

ABSTRACT
To survive and replicate within a host, many viruses have evolved strategies that target crucial components within the apoptotic cascade, leading to either inhibition or induction of cell apoptosis. Enterovirus 71 (EV71) infections have been demonstrated to impact the mitochondrial apoptotic pathway and induce apoptosis in many cell lines. However, the detailed mechanism of EV71-induced apoptosis remains to be elucidated. In this study, we report that EV71 2B protein (2B) localized to the mitochondria and induced cell apoptosis by interacting directly with and activating the proapoptotic protein Bax. 2B recruited Bax to the mitochondria and induced Bax conformational activation. In addition, mitochondria isolated from 2B-expressing cells that were treated with a recombinant Bax showed increased Bax interaction and cytochrome c (Cyt c) release. Importantly, apoptosis in cells with either EV71 infection or 2B expression was dramatically reduced in Bax knockdown cells but not in Bak knockdown cells, suggesting that Bax played a pivotal role in EV71- or 2B-induced apoptosis. Further studies indicate that a hydrophobic region of 18 amino acids (aa) in the C-terminal region of 2B (aa 63 to 80) was responsible for the location of 2B in the mitochondria. A hydrophilic region of 14 aa in the N-terminal region of 2B was functional in Bax interaction and its subsequent activation. Moreover, overexpression of the antiapoptotic protein Bcl-XL abrogates 2B-induced release of Cyt c and caspase activation. Therefore, this study provides direct evidence that EV71 2B induces cell apoptosis and impacts the mitochondrial apoptotic pathway by directly modulating the redistribution and activation of proapoptotic protein Bax.
IMPORTANCE EV71 infections are usually accompanied by severe neurological complications. It has also been postulated that the induction of cell apoptosis resulting from tissue damage is a possible process of EV71-related pathogenesis. In this study, we report that EV71 2B protein (2B) localized to the mitochondria and induced cell apoptosis by interacting directly with and activating the proapoptotic protein Bax. This study provides evidence that EV71 induces cell apoptosis by modulating Bax activation and reveals important clues regarding the mechanism of Cyt c release and mitochondrial permeabilization during EV71 infection.
INTRODUCTION
Enterovirus 71 (EV71) is an RNA icosahedral virus that belongs to the human enterovirus species A of the genus Enterovirus within the Picornaviridae family (1, 2). EV71 is thought to be one of the main pathogens that cause foot, hand, and mouth disease (HFMD) in young children (3, 4). In recent years, outbreaks of EV71-related HFMD have been reported in Southeast and East Asia, including Taiwan, Malaysia, Singapore, Japan, and China (5, 6). Particularly, over one million EV71-related HFMD cases were reported each year in China since 2008, including hundreds of fatal cases per year (7). EV71 infections are usually accompanied by severe neurological complications such as aseptic meningitis, acute flaccid paralysis, encephalitis, and other rarer manifestations (4, 8). It has also been postulated that toxic inflammatory cytokines in conjunction with the induction of cell apoptosis resulting from tissue damage are possible processes of pathogenesis (9–11).
Apoptosis can be triggered by two distinct signaling cascades: the mitochondrial apoptosis pathway, which needs the disruption of the mitochondrial transmembrane (TM) potential, and the extrinsic cell apoptosis pathway, which is initiated by the activation of cell death receptors (12, 13). The extrinsic cell death pathway involves the activation of caspase-8 through binding to the adaptor protein Fas-associated protein, which in turn activates caspase-3 to facilitate cell death (14, 15). The mitochondrial apoptotic pathway usually involves a variety of pro- and antiapoptotic proteins of the Bcl-2 family which act via at least one of four conserved Bcl-2 homology domains present (16, 17). The antiapoptotic proteins Bcl-2, Bcl-w, Mcl-1, Bfl-1, and Bcl-XL contain all four Bcl-2 homology domains (BH1 to -4) (17, 18). The proapoptotic Bcl-2 proteins Bim, Bid, Bad, Bik, Noxa, and Bmf contain only the BH3 domain (BH3-only proteins) and are often responsible for conveying the initial death signal (16, 19). The proapoptotic Bcl-2 proteins Bak and Bax possess BH1 to BH3 and are required for the induction of apoptosis via the mitochondrial pathway (20, 21). In most cells, Bax is normally localized in the cytosol or loosely associated with the outer mitochondrial membrane (OMM), whereas Bak is localized mostly in the OMM and remains inactive in nonapoptotic cells (22). The BH3 domain of Bax, which is essential for its proapoptotic activity and interaction with Bcl-2, is masked in the hydrophobic core of the protein as well as in the inactive Bax in the cytoplasm (23, 24). Following cytotoxic stimulation, Bax undergoes a series of conformational changes which lead to its translocation to the mitochondria, oligomerization, and integration into the mitochondrial membranes and eventually induce apoptosis (25, 26). Bak resides in the OMM in association with Mcl-1 and Bcl-XL, which occupy the dimerization and killing domain BH3 of Bak (25, 27). Upon activation, Bak is released from Mcl-1 and Bcl-XL, and the BH3 domain is displaced for oligomerization. This leads to the mitochondria releasing cytochrome c (Cyt c) and other key molecules that facilitate apoptosome formation, which activates caspase-9 and the downstream death programs (21, 28).
Viruses have evolved diverse strategies to mediate apoptosis to ensure their continued propagation and/or spread (29). Many viruses encode proapoptotic proteins that target crucial components within the apoptotic cascade to induce apoptosis at the end of their replication cycles (30, 31). Examples of virus-encoded proapoptotic proteins include the following: virus protein R (Vpr) of human immunodeficiency virus type 1 (HIV-1), which causes rapid dissipation of mitochondrial transmembrane potential (Δψm) and the release of inter membrane space proteins through direct interactions with the adenine nucleotide translocator (ANT) or voltage-dependent anion channel (VDAC), components of the mitochondrial permeability transition pore complex (32, 33); Bax, which binds to ANT and facilitates Vpr-mediated mitochondrial membrane potential but is inhibited by Bcl-2 overexpression and permeability transition pore complex (PTPC) inhibitors (34); HIV-1 protease, which processes caspase-8 and converts Bid into its activated form (tBid) (35); the NS4A protein of hepatitis C virus (HCV), which localizes to the mitochondria and alters the mitochondrial intracellular distribution, which is followed by the dissipation of Δψm and Cyt c release (36); and severe acute respiratory syndrome coronavirus (SARS-CoV) protein 7A, which induces apoptosis by inhibiting Bcl-XL (37). On the other hand, many viruses also encode antiapoptotic proteins to evade or delay the early onset of apoptosis. These include M11L of myxoma virus, E1B 19K of adenovirus, F1L and E1B-19K of vaccinia virus, and ORF125 of poxvirus, all of which inactivate Bak and/or Bax to inhibit apoptosis (38–41).
EV71 infection induces classic signs of apoptosis, such as the efflux of Cyt c from mitochondria and subsequent cleavage of caspase-9, in all infected cells (42, 43). A mitochondrial pathway of apoptosis mediated by the activation and cleavage of caspase-9 has been proven to be a main pathway in EV71-induced apoptosis (42). In this study, we showed that the EV71 2B protein localized to the mitochondria and activated the mitochondrial cell death pathway by directly interacting with and inducing conformational changes in the proapoptotic protein Bax. Cells lacking Bax but not Bak became resistant to the cytotoxic effect of 2B expression or EV71 infection. A hydrophilic region of 14 amino acids (aa) in the N-terminal region of 2B was crucial for Bax interaction and activation. Thus, EV71 2B represents a novel viral apoptotic protein that can interacts with Bax to induce Bax activation and trigger the apoptotic program.
MATERIALS AND METHODS
Virus isolates, cell lines, and virus infection.
The prototype enterovirus 71 (EV71) BrCr strain was a gift from Qi Jin (Institute of Pathogen Biology, Chinese Academy of Medical Sciences, Beijing, People's Republic of China). Human rhabdomyosarcoma (RD) cells and human cervical (HeLa) cells were propagated and maintained in either modified Eagle's medium or double modified Eagle's medium supplemented with antibiotics (penicillin and streptomycin) and 10% fetal bovine serum (Invitrogen, CA) at 37°C in the presence of 5% CO2.
To generate a HeLa cell line that stably overexpresses Bcl-XL–HA (HeLa-Bcl-XL), the full-length human cDNA for Bcl-XL was cloned into the restriction site of the pcDNA3.0-HA plasmid after digesting with EcoRI/NotI. The recombinant plasmid or pcDNA3.0-HA was then transfected into HeLa cells by using the Fu-GENE transfection reagent. Transfected cells were selected under the antibiotic G418 (800 μg ml−1). The expression of Bcl-XL–HA in stable transfectants was confirmed by Western blot analysis.
Virus infection was performed as described before (44). In short, semiconfluent monolayers of HeLa cells were infected with EV71. After adsorption for 30 min at 37°C, the cells were washed twice with phosphate-buffered saline (PBS) and overlaid with Dulbecco's modified Eagle's medium (DMEM) containing 10% calf serum. Cells were cultured at 37°C in the presence of 5% CO2.
Reagents and antibodies.
The protease inhibitor cocktail was obtained from Sigma-Aldrich, MO. Rabbit anti-Bak antibody and rabbit monoclonal antibodies against Bax6A7 (specifically recognizing activated Bax protein with an exposed N terminus), BaxNT (recognizing both native and activated Bax), Bid, Bim, Bcl-2, Bcl-XL, Cox IV, mitochondrial outer membrane translocator protein (TSPO), and Mcl-1 were obtained from Epitiomics, CA. Fu-GENE transfection reagent was obtained from Roche, IN. Rabbit anti-Cox IV polyclonal antibody, mouse anti-green fluorescent protein (anti-GFP), mouse anti-β-actin, mouse anti-His, and mouse anti-Flag monoclonal antibodies, fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG antibody, horseradish peroxidase-conjugated anti-mouse IgG, and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG, used in immunofluorescence studies and Western blot analyses, were all obtained from Santa Cruz Biotechnology, CA. Annexin V-FITC was obtained from Biyotime, China.
Cell viability analysis, apoptosis assay, and measurement of mitochondrial membrane potential.
Cell viability was analyzed as described before (45). Annexin V-FITC (Molecular Probes) was used to analyze cell apoptosis according to the manufacturer's instructions. In short, cells were harvested and resuspended in annexin V binding buffer at a concentration of 1 × 106 cells/ml. One hundred microliters of the suspension (105 cells) was mixed with annexin V-FITC (0.1 g/ml) and incubated for 15 min at room temperature (RT) in the dark. The annexin V-FITC fluorescence was measured with a BD FACSCalibur instrument (Becton Dickinson). A minimum of 30,000 cells were analyzed for each sample. Data were analyzed with CellQuest software (Becton Dickinson). Annexin V-FITC fluorescence was then determined as a percentage of the control value.
Changes in mitochondrial membrane potential in cells were quantified by staining the cells with tetramethylrhodamine ethyl ester (TMRE) (Molecular Probes). Cells were stained by incubating them in medium containing 0.2 μmol TMRE for 20 min at 37°C. After washing thrice with PBS, TMRE fluorescence was examined by flow cytometry.
Mitochondrion purification and fractionation, detection of Cyt c release, and Bax cross-linking.
Mitochondria were prepared as described before (46). In brief, the cells were washed three times with PBS, scraped from the dishes, and centrifuged at 500 × g for 5 min. The pellet was resuspended in 2 ml of cold mitochondrion isolation buffer consisting of 0.3 M sucrose, 1 mM EGTA, 5 mM morpholinepropanesulfonic acid (MOPS), 5 mM KH2PO4, and 0.1% (wt/vol) bovine serum albumin (BSA) (pH 7.5) in the presence of protease inhibitors and then homogenized in a glass homogenizer. Disrupted cells were centrifuged at 3,000 × g for 5 min to pellet the unlysed cells and nuclei. The supernatant was centrifuged further at 14,000 × g for 20 min at 4°C to obtain the crude mitochondrion fraction.
Subfractionation of mitochondria was conducted as described before (47). Briefly, the mitochondria were resuspended in 70 mM sucrose and 10 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES)-KOH (pH 7.5) and then incubated on ice for 20 min. The osmotic strength was adjusted to 0.3 M sucrose, and after a 20-min incubation on ice, the suspension was centrifuged at 18,000 × g for 15 min, yielding a supernatant containing the inner mitochondrial membrane, the outer mitochondrial membrane, and a mitoplast pellet. The supernatant was centrifuged at 200,000 × g for 10 min to separate the outer membranes (pellet). The resuspended pelleted mitoplasts were ruptured by freeze-thawing and then centrifuged at 18,000 × g for 15 min to obtain the inner membrane fractions (supernatant).
To monitor Cyt c release, purified mitochondria were incubated with the recombinant protein Bax with or without Bcl-XL for 30 min at 25°C. The mitochondria were pelleted by centrifugation at 8,000 × g for 10 min, solubilized in radioimmunoprecipitation assay (RIPA) buffer, and analyzed by immunoblotting with antibodies to Cyt c, Bax, or activated Bax.
For Bax cross-linking, pelleted mitochondria were resuspended in the conjugation buffer PBS (pH 7.2) and subjected to cross-linking using 0.2 mM bismaleimidohexane (BMH) for 2 h at 4°C. Cross-linking was quenched in a quenching solution at a final concentration of 30 mM by incubating for 15 min at room temperature. Protein samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blotting with an anti-Bax antibody.
siRNAs and transient transfection.
Small interfering RNAs (siRNAs) that target human Bax and Bak and nontargeting siRNAs (siControl) were chemically synthesized. Transient transfections using Lipofectamine 3000 (Invitrogen) were conducted according to the manufacturer's instructions. In brief, HeLa cells were plated in culture dishes and allowed to grow for 24 h to 90% confluence. A mixture of Opti-MEM medium and Lipofectamine 3000 was incubated for 5 min at RT and was then incubated with the siRNA (50 nM) or recombinant plasmid (2 μg/ml) for 10 min at RT. These mixtures containing siRNA or recombinant plasmid were then added to the wells. At 24 h after transfection, the medium was changed, and analyses were performed at the indicated time after transfection. Gene silencing and protein expression after transient transfection with siRNA were verified by detecting the appropriate proteins in Western blots.
Western blotting, confocal microscopy, and immunoprecipitation.
Western blotting and pulldown assays were performed as described before (48). For confocal microscopy, cells were grown on coverslips and either infected with EV71 or transfected with the recombinant plasmid pGFP-2B, pGFP-Bak, or pGFP. Cell mitochondria were stained using MitoTracker Red (50 nmol) according to the manufacturer's instructions. The stained cells were fixed in paraformaldehyde in phosphate buffer (pH 7.4) for 30 min. Following three PBS washes and then permeabilization with 0.1% NP-40 in PBS for 15 min, the cells were incubated with 5% BSA in PBS for 1 h at room temperature. After 3 washes with PBS, the cells were incubated for 1 h with the selected antibodies. After washing thrice with PBS, they were incubated for 45 min with the relevant secondary antibodies and examined under an inverted fluorescence or a confocal laser scanning microscope.
For coimmunoprecipitation experiments, compartments were immobilized on anti-Bax(NT), anti-Bax6A7, or anti-Flag monoclonal antibody-conjugated agarose beads. After washing three times with PBS and once with lysis buffer, the beads were incubated with HeLa cell lysate overnight at 4°C with gentle rocking. After washing with lysis buffer, the incubated beads were boiled and proteins in the supernatant were collected and subjected to Western blotting.
Plasmid construction and protein expression.
EV71 genomic RNA was extracted from the supernatant of virus-infected RD cells using a virus genome extraction kit. Single-stranded cDNA was then synthesized from the purified virus RNA by reverse transcription (Promega). EV71 2B or truncated 2B with a deletion from aa 1 to 22 [2B(Δ1–22)], aa 1 to 35 [2B(Δ1–35)], or aa 22 to 35 [2B(Δ22–35)] was amplified from the cDNA by PCR, separated by electrophoresis on 1% agarose gels, gel purified, and cloned into the EcoRI and SalI sites located behind the cytomegalovirus (CMV) promoter of the pCMV-Flag vector. A complementary oligonucleotide of 2B(Δ22–35) was chemically synthesized and used to produce DNA duplexes. These oligonucleotides were annealed and ligated to the EcoRI and SalI sites of the pCMV-Flag vector. To construct the vectors pGFP-2B, pGFP-2B(1–80), and pGFP-2B(1–63), which express the 2B protein and its truncation variants and with each containing an N-terminal GFP fusion, 2B, 2B aa 1 to 63, and 2B aa 1 to 80 were amplified from the pCMV-2B-Flag and cloned into the pGFP-C1 vector after digesting with EcoRI/SalI. Recombinant Bax and Bcl-XL were expressed and purified as described before (46).
Assay for mitochondrion-dependent caspase-3 activation.
Caspase-3 activity was assayed in triplicates by measuring the release of 7-amino-4-trifluoromethyl-coumarin (AFC) from DEVD-containing synthetic peptides using continuous-reading instruments according to the instructions of the manufacturer (Santa Cruz Biotechnology). In short, 40-μl aliquots of cell supernatants (in triplicate) were incubated with 200 μl of reaction buffer, 5 μl of EDVD-AFC substrate, and dithiothreitol (DTT) (final concentration of 10 mM). The reaction mixtures were incubated at RT for 0.5 h. Free AFC levels formed were analyzed by using a fluorescence microplate reader (Bio-Rad, CA, USA) at excitation/emission wavelength of 400/505 nm. EDVD-AFC values in assay buffer were subtracted from the fluorescence values of the samples. Caspase-3 activity was then determined as a percentage of the positive-control value.
Statistical analyses.
Data were subjected to one-way analysis of variance with factors of treatment and expressed as means ± standard deviations (SD). Comparisons between any two groups were performed by using unpaired Student t tests. ** indicates a significant difference at a P value of 0.02, and * indicates a significant difference at a P value of 0.05.
RESULTS
EV71 2B localizes to the mitochondria and activates the mitochondrial component of the apoptotic cascade.
HeLa cells were transfected with a vector expressing either EV71 2B or Bak with an N-terminal GFP fusion (pGFP-2B and pGFP-Bak, respectively) and then analyzed by confocal microscopy to determine the distribution of EV71 2B. As shown in Fig. 1A and B, transfection of HeLa cells with the pGFP vector alone resulted in a diffused fluorescence signal throughout the cell or cytoplasm that did not colocalize with the mitochondrion-specific dye MitoTracker Red. In contrast, cells transfected with the pGFP-2B or pGFP-Bak plasmid displayed a punctuated staining pattern that colocalized with MitoTracker Red, indicating that GFP-2B was localized to the mitochondria. Further confirmation of the mitochondrial localization of 2B was obtained in HeLa cells that were either infected with EV71 or transfected with pCMV-2B-Flag vector. The cytosol and mitochondrial fractions of these cells were separated by centrifugation, and equal amounts of proteins were resolved by SDS-PAGE followed by immunoblotting with an anti-Flag antibody or anti-2B antibody. Mitochondria purified from the pCMV-Flag-transfected cells showed the absence of 2B, as expected. In contrast, cells with EV71 infection or with 2B-Flag expression revealed the presence of 2B only in the mitochondrial pellet fraction, with undetectable levels in the supernatant (Fig. 1C). To further investigate the sublocation of 2B in the mitochondria, the inner and outer mitochondrial membranes were isolated by sucrose gradient centrifugation as described before (47). A band representing 2B was detected in the outer membrane fraction of the mitochondria isolated from cells with 2B-Flag expression, indicating that 2B localized in the outer membrane of the mitochondria (Fig. 1D).
FIG 1.

(A) Confocal microscopy analysis of the colocalization of 2B-GFP and Bak-GFP within the mitochondria in HeLa cells. HeLa cells were transfected with pGFP, pGFP-2B, or pGFP-Bak. At 24 h posttransfection, they were stained with MitoTracker Red and subjected to confocal microscopy analysis. An overlay of the MitoTracker Red (red) and GFP florescence (green) is also shown. Mito, mitochondria. (B) Colocalization index estimation for cells transfected with pGFP, pGFP-2B, or pGFP-Bak and labeled for MitoTracker Red as for panel A. To determine the percentage of colocalization, green and merged images were loaded into the Leica TCS SP8 platform, and the ratio of GFP-positive cells to counted cells (in a total of 100 cells) was determined. Cells with GFP and mitochondrion colocalization (Manders overlap coefficient of >0.8) were counted. (C) Immunoblotting analysis of 2B-Flag in the cytosol and mitochondrial fractions of HeLa cells infected with EV71 or transfected with pCMV-2B-Flag. Cytosolic and heavy membrane fractions were separated as described in Materials and Methods. Mitochondrial fractions were processed for immunoblotting with antibodies specific to EV71 2B, Bak, and Cox IV. Cytosolic fractions were immunoblotted with antibodies to EV71 2B and β-actin (internal control). Control, cells transfected with Tris-acetate-EDTA (TAE) buffer. Mock, cells with no EV71 infection. (D) Western blot analysis of the sublocation of 2B. Cells were transfected with pCMV-2B-Flag or pCMV-Flag. The mitochondrial fractions were isolated at 48 h posttransfection. The inner and outer mitochondrial membranes were isolated by sucrose gradient centrifugation and immunoblotted with an antibody to the Flag tag, Cox IV, and TSPO (internal control of outer mitochondrial membrane fraction). M, mitochondria of cells transfected with pCMV-Flag. I, inner mitochondrial membrane fraction. O, outer mitochondrial membrane fraction. (E) Analysis of cell apoptosis induced by 2B. Cells were treated with 30 nM staurosporine (positive control) or transfected with increasing amounts of pCMV-2B-Flag, pCMV-Flag, or pCMV-Bak. At 24 h posttransfection, cells were harvested and stained with annexin V-FITC. The percentages of apoptotic cells corresponding to the respective fluorescence intensities were calculated. (F) Analysis of the antiapoptosis effects of caspase inhibitors on 2B-induced apoptosis. Cells either infected with EV71 or transfected with pCMV-2B-Flag or pCMV-Bak-Flag (positive control) were treated with dimethyl sulfoxide (DMSO), zDEVD.fmk, or zLEHD.fmk for 24 h and then stained with annexin V-FITC and subjected to flow cytometry. NT, cells with no treatment. Control, cells transfected with pCMV-Flag.
The large number of viruses encoding mitochondrion-associated proteins with antiapoptotic or proapoptotic functions led us to hypothesize that 2B could also function in a similar capacity to modulate the mitochondrial apoptotic pathway. HeLa cells were treated with staurosporine or transfected with increasing amounts of either pCMV-2B-Flag, pCMV-Bax-Flag, or pCMV-Flag empty vector. By using annexin V-FITC staining, apoptotic cells were detected at 24 h posttransfection. As shown in Fig. 1E, a significant proportion of cells transfected with 2B showed an increased annexin V fluorescence intensity, while no obvious annexin V fluorescence was detected in the pCMV-Flag-transfected or control cells. The relative annexin V fluorescence signals also increased with the increasing amounts of pCMV-2B-Flag transfected. To determine whether caspases are crucial for 2B-induced apoptosis in HeLa cells, we examined the antiapoptotic effects of the caspase inhibitors zDEVD.fmk, a wide-spectrum caspase inhibitor that irreversibly inhibits caspase-3, and zLEHD.fmk, an irreversible inhibitor of caspase-9. As shown in Fig. 1F, EV71-infected cells and 2B- or recombinant Bak-transfected cells that were treated with zDEVD.fmk or zLEHD.fmk displayed very low levels of annexin V fluorescence signal. In contrast, strong annexin V fluorescence signals were detected in 2B- or recombinant Bak-EV71-infected cells that were treated with DMSO, indicating that zDEVD.fmk or zLEHD.fmk completely inhibited apoptosis induced by 2B or EV71 infection.
To further assess the ability of 2B to initiate apoptosis in HeLa cells, we monitored Cyt c released from mitochondria in cells expressing 2B or infected with EV71. The cytoplasmic and mitochondrial fractions were separated, and the levels of Cyt c were determined by Western blotting. Cells with 2B expression or EV71 infection displayed a clear loss of Cyt c from the mitochondrial fraction into the cytosol fraction (Fig. 2A). Apoptosis was also measured by quantifying loss of the inner mitochondrial membrane potential by fluorescence of TMRE, a hydrophobic cationic dye that is readily taken up by healthy respiring mitochondria. As shown in Fig. 2B, cells with 2B expression or EV71 infection showed an obvious loss of the inner mitochondrial membrane potential, and treatment with staurosporine resulted in 46% of mock-transfected cells losing their inner mitochondrial membrane potential. In addition, cells transfected with pCMV-Flag maintained high levels of TMRE fluorescence, demonstrating that 2B expression was independent of other EV71 proteins in inducing the loss of the inner mitochondrial membrane potential.
FIG 2.

(A) Western blot analysis showing the levels of Cyt c release induced by 2B. Cells were infected with EV71 or transfected with pCMV-2B-Flag. Cytosolic and mitochondrial fractions were separated at the indicated times and immunoblotted with an antibody specific to Cyt c. Control, cells transfected with TAE buffer. +EV71, EV71-infected cells. −EV71, uninfected control. (B) Analysis of the inner mitochondrial membrane potential in cells with EV71 infection or 2B expression. Cells were infected with EV71, transfected with pCMV-2B-Flag, or treated with staurosporine for 12 h; cells were then stained with TMRE and subjected to flow cytometry analysis. Control, cells with no EV71 infection and 2B expression. pCMV-Flag, cells transfected with pCMV-Flag.
EV71 2B specifically interacts with the proapoptotic protein Bax.
The above results indicate that 2B induced apoptosis by promoting the release of Cyt c and causing the loss of the inner mitochondrial membrane potential. To determine whether 2B functions by interacting with members of the Bcl-2 family which tightly regulate the mitochondrial checkpoint of apoptotic cells, a lysate of HeLa cells transfected with a plasmid expressing a Flag-tagged version of 2B was applied to an anti-Flag immunoaffinity column. Flag-tagged 2B protein was eluted and confirmed by immunoblotting with an anti-Flag antibody. The results showed that cells transfected with pCMV-2B-Flag or pCMV-Flag vector expressed similar levels of Bax, Bak, Bcl-2, Bcl-XL, Bid, Bim, Bad, and Mcl-1, whereas only the pCMV-2B-Flag-transfected cells expressed the Flag-tagged 2B. To determine if 2B interacted with members of the Bcl-2 family, the eluted 2B fractions were probed with antibodies directed against various Bcl-2 family members. The proapoptotic protein Bax consistently coeluted with the Flag-tagged version of 2B, while no interactions with Bak, Bcl-2, Bcl-XL, Bid, Bim, Bad, or Mcl-1 were detected (Fig. 3A). The total amount of Bak, Bcl-2, Bcl-XL, Bid, Bim, Bad, or Mcl-1 was not affected by 2B expression.
FIG 3.

Experiments showing specific interactions between 2B and Bax. (A) Detection of specific interactions between 2B and cellular Bax in cell lysates by immunoprecipitation (IP) assays and Western blotting (WB). Cells were transfected with pCMV-Flag (control) or pCMV-2B-Flag (2B). At 24 h posttransfection, they were harvested and subjected to immunoprecipitation with anti-Flag antibody. The eluted fractions were probed with anti-Flag, Bak, Bax, Bcl-2, Bcl-XL, Bid, Bim, Bad, and Mcl-1 antibodies. (B) Coimmunoprecipitation analysis of the specific interactions between 2B and Bax by using anti-BaxNT antibody. Cell lysates were incubated with anti-BaxNT antibody-conjugated agarose beads. The precipitated proteins were blotted with anti-Flag and anti-BaxNT antibodies. IgGL, immunoglobulin light chains. (C) Coimmunoprecipitation analysis of the specific interactions between 2B and Bax using anti-2B antibody. HeLa cells were transfected with pCMV-2B-Flag or infected with EV71 (multiplicity of infection [MOI] = 1), and at 12 h postinfection, cells were lysed and coimmunoprecipitation assays were performed. The precipitated proteins were blotted with anti-BaxNT, anti-2B, and anti-EV71 antibodies. Equal amounts of cell lysates were also blotted with anti-EV71 or anti-BaxNT antibodies. (D) Analysis of the interaction between recombinant Bax and 2B by pulldown assay. Recombinant Bax was immobilized on agarose beads conjugated with anti-His beads and incubated with cell lysates for 1 h at 4°C. The beads were washed with lysis buffer and heat denatured. The proteins were immunoblotted with anti-His and anti-2B antibodies. Input, Bax-His protein. Control, agarose beads incubated with the lysate of pCMV-Flag-transfected cells. 2B, agarose beads incubated with the lysate of pCMV-2B-Flag-transfected cells.
To confirm the interaction between 2B and Bax, we performed a series of coimmunoprecipitation assays. HeLa cells transfected with a plasmid expressing Flag-tagged 2B were lysed, and immunocomplexes were precipitated with an anti-BaxNT antibody. The results obtained showed that the anti-BaxNT coimmunoprecipitated Bax and Flag-2B (Fig. 3B). The band of IgGL shows an equal amount of anti-BaxNT conjugated to agarose beads. Reciprocal immunoprecipitations performed with an anti-2B antibody also showed that Flag-2B coprecipitated with Bax (Fig. 3C). To determine the ability of 2B to interact with endogenous Bax during EV71 infection, the interaction of 2B with Bax in EV71-infected cells was analyzed by coimmunoprecipitation assays. As expected, antibodies directed against Bax effectively immunoprecipitated Bax and coimmunoprecipitated 2B (Fig. 3C). The interaction of 2B with Bax was also analyzed by incubating cell lysates of Flag-tagged 2B with recombinant His-tagged Bax that was immobilized on agarose beads conjugated with anti-His beads. As shown in Fig. 3D, a 10-kDa band corresponding to 2B was detected by a Flag monoclonal antibody, whereas no 2B was detected in the IgG and control groups. These results provided strong evidence that 2B interacts directly and specifically with Bax.
EV71 2B induces translocation and activation of Bax.
Bax-induced apoptosis involves two key events: the translocation of Bax to the mitochondria and a subsequent conformational change that activates its oligomerization and integration into the mitochondrial membranes. To determine whether 2B could induce Bax translocation to the mitochondria and result in a conformational change in Bax, cytosol and mitochondrial fractions of cells transfected with pCMV-Flag-2B were separated by centrifugation, and equal amounts of proteins from each fraction were analyzed by Western blotting using an anti-BaxNT antibody. As shown in Fig. 4A, a low level of Bax was detected in the mitochondrial fraction from cells transfected with the control plasmid. This may represent a small amount of loosely attached Bax on the mitochondrial membrane. In contrast, we detected a significantly larger amount of Bax in the mitochondrial fraction from 2B-transfected cells than in that from mock-infected cells. Conversely, the amount of Bax in the cytosol fraction from 2B-transfected cells was notably less than that in the control cells, suggesting that Bax was translocated from the cytoplasm to the mitochondria in 2B-expressing cells. To confirm this, Bax translocation was monitored in cells transfected with pGFP-Bax, and the results showed that almost no immunofluorescence signal colocalized with the mitochondria in the pCMV-Flag-transfected cells, whereas in the pCMV-Flag-2B-transfected cells, a significant amount of Bax was detected in the mitochondria, indicating again that Bax was translocated to the mitochondria in cells with 2B expression (Fig. 4B and C). To confirm the ability of 2B to induce Bax activation, HeLa cells were transfected with the same panel of pCMV-Flag or pCMV-2B plasmids, and activated Bax was immunoprecipitated from the cell lysates using anti-Bax6A7. Activated Bax was precipitated from cells transfected with pCMV-2B but not from cells transfected with pCMV-Flag, thus confirming the ability of 2B to induce Bax activation (Fig. 4D). Bax activation was further illustrated by double immunofluorescence staining of activated Bax with anti-Bax6A7 and staining of mitochondria with MitoTracker Red. The merged yellow color confirmed the colocalization of activated Bax and mitochondria in pCMV-2B-transfected cells (Fig. 4E).
FIG 4.

Effects of EV71 2B on the translocation and activation of Bax. (A) Western blot analysis of Bax and activated Bax in the cytosol and mitochondrial fractions of cells transfected with pCMV-Flag or pCMV-2B-Flag. Cytosolic and mitochondrial fractions were separated, and equal amounts of proteins from each fraction were immunoblotted with anti-BaxNT, anti-Flag, or anti-Cox IV (internal control of mitochondrial fraction). Control, cells transfected with TAE buffer. Staurosporine, cells treated with staurosporine (30 nM). (B) An experiment indicating that 2B induces the translocation of Bax to the mitochondria. Cells were cotransfected with pGFP-Bax (green) and pCMV-Flag (control) or pGFP-Bax and pCMV-2B-Flag. At 12 h posttransfection, the cells were fixed and analyzed by confocal fluorescence microscopy. Red, MitoTracker Red. An overlay of the MitoTracker Red and GFP florescence is also shown (merged). (C) Colocalization indexes. Cells were cotransfected with pGFP-Bax and pCMV-Flag or pCMV-2B-Flag. At 24 h posttransfection, cells were labeled with MitoTracker Red and examined under a confocal laser scanning microscope. Cells with GFP and mitochondrial colocalization (Manders overlap coefficient of >0.8) were counted as described for Fig. 1. (D) Western blot analysis of the Bax activation in cells transfected with the same panel of pCMV-Flag or pCMV-2B-Flag plasmids. Cells were lysed and immunoprecipitated with anti-Bax6A7 antibody. Equal amounts of the precipitated protein and cell lysates were immunoblotted with antibody to Bax. (E) Immunofluorescence analysis of the Bax activation in cells transfected with pCMV-2B-Flag (2B). The cells were incubated with MitoTracker Red (red) for 10 min, fixed, and stained with anti-Bax6A7 (green). (F) An experiment indicating that 2B induces homo-oligomerization of Bax. Mitochondria from cells transfected with pCMV-Flag or pCMV-2B-Flag were cross-linked with BMH or DMSO as described in Materials and Methods. Equal amounts of proteins from each sample were immunoblotted with anti-Bax antibody. H, Bax homo-oligomers; I, monomeric intramolecularly cross-linked Bax species.
To determine if 2B induces Bax homo-oligomerization, mitochondria were isolated from 2B-transfected cells and the presence of higher-order Bax complexes was examined by chemical cross-linking of the mitochondrial lysates with BMH and then analysis by Western blotting. In the absence of BMH, Bax migrated at approximately 20 kDa; however, upon treatment with BMH, Bax displayed a reduced mobility as a result of intramolecular cross-linking. Thus, 2B expression resulted in an increase in intramolecular cross-linked Bax species and the formation of Bax oligomers, which is indicative of Bax activation as observed in the staurosporine-treated cells used as a positive control (Fig. 4F). Mitochondria purified from pCMV-Flag-transfected cells showed the retention of intramolecular cross-linked species of Bax and the absence of Bax homo-oligomers (Fig. 4F).
Apoptosis induced by EV71 2B is Bax dependent.
The proapoptotic proteins Bax and Bak in their active forms are the main proteins responsible for the destabilization of the mitochondrial membrane. Experiments were done to determine the relative importance of Bak and Bax in 2B-induced apoptosis. In the first experiment, HeLa cells were transfected with siRNA against either Bak, Bax, or both or with a nontargeting siRNA as a control. At 48 h posttransfection, cell lysates were prepared and analyzed by SDS-PAGE and immunoblotting, with β-actin as a loading standard. Figure 5A shows a representative immunoblot showing successful silencing of Bak or Bax in the presence of the corresponding siRNA. The levels of Bak and Bax were similarly very low at 48 h posttransfection compared to that of the nontargeting siRNA control. Next, to determine the impact of downregulation of Bak and Bax expression on 2B-induced apoptosis, cells were transfected with 2B or infected with EV71 at 48 h posttransfection with siRNA. Cell viability was determined 48 h posttransfection or 24 h postinfection. The graph shows that 2B or EV71 induced an obvious loss of cell viability in the control and nontargeting siRNA-transfected cells (Fig. 5B). A similar loss of cell viability was observed for the Bak siRNA-transfected cells. However, the loss of cell viability was remarkably lower in both the Bax siRNA-transfected cells and the Bak plus Bax siRNA-transfected cells after EV71 infection (Fig. 5B). The decrease in 2B- or EV71-induced apoptosis in the Bak siRNA-transfected cells was confirmed by an analysis of caspase-3-like activity in the cell lysates. siControl cells treated with staurosporine were used as a positive control. The results indicate that Bax and Bak siRNA-transfected cells both had caspase-3-like activity when treated with staurosporine. Control siRNA-transfected cells and Bak siRNA-transfected cells with 2B expression had relatively high levels of caspase-3-like activity, while the Bax siRNA-transfected cells and Bak plus Bax siRNA-transfected cells had significantly less active caspase-3 than the control siRNA-transfected cells (Fig. 5C). Cyt c was also detected in the cytosol fraction by immunoblotting with an anti-Cyt c antibody. The results showed that the supernatants of both the control siRNA-transfected cells and the Bak siRNA-transfected cells contained larger amounts of Cyt c than those of the Bax siRNA-transfected cells and Bak plus Bax siRNA-transfected cells (Fig. 5D). This indicates that the downregulation of Bax could prevent the loss of Cyt c from the mitochondria to the cytosol fraction induced by 2B. Collectively, the data shown in Fig. 5 showed that silencing of Bax expression lowered the rate of apoptosis induced by 2B, while silencing of Bak expression did not.
FIG 5.

Effect of downregulation of Bax expression on 2B-induced apoptosis in HeLa cells. (A) Western blot analysis of Bak and Bax expression in HeLa cells that were transfected with siRNA against either Bak (siBak), Bax (siBax), or both (siBax siBak) as described previously (45). Cells transfected with nontargeting siRNA (siControl) were used as a control. At 48 h posttransfection, cell lysates were prepared, analyzed by SDS-PAGE, and immunoblotted with antibodies to Bax, Bak, or β-actin (internal control). (B) Analysis of the impact of 2B expression or EV71 infection on the viability of HeLa cells transfected with siRNA. Cells were transfected with the siRNAs described in Materials and Methods. At 48 h transfection, they were either transfected with pCMV-2B-Flag for another 48 h or infected with EV71 (MOI = 0.5) for 24 h and then subjected to cell viability analysis. EV71 (UV), UV-inactivated EV71. Control, siControl cells treated with DMSO. (C) Analysis of the impact of 2B on caspase-3-like activity in cell lysates of siRNA-transfected cells. Caspase-3 activity was determined as described in Materials and Methods. Control, siControl-transfected cells treated with 30 nM staurosporine. (D) Western blot analysis of Cyt c in the cytosol fraction. The cytosol fraction of cells was separated and immunoblotted with an antibody to Cyt c. Control, cells transfected with pCMV-Flag.
Bax induces release of Cyt c from mitochondria in the presence of 2B.
To further examine the effects of 2B on the mitochondria, purified recombinant Bax protein was added to mitochondria isolated from cells that were transfected with either pCMV-Flag or pCMV-2B-Flag at 48 h after transfection with Bax siRNA. Soluble Bax protein without its last 20 C-terminal amino acids was produced as described before (34). After coincubating the mitochondria with either Bax or Bax diluent buffer control for 1.5 h, the mitochondria were pelleted by centrifugation, and the resulting supernatants were analyzed for the presence of Cyt c by immunoblotting. As shown in Fig. 6A, the supernatants from Bax-treated mitochondria contained larger amounts of Cyt c than that from the buffer control. However, the amounts of Cyt c released varied among the different preparations of mitochondria: there was relatively more Cyt c released from 2B mitochondria following Bax treatment than in the absence of 2B expression (Fig. 6A). Mitochondria that were stimulated by 150 μM Ca2+, a known inducer of the mitochondrial permeability transition, also displayed high levels of Cyt c release (Fig. 6A). Bax protein associated with the mitochondria was also analyzed by immunoblotting with an anti-His antibody. The results showed that the recombinant Bax could attach to the mitochondria from both pCMV-Flag- and pCMV-2B-tranfected cells (Fig. 6A). However, in comparison with the pCMV-Flag-transfected cells, a significantly larger amount of activated Bax was detected in mitochondria from pCMV-2B-transfected cells, indicating that mitochondria with 2B expression could combine more Bax and release more Cyt c.
FIG 6.
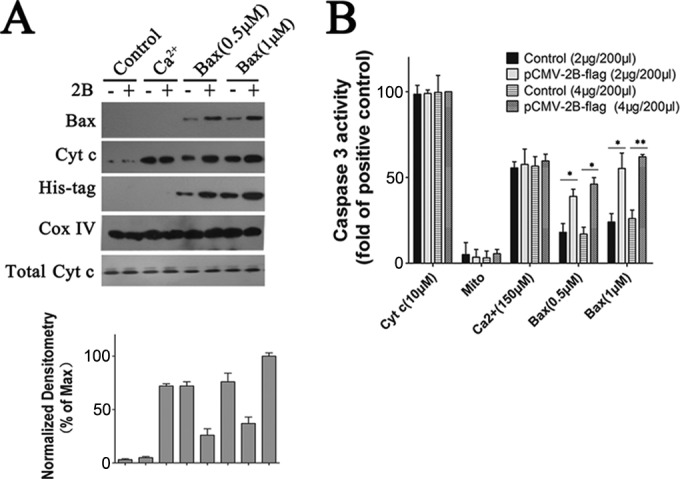
Experiments showing that Bax induces the release of Cyt c from isolated mitochondria in the presence of 2B. (A) Western blot analysis of the impact of recombinant Bax on Cyt c release in isolated mitochondria. The mitochondria were incubated for 1.5 h with either 1 μM recombinant Bax, 0.5 μM recombinant Bax, 150 μM Ca2+, or none of these reagents (control) at RT. They were then pelleted by centrifugation, and the resulting supernatants were immunoblotted with antibody to Cyt c. The pellet fractions were lysed and immunoblotted with antibody to His tag or Bax. Inputs of equivalent amounts of mitochondria and total Cyt c from the mitochondria were verified by immunoblot analysis of the mitochondrial fractions using an antibody to Cox IV or Cyt c, respectively. Densitometry of the Cyt c band normalized to Cox IV is presented as fold change compared with Cyt c release in isolated mitochondria incubated with 1 μM recombinant Bax, defined as 100. (B) Experiments showing that Bax induces the release of mitochondrial factors which trigger the processing and activation of cytosolic caspase-3 in mitochondria expressing 2B. Cells were transfected with 2 μg or 4 μg of pCMV-Flag or pCMV-2B-Flag. At 24 h posttransfection, mitochondrial fractions were separated as described in Materials and Methods. Mitochondria (Mito) or supernatants (400 μl) from mitochondria that had been incubated with either Bax or Ca2+ (150 μM) were incubated with purified cytosol (100 μl) at 25°C for 1.5 h. Aliquots were then analyzed for caspase-3 activity as described above. Positive control, purified cytosol treated with Cyt c (10 μM).
Cyt c has been shown to induce processing and activation of caspase-3 and some other caspases that cleave the peptide DEVD. To investigate whether Bax-treated mitochondria could release factors that induce the activation of DEVD-cleaving caspase-3, isolated mitochondria were treated in vitro for 1 h with either Bax protein or buffer control, followed by removal of the mitochondria by centrifugation and collection of the resulting supernatants. These supernatants were then added to the cytosol fractions from HeLa cells, and caspase-3 activity was measured 0.5 h later by spectrofluorimetric assays with DEVD-AFC as a substrate. As shown in Fig. 6B, addition of control supernatants caused only a modest elevation in DEVD-cleaving activity. In contrast, supernatants from Bax-treated 2B mitochondria consistently resulted in striking increases in caspase-3 activity, whereas no substantial elevations in caspase-3 activity were observed in cytosol fractions stimulated by mitochondria alone.
Bcl-XL prevents 2B-induced release of Cyt c from mitochondria.
When coexpressed in cells, Bcl-XL can abrogate the proapoptotic effects of Bax. We therefore examined the impact of Bcl-XL protein on 2B-induced cell apoptosis. HeLa cells that stably overexpress Bcl-XL–HA (HeLa-Bcl-XL cells) and control cells (HeLa cells transduced with the backbone vector) were transfected with pCMV-2B-Flag or pCMV-Flag. At 24 h posttransfection, cell apoptosis was analyzed by annexin V-FITC staining. As shown in Fig. 7A, HeLa-Bcl-XL cells transfected with pCMV-2B-Flag or pCMV-Flag displayed a very low annexin V fluorescence, indicating low levels of cell apoptosis. In contrast, control cells expressing 2B displayed a significant proportion of cells with increased annexin V fluorescence intensity, indicating that Bcl-XL could abrogate apoptosis induced by 2B. We also assayed the release of Cyt c from mitochondria in cells with recombinant Bcl-XL expression to confirm the above results. Overexpression of Bcl-XL completely inhibited the 2B-induced Cyt c release (Fig. 7B). The effects of Bcl-XL overexpression on caspase-3-like activity during 2B-induced apoptosis were also assessed. In contrast to the dramatic change observed in staurosporine-treated or pCMV-2B-transfected control cells, only a relatively modest change in the activated caspase-3 level was detected in HeLa-Bcl-XL cells with 2B expression (Fig. 7C). Together, the above results show that overexpression of the antiapoptotic protein Bcl-XL caused a dramatic reduction in the level of induction of apoptosis in cells with 2B expression.
FIG 7.

Experiments indicating that Bcl-XL prevents 2B-induced apoptosis and Cyt c release in HeLa cells. (A) Analysis of 2B- or EV71-induced apoptosis in HeLa-Bcl-XL cells by annexin V-FITC staining and flow cytometry. Staurosporine (control), cells treated with staurosporine for 12 h. EV71, cells infected for 24 h with EV71 (MOI = 0.5). HeLa, HeLa cells transduced with pcDNA3.0-HA. (B) Western blot analysis of Cyt c in the cytosol fraction of HeLa (HeLa cells transduced with the pcDNA3.0-HA) or HeLa-Bcl-XL cells transfected with pCMV-Flag (control) or pCMV-2B-Flag. Stau, cells treated with staurosporine (30 nM) for 12 h. (C) Analysis of caspase-3-like activity in cell lysates of HeLa-Bcl-XL or HeLa cells (HeLa cells transduced with the pcDNA3.0-HA) transfected with pCMV-Flag or pCMV-2B-Flag or infected with EV71. Caspase-3-like activity was determined as described in Materials and Methods. Staurosporine (positive control), cells treated with staurosporine for 12 h. (D) Quantification of Cyt c release and Bax binding in isolated mitochondria from siBax HeLa cells infected with EV71 (MOI = 1) and siBax HeLa cells transfected with pCMV-2B-Flag or pCMV-Flag by Western blotting. Mitochondria were isolated and incubated for 1 h with 1 μM recombinant Bax or/and 10 μM Bcl-XL at RT. After a short centrifugation at 4°C, the resulting supernatants were immunoblotted with antibody to Cyt c. Equal amounts of each pellet fraction were lysed, boiled, and immunoblotted with antibody to His tag.
To further confirm the role of Bcl-XL in 2B-induced apoptosis, we investigated the impact of purified recombinant Bcl-XL protein on Bax-induced release of Cyt c from mitochondria in vitro. Mitochondria purified from the EV71-infected siBax HeLa cells and siBax HeLa cells that were transfected with either pCMV-2B-Flag or pCMV-Flag were incubated with recombinant Bcl-XL and/or Bax protein and then pelleted by centrifugation. The resulting supernatants and pellets were analyzed by immunoblotting with antibody to Cyt c or His tag. As shown in Fig. 7D, compared to the pCMV-Flag mitochondria, a significantly larger amount of Bax was detected in the 2B-transfected mitochondria and EV71-infected mitochondria that were incubated with recombinant Bax. However, both the release of Cyt c from and binding of Bax to the mitochondria were almost completely inhibited by Bcl-XL in the pCMV-Flag-transfected, 2B-transfected, and EV71-infected mitochondria.
Identification of regions in the 2B protein that are crucial for mitochondrial localization and Bax activation.
Amino acid sequence analyses reveal that EV71 2B possesses a putative C-terminal hydrophobic α-helix TM region (aa 63 to 80), an amphipathic α-helix HR2 (aa 37 to 55), and an N-terminal hydrophilic helix HR1 (aa 22 to 35) (Fig. 8A). To determine whether the TM region is responsible for targeting 2B to the mitochondrial outer membrane, vectors expressing the 2B protein and truncation proteins containing an N-terminal GFP fusion [pGFP-2B(1–80) and pGFP-2B(1–63)] were constructed and transfected into HeLa cells. The cytoplasmic and mitochondrial fractions were separated, and equal amounts of proteins were resolved by SDS-PAGE followed by immunoblotting with GFP antibody. As shown in Fig. 8B, pGFP-2B(1–63)-transfected cells revealed an absence of GFP signal in the mitochondrial fraction. In contrast, cells transfected with pGFP-2B(1–80) showed the presence of pGFP-2B only in the mitochondrial pellet fraction, with undetectable levels in the supernatant cytoplasmic fraction, indicating that 2B without the TM region could not locate to the mitochondria. To further confirm the role of HR2 in the intracellular location of 2B, HeLa cells were transfected with vectors expressing either GFP, GFP-2B(1–63), GFP-2B(1–80), or GFP-2B. As shown in Fig. 8C and D, cells with GFP or GFP-2B(1–63) expression had a diffuse fluorescence signal throughout the cell cytoplasm that did not colocalize with the mitochondrion-specific dye MitoTracker Red. In contrast, cells transfected with pGFP-2B or pGFP-2B(1–80) displayed a punctuated staining pattern which colocalized with MitoTracker Red, indicating that GFP-2B and GFP-2B(1–80) localized to the mitochondria. All these results strongly suggest that the TM region in the C terminus may act as a transmembrane region that targets 2B to the mitochondrial outer membrane.
FIG 8.

Identification of regions that are responsible for targeting 2B to the mitochondrial outer membrane. (A) Schematic diagram of 2B indicating the following: a transmembrane region (TM), a putative C-terminal hydrophobic α-helix (aa 63 to 80); H2, an amphipathic α-helix (aa 37 to 55); and H1, an N-terminal hydrophilic helix (aa 22 to 35). The secondary structure and helix of 2B were predicted by using the Predict Protein and TMHMM server. (B) HeLa cells were transfected with pGFP-2B(1–63) (GFP fused to aa 1 to 63 of 2B), pGFP-2B(1–80) (GFP fused to aa 1 to 80 of 2B), or pGFP-2B for 24 h. The cytosol and mitochondrial fractions were separated and immunoblotted with GFP antibody. Cox IV and β-actin were used as the internal controls for the mitochondrial and cytosol fractions, respectively. (C) Confocal microscopy analysis of the colocalization of pGFP-2B(1–80) and pGFP-2B with the mitochondria. Cells were transfected with pGFP, pGFP-2B(1–63), pGFP-2B(1–80), or pGFP-2B as described above. At 24 h posttransfection, the cells were incubated with MitoTracker Red (red) for 10 min, fixed, and subjected to confocal microscopy analysis. (D) Colocalization indexes for cells transfected with pGFP, pGFP-2B, pGFP-2B(1–63), or pGFP-2B(1–80) and labeled for MitoTracker Red as for panel C. Cells with GFP and mitochondrial colocalization (Manders overlap coefficient of >0.8) were counted as described for Fig. 1.
To identify the domain of 2B that is responsible for Bax interaction, coimmunoprecipitation assays involving the Bax and 2B truncations were performed. HeLa cells were transfected with the respective vectors expressing either full-length 2B(1–99), 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35), lysed, and immunoprecipitated with anti-Flag antibody. The precipitated protein samples were then analyzed by Western blotting to detect 2B and Bax binding. The results showed that shortening the 2B polypeptide at the N terminus that included aa 22 to 35 abolished the Bax binding capacity of 2B, suggesting that aa 22 to 35 of 2B may contain the domain directly responsible for the specific binding of EV71 2B to Bax (Fig. 9A). To further analyze the influence of the 2B truncations on Bax activation, activated Bax was immunoprecipitated from cell lysates by using anti-Bax6A7 antibody. The eluates were then subjected to immunoblotting with anti-BaxNT antibody. As shown in Fig. 9B, a band representing Bax could be detected in the eluates from cells transfected with 2B(Δ1–22) and full-length 2B(1–99). In contrast, no such protein band was observed in eluates from cells transfected with 2B(Δ1–35), 2B(Δ22–35), and control resins, indicating that the N-terminal region from aa 23 to 35 of 2B plays a pivotal role in Bax activation.
FIG 9.

Experiments indicating that the N-terminal domain from aa 22 to 35 of 2B is crucial for Bax activation and the interaction between Bax and 2B. (A) Coimmunoprecipitation analysis of the region of 2B that is responsible for Bax interaction. HeLa cells were transfected with vectors expressing full-length 2B, 2B(Δ1–22) (2B with deletion of aa 1 to 22), 2B(Δ1–35) (2B with deletion of aa 1 to 35), or 2B(Δ22–35) (2B with deletion of aa 22 to 35). At 24 h posttransfection, the cells were harvested, and coimmunoprecipitation assay was performed using Flag antibody. The precipitated proteins were immunoblotted with antibodies to Bax and Flag. Control, cells transfected with pCMV-Flag. (B) Western blot analysis of Bax activation in cells expressing 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35). Activated Bax was immunoprecipitated from cell lysates by using anti-Bax6A7 antibody. The eluates were probed with antibody to Bax. Control, cells transfected with pCMV-Flag. (C) Analysis of cell apoptosis in cells expressing 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35). Cells were harvested and stained with annexin V-FITC as described in Materials and Methods. Cell apoptosis was then determined by flow cytometry. Control, cells transfected with pCMV-Flag. Staurosporine (positive control), = cells treated with staurosporine for 12 h. (D) Western blot analysis of Cyt c in the cytoplasmic fractions of cells expressing 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35). Equal amounts of each cytosol fraction were immunoblotted with antibody to Cyt c. (E) Analysis of the caspase-3-like activity in cells expressing 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35). Control, cells transfected with pCMV-Flag. Staurosporine (positive control), cells treated with staurosporine for 12 h.
To analyze the impact of the 2B truncations on cell apoptosis, HeLa cells were treated with staurosporine or transfected with increasing amounts of vector expressing the full-length 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35) or empty vector. Cell apoptosis was analyzed by using annexin V-FITC staining at 24 h posttransfection. The results showed that a significant proportion of the cells treated with staurosporine or transfected with vectors expressing 2B(Δ1–22) or full-length 2B displayed increased annexin V fluorescence intensity (Fig. 9C). In contrast, no obvious annexin V-FITC fluorescence was detected in the control cells or cells with 2B(Δ1–35) or 2B(Δ22–35) expression. Analyses of Cyt c levels in the cytoplasmic fractions by immunoblotting showed that in contrast to cells with 2B(Δ1–22) or full-length 2B expression, no Cyt c was observed in cell supernatants from 2B(Δ1–35)- or 2B(Δ22–35)-expressing cells (Fig. 9D). Additionally, we also monitored caspase-3 activation in cells expressing 2B truncations. Cells with 2B(Δ1–35) or 2B(Δ22–35) showed a very low level of caspase-3 activation, whereas cells expressing 2B(Δ1–22) or full-length 2B showed obvious caspase-3 cleavage at 48 h posttransfection (Fig. 9E).
Mitochondria were isolated from Bax siRNA cells transfected with 2B truncates and incubated with either recombinant Bax or buffer control as described above. After a short centrifugation, the resulting supernatants were analyzed for the presence of Cyt c. As shown in Fig. 10A, supernatants from Bax-treated mitochondria from cells transfected with vectors expressing 2B(Δ1–22) or full-length 2B contained large amounts of Cyt c. As a positive control, mitochondria stimulated by Ca2+ also released high levels of Cyt c, as expected. In contrast, low levels of Cyt c release were detected in supernatants from Bax-treated mitochondria isolated from cells with 2B(Δ1–35) or 2B(Δ22–35) expression. In addition, recombinant Bax protein associated with the mitochondria in the pellet was also analyzed by immunoblotting with anti-His antibody. As shown in Fig. 10A, more Bax was observed in the mitochondrial fractions from 2B(Δ1–22)- and full-length 2B-expressing cells than in those from 2B(Δ1–35)- or 2B(Δ22–35)-expressing cells, which displayed only a low level of recombinant Bax. To investigate release factors from Bax-treated mitochondria that activate the caspases, the supernatants were incubated with cytosol fractions derived from HeLa cells, and caspase activity was then measured. The results indicate that the supernatants derived from the Bax-treated mitochondria from 2B(Δ1–35)- or 2B(Δ22–35)-expressing cells activated caspase-3 only modestly (Fig. 10B). In contrast, more caspase-3 activation was observed in the corresponding supernatants from the Ca2+-treated and Bax-treated mitochondria from 2B(Δ1–35)- or 2B(Δ22–35)-expressing cells. Overall, the above results indicate that the 2B domain from aa 23 to 35 was responsible for cell apoptosis induced by EV71 2B.
FIG 10.

(A) Western blot analysis of the impact of recombinant Bax on Cyt c release in isolated mitochondria from cells expressing 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35). HeLa cells with Bax knockdown were transfected with full-length 2B, 2B(Δ1–22), 2B(Δ1–35), and 2B(Δ22–35). At 24 h posttransfection, the mitochondrial fractions were separated and incubated with 1 μΜ recombinant Bax, 100 μM Ca2+, 150 μM Ca2+, or neither of these reagents (control) for 1 h at RT. After centrifugation, the resulting supernatants were immunoblotted with antibody to Cyt c. The recombinant Bax that attached to the mitochondria was immunoblotted with antibody to His tag. (B) Analysis of caspase-3-like activity in cell lysates treated with supernatants from Bax-treated mitochondrial fractions of cells expressing 2B, 2B(Δ1–22), 2B(Δ1–35), or 2B(Δ22–35). Mitochondrial fractions were treated with Bax or Ca2+ as described for Fig. 6. The resulting supernatants were added to purified cell lysates. The caspase-3 activity was then determined as described above. Positive control, purified cytosol that was incubated with exogenous Cyt c (10 μM).
DISCUSSION
Viruses usually possess a variety of strategies to modulate apoptosis to ensure successful propagation. Mitochondria were thought to transduce key signals during the apoptotic response and thus to function as pivotal regulating organelles during apoptosis (49, 50). Members of the Bcl-2 family are key regulators of apoptosis via the mitochondrial pathway. The proapoptotic proteins Bax and Bak are the most downstream activator molecules known for Cyt c release (51, 52). Thus, it is not surprising that mitochondria are directly targeted by numerous virus proteins to modulate apoptosis. These virus proteins often induce or depress the effects of cellular proapoptotic Bcl-2 family proteins, especially Bax and Bak (53). EV71 has been shown to induce classic signs of mitochondrion-mediated apoptosis during infection. However, little detail is known about the mechanisms of activation of the mitochondrion-related cell death pathway induced by EV71. Here we show that cell apoptosis induced by EV71 was completely inhibited by pretreating HeLa cells with the caspase-3 inhibitor zDEVD.fmk, indicating that apoptosis induced by EV71 was directly dependent on caspase-3 activation. Importantly, we obtained evidence that assigns an essential functional role to the EV71 2B protein, which was localized to the mitochondria, where it interacted directly with Bax, the critical proapoptotic member of the Bcl-2 family, and resulted in Bax activation which was manifested by its conformational change as well as by its redistribution from the cytosol to the mitochondria, concomitant with the release of Cyt c.
Previous studies indicated that poliovirus (PV) 2B localizes partially in mitochondria and induces an anomalous perinuclear distribution of these organelles (54, 55). Detection of Cyt c release from mitochondria suggests involvement of the mitochondrial pathway in 2B-induced apoptosis (56, 57). Some studies have suggested that 2B induces cell apoptosis via a caspase-dependent pathway (58). Protein 2B from PV and coxsackievirus A (CAV) is thought to form homodimers and homotetramers that are located primarily in the membrane structure of host cells and leads to the formation of hydrophobic pores which increase the permeability of the host cell membrane and promote the budding of virus particles (56, 59). Another significant effect of PV and CAV 2B protein on host cells is the disruption of the Ca2+ balance in cells (60). Cells with PV or coxsackievirus B3 (CVB3) 2B expression displayed obvious cytosolic calcium elevation (59). However, the current induced by EV71 2B protein is carried mainly by chloride ions but not calcium (61). Little direct evidence has been provided to establish a link between changes in mitochondrion permeability and EV71 2B-induced apoptosis. The excessive mitochondrial uptake of Ca2+ has toxic effects on cells, and high Ca2+ concentrations can open the mitochondrial permeability transition pores, expand mitochondrial permeability, and rupture the mitochondrial outer membrane. Consequently, Cyt c and other proapoptotic factors are released, eventually leading to apoptosis (62). However, it was reported that the expression of PV as well as CAV 2B protein can decrease the Ca2+ levels in the mitochondria (63, 64). No obvious mitochondrial uptake of Ca2+ was observed in cells expressing 2B or cells infected with EV71 in our study (Fig. 11A). The mitochondrial apoptotic pathway activation and the subsequent cell apoptosis induced by EV71 2B could not be the consequences of Ca2+ overload in the mitochondria. Thus, we focused on members of the Bcl-2 family which tightly regulate the apoptotic cascade in the mitochondria. The data presented here suggest that EV71 2B-induced apoptosis was Bax dependent, as indicated by 2B-transfected cells that lacked Bax expression displaying no obvious mitochondrial apoptotic pathway activation or Cyt c release from mitochondria.
FIG 11.

(A) Analysis of the mitochondrial calcium in cells expressing 2B or cells infected with EV71. HeLa cells were transfected with pCMV-2B-Flag or infected with EV71 (MOI = 1). At 12 h and 24 h postinfection, the cells were probed with 1.5 μM Rhod-2 AM (for specific detection of intramitochondrial calcium) for 30 min at 37°C and then fixed in 4% paraformaldehyde at RT. Following three PBS washes, the cells were subjected to flow cytometry analysis using an excitation wavelength of 488 nm. The percentage of fluorescence-positive cells detected by flow cytometry upon staining with fluorescent dye was calculated. Control, unstained cells. pCMV-Flag, pCMV-Flag-transfected cells. EV71 (UV), cells infected with EV71 (UV). (B) Analysis of interactions between 2B and cellular Bcl-XL in cell lysates by immunoprecipitation (IP) assays and Western blotting. Cells were transfected with pCMV-Flag or pCMV-2B-Flag. At 24 h posttransfection, they were harvested and subjected to immunoprecipitation with mouse anti-Flag antibody or mouse IgG. The eluted fractions were probed with anti-Flag, Bax, and Bcl-XL. Input, cell lysate of cells with 2B expression.
In the absence of an apoptotic trigger, the majority of Bax was found in the cytoplasm or loosely associated with intracellular membranes (46). Following an apoptotic trigger, Bax undergoes a series of conformational changes which include the exposure of the N terminus and the liberation of the C-terminal transmembrane domain, resulting in mitochondrial membrane insertion, followed by subsequent homo-oligomerization and release of Cyt c (65, 66). As it is a proapoptotic member of the Bcl-2 family, how Bax induces these mitochondrion alterations is currently controversial. Bax has been shown to form channels in synthetic membranes and may even create very high conductance channels under some circumstances (26). It is possible that Bax creates pores in the mitochondrial outer membrane which are large enough to allow the escape of Cyt c (67). Alternatively, Bax might indirectly alter the permeability of the outer membrane through interactions with other proteins (68, 69). Thus, several potential mechanisms can be envisioned for 2B activation of Bax and the subsequent Cyt c release. The simplest explanation would be that 2B directly interacts with Bax, inducing its activation and forming a heterodimer or multidimer with Bax, leading to subsequent Cyt c release. Studies have suggested that the 2B proteins of some members of the Picornaviridae family may assemble into hydrophilic pores that are located primarily in the cell membrane system (70). Thus, it is conceivable that the interaction of Bax and 2B leads to formation of pores in the outer membrane of mitochondria that allow Cyt c to leak out. Alternatively, the interaction of 2B and Bax may result in mitochondrial membrane insertion and oligomerization of Bax, leading to the release of Cyt c and other key molecules from the mitochondria and facilitating caspase activation and apoptosis. Since overexpression of Bcl-XL caused a dramatic decrease in induced apoptosis in cells expressing 2B, 2B could function by sequestering and inhibiting the activity of the antiapoptotic protein Bcl-XL, as proposed for the BH3-only proteins that keep Bax in an active state. However, we were unable to detect an interaction between 2B and Bcl-XL (Fig. 11B).
The PV and CVB3 2B proteins have two hydrophobic domains, HR1 and HR2. The HR1 domain crosses the cell membrane via a cationic amphipathic α-helix, whereas the HR2 domain is a complete hydrophobic TM region (55, 71). Two hydrophobic domains and the stem-loop connecting them interact to form a “helix-angle-helix” structure (72). After the interaction, the protein could successfully embed into the membrane. Amino acid sequence comparisons between the EV71 2B, PV 2B, and CVB3 2B proteins suggest that there is a putative conserved TM domain in the 2B C terminus. We showed that the TM domain is crucial for the mitochondrial location of EV71 2B, as reflected by the absence of mitochondrial localization of 2B with a HR2 deletion. However, an obvious difference in the N-terminal amino acid sequences between the EV71 2B and the PV and CVB3 2B proteins may explain the functional differences between EV71 2B and PV or CVB3 2B. Our results indicate that a hydrophilic region of 14 amino acids in the N terminus of 2B may be responsible for the Bax interaction and its subsequent activation. It was reported that the activation of the apoptotic mitochondrial pathway in PV-infected neuronal IMR5 cells was Bax dependent (73). However, whether 2B is involved in the activation of Bax during PV infection is still unknown. Nevertheless, our results provide evidence that EV71 2B induces cell apoptosis by modulating Bax activation during EV71 infection.
In summary, we report that during infection, the EV71 2B protein localized to the mitochondria and induced mitochondrion-associated cell death signals by recruiting and directly interacting with the proapoptotic protein Bax. The interaction of 2B with Bax resulted in the translocation of the latter to the mitochondria and in its subsequent activation (Fig. 12). Cells lacking Bax expression showed resistance to 2B-induced apoptosis. Thus, this study provides evidence that EV71induces cell apoptosis by modulating Bax activation and reveals important clues regarding the mechanism of Cyt c release and mitochondrial permeabilization during EV71 infection.
FIG 12.
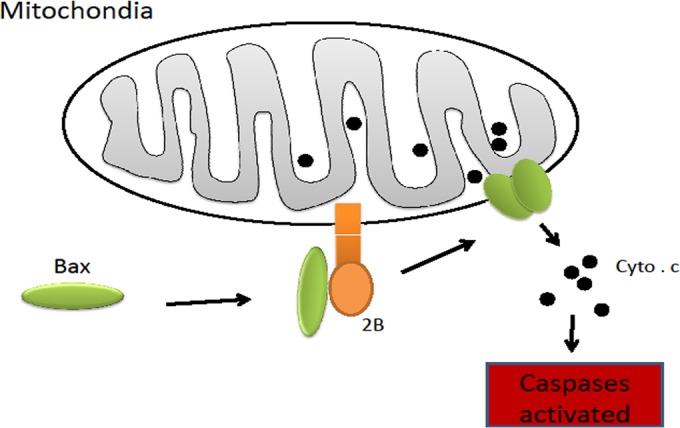
Schematic presentation of the mechanism of cell apoptosis induced by 2B. 2B encoded by EV71 is targeted to the outer membrane of the mitochondrion. Its N terminus is exposed on the membrane surface, interacts with Bax, and recruits Bax to the mitochondrion. The interaction between Bax and 2B induces the conformational activation of Bax and exposes its BH3 domain. Bax forms homo-oligomers that trigger the release of the caspase-activating protein Cyt c and induce cell apoptosis.
ACKNOWLEDGMENTS
This work is supported by grants from the National Natural Science Foundation of China (NSFC) (grants 81321063, 31270211, and 31370201) and the National Basic Research Program of China (973 Program) (grants 2011CB504703 and 2010CB530102).
We are grateful to Paul Chu, Guest Professor of the Institute of Microbiology, Chinese Academy of Sciences, for his help during the preparation of the manuscript. We also thank Xiaolan Zhang and Zhao Tong, Institute of Microbiology, CAS, for technical help with confocal microscopy and flow cytometry.
REFERENCES
- 1.Brown BA, Pallansch MA. 1995. Complete nucleotide sequence of enterovirus 71 is distinct from poliovirus. Virus Res 39:195–205. doi: 10.1016/0168-1702(95)00087-9. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt NJ, Lennette EH, Ho HH. 1974. Apparently new enterovirus isolated from patients with disease of central nervous-system. J Infect Dis 129:304–309. doi: 10.1093/infdis/129.3.304. [DOI] [PubMed] [Google Scholar]
- 3.Chang LY, Lin TY, Hsu KH, Huang YC, Lin KL, Hsueh C, Shih SR, Ning HC, Hwang MS, Wang HS, Lee CY. 1999. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet 354:1682–1686. doi: 10.1016/S0140-6736(99)04434-7. [DOI] [PubMed] [Google Scholar]
- 4.Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. 2010. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol 9:1097–1105. doi: 10.1016/S1474-4422(10)70209-X. [DOI] [PubMed] [Google Scholar]
- 5.Huang CC, Liu CC, Chang YC, Chen CY, Wang ST, Yeh TF. 1999. Neurologic complications in children with enterovirus 71 infection. N Engl J Med 341:936–942. doi: 10.1056/NEJM199909233411302. [DOI] [PubMed] [Google Scholar]
- 6.Tagaya I, Takayama R, Hagiwara A. 1981. A large-scale epidemic of hand, foot and mouth-disease associated with enterovirus-71 infection in Japan in 1978. Jpn J Med Sci Biol 34:191–196. doi: 10.7883/yoken1952.34.191. [DOI] [PubMed] [Google Scholar]
- 7.Yang F, Ren LL, Xiong ZH, Li JG, Xiao Y, Zhao R, He YQ, Bu G, Zhou SL, Wang JW, Qi J. 2009. Enterovirus 71 outbreak in the People's Republic of China in 2008. J Clin Microbiol 47:2351–2352. doi: 10.1128/JCM.00563-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan JJ, Wang JR, Liu CC, Yang HB, Su IJ. 2000. An outbreak of enterovirus 71 infection in Taiwan 1998: a comprehensive pathological, virological, and molecular study on a case of fulminant encephalitis. J Clin Virol 17:13–22. doi: 10.1016/S1386-6532(00)00067-6. [DOI] [PubMed] [Google Scholar]
- 9.Solomon T, Lewthwaite P, Perera D, Cardoso MJ, McMinn P, Ooi MH. 2010. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- 10.Lum LCS, Wong KT, Lam SK, Chua KB, Goh AYT. 1998. Neurogenic pulmonary oedema and enterovirus 71 encephalomyelitis. Lancet 352:1391–1391. [DOI] [PubMed] [Google Scholar]
- 11.Chang L, Huang L, Gau SS, Wu Y, Hsia S, Fan T, Lin K, Huang Y, Lu C, Lin T. 2007. Neurodevelopment and cognition in children after enterovirus 71 infection. N Engl J Med 356:1226–1234. doi: 10.1056/NEJMoa065954. [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Reed JC. 1998. Mitochondria and apoptosis. Science 281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 13.Brenner C, Kroemer G. 2000. Apoptosis—mitochondria—the death signal integrators. Science 289:1150–1151. doi: 10.1126/science.289.5482.1150. [DOI] [PubMed] [Google Scholar]
- 14.Li HL, Zhu H, Xu CJ, Yuan JY. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501. doi: 10.1016/S0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 15.Rokhlin OW, Glover RA, Cohen MB. 1998. Fas-mediated apoptosis in human prostatic carcinoma cell lines occurs via activation of caspase-8 and caspase-7. Cancer Res 58:5870–5875. [PubMed] [Google Scholar]
- 16.Martinou JC, Youle RJ. 2011. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gross A, McDonnell JM, Korsmeyer SJ. 1999. BCL-2 family members and the mitochondria in apoptosis. Gene Dev 13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 18.Tsujimoto Y. 1998. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells 3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- 19.Antonsson B. 2004. Mitochondria and the Bcl-2 family proteins in apoptosis signaling pathways. Mol Cell Biochem 256:141–155. doi: 10.1023/B:MCBI.0000009865.70898.36. [DOI] [PubMed] [Google Scholar]
- 20.Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. 2001. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol 153:1265–1276. doi: 10.1083/jcb.153.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dewson G, Kluck RM. 2009. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122:2801–2808. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei MC, Zong WX, Cheng EHY, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacCregor GR, Thompson CB, Korsmeyer SJ. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zha HB, AimeSempe C, Sato T, Reed JC. 1996. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BN3) distinct from BH1 and BH2. J Biol Chem 271:7440–7444. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]
- 24.Degenhardt K, Sundararajan R, Lindsten T, Thompson C, White E. 2002. Bax and Bak independently promote cytochrome c release from mitochondria. J Biol Chem 277:14127–14134. doi: 10.1074/jbc.M109939200. [DOI] [PubMed] [Google Scholar]
- 25.Zhou LY, Chang DC. 2008. Dynamics and structure of the Bax-Bak complex responsible for releasing mitochondrial proteins during apoptosis. J Cell Sci 121:2186–2196. doi: 10.1242/jcs.024703. [DOI] [PubMed] [Google Scholar]
- 26.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao SG, Robin AY, Smith BJ, Huang DCS, Kluck RM, Adams JM, Colman PM. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Qi YP, Xiao GF. 1999. Structure and function of bak gene and its apoptosis-inducing mechanism. Chin Sci Bull 44:605–609. doi: 10.1007/BF03182718. [DOI] [Google Scholar]
- 28.Antonsson B, Montessuit S, Sanchez B, Martinou JC. 2001. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 29.Cuconati A, White E. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Gene Dev 16:2465–2478. doi: 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- 30.Vives J, Juanola S, Cairo JJ, Prats E, Cornudella L, Godia F. 2003. Protective effect of viral homologues of bcl-2 hybridoma cells under apoptosis-inducing conditions. Biotechnol Prog 19:84–89. doi: 10.1021/bp0255715. [DOI] [PubMed] [Google Scholar]
- 31.Takayama S, Cazalshatem DL, Kitada S, Tanaka S, Miyashita T, Hovey LR, Huen D, Rickinson A, Veerapandian P, Krajewski S, Saito K, Reed JC. 1994. Evolutionary conservation of function among mammalian, avian, and viral homologs of the Bcl-2 oncoprotein. DNA Cell Biol 13:679–692. doi: 10.1089/dna.1994.13.679. [DOI] [PubMed] [Google Scholar]
- 32.Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HLA, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. 2000. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med 191:33–45. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart SA, Poon B, Jowett JBM, Chen ISY. 1997. Human immunodeficiency virus type 1 vpr induces apoptosis following cell cycle arrest. J Virol 71:5579–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart SA, Poon B, Song JY, Chen ISY. 2000. Human immunodeficiency virus type 1 Vpr induces apoptosis through caspase activation. J Virol 74:3105–3111. doi: 10.1128/JVI.74.7.3105-3111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nie Z, Phenix BN, Lum JJ, Alam A, Lynch DH, Beckett B, Krammer PH, Sekaly RP, Badley AD. 2002. HIV-1 protease processes procaspase 8 to cause mitochondrial release of cytochrome c, caspase cleavage and nuclear fragmentation. Cell Death Differ 9:1172–1184. doi: 10.1038/sj.cdd.4401094. [DOI] [PubMed] [Google Scholar]
- 36.Nomura-Takigawa Y, Nagano-Fujii M, Deng L, Kitazawa S, Ishido S, Sada K, Hotta H. 2006. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J Gen Virol 87:1935–1945. doi: 10.1099/vir.0.81701-0. [DOI] [PubMed] [Google Scholar]
- 37.Schaecher SR, Touchette E, Schriewer J, Buller RM, Pekosz A. 2007. Severe acute respiratory syndrome coronavirus gene 7 products contribute to virus-induced apoptosis. J Virol 81:11054–11068. doi: 10.1128/JVI.01266-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Douglas AE, Corbett KD, Berger JM, McFadden G, Handel TM. 2007. Structure of M11L: a myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci 16:695–703. doi: 10.1110/ps.062720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundararajan R, Cuconati A, Nelson D, White E. 2001. Tumor necrosis factor-alpha induces Bax-Bak interaction and apoptosis, which is inhibited by adenovirus E1B 19K. J Biol Chem 276:45120–45127. doi: 10.1074/jbc.M106386200. [DOI] [PubMed] [Google Scholar]
- 40.Marshall B, Puthalakath H, Caria S, Chugh S, Doerflinger M, Colman PM, Kvansakul M. 2015. Variola virus F1L is a Bcl-2-like protein that unlike its vaccinia virus counterpart inhibits apoptosis independent of Bim. Cell Death Dis 6:e1680. doi: 10.1038/cddis.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pogo BGT, Melana SM, Blaho J. 2004. Poxvirus infection and apoptosis. Int Rev Immunol 23:61–74. doi: 10.1080/08830180490265547. [DOI] [PubMed] [Google Scholar]
- 42.Chang SC, Lin JY, Lo LYC, Li ML, Shih SR. 2004. Diverse apoptotic pathways in enterovirus 71-infected cells. J Neurovirol 10:338–349. doi: 10.1080/13550280490521032. [DOI] [PubMed] [Google Scholar]
- 43.Liang CC, Sun MJ, Lei HY, Chen SH, Yu CK, Liu CC, Wang JR, Yeh TM. 2004. Human endothelial cell activation and apoptosis induced by enterovirus 71 infection. J Med Virol 74:597–603. doi: 10.1002/jmv.20216. [DOI] [PubMed] [Google Scholar]
- 44.Du N, Cong HL, Tian HC, Zhang H, Zhang WL, Song L, Tien P. 2014. Cell surface vimentin is an attachment receptor for enterovirus 71. J Virol 88:5816–5833. doi: 10.1128/JVI.03826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarosiek KA, Chi XK, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. 2013. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51:751–765. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jurgensmeier JM, Xie ZH, Deveraux Q, Ellerby L, Bredesen D, Reed JC. 1998. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A 95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun FC, Wei S, Li CW, Chang YS, Chao CC, Lai YK. 2006. Localization of GRP78 to mitochondria under the unfolded protein response. Biochem J 396:31–39. doi: 10.1042/BJ20051916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cong HL, Ning D, Tian HC, Yang Y, Zhang W, Zhang H, Zhang WL, Song L, Tien P. 2013. Enterovirus 71 VP1 activates calmodulin-dependent protein kinase II and results in the rearrangement of vimentin in human astrocyte cells. PLoS One 8:e73900. doi: 10.1371/journal.pone.0073900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G. 2004. Viral proteins targeting mitochondria: controlling cell death. Biochim Biophys Acta 1659:178–189. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 50.Ohta A, Nishiyama Y. 2011. Mitochondria and viruses. Mitochondrion 11:1–12. doi: 10.1016/j.mito.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann S, El Maadidi S, Faletti L, Haun F, Labib S, Schejtman A, Maurer U, Borner C. 2015. How do viruses control mitochondria-mediated apoptosis? Virus Res 209:45–55. doi: 10.1016/j.virusres.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Everett H, McFadden G. 2001. Viruses and apoptosis: meddling with mitochondria. Virology 288:1–7. doi: 10.1006/viro.2001.1081. [DOI] [PubMed] [Google Scholar]
- 53.O'Brien V. 1998. Viruses and apoptosis. J Gen Virol 79:1833–1845. doi: 10.1099/0022-1317-79-8-1833. [DOI] [PubMed] [Google Scholar]
- 54.Tolskaya EA, Romanova LI, Kolesnikova MS, Ivannikova TA, Smirnova EA, Raikhlin NT, Agol VI. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J Virol 69:1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez-Gil L, Bano-Polo M, Redondo N, Sanchez-Martinez S, Nieva JL, Carrasco L, Mingarro I. 2011. Membrane integration of poliovirus 2B viroporin. J Virol 85:11315–11324. doi: 10.1128/JVI.05421-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madan V, Sanchez-Martinez S, Carrasco L, Nieva JL. 2010. A peptide based on the pore-forming domain of pro-apoptotic poliovirus 2B viroporin targets mitochondria. Biochim Biophys Acta 1798:52–58. doi: 10.1016/j.bbamem.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 57.van Kuppeveld FJM, de Jong AS, Melchers WJG, Willems PHGM. 2005. Enterovirus protein 2B po(u)res out the calcium: a viral strategy to survive? Trends Microbiol 13:41–44. doi: 10.1016/j.tim.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 58.Madan V, Castello A, Carrasco L. 2008. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell Microbiol 10:437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, Melchers WJ. 1997. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J 16:3519–3532. doi: 10.1093/emboj/16.12.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brisac C, Teoule F, Autret A, Pelletier I, Colbere-Garapin F, Brenner C, Lemaire C, Blondel B. 2010. Calcium flux between the endoplasmic reticulum and mitochondrion contributes to poliovirus-induced apoptosis. J Virol 84:12226–12235. doi: 10.1128/JVI.00994-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie SQ, Wang K, Yu WJ, Lu W, Xu K, Wang JW, Ye B, Schwarz W, Jin Q, Sun B. 2011. DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res 21:1271–1275. doi: 10.1038/cr.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desagher S, Martinou JC. 2000. Mitochondria as the central control point of apoptosis. Trends Cell Biol 10:369–377. doi: 10.1016/S0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 63.Aldabe R, Irurzun A, Carrasco L. 1997. Poliovirus protein 2BC increases cytosolic free calcium concentrations. J Virol 71:6214–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campanella M, de Jong AS, Lanke KWH, Melchers WJG, Willems PHGM, Pinton P, Rizzuto R, van Kuppeveld FJM. 2004. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J Biol Chem 279:18440–18450. doi: 10.1074/jbc.M309494200. [DOI] [PubMed] [Google Scholar]
- 65.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. 1998. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci U S A 95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HLA, Prevost MC, Xie ZH, Matsuyama S, Reed JC, Kroemer G. 1998. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 67.Green DR, Chipuk JE. 2008. Apoptosis—stabbed in the BAX. Nature 455:1047–1049. doi: 10.1038/4551047a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belzacq AS, Vieira HLA, Verrier F, Vandecasteele G, Cohen I, Prevost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, Kroemer G. 2003. Bcl-2 and bax modulate adenine nucleotide translocase activity. Cancer Res 63:541–546. [PubMed] [Google Scholar]
- 69.Shimizu S, Tsujimoto Y. 2000. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci U S A 97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madan V, Sanchez-Martinez S, Vedovato N, Rispoli G, Carrasco L, Nieva JL. 2007. Plasma membrane-porating domain in poliovirus 2B protein. A short peptide mimics viroporin activity. J Mol Biol 374:951–964. [DOI] [PubMed] [Google Scholar]
- 71.Agirre A, Barco A, Carrasco L, Nieva JL. 2002. Viroporin-mediated membrane permeabilization—pore formation by nonstructural poliovirus 2B protein. J Biol Chem 277:40434–40441. doi: 10.1074/jbc.M205393200. [DOI] [PubMed] [Google Scholar]
- 72.Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462:55–70. doi: 10.1016/S0005-2736(99)00200-X. [DOI] [PubMed] [Google Scholar]
- 73.Autret A, Martin-Latil S, Mousson L, Wirotius A, Petit F, Arnoult D, Colbere-Garapin F, Estaquier J, Blondel B. 2007. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J Virol 81:7504–7516. doi: 10.1128/JVI.02690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]