

Abstract
The selective gas-phase oxidation of disulfide bonds to their thiosulfinate form using ion/ion reactions and subsequent cleavage is demonstrated here. Oxidizing reagent anions are observed to attach to all polypeptides, regardless of amino acid composition. Direct proton transfer yielding a charge-reduced peptide is also frequently observed. Activation of the ion/ion complex between an oxidizing reagent anion and a disulfide-containing peptide cation results in oxygen transfer from the reagent anion to the peptide cation to form the [M +H+O]+ species. This thiosulfinate derivative can undergo one of several rearrangements that result in cleavage of the disulfide bond. Species containing an intermolecular disulfide bond undergo separation of the two chains upon activation. Further activation can be used to generate more sequence information from each chain. These oxidation ion/ion reactions have been used to illustrate the identification of S-glutathionylated and S-cysteinylated peptides, in which low molecular weight thiols are attached to cysteine residues in peptides via disulfide bonds. The oxidation chemistry effectively labels peptide ions with readily oxidized groups, such as disulfide bonds. This enables a screening approach for the identification of disulfide-linked peptides in a disulfide mapping application involving enzymatic digestion. The mixtures of ions generated by tryptic and peptic digestions of lysozyme and insulin, respectively, without prior separation or isolation were subjected both to oxidation and proton transfer ion/ion chemistry to illustrate the identification of peptides in the mixtures with readily oxidized groups.
Graphical Abstract
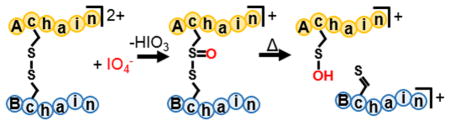
The formation of disulfide bonds is a post-translational modification (PTM) that plays critical roles in the formation and stabilization of peptide and protein native structures.1,2 Furthermore, peptides can form mixed disulfides with low molecular weight thiols, most commonly glutathione (i.e., glutathionylation) and cysteine (i.e., cysteinylation), during times of oxidative stress to prevent the irreversible oxidation of cysteine residues to their sulfinic and sulfonic acid derivatives.3–6 For these and other reasons, the identification and characterization of disulfide linkages is of widespread interest.7 Tandem mass spectrometry (MSn) has been used for the analysis of various PTMs in peptides and proteins.8–11 In the case of protonated peptides and proteins containing disulfide linkages, collision-induced dissociation (CID) often results in extensive backbone fragmentation with little to no evidence for cleavage of the disulfide bond.7 Additionally, intramolecular disulfide bonds can stabilize the regions of the peptide that are contained between the linked cysteine residues, thereby inhibiting detectable fragmentation within these regions and precluding the extraction of primary sequence information obtained for these oftentimes biologically relevant regions.12,13 For these reasons, a variety of approaches have been developed to favor disulfide bond cleavage over the many other competitive channels that may be available.
The selective cleavage of disulfide bonds in polypeptides can be done in a variety of ways. The most commonly employed methods are solution-phase oxidation with performic acid11,41,42 and solution-phase reduction with one of a variety of reagents, including dithiothreitol (DTT)14 and tris(2-carboxyethyl)phosphine (TCEP).15 Reduction is often coupled with alkylation to prevent recombination of the disulfide bonds.16 Enzymatic digestion and MS analysis can then be used to identify fragments containing disulfide bonds by comparison with the spectrum of the nonreduced sample.7,17 The electrochemical reduction of disulfide linkages in peptide and polypeptide ions just prior to electrospray ionization has also been described,18,19 while oxidation of disulfide linkages in polypeptides has been noted via exposure of species in a nanoelectrospray plume to hydroxyl radicals generated via a low-temperature plasma.20–22 Subsequent collisional activation of the oxidized disulfide-linked radical peptides resulted in cleavage of the disulfide bond. Several gas-phase approaches have also been shown to lead to cleavages along the disulfide linkage, either at the S–S bond or at an adjacent C–S bond. For example, irradiation at 266 nm has been observed to lead to selective disulfide bond cleavage,23 as has electron capture,24 electron transfer,25 and electron detachment26 dissociation. Furthermore, collisional activation of peptide or protein cations with low proton mobility,27 as well as peptide or protein anions,28 has been noted to result in predominant cleavage of the disulfide bonds. The “Route 66 Method” uses the highly selective ejection of hydrogen disulfide observed upon CID of sodium- and calcium-adducted peptides to identify and localize disulfide bonds.29 The incorporation of transition metal anions, such as Fe− and Co−, has been used to preferentially cleave the C–S bond in disulfide-containing molecules.30 The incorporation of Fe+ into insulin anions via ion/ion reactions resulted in enhanced cleavage near cysteine residues, either next to the disulfide-linked cysteine residue or at the disulfide bond.31 The generation of aurated species, viz., [M+Au(I)]+ and [M +Au(III)-2H]+, from multiply protonated disulfide-containing peptides and proteins via gas-phase cation switching ion/ion reactions has been used to selectively cleave the disulfide bonds in somatostatin and insulin upon CID.32,33 In this work, we demonstrate the gas-phase oxidation of disulfide bonds to thiosulfinate esters (e.g., S(O)-S linkages) as a means for labeling disulfide-linked polypeptide chains. Subsequent collisional activation of these oxidized species results in dominant cleavage of disulfide bonds.
Ion/ion reactions have recently been used to carry out some common solution-phase derivatizations in the gas-phase.34 These gas-phase reactions have several benefits over their solution-phase counterparts including faster reaction times, greater control over the extent of modification via control of both reactant concentrations and reaction times, and, perhaps most significantly, the avoidance of complicating the sample mixture due to unwanted side reactions or the introduction of species that adversely affect ionization yields.34 Recent examples of the manipulation of ion-type via ion/ion reactions include the selective removal of alkali metal adducts from multiply charged peptides,35 the charge-inversion of phosphatidylethanolamine and phosphatidylcholine lipid cations to form structurally informative anions36 and to differentiate isomers,37 and “Click” chemistry reactions between azides and alkynes.38
In addition to the reactions described above, we have reported two methods for generating oxidized peptides via ion/ion reactions. Both the selective oxidation of methionine residues to the sulfoxide form via ion/ion reactions with periodate anions39 and the oxidation of multiply protonated polypeptides to various forms, including [M+H+O]+, [M-H]+, and M•+, via ion/ion reactions with a suite of anionic reagents derived from persulfate40 have been described. Here, we show disulfide bonds are also readily susceptible to oxidation via ion/ion reactions with periodate and persulfate anions. While oxidation is a common solution-phase method used to cleave disulfide bonds, the thiosulfinate form is rarely seen.16,41,42 Instead, multiple oxygen atoms are added to the cysteine residues involved in the disulfide bond to form the cysteic acid derivatives.41 In the gas-phase ion/ion reactions described here, the addition of one oxidizing reagent anion results in the transfer of precisely one oxygen atom, thus limiting the oxidation to the thiosulfinate form. The thiosulfinate can then undergo rearrangement to cleave the disulfide bond upon CID. Whereas one of the most common conventional approaches to disulfide mapping, i.e., enzymatic digestion of disulfide-linked species followed by LC-MS comparison of reduced and nonreduced samples, requires multiple injection steps, the ion/ion reaction approach described here enables the screening of polypeptide cations for the presence of readily oxidized structural features, such as disulfide bonds, within the context of an MSn experiment.
EXPERIMENTAL SECTION
Materials
Methanol and glacial acetic acid were purchased from Mallinckrodt (Phillipsburg, NJ, USA). Sodium periodate, sodium persulfate, lysozyme, somatostatin, trypsin, pepsin, insulin, cysteine, methionine, S-nitrosoglutathione, and ammonium bicarbonate were purchased from Sigma-Aldrich (St. Louis, MO, USA). ARACAKA was synthesized by Pepnome Ltd. (Shenzhen, China). S-Glutathionylated ARACAKA was prepared by incubating 1 mg of ARACAKA with 1 mg of S-nitrosoglutathione in 1 mL of water at 37 °C for 1 h. S-cysteinylated ARACAKA was prepared by combining 1 mg of ARACAKA with 1 mg of cysteine in water for 1 week at room temperature. All peptide stock solutions for positive nanoelectrospray (nESI) were prepared in a 49.5/49.5/1 (vol/vol/vol) solution of methanol/water/acetic acid at an initial concentration of ~1 mg/mL and diluted 100-fold prior to use. Solutions of sodium persulfate (aqueous) and sodium periodate (50/50 vol/vol methanol/water) anions were prepared at concentrations of ~1 mg/mL and diluted 10-fold prior to use. Sodium periodate was incubated with 1 mg/mL of methionine (50/50 vol/vol methanol/water) to yield the iodate (IO3−) anion.
Enzymatic Digestions
Similar to a previously published procedure,43 1 mg of lysozyme or somatostatin was dissolved in 0.5 mL of 200 mM aqueous ammonium bicarbonate. One milligram of TPCK-treated trypsin was dissolved in 1 mL of water. Twenty microliters of this solution was added to the lysozyme or somatostatin solution. The lysozyme reaction mixture was incubated at 38 °C for ~18 h, while the somatostatin mixture was incubated at 38 °C for ~30 min. The peptic digest was performed by combining 0.1 mg of insulin with 0.025 mg of pepsin in 1% acetic acid at 37 °C for 6 h.29 The digestion mixtures were diluted to 1–5 μM in 49/49/2 vol/vol/vol methanol/water/acetic acid and used without further purification.
Mass Spectrometry
Experiments were performed on either a QTRAP 4000 hybrid triple quadrupole/linear ion trap or a TripleTOF 5600 mass spectrometer (Sciex, Concord, ON, Canada), both previously modified for ion/ion reactions.44 For experiments using the QTRAP platform multiply protonated peptides were isolated in the Q1-mass filter and injected into the q2 reaction cell followed by singly charged reagent anions via alternately pulsed nESI.45 The peptide cations and reagent anions were allowed to react for a mutual storage reaction time of 20 ms–1000 ms. The ion/ion reaction products were then transferred to Q3, where the complex was subjected to further characterization via MSn and mass analysis using mass-selective axial ejection (MSAE).46
For experiments performed in the TripleTOF 5600 mass spectrometer the tryptic and peptic digestion mixtures of lysozyme and insulin, respectively, were subjected to positive nESI and accumulated in the q2 reaction cell without any isolation. The reagent anions (either IO4− or IO3−) were isolated in Q1 prior to injection into q2 where they were allowed to react for <100 ms. The ion/ion reaction products were then subjected to activation via dipolar direct current (DDC) CID, a broadband heating technique where the ions are moved from the center of the trap toward one of the rods to induce RF heating, to remove neutral HIO4 and HIO3 adducts prior to mass analysis in the TOF.47 The spectrum resulting from DDC of the post-ion/ion products from reaction with IO3− was subtracted from the analogous IO4− spectrum using the subtraction tool in Sciex PeakView software (version 1.1.1.0), and the remaining peaks with positive abundance were plotted as a function of mass to create the difference spectrum.
RESULTS AND DISCUSSION
Periodate anion and intact persulfate anion have both been shown to oxidize polypeptides in the gas-phase, with persulfate anion being the stronger oxidizing agent.39,40 For the reactions shown here, these reagents are, for the most part, interchangeable. As the stronger oxidizing agent of the two, persulfate tends to lead to more reactive pathways than periodate. For this reason, the periodate anion was used here for most of the reactions, but some data using persulfate are included to demonstrate that it also can be used for disulfide oxidation.
Demonstration of Phenomenology: Peptides Resulting from a Tryptic Digest of Somatostatin
The gas-phase oxidation of intermolecularly disulfide-linked polypeptide chains is illustrated with ions derived from the tryptic digestion of somatostatin, a 14-residue peptide with the majority of the amino acids contained within a disulfide bridge between Cys-3 and Cys-14. The product generated from complete tryptic digestion (i.e., no missed cleavages) is indicated as AGCK/TFTSC, where the slash indicates the presence of separate peptides connected by a disulfide bond. Two isomeric products can arise from tryptic digestion with one missed cleavage, depending upon which lysine is cleaved: AGCK/NFFWKTFTSC or AGCKNFFWK/TFTSC. Formation of the AGCKNFFWK/TFTSC peptide appears to be favored as CID of the [M+H]+ species shows very few fragments corresponding to the AGCK/NFFWKTFTSC peptide (Supplemental Figure S-1). The [M+2H]2+ species from both AGCK/TFTSC and AGCKNFFWK/TFTSC were subjected to ion/ion reactions with oxidizing anions. A greater degree of oxidation was observed for the smaller peptide with persulfate than with periodate, whereas both reagents lead to highly efficient oxidation of the larger peptide.
The spectrum obtained following the ion/ion reaction between doubly protonated AGCKNFFWK/TFTSC and periodate anion is shown in Figure 1(a). In general, direct proton transfer and complex formation (i.e., attachment of the reagent anion to the cation) are the two main competing reaction channels observed upon ion/ion reaction with these reagents. Generation of the electrostatic complex is the dominant pathway observed in this case, with proton transfer observed at a very minor abundance. Collisional activation of this complex (Figure 1(b)) results in both proton transfer from the peptide to the reagent and oxidation of the peptide to form the [M+H+O]+ species. Loss of carbonic acid from the complex and from the [M+H+O]+ species, which could be distinguished from loss of CH2SO on the basis of exact mass, is also observed in Figure 1(b). The loss of carbonic acid was observed only when a C-terminal disulfide-linked cysteine was present. A plausible mechanism for this loss is provided in Scheme S-1. Additionally, activation of the complex results in cleavage of the disulfide bond as the protonated A-chain (AGCKNFFWK) is observed. The protonated B-chain is not observed, likely because of the expected lesser proton affinity of the B-chain due to the absence of a basic side chain. Cleavage of the disulfide bond becomes the dominant pathway when the oxidized [M+H+O]+ species is isolated and subjected to CID (Figure 1(c)). Some minor fragments with the disulfide bond still intact are observed (i.e., Ab7B, Ay5+O, Ay7B+O, nomenclature: capital letters refer to the peptide chain while lowercase letters next to them refer to peptide backbone fragments along that chain, e.g., Ab7B corresponds to the b7 ion from the A-chain connected to the intact B-chain via disulfide bond); these may well be derived from oxidation of the tryptophan residue instead of the disulfide bond. The peptide corresponding to no missed cleavages from the tryptic digest of somatostatin, AGCK/TFTSC, was also subjected to ion/ion reactions with oxidizing anions (persulfate, in this case). Activation of the [M+H+O]+ species from AGCK/TFTSC is shown in Figure 1(d) and predominantly yields peaks corresponding to cleavage of the disulfide bond. An additional stage of isolation and activation of the [A-H]+ ion was done to confirm the sequence and identity of the peak (Supplemental Figure S-2).
Figure 1.

Oxidation of two disulfide-linked peptides derived from a tryptic digest of somatostatin. (a) Ion/ion reaction between AGCKNFFWK/TFTSC [M+2H]2+ and IO4−. Activation of the (b) [M+2H+IO4]+ and (c) [M+H+O]+ from the ion/ion reaction between doubly protonated AGCKNFFWK/TFTSC and IO4−. (d) Activation of the [M+H+O]+ derived from the reaction between doubly protonated AGCK/TFTSC and HS2O8−. Asterisks indicate ammonia losses, degree signs indicate water losses, and lightning bolts indicate species subjected to CID.
Plausible mechanisms for the oxidation of a disulfide bond with periodate and the resulting rearrangements that result in cleavage of the disulfide bond are shown in Scheme 1(a)–(d). Similar to the mechanisms proposed for the oxidation of methionine residues via gas-phase ion/ion reactions with periodate anion,39 and the solution-phase oxidation of disulfides with periodate,48 the reaction is presumed to proceed via nucleophilic attack by one of the sulfur atoms in the disulfide bond on one of the oxygen atoms on the periodate reagent (Scheme 1(a)). Activation of post-ion/ion oxidized species of peptides containing intermolecular disulfide bonds results in a cluster of peaks arising from cleavage of the S–S(O) and C–S bonds (Figure 1(b)–(d)). For two identical peptides, M, linked by a disulfide bond, the cluster of peaks consists of several complementary ions, including the [M-H]+/[M+H +O]+, [M-H-S]+/[M+H+S+O]+, and [M+H]+/[M-H+O]+ species, and results from the abstraction of hydrogen atoms at different locations by one of the sulfur atoms in the oxidized disulfide bond. A mechanism for the formation of the [M-H]+/[M+H+O]+ pair is proposed in Scheme 1(b) and results from abstraction of the hydrogen atom on the carbon adjacent to the nonoxidized sulfur atom. It is also likely that the [M+H+O]+ species undergoes facile water loss to generate the [M-H]+ species, as supported by the high abundance of the [M-H]+ species relative to the other possible fragments. A mechanism for the formation of the [M-H-S]+/[M+H+S+O]+ pair is proposed in Scheme 1(c) and results from abstraction of the hydrogen atom on the α carbon of the oxidized sulfur atom. A mechanism for the formation of the [M+H]+/[M-H+O]+ pair is proposed in Scheme 1(d) and results from abstraction of the hydrogen atom on the carbon adjacent to the oxidized sulfur atom. The pattern of complementary cleavages associated with each of the initially linked peptide chains makes de novo identification of a disulfide-containing peptide straightforward.
Scheme 1.

Proposed Mechanisms for the Oxidation of Disulfide Bonds and Cleavage of the Oxidized Disulfide Bonds
Identification of S-Glutathionylated and S-Cysteinylated Peptides via Oxidation Ion/Ion Reactions
Oxidative stress in cells can lead to the reversible oxidation of cysteine thiols to the sulfenic acid derivative (–SOH), which can be further, irreversibly, oxidized to the sulfinic (–SO2H) and sulfonic (–SO3H) acid derivatives.3,4 Cysteine thiols play important roles in the structure and stability of proteins, and, thus, changes in their oxidation state can greatly modify protein structure and physiological activity.5,6 In order to prevent the irreversible oxidation of cysteine, free thiols can form mixed disulfides with low molecular weight thiols, most commonly glutathione (S-glutathionylation) and free cysteine (S-cysteinylation). 5,6 Glutathionylation has also been implicated in the regulation of various proteins, including signaling molecules, cytoskeletal proteins, transcription factors, and enzymes.49–51 Here, we show that gas-phase oxidation of S-glutathionylated and S-cysteinylated peptides yields dominant cleavage of the disulfide bond and ejection of the glutathione or cysteine moieties resulting in easy to identify signature losses.
The model peptide ARACAKA was incubated with S-nitrosoglutathione to make S-glutathionylated ARACAKA (structure shown in the inset of Figure 2(b)). Activation of protonated S-glutathionylated ARACAKA is shown in Figure 2(c) and consists of several neutral losses characteristic of glutathionylated species (e.g., neutral loss of 129 Da corresponding to pyroglutamic acid).52,53 While these neutral losses are often used to identify S-glutathionylated peptides, these losses are not always present and can lead to false negatives when trying to identify S-glutathionylation.54 The doubly protonated S-glutathionylated peptide was subjected to ion/ion reactions with periodate anion. Generation of both the proton transfer product and the oxidized species is seen upon activation of the ion/ion complex (Figure 2(a)). Additionally, cleavage of the disulfide bond is already observed as indicated by the presence of the [A-H]+ peak, where A is ARACAKA. Isolation and activation of the oxidized [M+H+O]+ species results almost exclusively in peaks corresponding to cleavage of the disulfide bond (Figure 2(b)).
Figure 2.

CID of ion/ion complex between periodate anion and doubly protonated (a) S-glutathionylated ARACAKA and (d) S-cysteinylated ARACAKA. (b) CID of [M+H+O]+ species of S-glutathionylated ARACAKA. (c) Control CID of the [M+H]+ species of S-glutathionylated ARACAKA. Structures of S-glutathionylated and S-cysteinylated ARACAKA inset in (b)/(c) and (d), respectively. ‘A’ refers to the peptide ARACAKA; “Glu”, and “Cys” refer to glutathione and cysteine, respectively. Lightning bolts indicate the species subjected to CID, while degree signs indicate water losses. Pyroglutamic acid loss indicated by -pyr.
Similarly, ARACAKA was incubated with free cysteine to produce S-cysteinylated ARACAKA (structure shown in the inset of Figure 2(d)). Doubly protonated S-cysteinylated ARACAKA was subjected to ion/ion reaction with periodate anion, and the resulting ion/ion complex was subjected to CID. Both the proton transfer [M+H]+ and oxidation [M+H+O]+ species are produced upon activation of the [M+2H+IO4]+, along with the [A-H]+ species (A indicates ARACAKA) indicating cleavage of the disulfide bond. It is important to note that activation of oxidized S-glutathionylated and S-cysteinylated peptides exclusively results in neutral loss of the glutathione or cysteine modification with water (323 and 137 Da, respectively) due to the low proton affinities of the modifications compared to the peptide. Several additional model peptides were modified via S-glutathionylation or S-cysteinylation, a few of which are shown in Supplemental Figure S-3, and subjected to ion/ion reactions with oxidizing reagent anions. All peptides studied reacted similarly to S-glutathionylated/cysteinylated ARACAKA as they underwent cleavage of the disulfide bond (as indicated by the signature losses of 323 and 137 Da for S-glutathionylation and S-cysteinylation, respectively, to return the [Peptide-H]+ species) upon activation of the oxidized species. These signature losses can be used to identify doubly protonated S-glutathionylated and S-cysteinylated peptides without prior knowledge of the sequence.
Identification of Disulfide-Linked Peptides in a Tryptic Digest of Lysozyme
Enzymatic digestion of proteins leads to another common scenario for the observation of disulfide-linked polypeptide chains. The behavior of such peptides is illustrated here with tryptic peptides generated from lysozyme, a ~14.3 kDa protein with four disulfide bonds. Tryptic digestion of lysozyme results in three peptides containing disulfide bonds with one peptide containing an intermolecular and an intramolecular disulfide bond, as well as peptides that do not contain disulfide linkages. All of the peptides obtained from the tryptic digestion that were observed as multiply protonated species (all peptides greater than five residues in length were observed, 88% sequence coverage) were isolated and subjected to ion/ion reactions with oxidizing reagent anions.
Product ion spectra resulting from CID of adducted ions generated from periodate anion reactions with the three digestion products containing disulfide linkages are shown in Figure 3. Due to the facile nature of the disulfide bond cleavages, cleavage of disulfide bonds was observed upon CID of the ion/ion complexes, without the need for further activation of the [M+H+O]+ ions that were generated as first generation products. The peptide of Figure 3(a), GYSLGNWVCAAK/CK, contains a tryptophan residue, which has been shown to be marginally susceptible to gas-phase oxidation with periodate anion.39 Here, however, the disulfide bond is a more reactive site as it is preferentially oxidized upon the addition of one periodate anion (Figure 3(a)). The second peptide shown, CELAAAMK/GCR, contains a methionine residue, which has the most readily oxidized side chain among the amino acids. In this case, the methionine residue is more reactive than the disulfide bond and is preferentially oxidized upon the addition of one periodate anion, as reflected by products that show loss of 64 Da corresponding to the loss of methanesulfenic acid from oxidized methionine side chains upon CID of the ion/ion complex with one periodate anion (Supplemental Figure S-4). There is a minor population where the disulfide bond undergoes oxidation, however, as some separated A- and B-chains are observed. For more complete cleavage of the disulfide bond, a complex between the peptide and two periodate anions was generated. In order to add two periodate anions the triply charged peptide precursor must be used to avoid neutralization. The activation of the complex between triply charged CELAAAMK/GCR and two periodate anions is shown in Figure 3(b). Ramped CID, where the amplitude of the rf is ramped to bring a range of m/z values into resonance with the auxiliary waveform used for ion trap CID, was done in order to activate each loss of iodic or periodic acid to yield the oxidized peptide. Cleavage of the disulfide bond is observed, as indicated by the presence of the separated A-chain. The complementary ion pairs (viz., [A-H+O]+/[A+H+S+2(O)]+ and [A-H-S+O]+/[A+H+S+2(O)]+) indicating the cleavage of a disulfide bond are observed. Different from before, however, is the addition of oxygen to each of the peaks in the motif. This is because the A-chain contains the methionine residue, which was also oxidized by one of the periodate anions.
Figure 3.

Activation of the ion/ion complexes between periodate anion and (a) doubly protonated GYSLGNWVCAAK/CK, (b) triply protonated CELAAAMK/GCR, and (c) triply protonated WWCNDGR/NLCNIPCSALLSSDITASVNCAK. Degree signs indicate water losses, and lightning bolts indicate species subjected to CID.
The third disulfide-linked peptide, WWCNDGR/NLCNIPCSALLSDITASVNCAK, contains one intramolecular (Cys-B3 to Cys-B21) and one intermolecular (Cys-A3 to Cys-B7) disulfide bond. While the doubly protonated peptide was observed upon positive nESI, the singly charged post-ion/ion product would have been outside of the mass range of the instrument so the triply protonated peptide was used instead. Activation of the ion/ion complex shows separation of the peptide chains into clusters of peaks consistent with the pattern previously described (Figure 3(c)). Further CID of the [A-H]+ and [B-H]+ peaks is consistent with the presence of an additional intramolecular disulfide bond containing most of the residues in the B-chain, as there are no peaks corresponding to cleavage within this region (Supplemental Figure S-5). It is possible that the addition of two periodate anions to the triply protonated species might show evidence for cleavage of both disulfide linkages, but this product was outside of the upper mass/charge range of the instrument used for these experiments.
The nondisulfide containing peptides resulting from the tryptic digest of lysozyme were also subjected to ion/ion reactions with persulfate anions to contrast their behaviors with those of the disulfide-linked peptides. Three of these peptides, FESNFNTQATNR (Supplemental Figure S-6(a)), HGLDNYR, and NTDGSTDYGILQINSR (data not shown), did not contain any readily oxidized residues (methionine or tryptophan) and did not undergo any oxidation upon activation of the ion/ion complex. Proton transfer was observed exclusively. One of the peptides, GTDVQAWIR (Supplemental Figure S-6(b)), contained a tryptophan residue and underwent very minor oxidation upon activation of the ion/ion complex. The minor oxidation and lack of the product ion patterns observed for disulfide-containing peptides reflects the lack of a disulfide bond in this peptide in de novo sequencing applications. The final peptide, IVSDGNGMNAWVAWR (Supplemental Figure S-6(c) and (d)), contained one methionine residue and two tryptophan residues. This peptide almost exclusively yielded oxidation upon CID of the ion/ion complex (Supplemental Figure S-6(c)). Further activation of the [M+H+O]+ species yielded a dominant 64 Da loss indicating that the oxidation occurred at a methionine side chain (Supplemental Figure S-6(d)). No evidence for a disulfide bond upon activation of the ion/ion complex or the [M+H+O]+ species was noted for this peptide. Based on the behavior of the singly oxidized CELAAAMK/GCR product ion mentioned above, a minor contribution of CID products showing the pattern for disulfide bond cleavage (see Supplemental Figure S-4) would be expected if it were present. Further evidence could be obtained by the addition of more oxidizing anions to more highly charged species. These three classes of peptides (lacking easily oxidized residues, containing a tryptophan, and containing a methionine) can be easily distinguished from disulfide bond-containing peptides via the absence of the pattern derived from symmetric and asymmetric cleavage of a disulfide bond.
Labeling Experiments Using Entire Enzymatic Digestion Mixtures without Separation or Isolation
An interesting characteristic of the gas-phase oxidation chemistry is that it effectively labels polypeptide ions that contain readily oxidized groups, with methionine residues and disulfide linkages being the most reactive species. The tryptophan side chain is only modestly reactive. Peptide ions that lack these sites undergo proton transfer exclusively. By comparing postion/ion reaction data using an oxidizing reagent anion with results generated using a reagent that leads exclusively to proton transfer, it is straightforward to identify ions that have undergone oxidation via the mass increase of 16 Da. To explore the utility of this method as a rapid method to identify analytes of interest (viz., disulfide-linked peptides) via oxidative labeling, the entire tryptic digest of lysozyme was subjected to ion/ion reactions with periodate and iodate anions. The positive nESI spectrum of the digest can be seen in Figure 4(a). The three disulfide-linked peptides are all observed; each linked peptide is indicated with a different color. The entire ion population shown in Figure 4(a) was then subjected to ion/ion reaction with periodate anion to generate the spectrum shown in Figure 4(b). Some proton transfer species (i.e., HGLDNYR+ and GTDVQAWIR+) are observed along with ion/ion complexes. The post-ion/ion spectrum was then subjected to DDC CID to activate all of the ions simultaneously (Figure 4(c)).
Figure 4.

(a) Positive nESI spectrum of the tryptic digest of lysozyme, (b) ion/ion reaction between the entirety of (a) and IO4−, and (c) DDC CID of the entirety of (b). The peptide WWCNDGR/NLCNIPCSALLSDITASVNCAK is indicated in the figure by the shortened form, WWCNDGR/NLC-C-CAK.
This process was then repeated using IO3− instead of IO4−, and the postion/ion DDC CID spectrum generated using IO3− was subtracted from the analogous spectrum generated using IO4− to generate the spectrum shown in Figure 5. Since IO3− is incapable of oxidation chemistry, the oxidized analytes of interest should remain in the difference spectrum, while species resulting from the proton transfer pathway are present in both postion/ion DDC CID spectra and, as such, are not observed in the difference spectrum. Indeed, the peaks remaining in the Figure 5 difference spectrum are of three main types: (1) residual ion/ion complexes with IO4− that survived the DDC CID step, (2) disulfide-linked peptides that have undergone oxidation (i.e., the [M+H+O]+ species generated from HIO3 loss from the complex), and (3) peptides that were originally involved in disulfide linkages and underwent oxidative disulfide cleavage to separate the peptides upon DDC CID (i.e., ions that arise from complexes that underwent successive loss of HIO3 and disulfide bond cleavage). Of the three disulfide-linked peptides, two, CELAAAMK/GCR and WWNCDGR/NLCNIPCSALLSDITASVNCAK, are observed in Figure 5 in their oxidized form. Evidence for oxidative cleavage of the disulfide bonds in both of these peptides is also observed (e.g., [CELAAAMK-H+O]+, [WWCNDGR-H]+, and [WWCNDGR +H+S+O]+). The species corresponding to [WWNCDGR/NLCNIPCSALLSDITASVNCAK+H+O]2+ overlaps with the [FESNFNTQAWIR+H+Na+IO4]+ species. The isotopic distribution at this mass indicates the presence of both doubly charged and a singly charged species and CID results in both loss of HIO4 from the [FESNFNTQAWIR+H+Na+IO4]+ species and oxidative cleavage of the disulfide bond in WWCNDGR/NLCNIPCSALLSDITASVNCAK, thus confirming the presence of both species. While the third disulfide-linked peptide, GYSLGNWVCAAK/CK, is not observed in its intact oxidized form, peaks corresponding to the oxidative cleavage of its disulfide bond are apparent (e.g., [GYSLGNWVCAAK-H]+ and [GYSLGNWVCAAK+H+O]+). In this case, all of the initially formed complexes underwent sequential cleavages. (The DDC CID conditions can be tuned to decrease the amount of secondary fragmentation that occurs to increase the amount of intact oxidized peptide in the spectrum and, thus, increase the likelihood of the [M+H+O]+ appearing in the difference spectrum.) The remaining peaks correspond to ion/ion complexes that did not undergo dissociation (i.e., [HGLDNYR+H+Na+IO4]+, [GTDVQAWIR+H+Na+IO4]+, etc.). Once again, the DDC CID conditions could be tuned to enhance dissociation of these complexes; it is apparent that there is a compromise between increased dissociation of ion/ion complexes and the prevention of secondary fragmentation of species containing oxidized disulfide bonds. The peaks present in the difference spectrum indicate analytes of interest that should be subjected to further analysis. Activation of the remaining ion/ion complexes will result in loss of the periodate adduct. Activation of the oxidized, mass-shifted peaks will result in oxidative cleavage of the disulfide bond, and intermolecularly linked peptides will separate. Activation of the peaks corresponding to oxidative cleavage of the disulfide bond will result in cleavage of the newly separated peptide along the peptide backbone to provide further sequence information.
Figure 5.

Difference spectrum where the DDC CID spectrum resulting from the ion/ion reaction with IO3− is subtracted from that with IO4−. The peptide WWCNDGR/NLCNIPCSALLSDITASVNCAK is indicated in the figure by the shortened form, WWCNDGR/NLC-C-CAK.
This procedure for labeling disulfide linked peptides in digest mixtures was repeated with a peptic digest of insulin, a peptide hormone containing one intramolecular and two intermolecular disulfide bonds. The disulfide bonds in insulin are notoriously difficult to characterize due to the close proximity of Cys-A6 and Cys-A7 (capital letters indicate peptide chain while number indicates location within the specified chain, e.g., Cys-A6 is the sixth residue on the A-chain of insulin), where Cys-A6 is intramolecularly disulfide-linked to Cys-A11 and Cys-A7 is intermolecularly disulfide-linked to Cys-B7 (see the structure in Supplemental Figure S-7). The difference spectrum generated for the peptic digest of insulin is shown in Supplemental Figure S-7 and is dominated by oxidized disulfide-linked species and species derived from the oxidative cleavage of disulfide bonds. Table S-1 lists the sequences associated with the indicated ions in Figure S-7. The insulin data further illustrates the straightforward nature of this method of labeling peaks of interest in enzymatic digestion mixtures.
CONCLUSIONS
The gas-phase oxidation of intermolecular disulfide bonds to the thiosulfinate ester form is demonstrated here using gas-phase ion/ion reactions with periodate and persulfate anions. Ion trap CID of the long-lived complex generated upon ion/ion reaction between disulfide-containing species and oxidizing reagent monoanions results in oxygen transfer from the reagent to the peptide to form the [M+H+O]+ species. The oxidized thiosulfinate species undergoes rearrangement to cleave the oxidized disulfide bond, either at the S(O)–S bond or the C– S(O) bond. These cleavages result in the generation of a pattern that can easily be recognized to indicate the presence and cleavage of a disulfide bond. This phenomenology was demonstrated here with S-glutathionylated and S-cysteinylated peptides as well as intermolecularly linked peptides derived from tryptic digestion of somatostatin and lysozyme. Relative to other approaches for the selective gas-phase cleavage of disulfide linkages, the oxidation approach is unique in that it can be used to label species with readily oxidized sites. This characteristic enables an approach for screening mixtures of ions for disulfide linkages, without multiple injection steps or solution-phase reduction. Ions derived from the entire tryptic digest of lysozyme were subjected to oxidation ion/ion reactions without prior separation or isolation of peaks, resulting in oxidative labeling of the disulfide-linked peptides that could be easily identified by the 16 Da mass shift or by comparison with the analogous spectrum derived from ion/ion reaction with a nonoxidizing reagent anion. This provides a novel means for disulfide mapping studies that can be executed exclusively via MSn, thereby avoiding sample manipulation in solution other than digestion. The application of oxidation via ion/ion reactions to species with intramolecular disulfide bonds, which might be useful within the context of top-down structural characterization, is currently under investigation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health under Grant GM R37-45372. Graduate student support for A.L.P. provided by Emerson Kampen and Bilsland Dissertation Fellowships.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem. 6b01043.
Figures S1–S7 (PDF)
References
- 1.Pantoliano MW, Ladner RC, Bryan PN, Rollence ML, Wood JF, Poulos TL. Biochemistry. 1987;26:2077–2082. doi: 10.1021/bi00382a002. [DOI] [PubMed] [Google Scholar]
- 2.Cook KM, Hogg PJ. Antioxid Redox Signaling. 2013;18:1987–2015. doi: 10.1089/ars.2012.4807. [DOI] [PubMed] [Google Scholar]
- 3.Leonard SE, Carroll KS. Curr Opin Chem Biol. 2011;15:88–102. doi: 10.1016/j.cbpa.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Circ Res. 2013;112:382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reynaert NL, Ckless K, Guala AS, Wouters EFM, van der Vliet A, Janssen-Heininger YMW. Biochim Biophys Acta, Gen Subj. 2006;1760:380–387. doi: 10.1016/j.bbagen.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Hochgräfe F, Mostertz J, Pöther DC, Becher D, Helmann JD, Hecker M. J Biol Chem. 2007;282:25981–25985. doi: 10.1074/jbc.C700105200. [DOI] [PubMed] [Google Scholar]
- 7.Gorman JJ, Wallis TP, Pitt JJ. Mass Spectrom Rev. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 8.Biemann K, Scoble HA. Science. 1987;237:992–998. doi: 10.1126/science.3303336. [DOI] [PubMed] [Google Scholar]
- 9.Qin J, Chait BT. Anal Chem. 1997;69:4002–4009. doi: 10.1021/ac970489n. [DOI] [PubMed] [Google Scholar]
- 10.Mann M, Jensen ON. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 11.Larsen MR, Trelle MB, Thingholm TE, Jensen ON. BioTechniques. 2006;40:790–798. doi: 10.2144/000112201. [DOI] [PubMed] [Google Scholar]
- 12.Stephenson JL, Cargile BJ, McLuckey SA. Rapid Commun Mass Spectrom. 1999;13:2040–2048. doi: 10.1002/(SICI)1097-0231(19991030)13:20<2040::AID-RCM754>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 13.Hogan JM, McLuckey SA. J Mass Spectrom. 2003;38:245–246. doi: 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- 14.Bewley TA, Li CH. Int J Protein Res. 1969;1:117–124. doi: 10.1111/j.1399-3011.1969.tb01633.x. [DOI] [PubMed] [Google Scholar]
- 15.Burns JA, Butler JC, Moran J, Whitesides GM. J Org Chem. 1991;56:2648–2650. [Google Scholar]
- 16.Wall JS. J Agric Food Chem. 1971;19:619–625. doi: 10.1021/jf60176a003. [DOI] [PubMed] [Google Scholar]
- 17.Lu S, Fan SB, Yang B, Li YX, Meng JM, Wu L, Li P, Zhang K, Zhang MJ, Fu Y, Luo J, Sun RX, He SM, Dong MQ. Nat Methods. 2015;12:329–336. doi: 10.1038/nmeth.3283. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Cui W, Zhang H, Dewald HD, Chen H. Anal Chem. 2012;84:3838–3842. doi: 10.1021/ac300106y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mysling S, Salbo R, Ploug M, Jørgensen Anal Chem. 2014;86:340–345. doi: 10.1021/ac403269a. [DOI] [PubMed] [Google Scholar]
- 20.Xia Y, Cooks RG. Anal Chem. 2010;82:2856–2864. doi: 10.1021/ac9028328. [DOI] [PubMed] [Google Scholar]
- 21.Ma X, Love CB, Zhang X, Xia Y. J Am Soc Mass Spectrom. 2011;22:922–930. doi: 10.1007/s13361-011-0104-1. [DOI] [PubMed] [Google Scholar]
- 22.Tan L, Xia Y. J Am Soc Mass Spectrom. 2012;23:2011–2019. doi: 10.1007/s13361-012-0465-0. [DOI] [PubMed] [Google Scholar]
- 23.Agarwal A, Diedrich JK, Julian RR. Anal Chem. 2011;83:6455–6458. doi: 10.1021/ac201650v. [DOI] [PubMed] [Google Scholar]
- 24.Zubarev RA, Kruger NA, Fridriksson MA, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. J Am Chem Soc. 1999;121:2857–2862. [Google Scholar]
- 25.Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. J Am Soc Mass Spectrom. 2005;16:1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalli A, Hakansson K. Int J Mass Spectrom. 2007;263:71–81. [Google Scholar]
- 27.Wells JM, Stephenson JL, Jr, McLuckey SA. Int J Mass Spectrom. 2000;203:A1–A9. [Google Scholar]
- 28.Chrisman PA, McLuckey SA. J Proteome Res. 2002;1:549– 557. doi: 10.1021/pr025561z. [DOI] [PubMed] [Google Scholar]
- 29.Kim HI, Beauchamp JL. J Am Soc Mass Spectrom. 2009;20:157–166. doi: 10.1016/j.jasms.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Sallans L, Lane KR, Freiser BS. J Am Chem Soc. 1989;111:865–873. [Google Scholar]
- 31.Payne AH, Glish GL. Int J Mass Spectrom. 2001;204:47–54. [Google Scholar]
- 32.Mentinova M, McLuckey SA. Int J Mass Spectrom. 2011;308:133–136. doi: 10.1016/j.ijms.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunawardena HP, O’Hair RAJ, McLuckey SA. J Proteome Res. 2006;5:2087–2092. doi: 10.1021/pr0602794. [DOI] [PubMed] [Google Scholar]
- 34.Prentice BM, McLuckey SA. Chem Commun. 2013;49:947–965. doi: 10.1039/c2cc36577d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luongo CA, Bu J, Burke NL, Gilbert JD, Prentice BM, Cummings S, Reed CA, McLuckey SA. J Am Soc Mass Spectrom. 2015;26:404–414. doi: 10.1007/s13361-014-1052-3. [DOI] [PubMed] [Google Scholar]
- 36.Stutzman JR, Blanksby SJ, McLuckey SA. Anal Chem. 2013;85:3752–3757. doi: 10.1021/ac400190k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rojas-Betancourt S, Stutzman JR, Londry FA, Blanksby SJ, McLuckey SA. Anal Chem. 2015;87:11255–11262. doi: 10.1021/acs.analchem.5b02243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bu J, Pilo AL, McLuckey SA. Int J Mass Spectrom. 2015;390:118–123. [Google Scholar]
- 39.Pilo AL, McLuckey SA. J Am Soc Mass Spectrom. 2014;25:1049–1057. doi: 10.1007/s13361-014-0861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilo AL, Bu J, McLuckey SA. J Am Soc Mass Spectrom. 2015;26:1103–1114. doi: 10.1007/s13361-015-1125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y, Smith DL. Anal Biochem. 1988;172:130–138. doi: 10.1016/0003-2697(88)90421-6. [DOI] [PubMed] [Google Scholar]
- 42.Sanger F. Biochem J. 1949;44:126–128. doi: 10.1042/bj0440126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han H, Xia Y, McLuckey SA. J Proteome Res. 2007;6:3062–3069. doi: 10.1021/pr070177t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Y, Wu J, Londry FA, Hager JW, McLuckey SA. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 45.Liang W, Xia Y, McLuckey SA. Anal Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Londry FA, Hager JW. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 47.Webb IK, Londry FA, McLuckey SA. Rapid Commun Mass Spectrom. 2011;25:2500–2510. doi: 10.1002/rcm.5152. [DOI] [PubMed] [Google Scholar]
- 48.Evans BJ, Doi JT, Musker WK. J Org Chem. 1990;55:2580–2586. [Google Scholar]
- 49.Chen HJC, Lin WP, Chiu SD, Fan CH. Chem Res Toxicol. 2014;27:864–872. doi: 10.1021/tx5000359. [DOI] [PubMed] [Google Scholar]
- 50.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. Free Radical Biol Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 51.Klatt P, Lamas S. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 52.Baillie TA, Davis MR. Biol Mass Spectrom. 1993;22:319–325. doi: 10.1002/bms.1200220602. [DOI] [PubMed] [Google Scholar]
- 53.Haroldsen PE, Reilly MH, Hughes H, Gaskell SJ, Porter CJ. Biomed Environ Mass Spectrom. 1988;15:615–621. doi: 10.1002/bms.1200151107. [DOI] [PubMed] [Google Scholar]
- 54.Brink A, Fontaine F, Marschmann M, Steinhuber B, Cece EN, Zamora I, Pahler A. Rapid Commun Mass Spectrom. 2014;28:2695–2703. doi: 10.1002/rcm.7062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.