

ABSTRACT
Mouse hepatitis virus strain A59 infection of mice is a useful tool for studying virus-host interaction during hepatitis development. The NS2H126R mutant is attenuated in liver replication due to loss of phosphodiesterase activity, which the wild-type (WT) virus uses to block the 2′,5′-oligoadenylate synthetase (OAS)-RNase L (RNase L) antiviral pathway. The activation of RNase L by NS2H126R is cell type dependent and correlates with high basal expression levels of OAS, as found in myeloid cells. We tested the hypothesis that the resident liver macrophages, Kupffer cells (KC), represent the cell type most likely to restrict NS2H126R and prevent hepatitis. As found previously, A59 and NS2H126R replicate similarly in hepatocytes and neither activates RNase L, as assessed by an rRNA degradation assay. In contrast, in KC, A59 exhibited a 100-fold-higher titer than NS2H126R and NS2H126R induced rRNA degradation. Interestingly, in liver sinusoidal endothelial cells (LSEC), the cells that form a barrier between blood and liver parenchymal cells, NS2H126R activates RNase L, which limits viral replication. Similar growth kinetics were observed for the two viruses in KC and LSEC from RNase L−/− mice, demonstrating that both use RNase L to limit NS2H126R replication. Depletion of KC by gadolinium(III) chloride or of LSEC by cyclophosphamide partially restores liver replication of NS2H126R, leading to hepatitis. Thus, during mouse hepatitis virus (MHV) infection, hepatitis, which damages the parenchyma, is prevented by RNase L activity in both KC and LSEC but not in hepatocytes. This may be explained by the undetectable levels of RNase L as well as by the OASs expressed in hepatocytes.
IMPORTANCE Mouse hepatitis virus infection of mice provides a useful tool for studying virus-host interactions during hepatitis development. The NS2H126R mutant is attenuated in liver replication due to loss of phosphodiesterase activity, by which the wild-type virus blocks the potent OAS-RNase L antiviral pathway. RNase L activation by NS2H126R is cell type dependent and correlates with high basal expression levels of OAS, as found in myeloid cells. We showed that the hepatocytes that comprise the liver parenchyma do not activate RNase L when infected with NS2H126R or restrict replication. However, both Kupffer cells (KC) (i.e., the liver-resident macrophages) and the liver sinusoidal endothelial cells (LSEC) which line the sinusoids activate RNase L in response to NS2H126R. These data suggest that KC and LSEC prevent viral spread into the parenchyma, preventing hepatitis. Furthermore, hepatocytes express undetectable levels of OASs and RNase L, which likely explains the lack of RNase L activation during NS2H126R infection.
INTRODUCTION
Mouse hepatitis virus (MHV) belongs to the order Nidovirales, family Coronaviridae, and genus Betacoronavirus. Coronaviruses are enveloped, nonsegmented positive-strand RNA viruses (1). MHV strain A59 induces moderate to severe hepatitis in infection of mice (2); replication and the consequent liver pathogenesis are dependent on the 2′,5′-phosphodiesterase (PDE) activity of the viral accessary protein NS2 (3).
The 2′,5′-oligoadenylate (2-5A) synthetase (OAS)-RNase L pathway is involved in potent interferon (IFN)-induced antiviral activity (4). Upon infection by diverse viruses, including the human-liver-pathogenic hepatitis C virus (5, 6), double-stranded RNA (dsRNA) is detected by OAS proteins. Four mouse OAS species (OAS1a/g, OAS2, OAS3, and OASL2) and 3 human OAS species (OAS1, OAS2, and OAS3) can bind dsRNA in vitro and be activated to generate 2-5A (7), the latter binding to and promoting activation and dimerization of RNase L, which results in both cellular and viral RNA degradation and thus in inhibition of viral replication (7, 8).
The PDE (NS2) of MHV cleaves 2-5A and inhibits the activation of RNase L (3). An A59 mutant, NS2H126R, which encodes a catalytically inactive PDE, replicates to a minimal extent in the mouse liver and causes minor to no liver damage during infection (3). Previous studies have illuminated that the activation of the OAS-RNase L antiviral pathway is cell type dependent. Bone marrow-derived macrophages (BMM) and bone marrow-derived dendritic cells (BMDC) as well as microglia, brain-resident macrophages, limit NS2H126R replication by activating the OAS-RNase L pathway. The basal mRNA expression levels of OASs differ among the cell types, and high levels of OAS correlate with activation of the pathway (9, 10).
While the liver is known as a digestive organ, it also serves as an immunological organ (11, 12). The liver is composed of two populations of cells, including approximately 80% liver parenchymal cells (hepatocytes) and 20% nonparenchymal cells (NPC). About 10% of NPC are Kupffer cells (KC), or liver macrophages, and 50% of NPC are liver sinusoidal endothelial cells (LSEC) (11). We found previously that NS2H126R infection failed to induce RNase L activation in hepatocytes and that NS2H126R replicated to an extent similar to that seen with A59 in that cell type (9). However, little is known about the activation of the OAS-RNase L pathway in the other liver cell types and, specifically, in which cell type RNase L activation limits NS2H126R replication, although previous studies implicated a protective role of KC (3). Here, we isolated the various primary liver cells and infected them with WT A59 and NS2H126R to assess the activation of RNase L as well as to quantify viral replication from these cells. We found that KC and LSEC but not hepatocytes can limit NS2H126R replication, indicating they have an active OAS-RNase L pathway, and that this is likely due to differential levels of expression of RNase L as well as OAS among these liver cell types. To further study the protective role of KC and LSEC, we performed in vivo cell depletion in mice and showed that depletion of KC or LSEC enhances NS2H126R liver replication and pathogenicity.
MATERIALS AND METHODS
Cell lines, mice, and viruses.
Mouse L2 fibroblasts were cultured as described previously (14). Recombinant coronaviruses inf-MHV-A59 (A59) and inf-NS2H126R (NS2H126R) have been described previously (14, 15). C57BL/6 (B6) mice were purchased from the National Cancer Institute (Frederick, MD) and bred in the University of Pennsylvania animal facility. All procedures were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
Isolation of primary liver cells.
Mice were euthanized with CO2, and the livers were perfused. Briefly, the animal was opened to expose the inferior vena cava (IVC) and portal vein and a catheter was inserted into the IVC and connected to a pump filled with dissociation buffer (NaCl [8 g/liter], KCl [0.4 g/liter], NaH2PO4·H2O [78 mg/liter], Na2HPO4 [120.45 mg/ml], HEPES [2,380 mg/liter], Na2CO3 [350 mg/liter], EGTA [190 mg/liter], glucose [900 mg/liter], pH 7.4). The portal vein was cut immediately to allow the buffer (20 to 30 ml) to flow through the liver.
For hepatocytes, the liver was digested with collagenase IV (0.05%) (Sigma-Aldrich) in liver digestion buffer (NaCl [8 g/liter], KCl [0.4 g/liter], NaH2PO4·H2O [78 mg/liter], Na2HPO4 [120.45 mg/ml], HEPES [2,380 mg/liter], Na2CO3 [350 mg/liter], phenol red [6 mg/liter], CaCl2·2H2O [560 mg/liter], pH 7.4). After digestion, the liver was removed from the mouse and cells were dissociated from Glisson's capsule and were filtered through a 100-μm-pore-size cell strainer (Becton Dickinson [BD]). The cells were centrifuged at 40 × g for 3 min at 4°C, and the pellet was resuspended in digestion buffer containing 1 mg/ml DNase I (Roche). The cells were centrifuged and washed with minimum essential medium Eagle (MEM; Sigma-Aldrich) three times and were resuspended in William's E media (Gibco) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine. The cells were placed on plates coated with rat tail collagen (Sigma-Aldrich). Four hours after plating, the cells were washed with phosphate-buffered saline (PBS) and used for experimentation.
Kupffer cells (KC) were isolated from mixed primary liver cell cultures using previously described methods (16). Briefly, after perfusion with dissociation buffer, the liver was digested with collagenase I (Sigma-Aldrich) (0.05%) in digestion buffer supplemented with 50 μg/ml trypsin inhibitor (Sigma-Aldrich). Cells were harvested and washed three times with MEM and cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco; 10566) supplemented with 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, 100 μM β-mercaptoethanol, 10 μg/ml insulin, 10 mM HEPES, and 50 μg/ml gentamicin, in a flask coated with rat tail collagen (1 × 107 cells per T175 flask). The next day, the flasks were shaken to remove the dead cells; on day 4 and day 7, fresh medium was added; and at days 9 to 12 postplating, the KC were harvested by shaking and then selected by binding to a petri dish (BD). KC were recovered by TrpLE select enzyme (Invitrogen) digestion and plated for experiments.
The isolation protocol used for the liver sinusoidal endothelial cells (LSEC) was adapted from previously described methods (17). Briefly, after perfusion with dissociation buffer, the liver was digested with collagenase I (0.025%) and collagenase II (0.025%) (Sigma-Aldrich) in digestion buffer supplemented with 50 μg/ml trypsin inhibitor (Sigma-Aldrich). After digestion, the liver was removed and cells were dissociated from Glisson's capsule in preservation buffer (NaCl [4.15 g/liter], KCl [0.25 g/liter], HEPES [1.12 g/liter], NaOH [0.24 g/liter], bovine serum albumin [BSA] [10 g/liter]) and filtered through a 100-μm-pore-size cell strainer (BD). The supernatant was centrifuged at 60 × g for 2 min three times to remove parenchymal cells. The supernatant was further centrifuged at 1,350 × g and 4°C for 10 min to harvest the nonparenchymal cells (NPC). The NPC were resuspended in preservation buffer and were applied on top of a 25%/50% Percoll (GE Amersham) bilayer. The cells were centrifuged at 1,350 × g and 4°C for 10 min. LSEC and KC were collected from the interface between the two density layers. Cells were washed with preservation buffer and resuspended in RPMI 1640 medium (Gibco) supplemented with100 U/ml penicillin and 100 mg/ml streptomycin. Cells then were plated onto a petri dish and incubated for 8 min to remove the KC which attached to the surface; the removal of KC was repeated once more. The supernatant containing the LSEC was harvested and centrifuged at 1,350 × g and 4°C for 10 min. The pellet was resuspended in RPMI 1640 medium (Gibco) supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin and placed onto rat tail collagen-coated plates. Two hours after plating, the cells were washed vigorously with ice-cold PBS twice. The cells remaining on the plates were used for further experiments.
Antibodies.
Mouse anti-human hepatocyte nuclear factor 4α (HNF-4α) (cross-reactive with mouse HNF-4α) monoclonal antibody (MAb) (R&D) (5 μg/ml) and goat anti-mouse IgG conjugated to Alexa Fluor 488 (Invitrogen) (1:400) were used to detect HNF-4α by immunofluorescence assay (IFA); rat anti-mouse CD68 MAb (Serotech) (1:100) conjugated to Alexa Fluor 488 was used to detect CD68 by IFA. OAS1A MAb (clone E-2; Santa Cruz) (1:200) and OAS2 (clone G-9; Santa Cruz) (1:200), OAS3 (clone D-7; Santa Cruz) (1:200), and goat anti-RNase L (clone T-16; Santa Cruz) (1:200) as well as anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) (Thermo-Fisher) (1:1,000), goat anti-mouse IgG-horseradish peroxidase (IgG-HRP) (Santa Cruz) (1:5,000), and donkey anti-goat IgG-HRP (1:5,000) secondary antibodies were used to detect the primary antibodies of the appropriate species.
Immunolabeling.
Cells were fixed in PBS containing 4% paraformaldehyde (Bio-Rad) and blocked with 2% BSA, and immunolabeling with anti-HNF-4α antibodies was used to detect hepatocytes; immunolabeling with anti-CD68 antibodies conjugated with Alexa Fluor 488 was used to distinguish KC (CD68 positive) and LSEC (CD68 negative). Primary anti-HNF-4α antibody was detected with goat anti-mouse Alexa Fluor 488 (Invitrogen). DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) (Invitrogen) was used to stain the nucleus. Fluorescence was visualized with a Nikon Eclipse 2000E-U fluorescence microscope, and images were acquired using Nikon NIS-Element BR software (Nikon).
Low-density lipoprotein (LDL) uptake assay of LSEC.
After isolation, cells were cultured in RPMI 1640 without FBS, medium containing 5 μg/ml of LDL conjugated with Alexa Fluor 488 (Invitrogen) was added, the cells were incubated at 37°C for 4 h, and, at 30 min before imaging, 1 μg/ml of Hoechst stain solution (Sigma-Aldrich) was added to the medium. Fluorescence was visualized with a Nikon Eclipse 2000E-U fluorescence microscope, and images were acquired using Nikon NIS-Element BR software (Nikon).
Virus growth kinetics.
Hepatocytes (1.5 × 105 cells per well in 24-well plates), KC (1 × 105 cells per well in 48-well plates), or LSEC (5.0 × 105 cells per well in 48-well plates) were infected with A59 or NS2H126R (multiplicity of infection [MOI] = 1 PFU/cell); at the indicated time points, cells and supernatants were harvested together and then frozen (−80°C) and thawed three times; and, finally, viruses were titrated by L2 cell plaque assay (18).
KC and LSEC depletion and MHV infection.
Gadolinium(III) chloride was used deplete KC from the livers of mice. B6 mice were intravenously injected with 20 mg/kg of body weight of gadolinium(III) chloride–100 μl of saline solution (19). Cyclophosphamide was used to deplete LSEC from the livers of mice. B6 mice were intraperitoneally (i.p.) injected with 300 mg/kg of cyclophosphamide–200 μl saline solution. At 24 h postinjection, mice were infected intrahepatically (i.h.) with 200 PFU of virus. On day 5 postinfection, mice were sacrificed and livers were harvested for virus titration (18) and histological analysis (3).
Western blotting.
Cells were mock treated or treated with 1,000 U of alpha interferon (IFN-α) (PBL) overnight, harvested, washed in PBS, and lysed with NP-40 buffer (1% NP-40, 2 mM EDTA, 10% glycerol, 150 mM NaCl, 50 mM Tris [pH 8.0] with protease inhibitor cocktail [Roche]). Cell lysates were mixed with 4× Laemmli buffer, boiled at 95°C for 5 min, and analyzed by electrophoresis on 4% to 15% gradient SDS gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes, which were treated with 5% nonfat milk–TBST (Tris-buffered saline with 0.5% Tween 20) blocking buffer for 1 h, followed by incubation overnight at 4°C with antibodies diluted into TBST. Membranes were then washed three times with TBST and incubated with secondary antibodies for 1 h at room temperature, washed three times with TBST, and then incubated with SuperSignal West Dura Extended Duration substrate (Thermo), and the signal was detected using an Amersham Imager 600 (GE).
Histology.
Livers were isolated and fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned. Sections were stained with hematoxylin and eosin (H&E) as described previously (3).
Statistical analysis.
The two-tailed Student t test was performed to determine statistically significant differences for the results of in vitro experiments. The Mann-Whitney test was used to analyze differences in virus titers in different mouse tissues. Any undetectable titers from in vitro and in vivo infections were entered as the limit of detection value for each experiment. All data were analyzed by using Prism software (GraphPad Software, Inc., CA).
RESULTS
Isolation and purification of Kupffer cells (KC), liver sinusoidal endothelial cells (LSEC), and hepatocytes from mouse liver.
To study the role of individual liver cell types in limiting NS2H126R replication, we purified each of the three major cell types, representing >95% of liver cells, and cultured them in vitro. Specifically, we focused on the parenchymal cells (the hepatocytes) and the nonparenchymal Kupffer cells (KC) and liver sinusoidal endothelial cells (LSEC), using established isolation and culture methods, as described elsewhere (16, 17). To assess the purity of the liver cell preparations, we used the nonspecific stain DAPI in all the tests to identify the nuclei. To identify hepatocytes, we stained with an antibody that recognizes hepatocyte nuclear factor 4α (HNF4α), a marker for hepatocytes (green) (20). Over 99% of the cells in the hepatocyte preparation were HNF-4α positive, as seen in Fig. 1A to C, which show the blue and green stains and a merged image. KC cells, the resident liver macrophages, were detected by immunostaining for CD68 (Fig. 1D), a KC-specific marker (21). In the KC-purified preparation, we found that >95% of the cells were CD68-positive cells. LSEC were isolated from total liver nonparenchymal cells. LSEC readily take up LDL (low-density lipoprotein) (22). Therefore, we incubated the LSEC with Alexa Fluor 488-conjugated LDL for 4 h and then monitored the cells. Over 95% of the cells were Alexa Fluor 488 positive (Fig. 1F). To confirm there was little to no KC cell contamination, the LSEC were stained with anti-CD68 antibodies; less than 1% of the cells were CD68-positive cells (Fig. 1E). These results indicate that our isolation methods for hepatocytes, KC, and LSEC yielded highly purified products.
FIG 1.
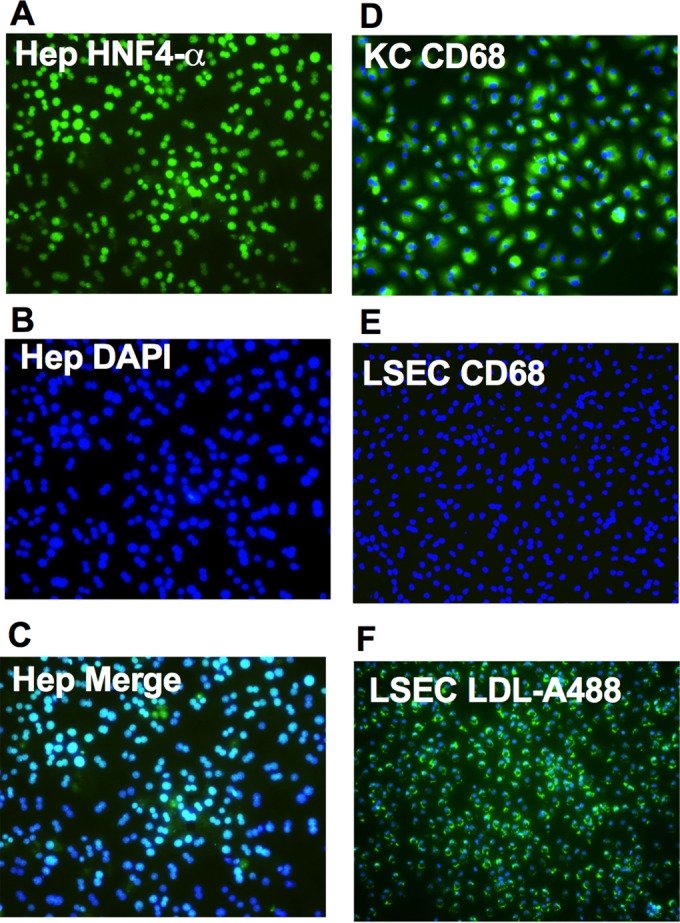
Isolation and characterization of hepatocytes (Hep), KC, and LSEC. Hep, KC, and LSEC were isolated as described in Materials and Methods. (A) Hep were stained with mouse anti-human HNF-4α followed by secondary Alexa Fluor 488-conjugated goat anti-mouse antibody (green). (B) DAPI staining (blue). (C) The two stains merged. (D and E) KC (D) and LSEC (E) were stained with Alexa Fluor 488-conjugated anti-CD68 antibodies (green) and with DAPI (blue) to identify the nuclei. (F) LSEC were incubated with Alexa Fluor 488-conjugated LDL (green) in serum-free media at 5 μg/ml for 4 h, and Hoechst (blue) was added into the media to stain the nucleus 30 min before imaging.
Kupffer cells (KC) and liver sinusoidal endothelial cells (LSEC) but not hepatocytes limit NS2H126R replication in vitro through activation of RNase L.
To determine which cells limit NS2H126R replication, hepatocytes, KC, and LSEC derived from B6 (WT) and RNase L knockout (KO) mice were infected in vitro with WT A59 and mutant NS2H126R virus. As observed previously in B6 hepatocytes, A59 and NS2H126R replicated similarly (9). However, in B6 KC and LSEC, A59 virus was measured at 10- to 100-fold-higher titers than NS2H126R at 15 and 24 h postinfection (Fig. 2). When KC and LSEC from RNase L KO mice were infected with NS2H126R, the virus replicated to a titer similar to that seen with A59. These results suggest that both KC and LSEC limit NS2H126R virus replication by activation of the OAS-RNase L pathway, as evidenced by the fact that replication in both these cell types was robust in cells from RNase L KO mice and minimal in cells from B6 animals. To directly determine whether RNase L is activated in KC and LSEC, cultures of each liver cell type, as well as BMM, were infected with A59 and NS2H126R and, at 12 h (KC and BMM), 13 h (LSEC), or 15 h (hepatocytes) postinfection, cells were lysed and total cellular RNA was harvested and analyzed for degradation by the use of a Bioanalyzer, as shown in Fig. 3. Degradation of rRNA was observed in KC, LSEC, and BMM but not in hepatocytes, consistent with the replication phenotype (Fig. 2).
FIG 2.
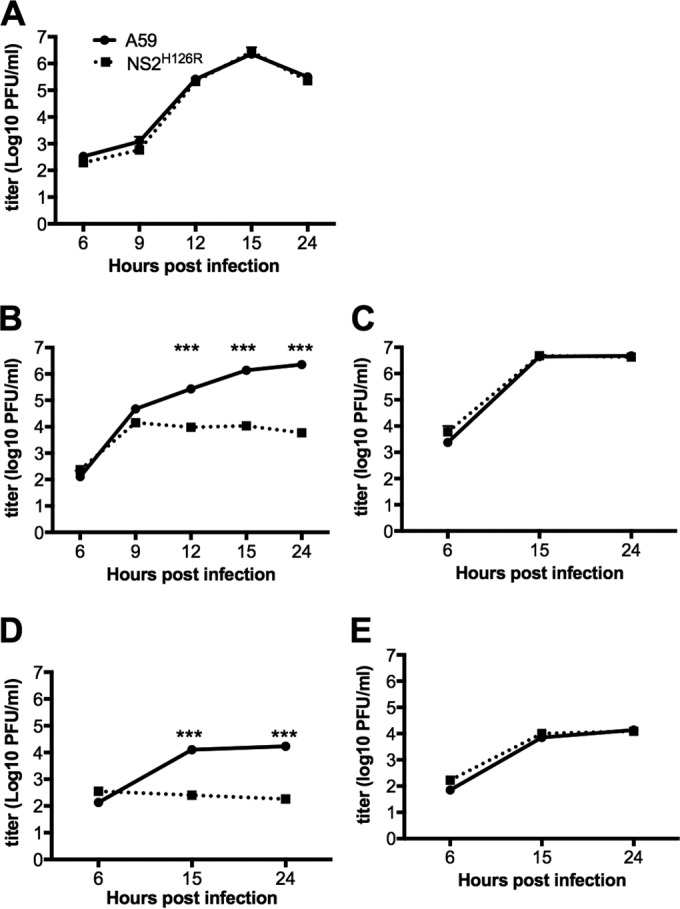
The replication of NS2H126R is limited in KC and LSEC isolated from B6 mice but not from RNase L−/− mice. Hepatocytes (Hep) from B6 mice (A), KC from B6 mice (B), KC from RNase L−/− mice (C), LSEC from B6 mice (D), and LSEC from RNase L−/− mice (E) were infected with A59 and NS2H126R (MOI = 1, n = 3). At the indicated time points, infectious virus was quantified by plaque assay on L2 cells from the combined cell lysates and supernatants. Data shown were analyzed by the two-tailed Student t test and expressed as means ± standard deviations (SD) (***, P<0.001) and are from one representative of two independent experiments.
FIG 3.
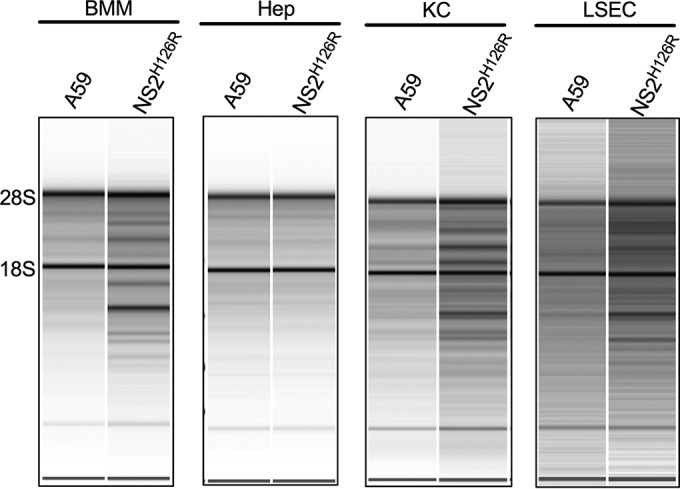
RNase L is activated in BMM, KKC, and LSEC but not in hepatocytes (Hep) during infection with NS2H126R. BMM, hepatocytes, KC, and LSEC were infected with MHV A59 and ns2H126R (MOI = 1). At 12 h (BMM and KC), 13 h (LSEC), or 15 h (Hep) postinfection, RNA was harvested and analyzed by a Bioanalyzer for rRNA integrity. The positions of 28S and 18S rRNA are indicated. Data shown are from one representative of two independent experiments.
Depletion of either KC or LSEC from mouse liver partially restores NS2H126R replication and liver pathogenicity.
To determine the role of KC and LSEC during MHV infection of intact animals, KC or LSEC were depleted from the livers of B6 mice by intravenous injection of gadolinium(III) chloride (KC depletion) (19) or by intraperitoneal injection of cyclophosphamide (LSEC depletion) (19, 23). The depletion of KC was monitored by India ink uptake, which occurs only with KC (24), and the disruption of the endothelial barrier was monitored by morphology as assessed by electronic microscopy (23). We observed that >80% of KC or >50% of LSEC were depleted from mouse liver (data not shown). Following depletion of KC from B6 mice, NS2H126R replicated to a 100- to 500-fold-higher level than in the nondepleted livers. The depletion of LSEC showed a less dramatic increase in virus titer than KC depletion, but the titers were still significantly higher than that in the control mice (Fig. 4A). Liver pathogenicity was monitored by H&E staining of liver sections (Fig. 4B). Necrosis was readily apparent in livers from KC- and LSEC-depleted samples but not in those from the nondepleted animals. These results suggest that both the KC and LSEC protect the liver from coronavirus infection in vivo.
FIG 4.
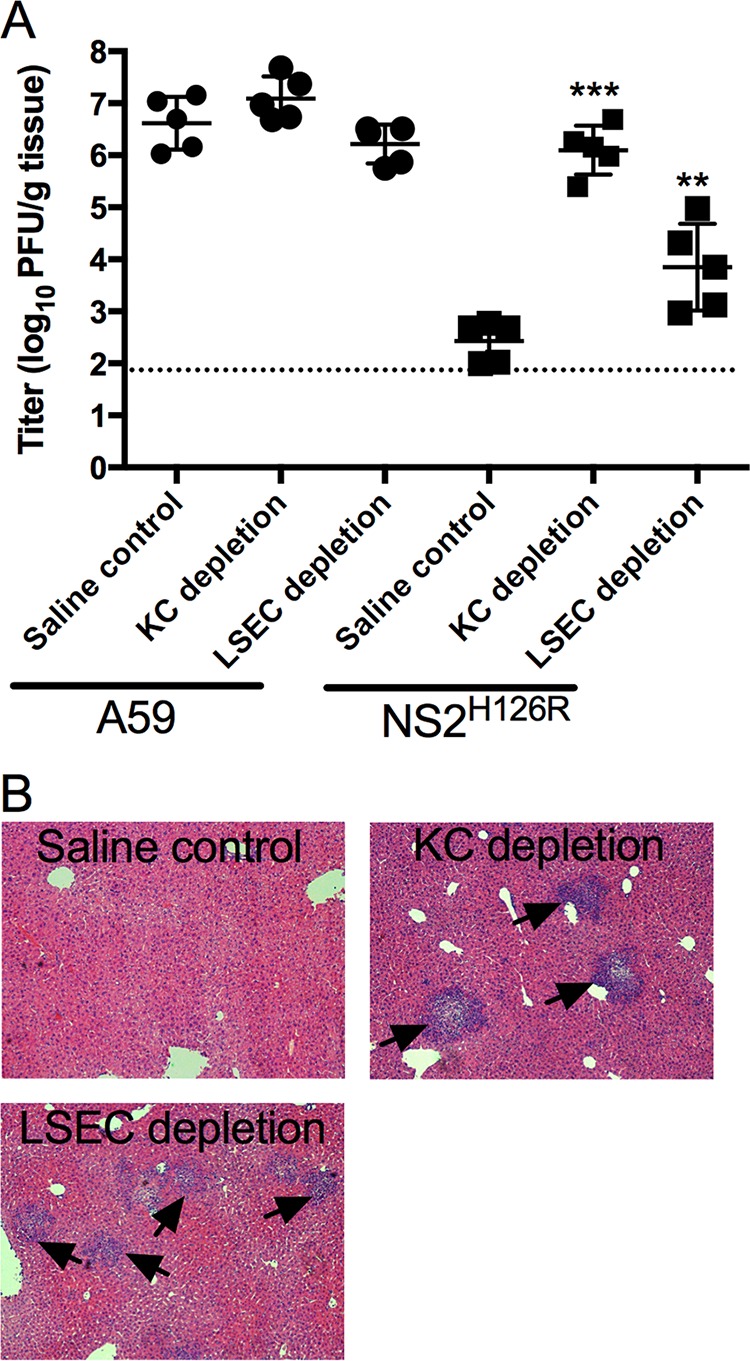
Depletion of KC or LSEC partially restores NS2H126R replication and liver pathogenicity. B6 mice were intravenously injected with 20 mg/kg of gadolinium(III) chloride (KC depletion) or intraperitoneally injected with 300 mg/kg of cyclophosphamide (LSEC depletion) or with saline solution as a control (n = 5 for each group). Mice were infected with 200 PFU of virus intrahepatically at 24 h postinjection and sacrificed 5 days postinfection. (A) Virus was quantified from liver lysates by plaque assay on L2 cells. The data were analyzed by the Mann-Whitney test and expressed as means ± SD (**, P < 0.01; ***, P < 0.001 [titers in depleted mice compared to the saline control]). (B) Liver sections from NS2H126R-infected mice were stained with hematoxylin and eosin for histological analysis. Arrows indicate foci of pathology. Data shown are from one representative of two independent experiments.
Hepatocytes express undetectable levels of RNase L and OAS.
Previous studies have indicated that the activation of the OAS-RNase L pathway by NS2H126R is cell type dependent and independent of virus-induced IFN (3, 9). High basal levels of OAS mRNA and protein expression correlate with activation of RNase L, while RNase L expression levels were similar across the cell types examined, including several primary central nervous system (CNS)-derived primary cells as well as BMM and BMDC (10). Since we found that, among liver cells, the OAS-RNase L pathway can be activated in KC and LSEC but not in hepatocytes, we hypothesized that KC and LSEC might have higher basal expression levels of OAS genes than hepatocytes. Thus, we assessed the basal and IFN-induced expression levels of OAS1a, OAS2, OAS3, and RNase L protein in hepatocytes, KC, and LSEC by Western blotting. OAS1a was detected in KC and LSEC with or without IFN treatment (Fig. 5) but not in hepatocytes. OAS2 was not detected in any cell type without IFN treatment; however, OAS2 was induced to a detectable level by IFN in KC. Basal expression of OAS3 was detected in KC but not in hepatocytes and LSEC; however, upon IFN treatment, OAS3 was further induced in KC and detected in LSEC but not hepatocytes. The results are consistent with our previous data, in that myeloid cells express higher basal levels of OAS genes and murine OAS mRNA is induced by IFN treatment (9, 10, 25). RNase L is constitutively expressed in many types of the cells and is not generally induced by IFN treatment (9, 10, 25). Surprisingly, while we detected RNase L in KC and LSEC, RNase L expression was below the level of detection in hepatocytes. Consistent with the immunoblot data, RNase L mRNA levels as quantified by quantitative reverse transcription PCR (qRT-PCR) were also 100- to 1,000-fold lower in hepatocytes than in KC and LSEC (data not shown), suggesting that there is very tight control of this pathway in this cell type.
FIG 5.
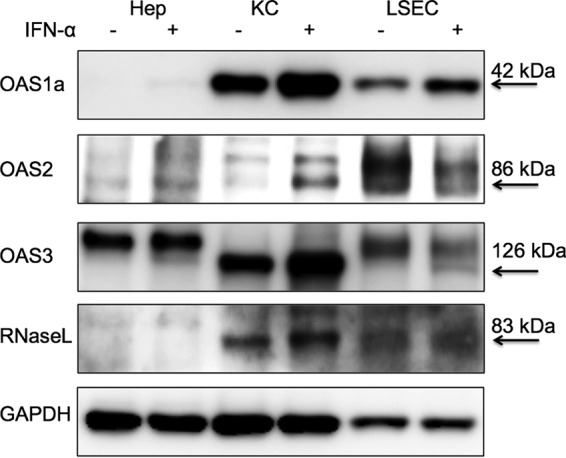
Expression levels of OAS1a, OAS2, OAS3, and RNase L were below the level of detection in hepatocytes. Hepatocytes (Hep), KC, and LSEC were mock treated or treated with IFN-α (1,000 U/ml) overnight. Cells were lysed, and proteins were analyzed by immunoblotting with antibodies against OAS1a, OAS2, OAS3, RNase L, and GAPDH. The positions and molecular masses of the indicated proteins are indicated with arrows. Data shown are from one representative of two independent experiments. The additional bands detected with OAS2 antibody in all cell types and with OAS3 antibody in Hep and LSEC are not induced by IFN as expected for OAS proteins and are nonspecific (10).
DISCUSSION
Based on previous findings showing that the OAS-RNase L pathway can be activated in BMM and BMDC as well as microglia during NS2 H126R infection (9, 10), it is not surprising that the KC also activate RNase L in response to viral infection. KC can be divided into two populations; one population consists of cells that are yolk sac derived and develop in the liver before the formation of bone marrow, and the other population consists of cells that are myeloid derived (26). A previous study showed that the yolk sac-derived KC play roles in immunosuppression whereas myeloid-derived KC are more immune active (26). We would predict that the myeloid-derived cells would be more readily activated to degrade RNA during viral infection, and future studies will include separating these types of KC and determining whether the two populations show differences in the activation of the OAS-RNase L pathway during infection.
LSEC and KC form a barrier between the parenchymal hepatocytes and the blood (11, 12). Previous studies showed that depletion of KC from rat or mouse liver improves the efficacy of gene therapy mediated by lentivirus and adenovirus vectors (24). We have shown here that depletion of KC or LSEC leads to a partial restoration of NS2H126R liver replication, demonstrating that KC and LSEC protect the liver from infection by limiting viral replication through activation of OAS-RNase L antiviral pathways. These data are consistent with a previous publication that reported that IFN signaling in myeloid cells was crucial for protection from MHV-induced hepatitis (27) and extend that finding to show that RNase L activity specifically can restrict MHV NS2 mutant replication in the liver. Indeed, this was the first study to have found that KC and LSEC use the OAS-RNase L pathway to protect the mouse from viral liver infection. It will be important to determine if this pathway limits other viral infections of the liver.
A key characteristic of LSEC is fenestration; these cells form pores with diameters of 100 to 200 nm (28). Our results suggest that, while viruses need to cross the fenestration to infect hepatocytes, MHV either cannot cross the fenestration or can cross only inefficiently. Our results suggest that LSEC limit MHV NS2H126R liver spread by activation of the OAS-RNase L antiviral pathway as well as by formation of a physical barrier. We previously observed in the primary cells derived from the central nervous system that, while activation of the OAS-RNase L pathway occurs in microglia, which are cells of myeloid lineage, nonmyeloid cells such as neurons or astrocytes could not be activated (9). LSECs represent the first example of nonmyeloid cells in which rRNA degradation and inhibition of viral replication are observed in NS2H126R infection. This may reflect their role in protecting the liver parenchyma from viral invasion.
RNase L is constitutively expressed in many types of cells, including myeloid and nonmyeloid cells, and expression is not induced by IFN treatment (10, 25). While OAS genes are induced by IFN, the activation of the OAS-RNase L pathway in myeloid cells is not dependent on induction of IFN during viral infection (10, 25). Rather, it is the high basal expression level of OAS proteins that plays a key role in activation of the pathway (10). We found that, by reducing the levels of basal OAS gene expression in BMM, we were able to prevent RNase L activation (10). Thus, we hypothesized that lack of RNase L activation in hepatocytes would be due to low basal expression levels of OAS genes. We were surprised to find undetectable levels of RNase L as well as of OAS1a, OAS2, and OAS3 in hepatocytes, which would provide an explanation for the lack of RNA degradation observed in virus-infected cells. This was the first cell type in which we found that a lack of activation of the OAS-RNase pathway correlated with low expression of RNase L as well as low levels of basal expression of OAS. We speculate that, since the liver has many pathogens and various forms of debris flowing though the sinusoids, this may induce type I IFN expression and, subsequently, activation of RNase L, which can lead to apoptosis (29). Thus, parenchymal hepatocytes tightly control the OAS-RNase L pathway, which may protect them from pathogen-induced cell death, which can result from RNase L activation.
ACKNOWLEDGMENTS
We thank Ekihiro Seki, Maryna Perepelyuk, and Rebecca Wells for advice on isolating and culturing primary liver cells and Yan Wang for help with the intravenous catheterization.
REFERENCES
- 1.Cavanagh D. 1997. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol 142:629–633. [PubMed] [Google Scholar]
- 2.Weiss SR, Leibowitz JL. 2011. Coronavirus pathogenesis. Adv Virus Res 81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong B, Silverman RH. 1995. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem 270:4133–4137. doi: 10.1074/jbc.270.8.4133. [DOI] [PubMed] [Google Scholar]
- 5.Han JQ, Barton DJ. 2002. Activation and evasion of the antiviral 2′-5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA 8:512–525. doi: 10.1017/S1355838202020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Washenberger CL, Han JQ, Kechris KJ, Jha BK, Silverman RH, Barton DJ. 2007. Hepatitis C virus RNA: dinucleotide frequencies and cleavage by RNase L. Virus Res 130:85–95. doi: 10.1016/j.virusres.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kakuta S, Shibata S, Iwakura Y. 2002. Genomic structure of the mouse 2′,5′-oligoadenylate synthetase gene family. J Interferon Cytokine Res 22:981–993. doi: 10.1089/10799900260286696. [DOI] [PubMed] [Google Scholar]
- 8.Justesen J, Hartmann R, Kjeldgaard NO. 2000. Gene structure and function of the 2′-5′-oligoadenylate synthetase family. Cell Mol Life Sci 57:1593–1612. doi: 10.1007/PL00000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao L, Birdwell LD, Wu A, Elliott R, Rose KM, Phillips JM, Li Y, Grinspan J, Silverman RH, Weiss SR. 2013. Cell-type-specific activation of the oligoadenylate synthetase-RNase L pathway by a murine coronavirus. J Virol 87:8408–8418. doi: 10.1128/JVI.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birdwell LD, Zalinger ZB, Li Y, Wright PW, Elliott R, Rose KM, Silverman RH, Weiss SR. 2016. Activation of RNase L by murine coronavirus in myeloid cells is dependent on basal Oas gene expression and independent of virus-induced interferon. J Virol 90:3160–3172. doi: 10.1128/JVI.03036-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racanelli V, Rehermann B. 2006. The liver as an immunological organ. Hepatology 43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 12.Crispe IN. 2009. The liver as a lymphoid organ. Annu Rev Immunol 27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 13.Reference deleted.
- 14.Roth-Cross JK, Stokes H, Chang G, Chua MM, Thiel V, Weiss SR, Gorbalenya AE, Siddell SG. 2009. Organ-specific attenuation of murine hepatitis virus strain A59 by replacement of catalytic residues in the putative viral cyclic phosphodiesterase ns2. J Virol 83:3743–3753. doi: 10.1128/JVI.02203-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarz B, Routledge E, Siddell SG. 1990. Murine coronavirus nonstructural protein ns2 is not essential for virus replication in transformed cells. J Virol 64:4784–4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitani H, Takenouchi T, Sato M, Yoshioka M, Yamanaka N. 24 May 2011. A simple and efficient method to isolate macrophages from mixed primary cultures of adult liver. J Vis Exp doi: 10.3791/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Øie CI, Appa RS, Hilden I, Petersen HH, Gruhler A, Smedsrød B, Hansen JB. 2011. Rat liver sinusoidal endothelial cells (LSECs) express functional low density lipoprotein receptor-related protein-1 (LRP-1). J Hepatol 55:1346–1352. doi: 10.1016/j.jhep.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Gombold JL, Hingley ST, Weiss SR. 1993. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J Virol 67:4504–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. 2005. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65. doi: 10.1172/JCI200522675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Ning G, Duncan SA. 2000. Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev 14:464–474. [PMC free article] [PubMed] [Google Scholar]
- 21.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, Crispe IN. 2007. Kupffer cell heterogeneity: functional properties of bone marrow-derived and sessile hepatic macrophages. Blood 110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling W, Lougheed M, Suzuki H, Buchan A, Kodama T, Steinbrecher UP. 1997. Oxidized or acetylated low density lipoproteins are rapidly cleared by the liver in mice with disruption of the scavenger receptor class A type I/II gene. J Clin Invest 100:244–252. doi: 10.1172/JCI119528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malhi H, Annamaneni P, Slehria S, Joseph B, Bhargava KK, Palestro CJ, Novikoff PM, Gupta S. 2002. Cyclophosphamide disrupts hepatic sinusoidal endothelium and improves transplanted cell engraftment in rat liver. Hepatology 36:112–121. doi: 10.1053/jhep.2002.33896. [DOI] [PubMed] [Google Scholar]
- 24.van Til NP, Markusic DM, van der Rijt R, Kunne C, Hiralall JK, Vreeling H, Frederiks WM, Oude-Elferink RP, Seppen J. 2005. Kupffer cells and not liver sinusoidal endothelial cells prevent lentiviral transduction of hepatocytes. Mol Ther 11:26–34. doi: 10.1016/j.ymthe.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Li YZ, Banerjee S, Wang YY, Goldstein SA, Dong BH, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A 113:2241–2246. doi: 10.1073/pnas.1519657113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. 2012. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 27.Cervantes-Barragán L, Kalinke U, Züst R, König M, Reizis B, López-Macías C, Thiel V, Ludewig B. 2009. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J Immunol 182:1099–1106. doi: 10.4049/jimmunol.182.2.1099. [DOI] [PubMed] [Google Scholar]
- 28.Elvevold K, Smedsrod B, Martinez I. 2008. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol 294:G391–G400. [DOI] [PubMed] [Google Scholar]
- 29.Castelli JC, Hassel BA, Maran A, Paranjape J, Hewitt JA, Li XL, Hsu YT, Silverman RH, Youle RJ. 1998. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ 5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]