments, and wrote and revised the manuscript.
REFERENCES
- 1.Brioen P, Dekegel D, Boeyé A. 1983. Neutralization of poliovirus by antibody-mediated polymerization. Virology 127:463–468. doi: 10.1016/0042-6822(83)90159-9. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi K, Morita Y, Urasawa T, Urasawa S. 1987. Cross-reactive neutralization epitopes on VP3 of human rotavirus: analysis with monoclonal antibodies and antigenic variants. J Virol 61:1726–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Che Z, Olson NH, Leippe D, Lee WM, Mosser AG, Rueckert RR, Baker TS, Smith TJ. 1998. Antibody-mediated neutralization of human rhinovirus 14 explored by means of cryoelectron microscopy and X-ray crystallography of virus-Fab complexes. J Virol 72:4610–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hewat EA, Blaas D. 1996. Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J 15:1515. [PMC free article] [PubMed] [Google Scholar]
- 5.Hewat EA, Marlovits TC, Blaas D. 1998. Structure of a neutralizing antibody bound monovalently to human rhinovirus 2. J Virol 72:4396–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katpally U, Wobus CE, Dryden K, Virgin HW, Smith TJ. 2008. Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J Virol 82:2079–2088. doi: 10.1128/JVI.02200-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith TJ, Olson NH, Cheng RH, Chase ES, Baker TS. 1993. Structure of a human rhinovirus-bivalently bound antibody complex: implications for viral neutralization and antibody flexibility. Proc Natl Acad Sci U S A 90:7015–7018. doi: 10.1073/pnas.90.15.7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lok S-M, Kostyuchenko V, Nybakken GE, Holdaway HA, Battisti AJ, Sukupolvi-Petty S, Sedlak D, Fremont DH, Chipman PR, Roehrig JT, Diamond MS, Kuhn RJ, Rossmann MG. 2008. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat Struct Mol Biol 15:312–317. doi: 10.1038/nsmb.1382. [DOI] [PubMed] [Google Scholar]
- 9.Lee H, Brendle SA, Bywaters SM, Guan J, Ashley RE, Yoder JD, Makhov AM, Conway JF, Christensen ND, Hafenstein S. 2015. A cryo-electron microscopy study identifies the complete H16.V5 epitope and reveals global conformational changes initiated by binding of the neutralizing antibody fragment. J Virol 89:1428–1438. doi: 10.1128/JVI.02898-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strassheim ML, Gruenberg A, Veijalainen P, Sgro JY, Parrish CR. 1994. Two dominant neutralizing antigenic determinants of canine parvovirus are found on the threefold spike of the virus capsid. Virology 198:175–184. doi: 10.1006/viro.1994.1020. [DOI] [PubMed] [Google Scholar]
- 11.Parrish CR. 1995. Pathogenesis of feline panleukopenia virus and canine parvovirus. Baillieres Clin Haematol 8:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hueffer K, Parrish CR. 2003. Parvovirus host range, cell tropism and evolution. Curr Opin Microbiol 6:392–398. doi: 10.1016/S1369-5274(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 13.Hafenstein S, Palermo LM, Kostyuchenko VA, Xiao C, Morais MC, Nelson CDS, Bowman VD, Battisti AJ, Chipman PR, Parrish CR, Rossmann MG. 2007. Asymmetric binding of transferrin receptor to parvovirus capsids. Proc Natl Acad Sci U S A 104:6585–6589. doi: 10.1073/pnas.0701574104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llamas-Saiz AL, Agbandje-McKenna M, Parker JS, Wahid AT, Parrish CR, Rossmann MG. 1996. Structural analysis of a mutation in canine parvovirus which controls antigenicity and host range. Virology 225:65–71. doi: 10.1006/viro.1996.0575. [DOI] [PubMed] [Google Scholar]
- 15.Tsao J, Chapman MS, Agbandje M, Keller W, Smith K, Wu H, Luo M, Smith TJ, Rossmann MG, Compans RW. 1991. The three-dimensional structure of canine parvovirus and its functional implications. Science 251:1456–1464. doi: 10.1126/science.2006420. [DOI] [PubMed] [Google Scholar]
- 16.Parker JS, Murphy WJ, Wang D, O'Brien SJ, Parrish CR. 2001. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J Virol 75:3896–3902. doi: 10.1128/JVI.75.8.3896-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Govindasamy L, Hueffer K, Parrish CR, Agbandje-McKenna M. 2003. Structures of host range-controlling regions of the capsids of canine and feline parvoviruses and mutants. J Virol 77:12211–12221. doi: 10.1128/JVI.77.22.12211-12221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson CDS, Palermo LM, Hafenstein SL, Parrish CR. 2007. Different mechanisms of antibody-mediated neutralization of parvoviruses revealed using the Fab fragments of monoclonal antibodies. Virology 361:283–293. doi: 10.1016/j.virol.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hafenstein S, Bowman VD, Sun T, Nelson CDS, Palermo LM, Chipman PR, Battisti AJ, Parrish CR, Rossmann MG. 2009. Structural comparison of different antibodies interacting with parvovirus capsids. J Virol 83:5556–5566. doi: 10.1128/JVI.02532-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitelegg NR, Rees AR. 2000. WAM: an improved algorithm for modelling antibodies on the WEB. Protein Eng 13:819–824. doi: 10.1093/protein/13.12.819. [DOI] [PubMed] [Google Scholar]
- 21.Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JLS, Subramaniam S. 2015. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science 348:1147–1151. doi: 10.1126/science.aab1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, Stark H. 2015. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520:567–570. doi: 10.1038/nature14275. [DOI] [PubMed] [Google Scholar]
- 23.Campbell MG, Veesler D, Cheng A, Potter CS, Carragher B. 2015. 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. eLife 4:e06380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rohou A, Grigorieff N. 2015. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol 192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheres SHW. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol 157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Organtini LJ, Makhov AM, Conway JF, Hafenstein S, Carson SD. 2014. Kinetic and structural analysis of coxsackievirus B3 receptor interactions and formation of the A-particle. J Virol 88:5755–5765. doi: 10.1128/JVI.00299-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wriggers W, Chacón P. 2001. Modeling tricks and fitting techniques for multiresolution structures. Structure 9:779–788. doi: 10.1016/S0969-2126(01)00648-7. [DOI] [PubMed] [Google Scholar]
- 34.Simpson AA, Chandrasekar V, Hébert B, Sullivan GM, Rossmann MG, Parrish CR. 2000. Host range and variability of calcium binding by surface loops in the capsids of canine and feline parvoviruses. J Mol Biol 300:597–610. doi: 10.1006/jmbi.2000.3868. [DOI] [PubMed] [Google Scholar]
- 35.Xiao C, Rossmann MG. 2007. Interpretation of electron density with stereographic roadmap projections. J Struct Biol 158:182–187. doi: 10.1016/j.jsb.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Concepcion J, Witte K, Wartchow C, Choo S, Yao D, Persson H, Wei J, Li P, Heidecker B, Ma W, Varma R, Zhao L-S, Perillat D, Carricato G, Recknor M, Du K, Ho H, Ellis T, Gamez J, Howes M, Phi-Wilson J, Lockard S, Zuk R, Tan H. 2009. Label-free detection of biomolecular interactions using BioLayer interferometry for kinetic characterization. Comb Chem High Throughput Screen 12:791–800. doi: 10.2174/138620709789104915. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu H, Rossmann MG. 1993. The canine parvovirus empty capsid structure. J Mol Biol 233:231–244. doi: 10.1006/jmbi.1993.1502. [DOI] [PubMed] [Google Scholar]
- 40.Organtini LJ, Allison AB, Lukk T, Parrish CR, Hafenstein S. 2015. Global displacement of canine parvovirus by a host-adapted variant: structural comparison between pandemic viruses with distinct host ranges. J Virol 89:1909–1912. doi: 10.1128/JVI.02611-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie Q, Chapman MS. 1996. Canine parvovirus capsid structure, analyzed at 2.9 A resolution. J Mol Biol 264:497–520. doi: 10.1006/jmbi.1996.0657. [DOI] [PubMed] [Google Scholar]
- 42.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. 2007. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]