

Abstract
Expansion microscopy (ExM) enables imaging of preserved specimens with nanoscale precision on diffraction limited instead of specialized super-resolution microscopes. ExM works by physically separating fluorescent probes after anchoring them to a swellable gel. The first expansion microscopy method was unable to retain native proteins in the gel and used custom made reagents not widely available. Here, we describe protein retention ExM (proExM), a variant of ExM that anchors proteins to the swellable gel allowing the use of conventional fluorescently labeled antibodies and streptavidin, and fluorescent proteins. We validate and demonstrate utility of proExM for multi-color super-resolution (~70 nm) imaging of cells and mammalian tissues on conventional microscopes.
We recently developed a technology, named expansion microscopy (ExM) that enables imaging of thick preserved specimens with ~70 nm lateral resolution1. Using ExM the optical diffraction limit is circumvented by physically expanding a biological specimen before imaging, thus bringing sub-diffraction limited structures into the size range viewable by a conventional diffraction-limited microscope. We showed that ExM can image biological specimens at the voxel rates of a diffraction limited microscope, but with the voxel sizes of a super-resolution microscope. Expanded samples are transparent, and index-matched to water, as the expanded material is >99% water. The original ExM protocol worked by labeling biomolecules of interest with a gel-anchorable fluorophore. Then, a swellable polyelectrolyte gel was synthesized in the sample, so that it incorporated the labels. Finally, the sample was treated with a nonspecific protease to homogenize its mechanical properties, followed by dialysis in water to mediate uniform physical expansion of the polymer-specimen composite. All of the chemicals required for ExM can be purchased except for the gel-anchorable label, which requires custom synthesis and raises the barrier for researchers to adopt the method. Another drawback of the ExM protocol is that genetically encoded fluorophores cannot be imaged without antibody labeling.
Here, we report the development of a variant of ExM, named protein retention ExM (proExM), in which proteins, rather than labels, are anchored to the swellable gel, using a commercially available cross-linking molecule. We demonstrate that fluorescent signals from genetically encoded fluorescent proteins and conventional fluorescently labeled secondary antibodies and streptavidin that are directly anchored to the gel are preserved even when subjected to the nonspecific proteolytic digestion from the original ExM protocol. proExM is a simple extension of standard histological methods used to prepare samples for imaging that should encourage more widespread adoption.
Strong protease digestion (i.e., with proteinase K) enabled isotropic expansion in the original ExM protocol. We asked whether native proteins could be chemically anchored to the ExM gel and stained with antibodies in the expanded state. As a first experiment, we used a modified approach with reduced proteolysis to preserve epitopes. To incorporate proteins into the polymeric gel we used the succinimidyl ester of 6-((Acryloyl)amino)hexanoic acid (Acryloyl-X, SE, abbreviated AcX, Life Technologies), which modifies amines on proteins with an acrylamide functional group. Borrowing from denaturing SDS-PAGE2 and antigen retrieval protocols3, we treated gel-embedded tissues in an alkaline detergent-rich buffer for one hour in an autoclave, and found ~4× expansion of Thy1-YFP mouse brain samples (Supplementary Fig. 1a, showing endogenous YFP pre-treatment; Supplementary Fig. 1b, showing post-expansion labeling with anti-GFP). We found that antibodies could indeed be delivered successfully post-expansion (Supplementary Fig. 1c–e). As a second treatment strategy, we exposed gel-embedded tissues to LysC, which cuts proteins at Lys residues (in contrast to nonspecific proteinase K)4,5 (Supplementary Fig. 2). Post-expansion staining in both cases was highly variable depending upon antibody identity (e.g., compare lamin A/C examined with three different protocols, Supplementary Fig. 1f(i–iii), to images obtained in the original ExM protocol, Supplementary Fig. 4 of ref. 1; additional examples, Supplementary Fig. 3). For some antibodies, post-expansion staining appeared to result in brighter signal compared to pre-gelation staining (Tom20, Supplementary Fig. 1g(i) vs h(i) (autoclaved); GFP, Supplementary Fig. 1g(ii) vs. h(ii) (autoclaved); PSD-95, Supplementary Fig 1g(iii) vs. h(iii) (LysC)). However, the variability (Supplementary Fig. 3) and incomplete homogenization (Supplementary Fig. 4) suggested that the strong proteolysis of the original ExM protocol was necessary for reliable expansion.
We next sought to devise a strategy that would combine the convenience of direct protein anchoring with strong proteinase K treatment. It is known that green fluorescent protein (GFP) exhibits extraordinary stability to proteases6,7. We hypothesized that GFP and GFP-like fluorescent proteins (FPs) might retain their fluorescence after the proteolytic digestion of the original ExM method, if they were retained in the polymer-specimen composite using AcX. We discovered that treatment with AcX followed by the standard ExM workflow, including proteinase K digestion, can preserve GFP fluorescence in the expanded gel with high efficiency (65 ± 5% preservation; mean ± std. dev.; n = 4; Fig. 1a, 2b and Supplementary Fig. 5). Because of the utility of this protocol, we termed the process of AcX treatment of a fixed specimen, followed by gelation, strong digestion, expansion, and imaging as protein retention expansion microscopy (proExM).
Figure 1.
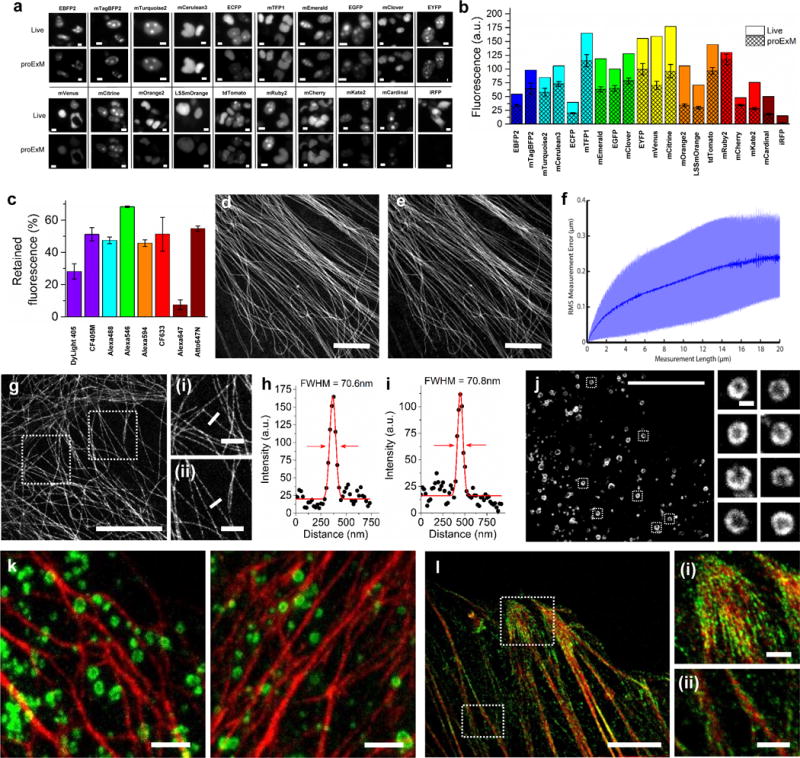
Retention of fluorescent protein (FP) and antibody fluorescence signals in proExM and proExM of FP fusions. (a) Representative images of selected FP-histone fusion proteins in live HEK293FT cells (upper row) and in the same cells after proExM treatment (lower row); iRFP was expressed as N-terminal fusion with nuclear localization sequence (NLS). (b) Quantified fluorescence of experiments as in panel a, after proExM treatment (crosshatched bars; mean ± standard deviation; n = 4 transfection replicates each). Open bars, literature values of the brightnesses of these fluorophores, normalized to the brightness of EGFP. (c) Retention of fluorescence for selected dyes conjugated with antibodies, after proExM treatment (mean ± standard deviation, n = 3 samples each), in mouse brain slice. (d) Super-resolution structured illumination microscopy (SR-SIM) image of immunostained microtubules after the anchoring step vs. (e) post-expansion image of the same sample acquired with a spinning disk confocal microscope. (f) Root mean square (RMS) length measurement error as a function of measurement length for proExM vs SIM images (blue line, mean; shaded area, standard deviation; n = 4 samples). (g) Confocal image of mClover-α-tubulin fusion. HeLa cells are used throughout the rest of this figure. Panels (i and ii) are magnified views of boxed regions in (g). Linecuts are quantified in panels h, i. Solid red lines in (h, i) indicate the Gaussian fit used to determine the full width at half maximum (FWHM; illustrated with red arrows). (j) Confocal image of mEmerald-clathrin fusion (left) and magnified views of single CCPs in the boxed regions (right). (k) Dual color proExM of clathrin (fused to mEmerald, green) and keratin (mRuby2, red). (l) Dual color proExM image of actin (mRuby2, red) and paxillin (mEmerald, green) fusions. Panels (i and ii) are magnified views of boxed regions in (f). Scale bars: (a) 5 μm, (d) 5 μm, (e) 5 μm (physical size post-expansion, 20.5 μm), (g) 5 μm (21.5 μm), (i–ii) 1 μm; (j) 10 μm (42.6 μm), insets 200 nm; (k) 1 μm (4.3 μm), (l) 5 μm (21.5 μm), (i–ii) 1 μm.
Figure 2.
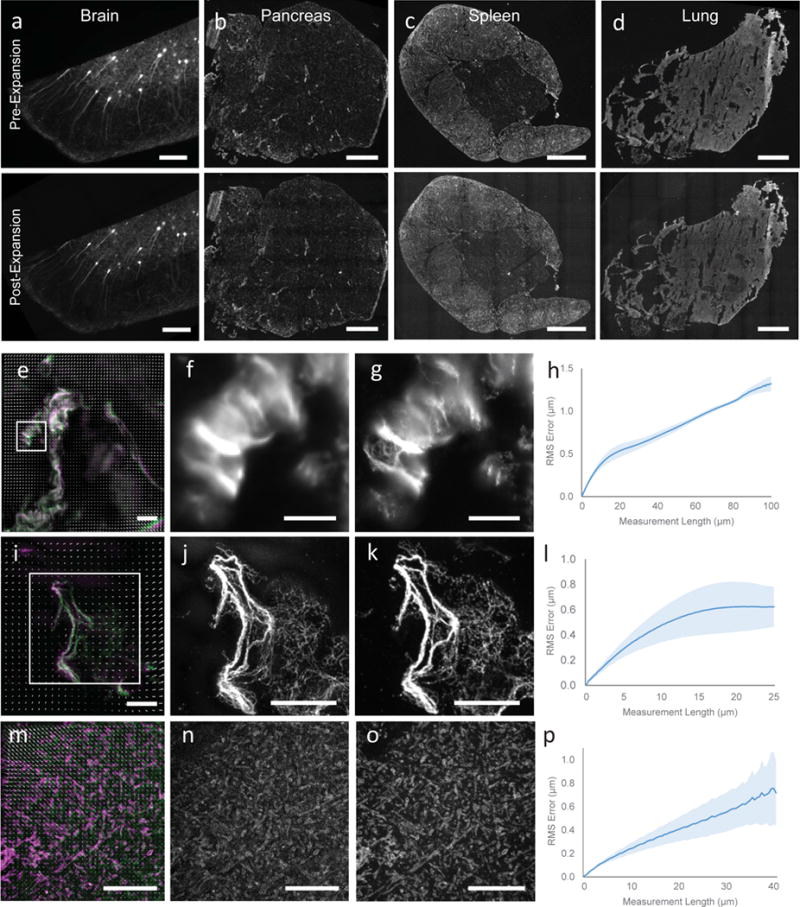
Validation of proExM in different mammalian tissue types. (a–d) Low magnification, wide-field images of pre-expansion (top) and post-expansion (bottom) samples of Thy1-YFP mouse brain (a) and vimentin-immunostained mouse pancreas (b), spleen (c), and lung (d). (e) Composite fluorescence image of Tom20 in Thy1-YFP mouse brain imaged with super-resolution structured illumination microscopy (SR-SIM) (green) and proExM (purple) with conventional confocal microscopy with distortion vector field overlaid (white arrows). (f) Pre-expansion SR-SIM image showing boxed region in (a). (g) Post-expansion confocal image of (f). (h) RMS length measurement error as a function of measurement length for proExM vs SR-SIM pre-expansion for Tom20 staining in Thy1-YFP mouse brain (blue line, mean; shaded area, standard deviation; n = 3 mouse brain cortex samples). (i) High magnification, wide-field fluorescence composite image of vimentin in mouse pancreas before (green) and after (purple) expansion with distortion vector field overlaid (white arrows, see methods). (j) Pre-expansion wide-field image showing boxed region in (i). (k) Post-expansion image of (j). (l) Root mean square (RMS) length measurement error as a function of measurement length for proExM vs widefield pre-expansion images for the different tissue types in (b–d) (blue line, mean; shaded area, standard deviation; n = 3 samples from pancreas, spleen, and lung). (m) Composite fluorescence image of vimentin in mouse pancreas imaged with super-resolution structured illumination microscopy (SR-SIM) (green) and proExM (purple) with conventional confocal microscopy with distortion vector field overlaid (white arrows). (n) Pre-expansion SR-SIM image showing boxed region in (m). (o) Post-expansion confocal image of (n). (p) RMS length measurement error as a function of measurement length for proExM vs SR-SIM pre-expansion for vimentin staining in pancreas (blue line, mean; shaded area, standard deviation; n = 4 fields of view from 2 samples). Scale bars: (a) top 200 μm, bottom 200 μm (physical size post-expansion, 800 μm), (b–d) top 500 μm, bottom 500 μm (2.21 mm, 2.06 mm, 2.04 mm, respectively), (e, f) 10 μm, (g) 10 μm (40 μm), (i) 10 μm, (j) 5 μm, (k) 5 μm (20.4 μm), (m) 5 μm, (n) 5 μm, (o) 5 μm (20.65 μm).
We systematically examined persistence of fluorescence for various FPs in the proExM workflow. We selected 20 widely used FPs with spectra ranging from the blue to the near-infrared (Supplementary Table 1). Selected FPs were fused to histone proteins and expressed in human embryonic kidney (HEK293FT) cells. We compared images of live cultures vs. after-proExM images of the same cells (Fig. 1a). Most FPs retained more than 50% of their live fluorescence intensity after proExM (n = 4 samples each; Fig. 1a, 1b, Supplementary Table 1), comparable to the persistence of small-molecule fluorophores in the original ExM protocol1.
Having seen that FPs could persist sufficiently to report signals even after a strong digestion process, we next sought to determine if other signals might persist. We discovered that proExM anchors, and preserves the fluorescence of, commercial fluorescently conjugated secondary antibodies. Following gelation and digestion, specimens labeled with secondary antibodies bearing a variety of small-molecule fluorophores retained ~50% of their initial brightness (n = 3 samples each; Fig. 1c; Supplementary Table 2). In the original ExM protocol, custom conjugation of secondary antibodies to enable labeling with a gel-anchorable fluorophore was required1. proExM allows commercial secondary antibodies to be used in place of these custom formulations.
In addition to antibodies, we also observed preservation of signals from fluorescently labeled streptavidin. Using streptavidin, we imaged probes designed to capture cysteine-S-nitrosation using a previously developed chemical method, SNOTRAP8, thus demonstrating the imaging of S-nitrosation signals with proExM (Supplementary Fig. 6). This protocol also points towards the possibility of anchoring other protease-resistant tags to the polymer, followed by gelation, digestion, expansion, and imaging, as a potentially generalized strategy.
Although the digestion step was the same as for our originally validated ExM protocol, suggesting that nanoscale isotropy would be preserved, we further validated proExM by performing imaging of immunostained microtubules in cultured cells with super-resolution structured illumination microscopy (SR-SIM) (Fig. 1d) before proExM and confocal imaging after proExM (Fig. 1e), as we did to validate our original ExM protocol1. We quantified the root-mean-square (RMS) error of feature measurements after proExM over length scales between 0 and 20 microns, and found that RMS errors were ~1–2% of the measurement distance (Fig. 1f).
We performed proExM followed by confocal microscopy to image several fusion proteins bearing genetically encoded fluorophores (i.e., unstained) in cultured HeLa cells. We first examined fusions of tubulin, clathrin and keratin (Fig. 1g–k), which we and others have commonly used as stereotyped structures to demonstrate super-resolution imaging of cells9–12. The tubulin-mClover fusion presented a microtubule full-width at half-maximum (FWHM) of 67±8 nm (n = 16 microtubules in 3 samples) (Fig. 1h, i), suggesting a resolution of better than 70 nm11. We also imaged clathrin-mEmerald in HeLa cells obtaining excellent definition of the nulls in the middle of the pits (Fig. 1j). Dual-color proExM imaging of fusion proteins containing mEmerald and mRuby2, two of the genetically encoded fluorophores in our screen, yielded excellent image quality as expected (keratin-mRuby2 and clathrin-mEmerald, Fig. 1k; paxillin-mEmerald and actin-mRuby2, Fig. 1l). We examined the stability of four photoswitchable FPs during proExM (Supplementary Fig. 7, Supplementary Table 3). We imaged cells expressing histone 2B-Dendra and mEos2-tubulin fusions with PALM microscopy (Supplementary Fig. 7), demonstrating preservation of photoswitching fluorophores compatible with PALM (although validating resolution claims for post-ExM PALM is beyond the scope of this paper).
To assess the performance of proExM in various three-dimensional tissues, we performed proExM on four different mouse tissue types (brain, pancreas, lung and spleen, Fig. 2a–d). Mouse brain expressing YFP under the Thy1 promoter (Thy1-YFP) in a sparse subset of neurons expands without distortion at the millimeter scale following treatment with proteinase K as described for cultured cells (Fig. 2a, top vs. bottom). Pancreas, spleen and lung have different mechanical properties than brain (e.g., more connective tissue), which hinders expansion following room temperature proteinase K digestion. We antibody stained the intermediate filament vimentin as a marker of connective tissue to examine the isotropy of expansion in these diverse tissue types. We observed that, with a slight modification in the digestion temperature to the optimum of the proteinase K enzyme (60° C for 4 hours), proExM allows for expansion of pancreas, lung, and spleen tissue, with excellent preservation of tissue morphology at the millimeter length scale (Fig. 2b–d, top vs. bottom). High-resolution diffraction-limited microscopy of the tissue before (Fig. 2e, 2f) vs after proExM (Fig. 2e, 2g) shows the resolution improvement of proExM. We quantified the isotropy of expansion by measuring the root-mean-square (RMS) error of feature measurements after proExM in the microscale (<100 μm) for pancreas, lung and spleen tissue. The RMS errors were small (1–3% of the measurement distance) and similar among all three of the tissue types (Fig. 2h) at this length scale.
To examine the isotropy of expansion at the nanoscale, we performed SR-SIM (Fig. 2i, 2j) and proExM confocal imaging (Fig. 2i, 2k) on vimentin staining in the pancreas. Again, we observed small RMS errors on the order of 1–5% of the measurement length for measurements between 0 and 25 microns (Fig. 2l, n = 4 fields of view from 2 samples). We performed a similar analysis on mouse brain cortical tissue stained with antibodies against Tom20, a mitochondrial marker, and imaged with SR-SIM before (Fig. 2m, 2n) and confocal after (2o) proExM processing using proteinase K digestion at room temperature. RMS errors for this tissue type were between 1–3% of the measurement length, between 0 and 40 microns (Fig. 2p, n = 3 specimens).
Transgenic animals expressing FPs, as well as animals expressing FPs after viral gene delivery, are routinely used in biology for labeling proteins and cells in intact tissues and organisms. We applied proExM for visualization of FPs expressed in intact mammalian brain tissue, including the brains of mice (Supplementary Fig. 8) and a rhesus macaque (Fig. 3a–c), obtaining images that, as expected, showed minimal macroscale distortion after expansion (e.g., compare Fig. 3a vs 3b). Using a high magnification lens on a conventional confocal microscope, dendritic spine morphology was easily resolved after expansion, with even thin spine necks visible (Fig. 3c inset, arrow).
Figure 3.
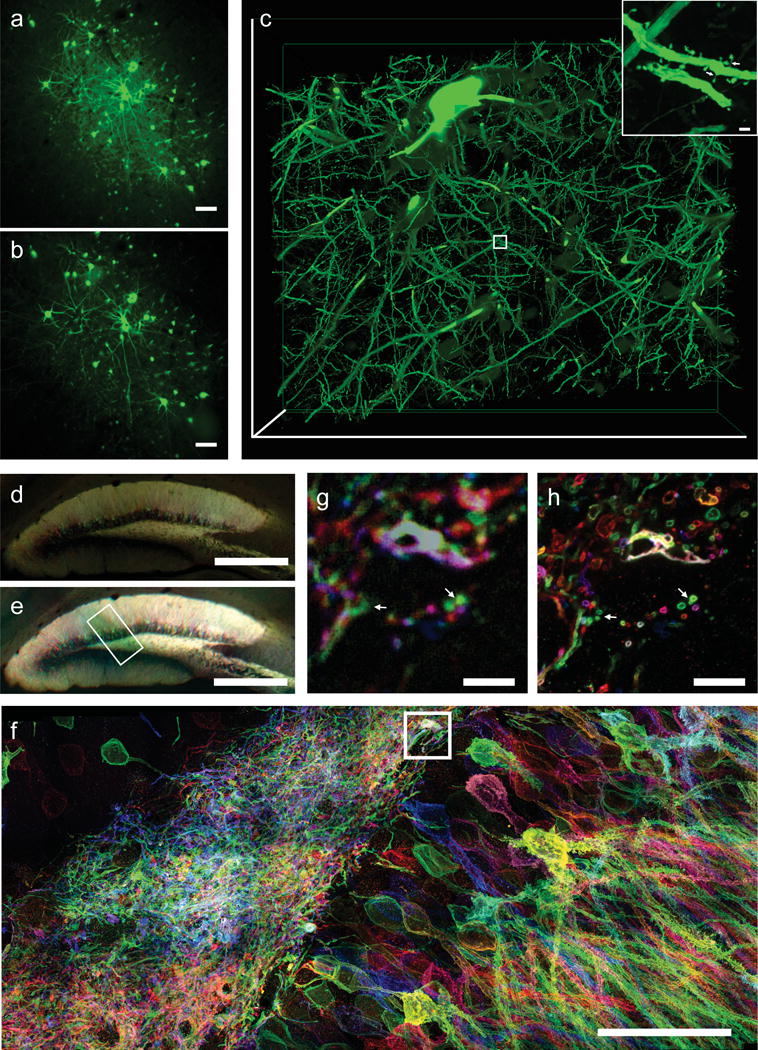
proExM of mammalian brain circuitry. (a) Wide-field image of GFP fluorescence in virally injected rhesus macaque cortex. (b) Post-expansion wide-field fluorescence image of (a). (c) Volume rendering of confocal microscopy images of subregion of (b). Inset shows a zoom-in of boxed region in (c) showing dendritic spines. (d) Low magnification widefield fluorescence imaging showing immunostained mouse hippocampus expressing virally delivered Brainbow3.0. (e) Post-expansion wide-field image of sample from (e). (f) MIP high resolution confocal microscopy image following expansion of membrane labeled Brainbow3.0 neurons from boxed region in (e). (g) Pre-expansion confocal image showing one optical section of boxed region in (f). (h) Post-expansion image of (g). Scale bars: (a) 100 μm, (b) 100 μm (physical size post-expansion, 413 μm); (c) 300 μm × 254 μm × 25 μm, (c) (i) 1 μm, (d) 500 μm, (e) 500 μm (1980 μm); (f) 5 μm, (g) 5 μm (19.8 μm); (h) 50 μm (198 μm).
We applied proExM for imaging of mouse brain circuitry expressing virally delivered Brainbow3.013,14, which marks neurons with random combinations of membrane anchored FPs. These FPs are antigenically distinct to allow for subsequent amplification via antibodies. Following proExM, antibody staining and morphology are preserved in brain tissues (Fig. 3d vs 3e). Confocal imaging allows for large volume, multi-color super-resolved imaging of the Brainbow sample (Fig. 3f). Side-by-side comparison of confocal images before (Fig. 3g) and after (Fig. 3h) expansion shows how axons and dendrites too close to resolve before expansion can be clearly resolved after expansion (Fig. 3g, h).
We here report that it is possible to directly anchor proteins to a swellable gel synthesized throughout a biological sample, by applying a commercially available small molecule cross-linker prior to gelation. This protein retention ExM (proExM) strategy can be used to perform nanoscale imaging of immunostained cells and tissues (Fig. 4, top), as well as samples expressing various FPs (Fig. 4, middle). ProExM variants can support post-expansion antibody delivery, potentially increasing brightness of staining and antibody access (Fig. 4, bottom).
Figure 4.
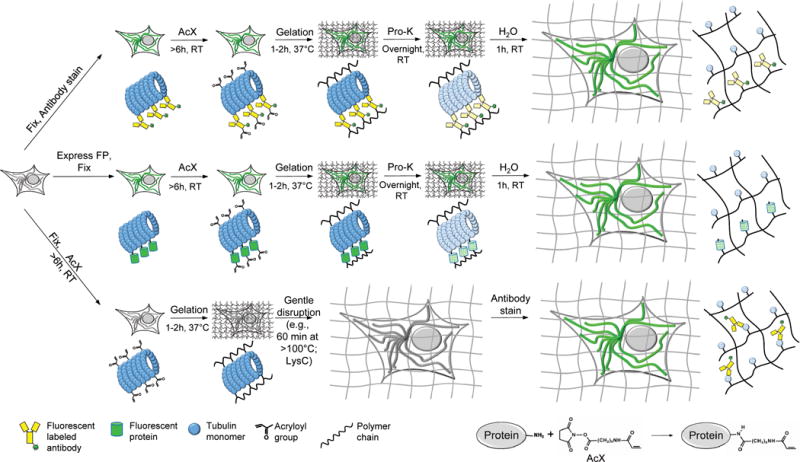
Workflows for expansion microscopy with protein retention. Three basic sample processing workflows were explored in this paper. Top, samples are chemically fixed and stained with antibodies, using conventional immunostaining protocols, before AcX treatment at room temperature and subsequent ExM processing (gelation, proteinase K treatment, and expansion in water). Middle, samples expressing fluorescent proteins (FPs) are chemically fixed (and optionally permeabilized) before AcX treatment, and subsequent ExM processing. Bottom, samples treated with AcX, followed by gelation, are then processed with a gentle homogenization procedure (e.g., alkaline hydrolysis and denaturation, or digestion with LysC), and finally antibody staining in the expanded state.
We show that native proteins anchored in this way can retain epitope functionality and be labeled post-expansion if the nonspecific proteolysis of ExM is replaced with modified post-gelation homogenization treatments. Such approaches may overcome the limitations inherent to delivering antibodies in the crowded environment of native tissue15–19. For example, closely packed epitopes may bind antibodies poorly in dense tissue, but better access antibodies after expansion (Supplementary Fig. 1)
We found that fluorescent proteins and fluorescent antibodies delivered using standard methods are also retained in the gel, and furthermore exhibit fluorescent signals following nonspecific proteolysis treatment. We demonstrate the multi-color, large volume capability of proExM by expanding Brainbow samples, which may be useful for circuit mapping. ProExM is simple and easily integrated into existing sample workflows. Preservation of endogenous fluorescence allows for the use of transgenic animals, viral expression vectors, and transfection of FPs, all without immunostaining.
As with our original ExM protocol, samples processed with proExM are optically clear and index matched to water1. This allows for super-resolution imaging deep into samples, on conventional fluorescence microscopes, limited only by working distance of the objective lens. As an effective sub-diffraction limited imaging modality, ExM protocols exhibit great speed of imaging. Although samples prepared by ExM protocols are larger in size than before expansion (and thus may require longer working distance objectives and tiled acquisition), there is a net win, since ExM protocols enable voxel sizes of super-resolution microscopy, but imaged at voxel acquisition rates of conventional microscopy. Like ExM, proExM does not require specialized hardware, extensive post-processing, or other enabling aspects of conventional super-resolution methods. As with any super-resolution method, because voxels are smaller they contain fewer fluorophores and are thus dimmer than unexpanded specimens. This effect is offset by the concomitant dilution of autofluorescence and reduced loss due to scattering, though longer exposure times are typically useful, depending on the brightness of the unexpanded specimen. proExM physically magnifies samples equally well along the axial as well as lateral dimensions. Expanded samples are compatible with downstream imaging and analysis strategies such as tomography, deconvolution, or even traditional super-resolution imaging (e.g., PALM, as shown here). Thus proExM will likely find many applications in supporting routine imaging of complex biological samples with nanoscale precision.
Online Methods
Fluorescent Protein Screening (Fig. 1a, b)
Most of the mammalian plasmids were obtained from Addgene (Supplementary Table 1 and 3). To construct the remaining ones, pmKate2-H2B-N1 and pPATagRFP-H2B-N1 plasmids the respective genes were PCR amplified as AgeI/NotI fragments and swapped with the LSSmOrange gene in pH2B-LSSmOrange-N1 (Addgene). To generate NLS-iRFP fusion protein, a PCR-amplified AgeI/NotI fragment encoding gene of iRFP was swapped with LSSmKate2 gene in pNLS-LSSmKate2-N1 (Addgene plasmid #31871). HEK293FT (Invitrogen) and HeLa (ATCC CCL-2) cells were cultured in DMEM medium (Cellgro) supplemented with 10% FBS (Invitrogen), 1% penicillin/streptomycin (Cellgro), and 1% sodium pyruvate (BioWhittaker). HEK293FT and HeLa cells were used for ease of transfection, cell lines were authenticated by STR-profiling and checked for mycoplasma contamination by the manufacturer. Cells were transfected using TransIT-X2 transfection reagent (Mirus Bio) according to the manufacturer’s protocol. Wide-field imaging of live HEK293FT cells was performed 24 h after transfection using a Nikon Eclipse Ti inverted microscope equipped with 10× NA 0.3 objective lens, a SPECTRA × light engine (Lumencor) with 390/22 nm, 438/24 nm, 475/28 nm, 510/25 nm, 585/29 nm, and 631/28 nm exciters (Semrock), and a 5.5 Zyla camera (Andor), controlled by NIS-Elements AR software. Immediately after live cell imaging cell cultures were fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.1% Triton-X for 15 min, washed 3 times for 5 minutes with PBS (Cellgro) and treated with 0.1 mg/ml AcX (LifeTechnologies) for at least 6 h, gelled and digested with proteinase K overnight as described below (see “AcX treatment” and “Gelation, digestion and expansion” sections).
Following digestion, the samples were processed by extensively washing with PBS, and then shrunk in 1 M NaCl and 60 mm MgCl2 (except for YFP, which is chloride sensitive20, and thus was measured in the expanded state). For control experiments shown on Supplementary Fig. 5 gels were washed only with PBS. Registration of pre- and post-sample processing images was carried out with an implementation of the SIFT/RANSAC algorithm, in MATLAB. Automatic Otsu thresholding via CellProfiler21 of fluorescent nuclei allowed for automated measurement of fluorescent intensity in the same set of cells before and after sample processing. Intensity measurements for each nucleus before and after sample processing were normalized by segmented area to account for fluorophore dilution (area was used since epifluorescent optical sectioning mitigates the axial expansion effect on brightness).
Quantification of fluorescent dye retention during ProExM
Fluorescent secondary antibodies (goat anti-rabbit, 10 μg/mL) were purchased from commercial vendors (see Supplementary Table 2 for list of fluorescent secondary antibodies). Retention (Fig. 1c) was quantified via before-after proExM imaging mouse cortex as described below. Cortical sections of wild type (only used for Alexa 488 due to Thy1-YFP crosstalk) and Thy1-YFP brain slices (50 μm thick) were stained with anti-Homer primary antibody (Synaptic Systems; see Supplementary Table 4), and different secondary antibodies described in Supplementary Table 2. Epifluorescence images of brain slices were taken with 4× 0.13 NA objective pre-gelation. Following proExM gelation and digestion, the brain slices were washed extensively with PBS (3 × 30 min), and epifluorescence images of the slice were taken again with identical imaging conditions. A region of interest in the cortex was used to determine the loss of fluorescence during proExM processing. Intensity measurements before and after sample processing were normalized by segmented area to account for fluorophore dilution.
Structured illumination microscopy pre-expansion imaging
HeLa cells were fixed with 4% paraformaldehyde for 10 min, washed 3 times for 5 minutes with PBS, and permeabilized with 0.1% Triton-X for 15 min. Microtubules in fixed HeLa were stained with primary antibodies (Sheep Anti-Tubulin, Cytoskeleton ATN02) in blocking buffer 1× PBS with 0.1% Triton X-100 and 2% normal donkey serum (PBT) at a concentration of 10 μg/mL for 1–4 hours and then washed in PBS three times for 5 minutes each. Specimens were then incubated with secondary antibodies (Donkey Anti-Sheep Alexa 488, Life Technologies, 10 μg/mL) in PBT for 1–4 hours and then washed in PBS three times for 5 minutes. 50 μm brain tissue slices were prepared and stained with primary and secondary antibodies (Rabbit Anti-Tom20, Santa Cruz Biotech sc-11415 and Goat Anti-Rabbit Alexa 568 (Life Technologies)) as described below. Super-resolution structured illumination microscope imaging was performed on a Deltavision OMX Blaze (GE healthcare) SIM microscope with 100× 1.40 NA (Olympus) oil objective. Stained cells were imaged with SlowFade Gold (Invitrogen) antifade reagent for suppression of photobleaching and refractive index matching for pre-expansion imaging.
Measurement Error Quantification
The same fields of view were imaged pre- and post-expansion. Post-expansion images were first registered to the corresponding pre-expansion images by rotation, translation and uniform scaling. In case the specimen tilt changed between pre- and post-expansion imaging, this was corrected using a 3D rotation without scaling using the Fiji 3D Viewer package. These scaled images were then registered again to the pre-expansion images, but this time with a B-spline-based non-rigid registration package in Matlab22 to capture any non-uniformities in the expansion process. Control points for registration were automatically generated using scale-invariant feature transform (SIFT) keypoints23. SIFT keypoints were generated using the VLFeat open source library24, and random sample consensus (RANSAC) was used to estimate a geometric transformation limited to rotation, translation, and scaling. The vector deformation field mapping the scaled post-expansion image to the pre-expansion image expresses the shift of each point in the post-expansion image relative to an ideal uniform expansion. By subtracting the resulting vectors at any two points, we find the relative localization error in using the post-expansion image to measure the distance between those two points. We sample the entire population of possible point-to-point measurements and find the root-mean-square error for such measurements as a function of measurement length.
Brainbow3.0 injection and antibody staining
Brainbow3.0 rAAV (University of Pennsylvania, Penn Vector Core) injections were performed as previously described13. Briefly, transgenic mice were anesthetized continuously with isoflurane and head-fixed to a stereotaxic apparatus. Surgery took place under sterile conditions with the animal lying on a heating pad. 2 μL AAV mix (7.5 × 1012 genome copy/mL) was injected at a rate of 0.2 μl/min through a 34-gauge injection needle into the brain (e.g., cortex, hippocampus), after which the needle was allowed to rest at the injection site for 5 min to allow viral diffusion. Animals expressed virus for 3–4 weeks, then were perfused (see “Mouse perfusion”).
Primary antibodies against Brainbow 3.0 fluorophores (chicken anti-GFP, guinea-pig anti-mKate2, rat anti-mTFP) were produced by the Cai lab. Slices were permeabilized and blocked with 1× PBS with 0.1% Triton X-100 and 2% normal donkey serum (PBT) for 30 minutes before antibody staining (all incubations at room temperature (RT)). Slices were incubated with primary antibodies for 3 days at 4°C in PBT, and then washed four times 30 minutes with PBT. Slices were incubated with secondary antibodies for 1 day at RT. Secondary antibodies used were: goat Anti-Chicken Alexa 488, goat Anti-Rat Alexa 546 (Life Technologies) and donkey Anti-Guinea Pig CF633 (Biotium), all at 10 μg/mL.
Mouse perfusion
All solutions below were made up in 1× phosphate buffered saline (PBS). Mice were anesthetized with isoflurane and perfused transcardially with ice cold 4% paraformaldehyde. Brains were dissected out, left in 4% paraformaldehyde at 4°C for one day, before moving to 100 mM glycine. Slices (50 μm, and 100 μm) were sliced on a vibratome (Leica VT1000S) and stored at 4°C until staining.
AcX treatment
Acryloyl-X, SE (6-((acryloyl)amino)hexanoic acid, succinimidyl ester, here abbreviated AcX; Thermo-Fisher) was resuspended in anhydrous DMSO at a concentration of 10 mg/mL, aliquoted and stored frozen in a desiccated environment. AcX prepared this way can be stored for up to 2 months. For anchoring, cells and tissue slices are incubated in AcX diluted in PBS at a concentration of 0.1 mg/mL for > 6 hours, at room temperature. For thick tissue (> 100 microns), AcX penetration depth and labeling uniformity can be improved by incubating the sample at lower pH, at lower temperature, and in a 2-(N-morpholino)ethanesulfonic acid (MES)-based saline (100 mM MES, 150 mM NaCl; Supplementary Fig. 9). Tissue slices can be incubated on a shaker or rocker to ensure mixing during the reaction.
Gelation, digestion and expansion
For AcX anchored fluorescent proteins and antibody staining, the following steps – gelation, digestion and expansion – can be performed as described previously1. Briefly, monomer solution (1× PBS, 2 M NaCl, 8.625% (w/w) sodium acrylate, 2.5% (w/w) acrylamide, 0.15% (w/w) N,N′-methylenebisacrylamide) was mixed, frozen in aliquots, and thawed before use. Monomer solution was cooled to 4°C before use. Concentrated stocks (10% w/w) of ammonium persulfate (APS) initiator and tetramethylethylenediamine (TEMED) accelerator were added to the monomer solution up to 0.2% (w/w) each. For slices, the inhibitor 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (4-hydroxy-TEMPO) was added up to 0.01% (w/w) from a 0.5% (w/w) stock to inhibit gelation during diffusion of the monomer solution into tissue sections. Cells or tissue slices were incubated with the monomer solution plus APS/TEMED (and 4-hydroxy-TEMPO for slices) at 4°C for one minute, 30 minutes for cultured cells, and brain slices respectively, and then transferred to a humidified 37°C incubator for two hours for gelation.
Proteinase K (New England Biolabs) was diluted 1:100 to 8 units/mL in digestion buffer (50 mM Tris (pH 8), 1 mM EDTA, 0.5% Triton X-100, 1 M NaCl) and incubated with the gels fully immersed in proteinase solution overnight at RT (this step can also be performed at 37° C for 4 hours). Digested gels were next placed in excess volumes of doubly de-ionized water for 0.25–2 hours to expand, with longer times for thicker gels. This step was repeated 3–5 times in fresh water, until the size of the expanding sample plateaued.
Fluorescence microscopy after expansion
Post-expansion confocal imaging of cells was performed on an Andor spinning disk (CSU-X1 Yokogawa) confocal system with a 60× 1.40 NA oil objective (Fig. 1). To quantify expansion factor for tissue slices and low-magnification before vs. after comparisons, specimens were imaged pre-ExM on a Nikon Ti-E epifluorescence microscope with a 4× 0.13 NA air objective (Fig 2. a–d, Supplementary Fig. 1a–b, Supplementary Fig. 3b, Supplementary Fig. 5a–g, and Supplementary Fig. 8a, 8b). For Fig. 3a–b, tissue slices were imaged on Nikon Ti-E epifluorescence microscope with a 10× 0.45 NA. Otherwise, all other tissues presented were imaged using an Andor spinning disk (CSU-X1 Yokogawa) confocal system with a 40× 1.15 NA water immersion objective (Nikon) with the exception of Supplementary Fig. 1, Supplementary Fig. 3a, Supplementary Fig. 4, and Supplementary Fig. 6, where a Zeiss LSM 710 with 40× 1.1 NA water objective. The Zeiss LSM 710 with 10× 0.3 NA air lens was used for Supplementary Fig. 9i.
To stabilize the gels against drift during imaging following expansion, gels were placed in glass bottom 6 well plates with all excess liquid removed. If needed for immobilization, liquid low melt agarose (2% w/w) was pipetted around the gel and allowed to solidify, to encase the gels before imaging.
PALM imaging
PALM data was recorded on a custom-built three-camera RAMM frame microscope (ASI) using an Olympus 1.4 NA PLAPON 60× OSC objective, and a custom tube lens (LAO-300.0, Melles Griot), resulting in 100× overall magnification25. A 2 mm thick quad-band excitation dichroic (ZT405/488/561/640rpc, Chroma), a 2 mm thick emission dichroic (T560lpxr, Chroma), and a band-pass emission filter (FF01-609/54-25, Semrock) filtered the emitted light. Dendra2 was photoconverted by 100 μs long excitation pulses of 405 nm (50 W/cm2) every 200 ms, which was ramped up to 1.2 ms long pulses every 200 ms during the course of image acquisition. Stroboscopic 405-nm excitation of the Stradus 405-100 laser (Vortran) was achieved using a NI-DAQ-USB-6363 acquisition board (National Instruments), Photoconverted Dendra2 molecules were excited with a 555-nm DPSS laser (CrystaLaser) at estimated sample power levels of 2 kW/cm2. Fluorescence was detected using μManager (v. 1.4.20)26 with a back-illuminated EMCCD camera (Andor Technology, Ixon Ultra DU-897_BV, 17 MHz EM amplifier, Gain 500, full-chip) at 20 frames/s.
Particle localization
Localizer27 was used for 8-way adjacency particle detection with 20 GLRT sensitivity and a PSF of 1.3 pixels. The resulting particles were drift corrected using ad-hoc fiducial markers. For each detected particle, integrated fluorescence intensities were converted to photon counts using analysis routines written in Igor Pro version 6.36. The mean and median localization errors were determined using equation 6 in reference28.
ProExM of different tissue types
Standard histology preparations of mouse normal fresh frozen tissue sections, postfixed with cold acetone, of pancreas, spleen and lung (5–10 μm) were obtained from US Biomax (MOFTS036, MOFTS051, and MOFTS031, respectively). Tissues were blocked with 1× PBS with 0.1% Triton X-100 and 2% normal donkey serum (PBT) for 30 minutes before antibody staining. Tissues were stained with primary chicken anti-vimentin (Abcam) for 4 hours at RT and then washed four times 30 minutes with PBT. Slices were incubated with secondary antibodies for 2 hours at RT (Anti-Chicken Alexa 488, Life Technologies). Pre-expansion imaging was performed as described above. Tissues were incubated with 0.05 mg/mL AcX in PBS at RT overnight before gelation, digestion and expansion described above with the exception that digestion was performed at 60°C for 4 hours.
Antibody staining of endogenous proteins
Specimens, either before gelation or after autoclave or LysC treatment, were incubated in 1× PBS with 0.1% Triton X-100 and 2% normal donkey serum (PBT) at room temperature (RT) for 2 hours for blocking, and in the case of pre-gelation specimens, permeabilization. Specimens were incubated with primary antibodies at 3 μg/mL in PBT, for 4 hours (RT), and then washed four times 30 minutes with PBT. Specimens were incubated with secondary antibodies at 20 μg/mL in PBT, for 4 hours (RT), and then washed four times at least 30 minutes with PBT. Secondary antibodies used were: goat Anti-Chicken Alexa 488 (Life Technologies), goat Anti-Rabbit Alexa 546 (Life Technologies) and goat Anti-Mouse CF633 (Biotium), except that goat Anti-Chicken Alexa 546 (Life Technologies) was used for Supplementary Fig. 1e, g(ii), h(ii) and goat Anti-Rabbit Alexa 488 (Life Technologies) was used for Fig. 1e.
Specimen disruption using autoclave
After gelation, gels were recovered from gelation chambers and washed in 1M NaCl. Gels were washed for 15 minutes in Disruption Buffer (100mM Tris base, 5% Triton X-100, 1% SDS), then placed in fresh Disruption Buffer and treated by autoclave on liquid sterilization mode with a temperature of 121°C held for one hour. This treatment must be carried out in an autoclave-safe vessel such as polypropylene tubes. Gels were then transferred to well plates for antibody staining and imaging and washed in PBT (1×PBS, 2% normal donkey serum, 0.1% Triton X-100) to remove Disruption Buffer.
Mild digestion with LysC
After gelation, gels were pre-treated in HBSS buffer (with calcium and magnesium, ThermoFisher Scientific) with 600 U/ml collagenase type II (Life Technologies) in 37°C for 2–4 hours. Gels were then washed for 5 minutes in LysC digestion buffer (25 mM Tris-HCl, 1 mM EDTA, pH 8.5) and incubated with 33 μg/ml LysC (Promega) in 37°C for at least 8 hours. Finally, gels were washed in LysC digestion buffer 3× for 30 mins each and were subjected to immunostaining with identical steps that have been described above.
Synthesis of SNOTRAP-biotin
To a stirred 2-(diphenylphosphino)-benzenethiol (100mg, 0.34 mmol) in dry DMF (5 mL) was added biotin-PEG3-propionic acid (100 mg, 0.22 mmol, ChemPep, Inc,), N,N′-dicyclohexylcarbodiimide (DCC) (70 mg, 0.34 mmol) and dimethylaminopyridine (DMAP) (4 mg, 0.03 mmol) successively. The resulting mixture was stirred for 7 h at room temperature, and the resulting clear solution then concentrated under reduced pressure and purified by flash chromatography (hexane/EtOAc/MeOH gradient) to give the desired product (yield 30%). The SNOTRAP probe was repurified on an 1100 HPLC system with a photodiode array UV detector at 254 nm (Agilent Technologies, Wilmington, DE). HPLC columns and solvent systems were as follows: a semi-preparative Phenomenex Luna C18 (25 cm × 9.4 mm, 10 μm) column was eluted with a linear gradient of 0.1% formic acid in water (A) and acetonitrile (B) at a flow rate of 2.5 mL/min. Solvent composition was initially at 40% for 5 min, 70% at 10 min, 90% at 20 min, and then further to 95% B over 8 min. 1H NMR (500 MHz, CD3CN, δ) 7.42–7.38 (m, 9H), 7.23–7.18 (m, 4H), 7.84 (m, 1H), 4.60–4.51 (m, 2H), 3.67–3.51 (m, 12H), 3.2 (m, 3H), 2.8 (m, 2H), 2.55 (t, 2H), 2.15 (t, 2H), 1.57–3.51 (m, 6H); 13C NMR (125 MHz, CD3CN, δ) 199.19, 172.5, 164.5, 144.8, 138.1, 137.0, 134.8, 129.9, 129.6, 129.6, 118.3, 69.2, 63.1, 62.3, 45.9, 42.5, 38.2, 27.1, 23.1, 22.5; 31P NMR (202 MHz, CD3CN, δ) −10.3; HRMS-ESI+ (m/z): [M + H+]+ calculated for C37H47N3O6PS2, 724.2638; found, 724.2632.
ProExM of SNOTRAP staining
For SNOTRAP staining, primary neuron culture were washed 3 × 5 minutes using PBS and fixed using cold methanol at −20°C for 15 minutes. Neurons were incubated with 300nM N-ethylmaleimide (NEM) (Thermo Scientific) in PBS-Triton X100 (0.3% v/v) at 37°C for 30 minutes to block the free –SH group on proteins. Cells were then washed 3 × 5 minutes using PBS and incubated with SNOTRAP probe (250uM) in acetonitrile-PBS-triton (50% : 50% v/v) at R.T. for 1 hour, and then further incubated with streptavidin-Alexa 488 (Thermo Scientific) in 1/500 dilution (PBS-Triton) at R.T. for 1 hour and afterwards washed 5 × 5 minutes. Antibody staining for anti-tubulin (Alexa 546 secondary) and proExM was performed as described above.
Animal care
All methods for animal care and use were approved by the Massachusetts Institute of Technology Committee on Animal Care and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. One adult male rhesus macaque (Macaca mulatta) weighing 12kg was used for this study, as well as 1 C57BL/6 mouse, 4 Emx1-Cre mice, and 10 Thy1-YFP mice, ages ~1–3 months old. Mice were used without regard for gender.
Macaque procedures
Virus injections were performed with sevoflurane anesthesia using stereotactic coordinates to target 8 injection sites. Viruses (AAV8,) were centrifuged and loaded into 10μL gas-tight syringes (Hamilton) that had been back-filled with silicone oil (Sigma). A total of 3μL of virus was infused into the brain at two locations (deep then 500 μm superficial) at a rate of 100–200 nL/minute using stereotactic micromanipulator arms (David Kopf Instruments) and UMP3 micro-syringe injector pumps (World Precision Instruments). After each injection, the needle and syringe were left in place for 10 minutes before withdrawal. Blunt 33G needles were used for all injections. 1mg Dexamethasone was also administered to prevent brain swelling. Euthanasia took place 4 weeks after viral injection. An overdose of pentobarbital was administered prior to perfusion with phosphate buffered saline (PBS) and 4% paraformaldehyde (PFA). The brain was then extracted, blocked, and stored in a 20% glycerol with 0.1% sodium azide solution, and finally cut into 40μm microtome sections.
Supplementary Material
Acknowledgments
We acknowledge N. Pak for assistance with perfusions. For funding, ESB acknowledges the MIT Media Lab, the MIT Brain and Cognitive Sciences Department, the New York Stem Cell Foundation-Robertson Investigator Award, NIH Transformative Award 1R01GM104948, NIH Director’s Pioneer Award 1DP1NS087724, NIH 1R01EY023173, and NIH 1U01MH106011. FC acknowledges the NSF Fellowship and Poitras Fellowship. AM was supported by T32 GM007484, Integrative Neuronal Systems, PWT acknowledges the Hertz Fellowship. BPE was supported by the Howard Hughes Medical Institute. The order of co-first author names was determined by a coin toss. ESB and SRT acknowledge the MIT McGovern Institute MINT program.
Footnotes
AUTHOR CONTRIBUTIONS
PWT, FC, KDP, YZ, CCY, and ESB all contributed key ideas, analyzed data, and wrote the paper. PWT designed and performed antigen retrieval experiments, FC designed and performed AcX-secondary antibody retention experiments and Brainbow experiments, KDP and FC designed and performed fluorophore preservation experiments, YZ designed and performed LysC experiments, and CCY and PWT designed and performed depth modulation of AcX experiments and preliminary characterizations of autoclave treatments. LG created code for data analysis. BE carried out PALM experiments, AM and H-JS carried out mouse surgeries, FY carried out primate surgeries under the supervision of RD with assistance from EMD, and DHR and DC assisted with Brainbow experiment design. GG, US, and SRT designed and performed the SNOTRAP experiment. FC and YZ designed and performed experiments for validation of proExM on non-brain mouse tissue. ESB supervised the project.
COMPETING FINANCIAL INTERESTS
PWT, FC, ESB, and CCY have filed for patent protection on a subset of the technologies here described. ESB has helped co-found a company to help disseminate kits to the community.
References
- 1.Chen F, Tillberg PW, Boyden ES. Expansion microscopy. Science (80-) 2015;347:543–548. doi: 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 3.Hunt NCA, Jasani B, A R. High temperature antigen retrieval and loss of nuclear morphology: a comparison of microwave\rand autoclave techniques. J Clin Pathol. 1996;49:767–770. doi: 10.1136/jcp.49.9.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jekel PA, Weijer WJ, Beintema JJ. Use of endoproteinase Lys-C from Lysobacter enzymogenes in protein sequence analysis. Anal Biochem. 1983;134:347–354. doi: 10.1016/0003-2697(83)90308-1. [DOI] [PubMed] [Google Scholar]
- 5.Wu CC, MacCoss MJ, Howell KE, Yates JR. A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–8. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 6.Sniegowski JA, Phail ME, Wachter RM. Maturation efficiency, trypsin sensitivity, and optical properties of Arg96, Glu222, and Gly67 variants of green fluorescent protein. Biochem Biophys Res Commun. 2005;332:657–63. doi: 10.1016/j.bbrc.2005.04.166. [DOI] [PubMed] [Google Scholar]
- 7.Bokman SH, Ward WW. Renaturation of Aequorea gree-fluorescent protein. Biochem Biophys Res Commun. 1981;101:1372–80. doi: 10.1016/0006-291x(81)91599-0. [DOI] [PubMed] [Google Scholar]
- 8.Seneviratne U, et al. S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc Natl Acad Sci U S A. 2016:1521318113. doi: 10.1073/pnas.1521318113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods. 2008;5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rego EH, et al. Nonlinear structured-illumination microscopy with a photoswitchable protein reveals cellular structures at 50-nm resolution. Proc Natl Acad Sci U S A. 2012;109:E135–43. doi: 10.1073/pnas.1107547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bossi M, et al. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species. Nano Lett. 2008;8:2463–8. doi: 10.1021/nl801471d. [DOI] [PubMed] [Google Scholar]
- 13.Cai D, Cohen KB, Luo T, Lichtman JW, Sanes JR. Improved tools for the Brainbow toolbox. Nat Methods. 2013;10:540–7. [PubMed] [Google Scholar]
- 14.Livet J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- 15.Schnell U, Dijk F, Sjollema KA, Giepmans BNG. Immunolabeling artifacts and the need for live-cell imaging. Nat Methods. 2012;9:152–158. doi: 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- 16.Hackstadt T. Steric hindrance of antibody binding to surface proteins of Coxiella burnetti by phase I lipopolysaccharide. Infect Immun. 1988;56:802–807. doi: 10.1128/iai.56.4.802-807.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiménez N, Post JA. A Novel Approach for Intracellular 3D Immuno-Labeling for Electron Tomography. Traffic. 2012;13:926–933. doi: 10.1111/j.1600-0854.2012.01363.x. [DOI] [PubMed] [Google Scholar]
- 18.Randall KJ, Pearse G. A dual-label technique for the immunohistochemical demonstration of T-lymphocyte subsets in formalin-fixed, paraffin-embedded rat lymphoid tissue. Toxicol Pathol. 2008;36:795–804. doi: 10.1177/0192623308322311. [DOI] [PubMed] [Google Scholar]
- 19.Kakimoto K, Takekoshi S, Miyajima K, Osamura RY. Hypothesis for the mechanism for heat-induced antigen retrieval occurring on fresh frozen sections without formalin-fixation in immunohistochemistry. J Mol Histol. 2008;39:389–399. doi: 10.1007/s10735-008-9177-y. [DOI] [PubMed] [Google Scholar]
- 20.Wachter RM, James Remington S. Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr Biol. 1999;9:R628–R629. doi: 10.1016/s0960-9822(99)80408-4. [DOI] [PubMed] [Google Scholar]
- 21.Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kroon DJ. B-spline Grid, Image and Point based Registration. Matlab Cent. at < http://www.mathworks.com/matlabcentral/fileexchange/20057-b-spline-grid–image-and-point-based-registration>.
- 23.Lowe DG. Distinctive Image Features from Scale-Invariant Keypoints. Int J Comput Vis. 2004;60:91–110. [Google Scholar]
- 24.Vedaldi A, Fulkerson B. Proc Int Conf Multimed – MM ’10. Vol. 1469. ACM Press; 2010. Vlfeat. [DOI] [Google Scholar]
- 25.English BP, Singer RH. A three-camera imaging microscope for high-speed single-molecule tracking and super-resolution imaging in living cells. In: Mohseni H, Agahi MH, Razeghi M, editors. SPIE Nanosci + Eng. International Society for Optics and Photonics; 2015. p. 955008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using μManager. Curr Protoc Mol Biol. 2010:20. doi: 10.1002/0471142727.mb1420s92. Chapter 14, Unit14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dedecker P, Duwé S, Neely RK, Zhang J. Localizer: fast, accurate, open-source, and modular software package for superresolution microscopy. J Biomed Opt. 2012;17:126008. doi: 10.1117/1.JBO.17.12.126008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 2010;7:377–81. doi: 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.