

Abstract
Actin and its ability to polymerize into dynamic filaments is critical for the form and function of cells throughout the body. While multiple proteins have been characterized as affecting actin dynamics through non-covalent means, actin and its protein regulators are also susceptible to covalent modifications of their amino acid residues. In this regard, oxidation-reduction (Redox) intermediates have emerged as key modulators of the actin cytoskeleton with multiple different effects on cellular form and function. Here, we review work implicating Redox intermediates in post-translationally altering actin and discuss what is known regarding how these alterations affect the properties of actin. We also focus on two of the best characterized enzymatic sources of these Redox intermediates – the NADPH oxidase NOX and the flavoprotein monooxygenase MICAL – and detail how they have both been identified as altering actin, but share little similarity and employ different means to regulate actin dynamics. Finally, we discuss the role of these enzymes and redox signaling in regulating the actin cytoskeleton in vivo and highlight their importance for neuronal form and function in health and disease.
Keywords: Actin, NADPH oxidase, MICAL, Redox, post-translational modification
Introduction
Actin filaments contribute to cellular (including neuronal) physiology in a ubiquitious context – including determining the shape and morphology of virtually all cells, promoting both chemotaxis and cell motility, regulating neurite outgrowth, synaptic connectivity, as well as vesicle trafficking, and orchestrating numerous other cellular functions and organismal behaviors (Fletcher and Mullins 2010; Kevenaar and Hoogenraad 2015; Pollard and Cooper 2009). The regulation of the actin filament (F-actin) cytoskeleton depends on a diverse array of molecular mechanisms that involve actin-associated proteins, including some of the best known such as the Arp 2/3 complex and ADF/cofilins, and well as signaling modulators such as Rho family small GTPases and assorted extracellular ligands and receptors (Gonzalez-Billault et al. 2012; Hung and Terman 2011; Pollard and Cooper 2009). Collectively, these molecular components control F-actin dynamics within both physiological and pathological contexts.
Many of the best characterized actin regulatory proteins use non-covalent means to control actin organization and dynamics (Blanchoin et al. 2014; Pollard and Cooper 2009), but actin and its regulatory enzymes, like other cellular proteins, are also targets for different types of post-translational modifications (PTMs) (Terman and Kashina 2013). One such PTM, which has emerged as a key means to affect multiple different biological processes, is through the oxidation-reduction (Redox) modification of amino acid residues (Walsh 2006). So too, Redox-mediated alterations have been found to regulate the dynamic properties of actin, as well as control specific actin regulatory proteins and signaling components (Terman and Kashina 2013; Wilson and Gonzalez-Billault 2015). Studies also indicate that Redox signaling plays important roles in vivo, including within neurons, to control the form and function of the actin cytoskeleton. Yet, similar to the best known PTM, phosphorylation, which plays disparate roles in regulating protein function and cellular behaviors, the functional consequences of Redox-mediated PTM of actin and its regulatory proteins depends on the particular protein affected. Furthermore, not all Redox signaling results in a similar effect, in that multiple residues on actin and its regulatory proteins have been found to be susceptible to Redox regulation with varying biochemical, cellular, and physiological consequences. Moreover, the Redox intermediates that control these effects range from diffusible oxidants that may have little ability to discriminate among different residues to enzymes that selectively target specific residues on actin. Herein, we provide an overview of the current understanding of the Redox regulation of actin. We also focus on two important Redox enzymes in this regard – NADPH oxidases and MICAL flavoprotein monooxygenases – and discuss how they both have been characterized as targeting actin, but use different mechanisms to exert their effects on the actin cytoskeleton. We also review the current understanding of the cellular role of these enzymes and pay particular attention to highlighting their effects on the form and function of the nervous system.
Actin – a target of Redox species
Following their identification and purification of actin over 70 years ago, Brunó Straub and his colleagues conducted biochemical studies that indicated that the presence of an oxidizing agent not only inhibited the polymerization of globular actin, but it also dismantled polymerized actin (Feuer et al. 1948). While the physiological relevence of these observations were unclear, a vast and dizzying array of literature has since emerged into this Redox-mediated regulation of actin, revealing much about actin’s susceptibility to Redox intermediates and making it clear that Redox agents don’t simply effect actin in a singular manner. Notably, a large number of physiologically and non-physiologically relevant reactive oxygen, nitrogen, and lipid reagents have been used over the years to test the Redox susceptibility of actin and this has revealed that actin is susceptible to Redox-mediated PTMs including oxidation, nitrosylation, nitration, as well as the addition of glutathione and carbonyl moieties (glutathionylation and carbonylation, respectively) (Terman and Kashina 2013). Collectively, these results have revealed that Redox-mediated post-translational effects on actin are widely variable and are critically dependent on the monomeric versus polymeric state of actin and the particular amino acid residues on actin that are modified.
Actin binds ADP/ATP and Mg2+/Ca2+ (among many other small molecules and proteins) and this binding regulates the polymerization properties of actin including the ability of single actin proteins (G-actin) to polymerize and form filaments. The effects that particular Redox reagents have on actin varies considerably depending on which of these small molecules are bound and/or the presence of filamentous versus globular forms of actin. For example, while treatment with hydrogen peroxide and other oxidants greatly reduces the ability of Ca2+-bound G-actin to polymerize (DalleDonne et al. 1995; Hung et al. 2011; Lassing et al. 2007; Milzani et al. 1997; Milzani et al. 2000), Mg2+-bound G-actin is largely resistant to the adverse effects of hydrogen peroxide and related oxidants (Balin and Barany 1967; Hung et al. 2011; Hung et al. 2010a; Muhlrad et al. 1968). These differences appear to be due to changes in the conformation and surface exposed residues present when actin is polymerized in the presence of Ca2+ versus Mg2+, and those residues that are exposed in G-actin versus F-actin (e.g., (Collins and Elzinga 1975; Dalle-Donne et al. 2002; Guan et al. 2003; Guan et al. 2005; Konno and Morales 1985; Lassing et al. 2007; Lin et al. 1990; Liu et al. 1990; Lusty and Fasold 1969; Takamoto et al. 2007)). These in vitro observations suggest that specific conditions might make actin more susceptibile to Redox regulation in vivo – and there are examples of these in vivo differences (e.g., (Duke et al. 1976; Gross and Lehman 2013; Lamb and Posterino 2003)). Along these lines, it is also important to note that some of these Redox modifications like oxidation and glutathionylation are reversible, while other modifications such as carbonylation are not (see below; (Castro et al. 2013; Terman and Kashina 2013)). In short, it has become clear that the effects of Redox-mediated post-translational modifications including oxidation, nitrosylation, nitration, glutathionylation and carbonylation of actin cannot be simply grouped together as causing one effect on the biochemical properties of actin – and so a full understanding of the sites on actin that are modified by Redox intermediates and how each of these modifications affects the properties of actin is an important research goal.
Actin – Redox Susceptible Residues and Redox Effectors
There are six different actin genes in mammals and in some lower organisms such as Drosophila, with a high-degree of conservation between them, including between muscle and non-muscle (including neuronal) actins (Bunnell and Ervasti 2011). Modification of cysteine (Cys) residues is the best characterized site of Redox regulation (Go and Jones 2013) and actin has 5–6 cysteine residues (depending on the specific actin) (Figure 1A, C). Both in vitro and in vivo studies have revealed that the cysteine residues on actin are highly susceptible to Redox intermediates – including reactive oxygen, nitrogen, and lipid species. In particular, the Redox-mediated post-translational modification of actin’s cysteine residues and their effects on the physicochemical properties of actin has been widely explored because of the availability and ease of use of diverse chemical oxidative reagents that target cysteine (Reviewed in (Dalle-Donne et al. 2001; Gergely 1966; Hung and Terman 2011; Kielley 1964; Mannherz and Goody 1976; Muhlrad et al. 1968)). Cys-374 is actin’s most Redox susceptible amino acid residue, undergoing oxidation, glutathionylation, carbonylation, and nitrosylation (Figure 1A, C; (reviewed in (Terman and Kashina 2013)). For example, oxidation of Cys-374 often occurs in purified actin samples with air-exposure, aging, and freeze-thawing (Balin and Barany 1967; Ishiwata 1976; Tang et al. 1999; Xu et al. 1998) and this results in disulfide bond formation between two actin monomers (Ishiwata 1976; Tang et al. 1999). Reducing agents such as DTT reverse these effects and are added to purified actin samples to prevent this abnormal dimerization. Likewise, subjecting purified actin to reactive oxygen, nitrogen, and lipid species results in modification of Cys-374 and induces both intermolecular disulfide bond-mediated actin aggregation (Farah et al. 2011; Gayarre et al. 2006; Ishiwata 1976) and intramolecular disulfide bond formation with Cys-285 (Farah et al. 2011). This Cys-374 modification also has other effects on actin including decreasing its polymerization and elongation rate, increasing its critical concentration, and weakening filaments (Aldini et al. 2007; Aldini et al. 2005; Dalle-Donne et al. 2003; Dalle-Donne et al. 2000; Dalle-Donne et al. 2002; DalleDonne et al. 1999; Gayarre et al. 2006; Pizarro and Ogut 2009; Shartava et al. 1995; Stournaras et al. 1990). Yet, it should also be noted that the nature of the Redox modification on Cys-374 is also important since its modification in some cases allows for relatively normal actin properties (Dalle-Donne et al. 2007; Tsapara et al. 1999) – and because of its reactivity, Cys-374 has been used to label actin, such as creating pyrene-labeled actin, and is widely used to characterize the biochemical properties of actin (e.g., (Cooper et al. 1983; Kouyama and Mihashi 1981; Tait and Frieden 1982)). In any case, further characterization of this Redox-modified Cys-374 residue is needed since Redox-mediated alterations of Cys-374 regulate actin dynamics under different conditions in cells (Aldini et al. 2007; Farah et al. 2011; Fiaschi et al. 2006; Gayarre et al. 2006; Sobierajska et al. 2014; Thom et al. 2008; Wang et al. 2001) and occur in irreversible sickled cells (Shartava et al. 1995). Likewise, future work should focus on the other cysteine residues within actin including Cys-10 (Berndt et al. 2014; Duke et al. 1976; Gross and Lehman 2013), Cys-217 (Fedorova et al. 2010a; Hamnell-Pamment et al. 2005; Su et al. 2013; Thom et al. 2008), Cys-257 (Berndt et al. 2014; Fedorova et al. 2010a; Su et al. 2013; Thom et al. 2008), Cys-272 (Lassing et al. 2007), and Cys-285 (Chen et al. 2008; Farah et al. 2011; Figueiredo-Freitas et al. 2015; Shartava et al. 1995; Su et al. 2013; Thom et al. 2008) that are susceptible to Redox regulation – including Cys modifications that have been associated with decreasing the ability of actin to polymerize and interact with specific regulatory proteins including profilin (Farah et al. 2011; Lassing et al. 2007) (Figure 1A, C).
Figure 1. Structural models of actin and F-actin denoting the individual amino acid residues modified by NOXs and MICALs.
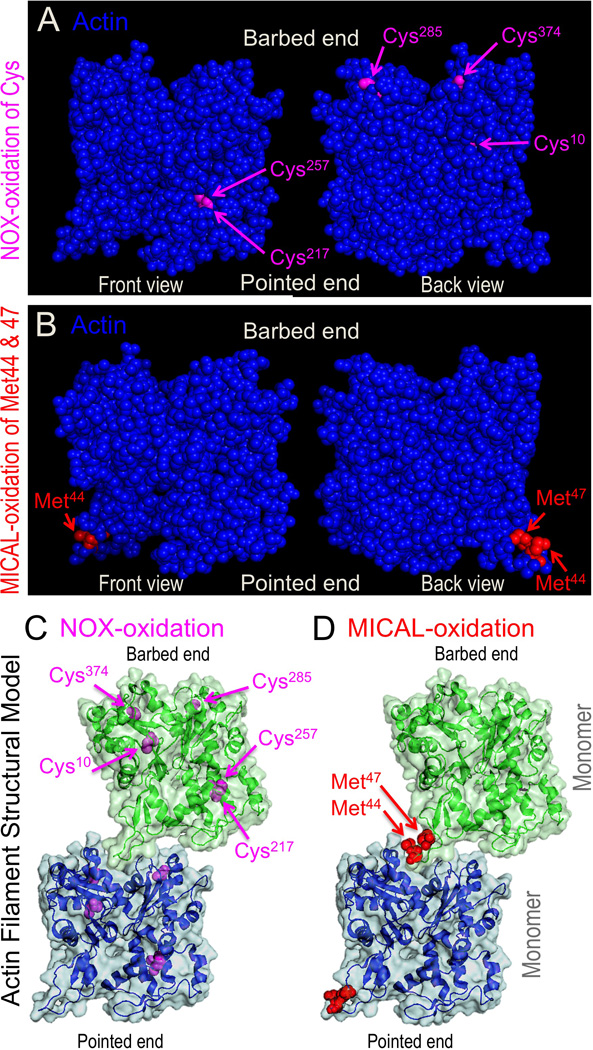
A) The cysteine (Cys)-374 residue is thought to be a major target of NOX-mediated oxidation of actin, while other cysteine residues in actin including Cys-10, Cys-217, Cys-257, Cys-285, and Cys-272 (not present in this actin) are also likely to be targets of NOX-mediated oxidation (see text). B) MICAL specifically oxidizes the Met-44 and Met-47 residues of actin (Hung et al. 2011) – with the oxidation of the Met-44 residue being the primary means by which MICAL induces the disassembly of F-actin (Hung et al. 2011). Front and back views of the same actin subunit are shown on the left and right sides, respectively. (C–D) Structural model of actin subunits and their fit within a structural model of a filament including general intersubunit interactions. (C) The amino acids susceptible to NOX oxidation and their position within the filament. (D) The amino acids oxidized by MICAL and their position within the filament. The residues oxidized by MICAL, Met-44 and Met-47, are at the critical interface between adjacent actin monomers. In particular, they are situated within the D-loop of actin at the pointed end of one actin monomer, where they associate with residues within the barbed end of another actin monomer (Galkin et al. 2015; Hung et al. 2011; von der Ecken et al. 2015). Structures are based on PDB identifier: 2ZWH (Oda et al. 2009).
In contrast to the extensive body of literature focusing on the Redox susceptibility of actin’s cysteine residues, far fewer studies have focused on the Redox regulation of actin’s other amino acid residues. For example, methionine residues, unlike cysteine residues, do not rapidly react with hydrogen peroxide in vitro and at physiological pH (Kim et al. 2014), and so other oxidative reagents such as hypochlorous acid have been used to test the susceptibility of actin’s methionine residues to Redox modification. These studies have revealed that several of the 16 methionine (Met) residues within actin including Met-44, Met-47, Met-176, Met-190, Met-227, Met-269, and Met-355 are susceptible to oxidation in vitro and their oxidation has been linked to functional impairment of actin (Dalle-Donne et al. 2002; Milzani et al. 2000). Moreover, recent work performed during the characterization of the Mical flavoprotein monooxygenase (see below; (Hung et al. 2011)) has revealed that the specific oxidation of the Met-44 residue of actin causes striking disassembly and decreased polymerization of actin filaments (Figure 1B, D; (Hung et al. 2011)). This Met-44 residue is within the D-loop of the subdomain 2 portion of actin that is crucial for actin subunit contacts (Figure 1B, D; (Galkin et al. 2015; Hung et al. 2011; von der Ecken et al. 2015). When this specific residue is oxidized, it brings a negative charge (i.e., a significant dipolar character with a negative charge centered on the oxygen) into the monomer-monomer contact region inducing severing of actin filaments (Figure 1D; (Hung et al. 2011)). Notably, similar to other Redox-affected residues on actin, the Redox susceptibility of these methionine residues including Met-44 varies depending on actin’s conformation, ionic state, and polymerization condition (Dalle-Donne et al. 2002; Guan et al. 2003; Guan et al. 2005; Hung et al. 2011; Takamoto et al. 2007). Therefore, it is possible that particular in vivo conditions and disease states may make some of these methionine residues more susceptible to Redox-regulation. It is also noteworthy that other actin amino acids, including specific tyrosine, histidine, and tryptophan residues, are also susceptible to Redox-induced modifications, although the functional significance of these modifications are unknown. For example, specific tyrosine residues have also been found to be nitrated in vivo (Tyr-294; (Zhan and Desiderio 2004)), while other tyrosine residues are nitrated following peroxynitrite anion treatment (Tyr-53, Tyr-198, and Tyr-362; (Aslan et al. 2003)), during aging (Tyr-218; (Kanski et al. 2005)), within amyotrophic lateral sclerosis (ALS) mouse models (Tyr53; (Casoni et al. 2005)) and within patients with sickle cell disease (Tyr-91, Tyr-198, and Tyr-362; (Aslan et al. 2003)). Likewise, treatment of purified actin with reactive lipid species induces the carbonylation of specific histidine residues (His-40, His-87, His-173; (Dalle-Donne et al. 2007)) and X-ray irradiation results in oxidation of Trp-79, Trp-86, Trp-340, and Trp-356 amino acid residues (Fedorova et al. 2010a; Fedorova et al. 2010b).
Identifying the cellular sources of Redox intermediates is also a crucial direction in characterizing the role and physiological importance of the Redox modifications of actin. In particular, while the effects that different chemicals and non-specific Redox intermediates have on actin have been well characterized, it has been unclear until more recently whether Redox processes that specifically target actin also exist. For instance, enzymes which produce reactive oxygen, nitrogen, and lipid species including NADPH oxidase (e.g., (Barth et al. 2009; Bhopale et al. 2015; Dailianis et al. 2009; Moldovan et al. 2000; Munnamalai and Suter 2009; Ullevig et al. 2012; Wojciak-Stothard et al. 2005)), lipoxygenases (e.g., (Fiaschi et al. 2006; Kang et al. 1999; Kang and Vanderhoek 1998; Lepley and Fitzpatrick 1994; Miller et al. 2001; Munnamalai and Suter 2009; Provost et al. 2001; Rice et al. 1998), xanthine oxidase (e.g., (Rosado et al. 2002)), myeloperoxidase (e.g, (Thom et al. 2008)), and nitric oxide synthase (NOS) (e.g., (Banan et al. 2003; Bhopale et al. 2015; Dailianis et al. 2009; Harris et al. 2008; Jaffrey et al. 2001; Su et al. 2007; Webb et al. 2001)) have been linked to the modification of actin. However, actin constitutes only one set of proteins that are modified by these enzymes since they diffusibly release reactive species, and so questions have arisen regarding the specificity of these effects. Likewise, actin is one of the most abundant cellular proteins, raising concerns that actin might be modified non-specifically by diffusible reactive oxygen, nitrogen, and lipid species produced by these enzymes. Moreover, it has been suggested that actin might even serve to “soak-up” these species and/or to protect the cell against redox intermediates (Dalle-Donne et al. 2007; Dalle-Donne et al. 2003; Farah et al. 2011). Therefore, many important directions remain to be explored related to the identity and specific mechanisms of action of the Redox effectors of actin dynamics and their physiological and pathological roles. However, recent work has provided a new understanding of these important concepts and below we focus on these emerging properties by highlighting two of actin’s Redox effectors: 1) NADPH oxidases, which are enzymes well-known to release reactive oxygen species to exert their effects, and 2) the recently characterized F-actin disassembly factor Mical, which is a Redox enzyme that specifically and directly oxidizes actin.
NADPH Oxidases: F-actin Effects and Cellular Functions (with a Focus on the Nervous System)
Pro-oxidation species such as superoxide, hydrogen peroxide and nitric oxide play critical roles as signaling molecules owing to the fact that their concentrations can be enzymatically regulated (Bigarella et al. 2014). In this context, NADPH oxidase (NOX) enzymes have important functions. The NOX enzymatic family comprises 7 members called NOX 1–5 and Duox 1–2 (Bedard and Krause 2007). NOXs catalyze the conversion of molecular oxygen (O2) into the superoxide radical (O2·−), a short-lived molecule that is rapidly converted both spontaneously or enzymatically into hydrogen peroxide (H2O2) (Nunez et al. 2012) (Figure 2). The superoxide and hydrogen peroxide are released extracellularly and can reach the intracellular compartment through either diffusion, as in the case of H2O2, or via membrane-enclosed vesicles, in the case of superoxide (Lassegue et al. 2012; Valdivia et al. 2015) (Figure 2). NOX proteins may also mediate signaling via ROS production in endosomal compartments, or “redoxosomes” (Oakley et al. 2009; Valdivia et al. 2015). NOX-derived ROS have been implicated in targeting intracellular signaling proteins in a paracrine, autocrine, and intracrine manner (Valdivia et al. 2015).
Figure 2. Main NOX complexes described in neurons.
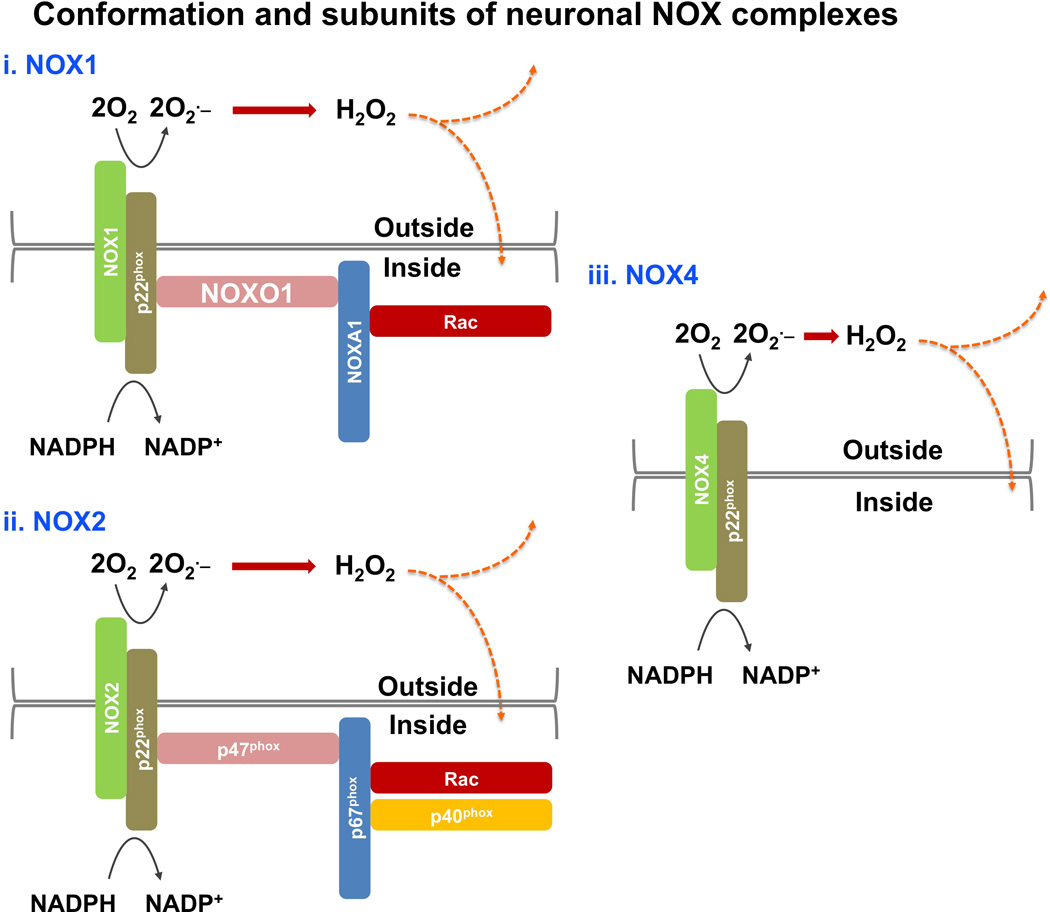
NADPH oxidases are a family of membrane-associated multisubunit enzymes that generate reactive oxygen species (ROS) to regulate diverse systems. Members of the Nox family include NOX1–5 and Duox1–2; with NOX1, NOX2 (also called gp91phox) and NOX4 being the main Nox family members described to date in neurons. Members of the Nox family catalyze the conversion of O2 into O2·− (superoxide), which can be converted into H2O2 (hydrogen peroxide) (Lassegue et al. 2012; Valdivia et al. 2015). These ROS are released extracellularly and can reach the intracellular compartment through either diffusion, as in the case of H2O2, or via membrane-enclosed vesicles, in the case of superoxide (Lassegue et al. 2012; Valdivia et al. 2015). Nox proteins may also mediate signaling via ROS production in endosomal compartments, or “redoxosomes” (not shown; reviewed in (Oakley et al. 2009)). NOX-derived ROS have been implicated in modifying intracellular signaling proteins in a paracrine, autocrine, and intracrine manner (Valdivia et al. 2015). It should also be noted that the distinguishing catalytic subunits (i.e., NOX 1–5, Duox 1–2) are also typically joined by five variable regulatory phox subunits to form the NADPH oxidase complex. (i–ii) The NOX1 and NOX2 complexes share common features, such that NOXO1 and p47phox play similar roles including assembling and organizing the plasma membrane subunits (i.e., NOXs and the stabilizing factor p22phox) with the cytosolic subunits (i.e., p67phox, NOXO1, Rac, and p40phox) of the complex. Note also that NOXA1, which is present in the NOX1 complex, is homologous to p67phox, the activator subunit of the NOX2 complex. (iii) The NOX4 complex lacks the cytosolic subunits and only displays the plasma membrane-associated proteins NOX4 and p22phox. Nox proteins with the exception of Nox4, which is constitutively active, are silent in resting cells (Lassegue et al. 2012).
The main NOXs expressed in the central nervous system of mammals are NOX 1, 2 and 4 (Sorce et al. 2012) (Figure 2). NOX2 is the best-characterized family member and appears to be the main NOX expressed in neurons (Bedard and Krause 2007; Lambeth 2004; Nayernia et al. 2014). NOX2 (also called gp91phox) is a plasma membrane protein of approximately 50 kDa spanning 6 transmembrane domains, with both NADPH and FAD binding sites towards the intracellular space (Groemping and Rittinger 2005; Harper et al. 1985; Paclet et al. 2004; Rotrosen et al. 1990). These distinguishing catalytic subunits (i.e., NOX 1–5, Duox 1–2) are also typically joined by up to five variable regulatory phox subunits to form the NADPH oxidase complex. For example, NOX2 forms a complex with p22phox, a 22 kDa plasma membrane protein that confers further stabilization to NOX2 (Ambasta et al. 2004; Dinauer et al. 1987; Parkos et al. 1988). The complex NOX2/p22phox interacts with 4 additional molecular partners: p40phox, p47phox, p67phox and the Rho GTPase Rac (Lambeth 2004) (Figure 2). The assembly of these interactors depends on signaling mechanisms involved in ROS production. To enhance ROS synthesis, protein kinase PKC phosphorylates p47phox, which allows for its translocation to the plasma membrane to meet the NOX2/p22phox complex (Groemping and Rittinger 2005). After phosphorylation, p47phox undergoes a conformational change that releases its own autoinhibitory domain, promoting the interaction with the NOX activator p67phox (Groemping and Rittinger 2005; Nauseef 2004; Sumimoto et al. 2005). p47phox is usually referred as the organizer of NOX complex assembly, which allows increasing ROS synthesis over basal levels in response to signaling mechanisms. Finally, Rac and p40phox bind the complex through a p67phox interaction (Bokoch and Zhao 2006; Glogauer et al. 2003). Rac, which is a well known regulator of F-actin dynamics, has 3 family members (Rac1, Rac2 and Rac3) (Heasman and Ridley 2008). In neutrophils, the knock down of Rac2 but not Rac1 decreases ROS production via NOX, suggesting a family member dependent modulatory effect (Ambruso et al. 2000; Kim and Dinauer 2001; Roberts et al. 1999). Rac1 is expressed ubiquitously, Rac2 is mainly expressed in hematopoietic cells (Didsbury et al. 1989), and Rac3 is present in developing neurons (de Curtis 2014). In neurons, both Rac1 and Rac3 are positively associated with F-actin polymerization, axonal growth and neuronal polarity (Gonzalez-Billault et al. 2012). Additional studies will be required to dissect which Rac family member is required for ROS production in neurons, which could clarify whether the pool of Rac required for ROS production is the same as that which regulates F-actin dynamics and neuronal polarization. Considering this, both Rac and NOX might be involved in a positive feedback loop that regulates the synthesis of ROS required for neuronal polarization through the regulation of the actin cytoskeleton.
NADPH oxidases have been found to have many targets in cells and regulate multiple different cellular events (reviewed in (Brandes et al. 2014; Katsuyama 2010; Sirokmany et al. 2016)). Among these, NOX family enzymes have been linked to regulating the organization of the actin cytoskeleton through different proteins in a number of contexts and cell types (reviewed in (Stanley et al. 2014; Terman and Kashina 2013; Valdivia et al. 2015)). In particular, since NADPH oxidases release ROS/H2O2 to exert their effects, they appear to target specific cysteine residues in proteins – and Cys374 is recognized as a predominant target for NOXs on actin, with effects that lead to oxidation, glutathionylation, and carbonylation of this residue (Figure 1A, C; reviewed in (Stanley et al. 2014; Valdivia et al. 2015)). Likewise, components of the NOX complex have been found to associate/co-localize with actin in different cells and evidence also indicates that the actin cytoskeleton plays an important role in facilitating the ability of NOX enzymes to be activated at distinct locations to result in focused ROS production (reviewed in (Stanley et al. 2014; Terman and Kashina 2013; Valdivia et al. 2015)). Work in neurons also supports the link between the NADPH oxidase complex and actin. In particular, although still poorly characterized in mammalian neurons, NOX proteins including NOX1, NOX5 and DUOX have a dynamic expression pattern during early development of the nervous system of zebrafish (as assayed in zebrafish [0–2 days post fertilization]), while cybb2/NOX2 is stably expressed during this period of development (Weaver et al. 2015). More indepth examination has revealed that components of the NOX complex such as p40phox colocalize with F-actin in growth cones (as assayed using the mollusc Aplysia; (Munnamalai et al. 2014)) (Figure 3A). NOX2, p47phox and p22phox have also been detected in growing axons of both mature and immature hippocampal cultured neurons (Serrano et al. 2003; Tejada-Simon et al. 2005; Wilson et al. 2015). These 3 proteins have also been detected within the axonal tips/growth cones of young polarizing neurons (2 days in vitro) (Wilson et al. 2015).
Figure 3. Contribution of NOX to actin-dependent neuronal processes in physiological and pathological contexts.
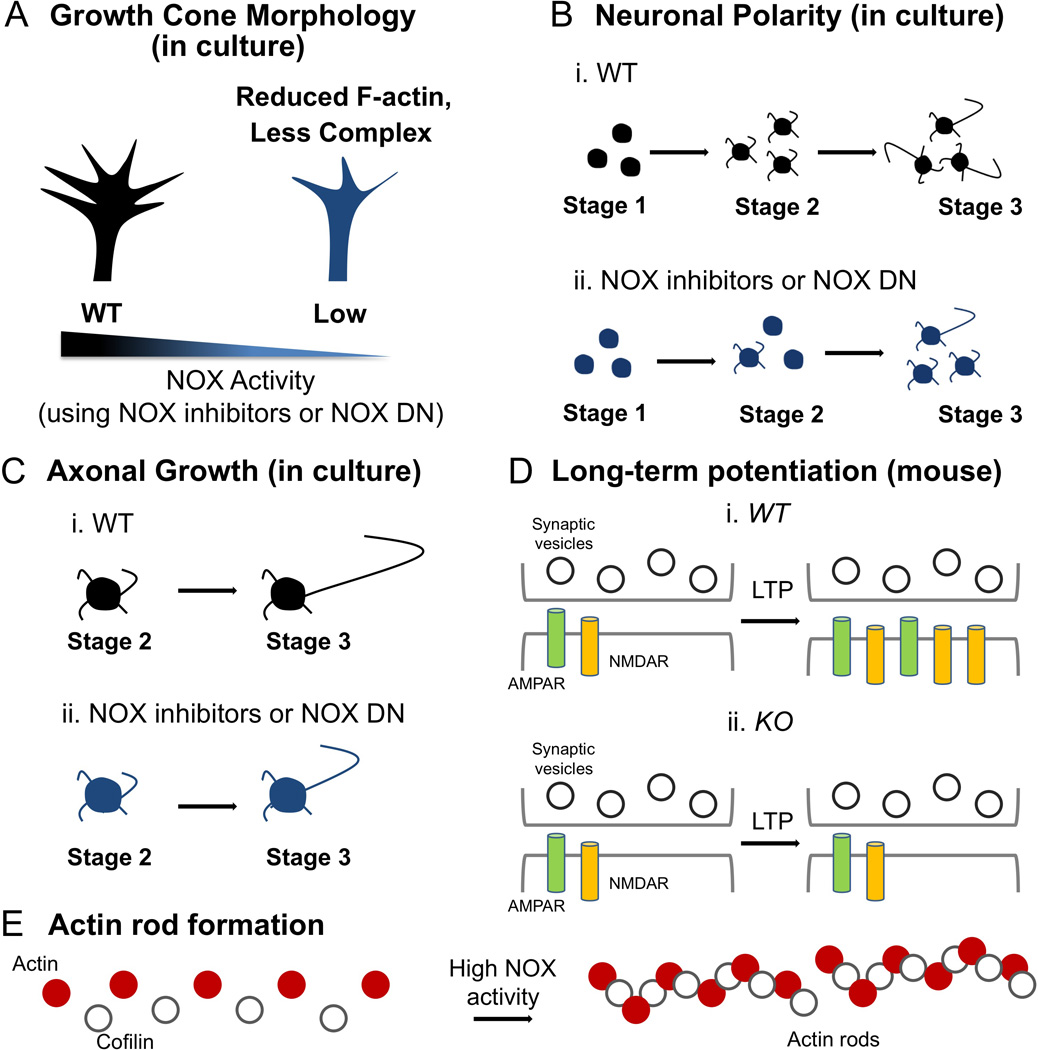
In A–D, WT designates wild-type (or no treatment), inhibitors designate inhibitors to NOX, DN designates dominant negative proteins that decrease NOX funtion, and KO designates NOX knockout. A) Inhibition of NOX2 activity using NOX inhibitors (e.g., apocynin, VAS2870, gp91 ds-tat) and other tools (e.g., ectopic expression of the mutant P156Q p22phox dominant negative [DN]) reduce the content of F-actin in the growth cone of both aplysia bag cells and hippocampal neurons in culture (Munnamalai and Suter 2009; Wilson et al. 2015). B–C) Loss of function of NOX2 (e.g., using VAS2870, gp91 ds-tat or the mutant P156Q p22phox dominant negative [DN]) blocks proper polarization of hippocampal neurons in culture, as well as the break of the symmetry of stage 2 neurons (B) and axonal growth (C) (Olguin-Albuerne and Moran 2015; Wilson et al. 2015). D) Analyses of NOX2 (gp91phox) and p47phox knockout mice, which serve as models for chronic granulomatous disease (CGD) that affect humans and carry point mutations in NOX proteins (NOX2, p47phox, p67phox and p22phox) (Cole et al. 2013; Pao et al. 2004), develop an impaired long-term potentiation (LTP) response, a method to evaluate synaptic plasticity characterized by an increase in the expression and translocation of AMPA and NMDA receptors (AMPAR and NMDAR, respectively) to the post-synaptic plasma membrane (Kishida et al. 2006). E) High ROS levels due to NOX over-activation in Alzheimer’s disease induce the aggregation of actin and cofilin in a 1:1 ratio in hippocampal neurons, leading to the formation of actin rods and neuronal dysfunction (Minamide et al. 2010; Walsh et al. 2014).
Functional studies have also provided a link between NADPH oxidase, physiological ROS, and actin dynamics in neurons. In particular, the acquisition of neuronal morphology occurs after the last post-mitotic division of the neural precursor, in a process called the establishment of neuronal polarity (Caceres et al. 2012). Here, axonal specification occurs approximately 1 day after neuronal differentiation and the break of the cellular symmetry occurs through an actin dependent mechanism (Bradke and Dotti 1999; Dotti et al. 1988). Inhibition of the NOX complex activity through both dominant negative proteins and pharmacological agents inhibits neuronal polarization and axonal growth (Wilson et al. 2015) (Figure 3B–C). Likewise, neuronal expression of a mutant isoform of p22phox that inhibits ROS production via NOX1, NOX2 and NOX3 (p22phox P156Q; (Kawahara et al. 2005)), decreases both the lamellar organization of actin in stage 1 cultured hippocampal neurons (the initial neuronal morphology before axonal specification) as well as growth cone filopodial dynamics within their elongating axons (e.g., Figure 3A; (Wilson et al. 2015)). Neurite outgrowth is also decreased after NOX inhibition in both cerebellar granule neurons of mice and Aplysia neurons, and work in Aplysia has linked this to changes in actin cytoskeletal structure and dynamics (Munnamalai and Suter 2009; Munnamalai et al. 2014; Olguin-Albuerne and Moran 2015). Interestingly, cultured cerebellar granule neurons isolated from NOX2 null mice exhibited normal neurite outgrowth compared to those from wild type mice. These animals also exhibited normal ROS levels, which may suggest compensative mechanisms and/or that mice also expresses other NOX forms, like NOX1 and NOX4, according to previous reports (Olguin-Albuerne and Moran 2015; Sorce et al. 2012). In short, the influence of NADPH oxidase/ROS production on the activity of actin and actin-related proteins has just begun to be explored in neurons. For instance, both Rac1 and Cdc-42 activities are decreased after NOX2 inhibition in cultured hippocampal neurons (Wilson et al. 2015), which suggests that molecular regulators of F-actin dynamics are also dependent on redox balance/signaling in neurons. These results further support the notion that the NOX complex is spatially correlated with the actin-rich sites of a neuron, like the periphery of axonal growth cones and emerging axons and neurites, supporting a functional relationship between NOX proteins and actin in neurons.
In addition to links between NOX during early neuronal development and axon extension, there are also emerging links between NOX and dendritic spine remodeling, synaptic plasticity, and synaptic function, which are also dependent on actin (Reviewed in (Hotulainen and Hoogenraad 2010; Kevenaar and Hoogenraad 2015)). In this regard, it is interesting that glutamate signaling activated by an NMDA receptor agonist induces superoxide synthesis via NOX2 (Brennan et al. 2009; Reyes et al. 2012). This raises the possibility of links between glutamatergic activity, synaptic plasticity, dendritic spine remodeling and actin cytoskeletal dynamics in a NOX/redox dependent manner. Previous findings also suggest that humans with deleterious point mutations in gp91phox, p47phox, p67phox and p22phox proteins develop cognitive impairments and lower intellectual coefficients compared to controls (Cole et al. 2013; Pao et al. 2004). These mutations trigger an inherited disease called Chronic Granulomatous Disease (CGD), in which 70% of the affected individuals have point mutations in the X-linked gp91phox (NOX2) gene, 25% have mutations in p47phox, and 5% have mutations in either p67phox or p22phox (Pao et al. 2004). Electrophysiological analyses of gp91phox (NOX2) and p47phox knockout mice also show that mutations in either of these genes results in decreased LTP responses compared to wild type animals and proposes that the lack of NOX proteins affects memory and learning consolidation processes in a murine model (Kishida et al. 2006) (Figure 3D).
Specific neurological disease processes have also been linked to NOX and its production of ROS. Alzheimer’s disease, for example, has been characterized by the development of actin rods within neurites, which consist of cofilin and actin aggregation in a stoichiometric ratio (1: 1 = actin: cofilin) (Minamide et al. 2010). These rods have been linked to inducing a loss of synaptic function by blocking vesicle trafficking across dendrites and by sequestering cofilin within the somato-dendritic compartment, affecting LTP responses (Cichon et al. 2012). The amyloid-β (Aβ) peptide, as well the proinflammatory cytokines TNF-α and IL-6, induce actin rod formation through a NOX dependent mechanism in both cortical and hippocampal cultured neurons (Walsh et al. 2014). Superoxide produced by NOX (and further interconversion into other ROS) oxidizes cofilin’s cysteines, triggering disulfide bridge formation and cofilin aggregation (Bamburg and Bernstein 2016; Bernstein and Bamburg 2010; Bernstein et al. 2012; Klamt et al. 2009; Walsh et al. 2014) (Figure 3E). In addition, sustained excitotoxic glutamatergic activity, as well as energy deprivation (i.e.: mithocondrial dysfunction), also induce rod formation but through a NOX independent (but perhaps not a ROS-independent; see for example, (Nunez et al. 2012; Rojas et al. 2015; Villegas et al. 2014)) mechanism (Walsh et al. 2014). NOX-derived ROS are also involved in neuronal degeneration of spinal cord neurons (Kuhn 2014). In particular, proinflammatory cytokines TNF-α and IL-1β induce the collapse of spinal cord neuronal growth cones through a NOX2 dependent mechanism (Kuhn 2014). Likewise, elevated ROS synthesis triggers aberrant actin reorganization in affected growth cones after spinal cord injury (Kuhn 2014) and the observed neurodegeneration after spinal cord injury has been hypothesized to be linked to NOX and alterations to the actin cytoskeleton (Walsh et al. 2014). Together, these findings support the notion that NADPH oxidase driven ROS production controls the physiology and pathology of neurons. Future work should focus on further analysis of these and other nervous system-related effects linked to NOX proteins (e.g., (Nayernia et al. 2014), as well as more-indepth analysis of NOX expression patterns in the nervous system (Nayernia et al. 2014) and the examination of NOX mutants for neuronal-related phenotypes (Sirokmany et al. 2016).
MICAL Flavoprotein Monooxygenases: F-actin Effects and Cellular Functions (with a Focus on the Nervous System)
The multi-domain flavoprotein monooxygenase Mical has recently provided an important new understanding of the Redox regulation of actin dynamics and its physiological importance. The MICAL family of enzymes (referred to as MICAL in vertebrates, Mical in invertebrates, and collectively called MICAL) and the presence of their Redox enzymatic moiety was first noted in conjunction with the identification of Mical as a binding partner of the transmembrane guidance receptor Plexin (Figure 4A; (Terman et al. 2002)). Mical was found to bind with Plexin, which serves as a receptor for members of the large Semaphorin family of axon/cellular guidance cues (Kolodkin and Tessier-Lavigne 2011), and regulate Semaphorin/Plexin effects on axon guidance and connectivity (Terman et al. 2002). These results became even more intriguing when it was determined that the integrity of Mical’s Redox (Flavoprotein Monooxygenase) enzymatic moiety was critical for its effects on these neuronal behaviors (Terman et al. 2002). However, the cellular role of Mical and its biochemical function was only recently characterized when it was determined that Mical uses it’s Redox enzymatic domain to bind to F-actin and that Mical modifies the organization of the actin cytoskeleton in vitro and in vivo in a Redox-dependent manner (Hung et al. 2010a). Provocatively, and in contrast to work with NADPH oxidases (see above), these studies also indicated that Mical was not simply releasing a diffusible oxidant such as H2O2 to cause effects on F-actin – since diffusible oxidants including cell-lethal levels of H2O2 had no effect on F-actin disassembly and H2O2 did not mimic the effects of Mical (Hung et al. 2011; Hung et al. 2010a). Moreover, the H2O2 scavenger catalase (and other types of reductants such as DTT and thioredoxin/thioredoxin reductase) did not affect Mical-mediated F-actin disassembly (Hung et al. 2011; Hung et al. 2013). Further analysis uncovered that Mical needed to be in close approximation with F-actin (Hung et al. 2011), and that binding to F-actin (but not G-actin) activates the enzymatic activity of Mical (Hung et al. 2011; McDonald et al. 2013; Zucchini et al. 2011), which then induces Mical to oxidize actin filament subunits (Hung et al. 2011). Turning to mass spectrometry analysis revealed that Mical selectively oxidizes two methionine residues on actin, Met-44 and Met-47 (Figure 1B, D; (Hung et al. 2011)) and mutagenesis analysis demonstrated that mutating the Met-44 residue on actin abolishes Mical’s effects on F-actin in vitro and in vivo (Figure 4B–D; (Hung et al. 2011)).
Figure 4. The MICAL and SelR/MsrB Reversible Redox Actin Regulatory System.
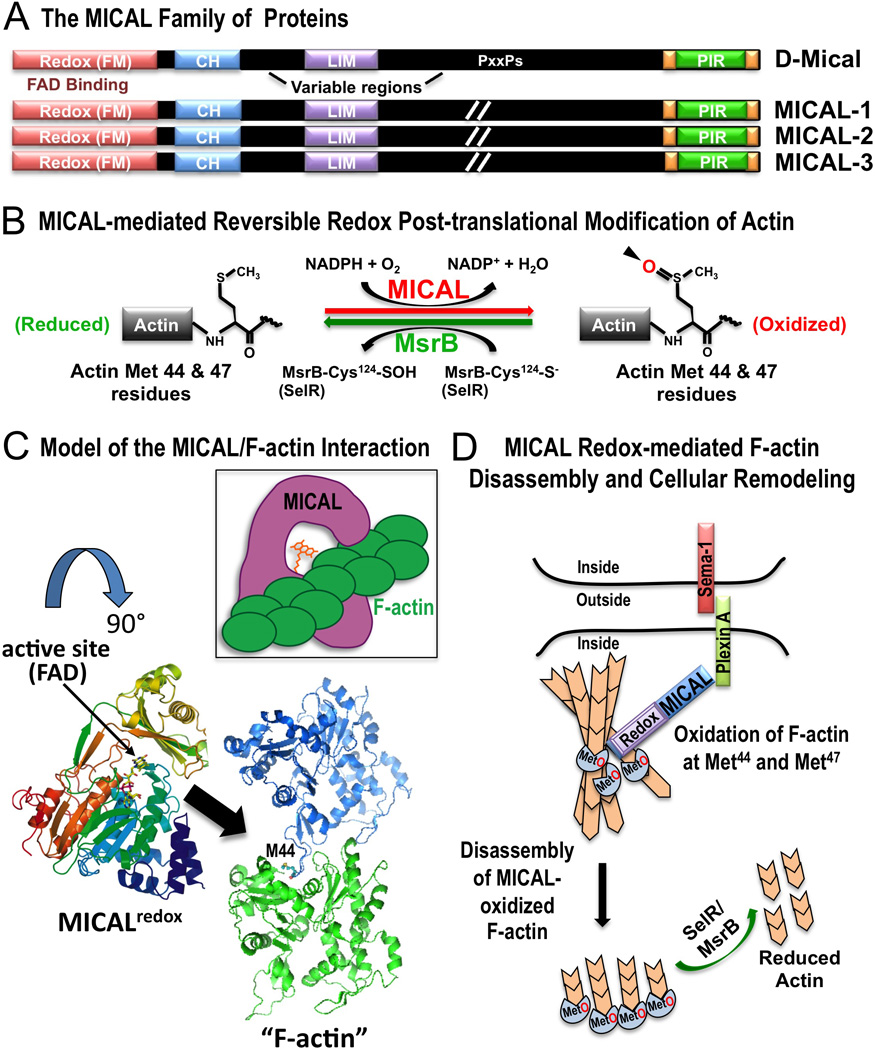
A) The MICAL family of proteins includes one Drosophila family member (D-Mical) and three vertebrate family members (MICALs 1–3) that share a conserved domain organization including the critical flavoprotein monooxygenase (Redox [FM]) domain. Each family member also contains a calponin homology (CH) domain, a LIM domain, a proline-containing (PxxPs) region, and the C-terminal plexin-interacting region (PIR). The length of the variable regions of MICAL family proteins varies and it has been designated as (//) in the diagram. (Terman et al. 2002). B) In the presence of its coenzyme NADPH, Mical adds an oxygen to both the Met-44 and Met-47 residues on F-actin. Mical oxidizes these two residues stereospecifically in the R conformation and generates actin Met-44-R-sulfoxide and Met-47-R-sulfoxide (actinMet(R)O-44 & 47) (Hung et al. 2011; Hung et al. 2013). SelR/MsrB family proteins specifically reduce MICAL-oxidized actin (Hung et al. 2013; Lee et al. 2013). C) Model of the interaction between MICAL and F-actin. The model suggests a possible mechanism by which the active site of Mical gains access to the poorly accessible Met-44 residue, which is buried within F-actin. In the model (based on published structures; PDB IDs are 2BRY, 2ZWH; (Nadella et al. 2005; Siebold et al. 2005)), rotating the Mical structure by 90 degrees (from the top of the page down) provides one mechanim to envision how the active site (small arrow) of Mical accesses (large arrow) the Met-44 (M44) residue – and as illustrated in the inset. D) Model of Mical Redox-mediated F-actin disassembly and cellular remodeling. Based on published data (Hung et al. 2011; Hung et al. 2013; Hung et al. 2010a), Semaphorin/Plexin association induces a conformational change in Plexin to allow Mical to bind to Plexin. Binding to Plexin activates Mical to associate with F-actin and oxidize the Met-44 and Met-47 residues on F-actin. Mical-mediated oxidation of F-actin induces actin filament disassembly. SelR/MsrB family proteins counteract Mical’s effects on F-actin disasembly and cellular remodeling.
Mical, therefore, is a new type of actin effector – a direct actin regulatory enzyme that uses F-actin as a specific substrate and employs a catalytic oxidation-dependent post-translational mechanism to selectively control actin dynamics. Moreover, Mical’s specific effects on actin lead to an instability of the F-actin cytoskeleton – such that Mical-mediated oxidiation of the Met-44 residue of actin triggers both F-actin depolymerization and aberrant F-actin reassembly (Figure 4B, D; (Hung et al. 2011; Hung et al. 2010a)). In this way, Mical’s ability to destabilize the F-actin cytoskeleton is also meaningful in light of its important role downstream of repulsive guidance cues like Semaphorins, which have long been characterized for their disruptive effects on the F-actin cytoskeleton (Figure 4D; (Hung and Terman 2011; Luo and Raper 1994)). While much remains to be learned about the enzymatic and biochemical properties of MICALs and their effects on actin, recent work also reveals that these Mical-mediated effects on actin dynamics are reversible by members of the SelR/MsrB family of methionine sulfoxide reductase enzymes (Hung et al. 2013; Lee et al. 2013). In particular, biochemical and mass spectrometric studies identified that SelR/MsrB proteins directly and specifically reverse the effects of MICALs on actin – by de-oxidizing actin – and restoring its normal polymerization properties (Figure 4B; (Hung et al. 2013; Lee et al. 2013)). Likewise, in vivo studies revealed that this MICAL-SelR system regulates actin dynamics in multiple cells including during axon guidance, synaptogenesis, mechanosensory organ formation, muscle development, and Semaphorin/Plexin repulsive signaling (Figure 4D; (Hung et al. 2013)). These results, therefore, reveal that the MICALS and SelR/MsrBs comprise a reversible Redox cellular signaling system that controls actin cytoskeletal organization (Figure 4D). Moreover, methionine has a unique oxidation pattern in that two stereoisomers (an R-isomer and an S-isomer) can be produced by oxidation. Actin was found to be stereo-specifically oxidized in the R-isomer conformation by the MICALs (and reduced by SelR/MsrB) (Hung et al. 2013; Lee et al. 2013) – providing a first demonstration that the stereospecific oxidation/de-oxidation of specific methionine residues (similar to the reversible phosphorylation of serine, threonine, and tyrosine residues) is a means to precisely modulate protein function.
Thus, unlike diffusible oxidants that induce random protein modifications (Kim and Gladyshev 2007; Shacter 2000; Stadtman et al. 2003), MICAL-mediated oxidation of actin is substrate, residue, and stereo-specific. It is also preferentially activated by one form of its actin substrate, the polymerized form, thereby further revealing unusual features of this new actin regulatory Redox enzyme. There is one Mical family member in Drosophila and three mammalian MICAL family members with a conserved domain organization, including their N-terminally located Redox enzymatic moiety (Figure 4A; (Terman et al. 2002)). An additional family of conserved proteins, the MICAL-Likes, have also been indentified that are similar to MICALs but lack the N-terminal Redox enzymatic region (Terman et al. 2002). The Met-44 and Met-47 residues that the MICALs oxidize are also conserved in all actins from invertebrates to humans, indicating that MICALs are likely to play prominent roles in modulating actin dynamics in both neuronal and non-neuronal tissues. Indeed, expression analysis reveals that MICALs exhibit widespread expression in both neuronal and non-neuronal tissues. For example, collectively, the MICALs appear to be ubiquitously expressed including being found in (among other tissues) brain, skeletal muscle, heart, fibroblasts, lung, kidney, bone marrow, thymus, spleen, liver, and testis – as well as in various cell lines and cancer cell lines (Reviewed in (Giridharan and Caplan 2014; Hung and Terman 2011; Zhou et al. 2011b)). More specifically, for instance, they are also collectively expressed in most, if not all areas of the central and peripheral (CNS and PNS) nervous systems (Reviewed in (Giridharan and Caplan 2014; Hung and Terman 2011; Zhou et al. 2011b) and see also (Hawrylycz et al. 2012; Lein et al. 2007; Miller et al. 2014)). Functional studies have also gone on to reveal prominent localization of MICALs with F-actin structures and important roles for MICALs in regulating multiple neuronal and non-neuronal actin dependent events. For instance, multiple different cell culture assays using primary cells and cell lines including neurons/neuronal-like cells (dorsal root ganglion neurons, hippocampal neurons, PC12 cells), fibroblasts (mouse embryonic fibroblasts [MEFs], NIH 3T3 cells), endothelial cells (rat brain capillary endothelial [RBEC] cells), kidney cells (podocytes, HEK 293 cells, COS-7 cells), and cancer cell lines (HeLa cells, 786-O kidney cancer cells, MERO-14 pleural cancer cells) have been used to characterize the effects of MICALs on F-actin dynamics and morphology (e.g., (Aggarwal et al. 2015; Giridharan et al. 2012; Grigoriev et al. 2011; Hou et al. 2015; Hung et al. 2011; Hung et al. 2013; Lee et al. 2013; Lundquist et al. 2014; Mariotti et al. 2015; Morinaka et al. 2011; Schmidt et al. 2008; Van Battum et al. 2014)). Likewise, both genetic and knockdown experiments in invertebrates and mammals indicate important roles for MICALs in non-neuronal functions including cell viability (Ashida et al. 2006; Loria et al. 2015; Mariotti et al. 2015; Zhou et al. 2011a), skeletal muscle morphology and function (Beuchle et al. 2007; Hung et al. 2013), immunity (Lee et al. 2013), podocyte (kidney) cell shape and function (Aggarwal et al. 2015), and cardiovascular integrity (Hou et al. 2015; Lundquist et al. 2014). Multiple different cellular, knockdown, and genetic experiments from invertebrates to mammals have also characterized roles for MICALs in a range of neuronal functions including axon growth, extension, and guidance (Hung et al. 2013; Hung et al. 2010a; Lundquist et al. 2014; Morinaka et al. 2011; Schmidt et al. 2008; Terman et al. 2002; Van Battum et al. 2014), synapse development (Beuchle et al. 2007; Hung et al. 2013; Van Battum et al. 2014), dendrite morphology (Kirilly et al. 2009; Rumpf et al. 2014), mechanosensory organ formation (Hung et al. 2011; Hung et al. 2013; Hung et al. 2010a), neuronal cell migration and positioning (Bron et al. 2007), and vesicle trafficking/fusion at the plasma membrane (Bachmann-Gagescu et al. 2015; Grigoriev et al. 2011; Van Battum et al. 2014) (Figure 5).
Figure 5. MICAL family Redox Enzymes are Essential for the Proper Form and Function of the Nervous System.
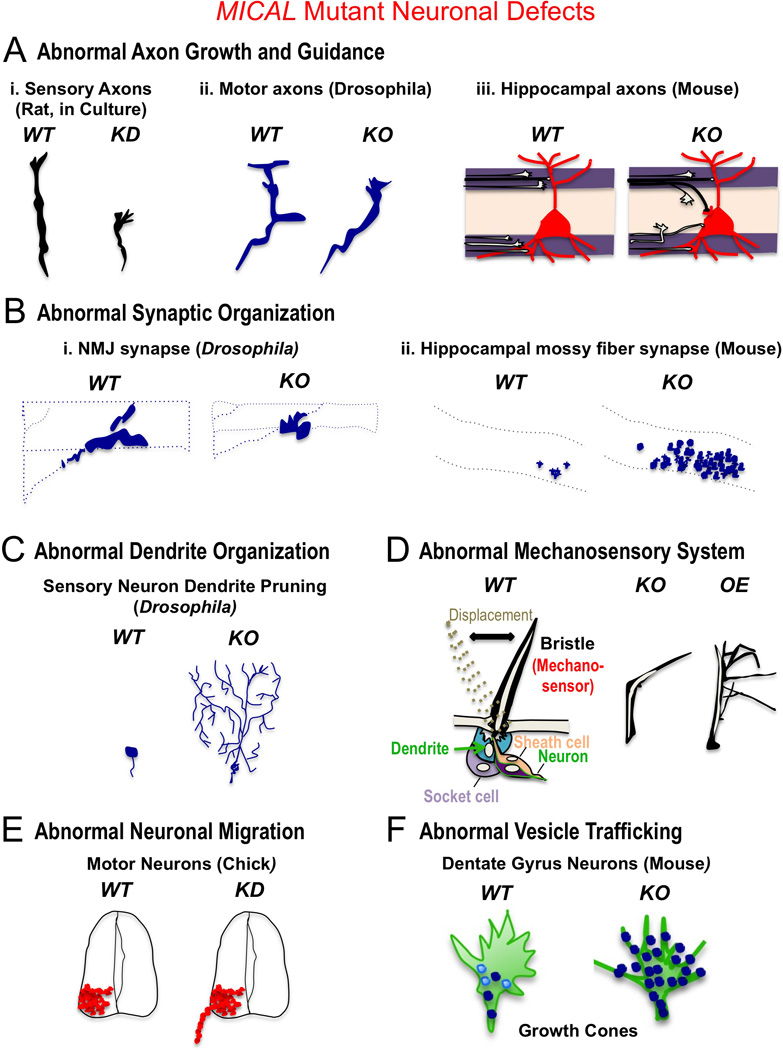
In A–F, WT designates wild-type, KD designates MICAL knockdown, KO designates Mical mutant or MICAL knockout, and OE designates increased MICAL expression. These defects and many in non-neuronal cells and tissues (see main text) are consistent with MICALs role in disassembling F-actin – including resulting in the presence of more stable F-actin in response to decreasing MICAL function, and less stable/disassembled F-actin in response to increasing MICAL function. A) MICALs are required for proper axon growth and guidance. i. Loss of MICAL (KD, MICAL-2 shRNA-mediated knockdown) in dorsal root ganglia (DRG) neurons decreases their rate of axon growth (Lundquist et al. 2014). ii. Loss of Mical (KO, Drosophila Mical−/− mutants) results in defective motor and CNS (not shown; (Hung et al. 2010a)) axon guidance in vivo (Terman et al. 2002). iii. Loss of MICAL (KO, Mouse MICAL-1−/− knockout mutants) results in defective hippocampal mossy fiber axon projections in vivo (Van Battum et al. 2014). B) MICALs are essential for proper synaptic organization. i. Loss of Mical (KO, Drosophila Mical−/− mutants) results in shorter and condensed synapses at nerve-muscle synapses (neuromuscular junction [NMJ]) in vivo (Beuchle et al. 2007). ii. Loss of MICAL (KO, Mouse MICAL-1−/− knockout mutants) generates abnormal (ectopic) synapses in the hippocampus in vivo (Van Battum et al. 2014). C) MICALs are required for proper dendrite organization, as depicted here where a loss of Mical (KO, Drosophila Mical−/− mutants) prevents the pruning and disassembly of dendrites that normally occurs in vivo (Kirilly et al. 2009). D) MICALs are required for proper formation of mechanosensory organs, as depicted here where a loss of Mical (KO, Drosophila Mical−/− mutants) generates mechanosensors (bristles) that do not form normally in vivo and contain more F-actin (Hung et al. 2010a). Increasing the levels of Mical in bristles (OE) also alters their morphology in a different manner and results in loss of F-actin (Hung et al. 2010a). E) MICALs are required for proper migration of neurons, as depicted here where a knockdown of MICAL (KD, MICAL-3 shRNA-mediated knockdown) generates abnormally positioned motor neuron cell bodies in vivo (Bron et al. 2007). F) MICALs are required for proper trafficking of vesicles in neurons, as depicted here where a loss of MICAL (KO, Mouse MICAL-1−/− knockout mutants) generates an abberant accumulation and distribution of vesicles to abnormal F-actin rich regions in the periphery of axonal growth cones (Van Battum et al. 2014).
An emerging body of work, therefore, is beginning to characterize the attributes of MICAL family Redox enzymes and these studies have revealed critical functions for these Redox enzymes in multiple different neuronal and non-neuronal physiological events. Yet, an extensive number of questions remain to be answered regarding the MICALs. For example, the different MICALs are heavily alternatively spliced (Hung and Terman 2011; Terman et al. 2002), so it is possible that different splice forms may dictate their localization and subcellular functions. In this regard, MICALs have been best characterized for their role in the cytoplasm, but MICAL splice variants have also been found to have functions in regulating the organization of actin in the nucleus (Lundquist et al. 2014). Likewise, MICALs were first functionally characterized as operating downstream of Semaphorin/Plexin repulsive signaling (Aggarwal et al. 2015; Hou et al. 2015; Hung et al. 2010a; Schmidt et al. 2008; Terman et al. 2002), but a growing body of data have also provided links between MICAL and growth factors such as nerve growth factor (NGF), vascular endothelial growth factor (VEGF), and hepatocyte growth factor (HGF) (Ashida et al. 2006; Hou et al. 2015; Lundquist et al. 2014), as well as cell adhesion molecules (Van Battum et al. 2014). In a related way, emerging evidence has begun to link MICAL with multiple neuronal and non-neuronal diseases and abnormalities including cancer (Ashida et al. 2006; Ho et al. 2012; Loria et al. 2015; Lundquist et al. 2014; Mariotti et al. 2015), diabetic nephropathy (Aggarwal et al. 2015), blood brain barrier dysfunction (Hou et al. 2015), muscular dystrophy (Marotta et al. 2009), liver disease (Chambers et al. 2011), prion susceptibility (Marbiah et al. 2014), epilepsy (Luo et al. 2011), Alzheimer’s disease (Muller et al. 2007), aging (Aenlle et al. 2009), skeletal anomalies (Bredrup et al. 2011), and obesity (Li et al. 2013). Likewise, dominant de novo mutations of MICALs substrate residues on actin, the Met-44 and Met-47 residues, have been characterized in both skeletal muscle and smooth muscle actins in human patients and they lead to actin accumulation defects that resemble the effects of mutating MICALs, as well as muscular dystrophies and vasculature disorders (Hoffjan et al. 2011; Hung et al. 2010b; Laing et al. 2009; Regalado et al. 2014). Furthermore, disruptions to the SelR/MsrB enzymes that reverse MICAL-mediated effects on actin dynamics have also been linked to neurodegenerative disorders, aging processes, and deafness (e.g., (Ahmed et al. 2011; Kim and Gladyshev 2007; Kim et al. 2016)). It is also possible that MICALs may have other direct substrates besides F-actin (i.e., proteins that activate the NADPH consumption activity of MICAL and are directly targeted by MICAL) --- but in any case, it is interesing to consider that MICALs may also have indirect substrates based on their ability to release oxidants such as H2O2 during the course of their enzyme reaction (reviewed in (Hung and Terman 2011)). For example, while MICAL does not release diffusible oxidants such as H2O2 to disassemble F-actin (as discussed above; (Hung et al. 2011; Hung et al. 2013; Hung et al. 2010a)), it is possible that in the course of directly oxidizing actin, MICAL might also influence the function of other nearby proteins through the release of difussible oxidants, such as has recently been reported for MICAL and the protein CRMP (Morinaka et al. 2011). Finally, as mentioned above, the Redox susceptibility of Met-44 and Met-47 varies depending on actin’s conformation, ionic state, and polymerization condition (Dalle-Donne et al. 2002; Guan et al. 2003; Guan et al. 2005; Hung et al. 2011; Takamoto et al. 2007), so it is possible that particular in vivo conditions and disease states may make these residues susceptible to abnormal Redox regulation. In particular, the identification of this new MICAL/SelR oxidant-based physiological signaling system raises the possibility that certain pathologies and traumas allow oxidants abnormal access to actin where they non-specifically work like MICALs and aberrantly oxidize Met-44 and/or Met-47 to pathologically disrupt F-actin-based cellular functions and behaviors. In this regard, it is interesting that Met-44, as well as Met-47, show increased oxidation in vivo in response to oxidative stress (Fedorova et al. 2010a; Ghesquiere et al. 2011). Collectively, therefore, the identification of MICAL and its reversible Redox actin regulatory system provides an unusal new system that directs cellular form and function with multiple implications for biomedicine.
Conclusions
The actin cytoskeleton is critical for many, if not all aspects of cellular behaviors and so a critical goal is to understand the factors that specify its organization and dynamic properties. It is now becoming clear that Redox-mediated effects are an important means to regulate actin dynamics and multiple different actin-dependent cellular functions. Yet, it is also clear that the Redox regulation of actin involves a diverse set of modifications – and these alter different residues on actin with distinct behavioral effects. Likewise, it has also become clear that there are different types of actin Redox effectors, and that they use different Redox species – and we describe herein the different mechanisms used by NADPH oxidases and MICAL flavoprotein monooxygenases, and the different residues on actin that they target. Thus, while exciting discoveries have been made, much remains to be investigated regarding the Redox regulation of actin and the action of the different Redox effectors. For example, it is still incompletely understood where and when the different Redox effectors of actin are acting. Likewise, it is poorly understood how the Redox-regulation of actin fits in with more classically studied actin regulatory proteins. Similarly, whether interactions exist among the different actin Redox effectors such as NADPH oxidase and MICAL is unknown – as are potential interactions with other Redox effectors that have been linked to actin regulation such as NOS and lipoxygenases. The range of specific celluar signaling pathways that impinge on these Redox effectors is also poorly understood, but is a critical step in our understanding of the Redox-mediated regulation of the actin cytoskeleton. Indeed, given the extensive links between oxidative conditions, Redox-related post-translational alterations to actin, and multiple diseases and pathological disorders within and without the nervous system that are associated with Redox-related modifications to actin including, among others, aging, Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), cardiovascular disease, Chagas’ disease, chronic obstructive pulmonary disease (COPD), chronic pain, diabetes, experimental autoimmune encephalomyelitis, Friedreich’s ataxia, inflammatory bowel disease, ischemia, macular degeneration, pulmonary tuberculosis, and sickle cell anemia/disease (Reviewed in (Terman and Kashina 2013)), it is a critical biomedical goal to understand the Redox regulation of actin.
Acknowledgments
This work was funded by CONICYT doctoral fellowship 21120221 to CW and by Fondecyt 1140325 and FONDAP 15150012 grants to CG-B and NIH (NS073968) and Welch Foundation (I-1749) grants to JRT.
Footnotes
The authors declare that they have no conflict of interests.
References
- Aenlle KK, Kumar A, Cui L, Jackson TC, Foster TC. Estrogen effects on cognition and hippocampal transcription in middle-aged mice. Neurobiol Aging. 2009;30(6):932–945. doi: 10.1016/j.neurobiolaging.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal PK, Veron D, Thomas DB, Siegel D, Moeckel G, Kashgarian M, Tufro A. Semaphorin3a promotes advanced diabetic nephropathy. Diabetes. 2015;64(5):1743–1759. doi: 10.2337/db14-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Yousaf R, Lee BC, Khan SN, Lee S, Lee K, Husnain T, Rehman AU, Bonneux S, Ansar M, et al. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet. 2011;88(1):19–29. doi: 10.1016/j.ajhg.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldini G, Carini M, Vistoli G, Shibata T, Kusano Y, Gamberoni L, Dalle-Donne I, Milzani A, Uchida K. Identification of actin as a 15-deoxy-Delta12,14-prostaglandin J2 target in neuroblastoma cells: mass spectrometric, computational, and functional approaches to investigate the effect on cytoskeletal derangement. Biochemistry. 2007;46(10):2707–2718. doi: 10.1021/bi0618565. [DOI] [PubMed] [Google Scholar]
- Aldini G, Dalle-Donne I, Vistoli G, Maffei Facino R, Carini M. Covalent modification of actin by 4-hydroxy-trans-2-nonenal (HNE): LC-ESI-MS/MS evidence for Cys374 Michael adduction. J Mass Spectrom. 2005;40(7):946–954. doi: 10.1002/jms.872. [DOI] [PubMed] [Google Scholar]
- Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279(44):45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, Gonzalez-Aller C, Hiester A, deBoer M, Harbeck RJ, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. 2000;97(9):4654–4659. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida S, Furihata M, Katagiri T, Tamura K, Anazawa Y, Yoshioka H, Miki T, Fujioka T, Shuin T, Nakamura Y, et al. Expression of novel molecules, MICAL2-PV (MICAL2 prostate cancer variants), increases with high Gleason score and prostate cancer progression. Clin Cancer Res. 2006;12(9):2767–2773. doi: 10.1158/1078-0432.CCR-05-1995. [DOI] [PubMed] [Google Scholar]
- Aslan M, Ryan TM, Townes TM, Coward L, Kirk MC, Barnes S, Alexander CB, Rosenfeld SS, Freeman BA. Nitric oxide-dependent generation of reactive species in sickle cell disease. Actin tyrosine induces defective cytoskeletal polymerization. J Biol Chem. 2003;278(6):4194–4204. doi: 10.1074/jbc.M208916200. [DOI] [PubMed] [Google Scholar]
- Bachmann-Gagescu R, Dona M, Hetterschijt L, Tonnaer E, Peters T, de Vrieze E, Mans DA, van Beersum SE, Phelps IG, Arts HH, et al. The Ciliopathy Protein CC2D2A Associates with NINL and Functions in RAB8-MICAL3-Regulated Vesicle Trafficking. PLoS Genet. 2015;11(10):e1005575. doi: 10.1371/journal.pgen.1005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balin G, Barany M. Studies on actin-actin and actin-myosin interaction. Biochim Biophys Acta. 1967;140:208–221. doi: 10.1016/0005-2795(67)90461-8. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Bernstein BW. Actin dynamics and cofilin-actin rods in Alzheimer disease. Cytoskeleton (Hoboken) 2016 doi: 10.1002/cm.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Key role of PLC-gamma in EGF protection of epithelial barrier against iNOS upregulation and F-actin nitration and disassembly. Am J Physiol Cell Physiol. 2003;285(4):C977–C993. doi: 10.1152/ajpcell.00121.2003. [DOI] [PubMed] [Google Scholar]
- Barth BM, Stewart-Smeets S, Kuhn TB. Proinflammatory cytokines provoke oxidative damage to actin in neuronal cells mediated by Rac1 and NADPH oxidase. Mol Cell Neurosci. 2009;41(2):274–285. doi: 10.1016/j.mcn.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Berndt C, Poschmann G, Stuhler K, Holmgren A, Brautigam L. Zebrafish heart development is regulated via glutaredoxin 2 dependent migration and survival of neural crest cells. Redox Biol. 2014;2:673–678. doi: 10.1016/j.redox.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20(4):187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Shaw AE, Minamide LS, Pak CW, Bamburg JR. Incorporation of cofilin into rods depends on disulfide intermolecular bonds: implications for actin regulation and neurodegenerative disease. J Neurosci. 2012;32(19):6670–6681. doi: 10.1523/JNEUROSCI.6020-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuchle D, Schwarz H, Langegger M, Koch I, Aberle H. Drosophila MICAL regulates myofilament organization and synaptic structure. Mech Dev. 2007;124(5):390–406. doi: 10.1016/j.mod.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Bhopale VM, Yang M, Yu K, Thom SR. Factors Associated with Nitric Oxide-mediated beta2 Integrin Inhibition of Neutrophils. J Biol Chem. 2015;290(28):17474–17484. doi: 10.1074/jbc.M115.651620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141(22):4206–4218. doi: 10.1242/dev.107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev. 2014;94(1):235–263. doi: 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal. 2006;8(9–10):1533–1548. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283(5409):1931–1934. doi: 10.1126/science.283.5409.1931. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbo M, Filhol E, Bole-Feysot C, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89(5):634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12(7):857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R, Vermeren M, Kokot N, Andrews W, Little GE, Mitchell KJ, Cohen J. Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev. 2007;2:21. doi: 10.1186/1749-8104-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell TM, Ervasti JM. Structural and functional properties of the actin gene family. Crit Rev Eukaryot Gene Expr. 2011;21(3):255–266. doi: 10.1615/critreveukargeneexpr.v21.i3.30. [DOI] [PubMed] [Google Scholar]
- Caceres A, Ye B, Dotti CG. Neuronal polarity: demarcation, growth and commitment. Curr Opin Cell Biol. 2012;24(4):547–553. doi: 10.1016/j.ceb.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, Salmona M, Bendotti C, Bonetto V. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem. 2005;280(16):16295–16304. doi: 10.1074/jbc.M413111200. [DOI] [PubMed] [Google Scholar]
- Castro JP, Jung T, Grune T, Almeida H. Actin carbonylation: from cell dysfunction to organism disorder. J Proteomics. 2013;92:171–180. doi: 10.1016/j.jprot.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, Sanna S, Kavousi M, Baumeister SE, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43(11):1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Huang B, Liu YC, Shyu KG, Lin PY, Wang DL. Acute hypoxia enhances proteins' S-nitrosylation in endothelial cells. Biochem Biophys Res Commun. 2008;377(4):1274–1278. doi: 10.1016/j.bbrc.2008.10.144. [DOI] [PubMed] [Google Scholar]
- Cichon J, Sun C, Chen B, Jiang M, Chen XA, Sun Y, Wang Y, Chen G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J Biol Chem. 2012;287(6):3919–3929. doi: 10.1074/jbc.M111.301911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TS, McKendrick F, Cant AJ, Pearce MS, Cale CM, Goldblatt DR, Gennery AR, Titman P. Cognitive ability in children with chronic granulomatous disease: a comparison of those managed conservatively with those who have undergone hematopoietic stem cell transplant. Neuropediatrics. 2013;44(4):230–232. doi: 10.1055/s-0033-1333875. [DOI] [PubMed] [Google Scholar]
- Collins JH, Elzinga M. The primary structure of actin from rabbit skeletal muscle. Completion and analysis of the amino acid sequence. J Biol Chem. 1975;250(15):5915–5920. [PubMed] [Google Scholar]
- Cooper JA, Walker SB, Pollard TD. Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J Muscle Res Cell Motil. 1983;4(2):253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- Dailianis S, Patetsini E, Kaloyianni M. The role of signalling molecules on actin glutathionylation and protein carbonylation induced by cadmium in haemocytes of mussel Mytilus galloprovincialis (Lmk) J Exp Biol. 2009;212(Pt 22):3612–3620. doi: 10.1242/jeb.030817. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Carini M, Vistoli G, Gamberoni L, Giustarini D, Colombo R, Maffei Facino R, Rossi R, Milzani A, Aldini G. Actin Cys374 as a nucleophilic target of alpha,beta-unsaturated aldehydes. Free Radic Biol Med. 2007;42(5):583–598. doi: 10.1016/j.freeradbiomed.2006.11.026. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34(1):23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Milzani A, Giustarini D, Di Simplicio P, Colombo R, Rossi R. S-NO-actin: S-nitrosylation kinetics and the effect on isolated vascular smooth muscle. J Muscle Res Cell Motil. 2000;21(2):171–181. doi: 10.1023/a:1005671319604. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic Biol Med. 2002;32(9):927–937. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med. 2001;31(12):1624–1632. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- DalleDonne I, Milzani A, Colombo R. H2O2-treated actin: assembly and polymer interactions with cross- linking proteins. Biophys J. 1995;69(6):2710–2719. doi: 10.1016/S0006-3495(95)80142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DalleDonne I, Milzani A, Colombo R. The tert-butyl hydroperoxide-induced oxidation of actin Cys-374 is coupled with structural changes in distant regions of the protein. Biochemistry. 1999;38(38):12471–12480. doi: 10.1021/bi990367k. [DOI] [PubMed] [Google Scholar]
- de Curtis I. Roles of Rac1 and Rac3 GTPases during the development of cortical and hippocampal GABAergic interneurons. Front Cell Neurosci. 2014;8:307. doi: 10.3389/fncel.2014.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264(28):16378–16382. [PubMed] [Google Scholar]
- Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos CA. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature. 1987;327(6124):717–720. doi: 10.1038/327717a0. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8(4):1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke J, Takashi R, Ue K, Morales MF. Reciprocal reactivities of specific thiols when actin binds to myosin. Proc Natl Acad Sci U S A. 1976;73(2):302–306. doi: 10.1073/pnas.73.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, Amberg DC. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (Hoboken) 2011;68(6):340–354. doi: 10.1002/cm.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova M, Kuleva N, Hoffmann R. Identification of cysteine, methionine and tryptophan residues of actin oxidized in vivo during oxidative stress. J Proteome Res. 2010a;9(3):1598–1609. doi: 10.1021/pr901099e. [DOI] [PubMed] [Google Scholar]
- Fedorova M, Todorovsky T, Kuleva N, Hoffmann R. Quantitative evaluation of tryptophan oxidation in actin and troponin I from skeletal muscles using a rat model of acute oxidative stress. Proteomics. 2010b;10(14):2692–2700. doi: 10.1002/pmic.201000147. [DOI] [PubMed] [Google Scholar]
- Feuer G, Molnar F, Pettko E, Straub FB. Studies on the composition and polymerization of actin. Hung. Acta Physiol. 1948;1:150–163. [PubMed] [Google Scholar]
- Fiaschi T, Cozzi G, Raugei G, Formigli L, Ramponi G, Chiarugi P. Redox regulation of beta-actin during integrin-mediated cell adhesion. J Biol Chem. 2006;281(32):22983–22991. doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- Figueiredo-Freitas C, Dulce RA, Foster MW, Liang J, Yamashita AM, Lima-Rosa FL, Thompson JW, Moseley MA, Hare JM, Nogueira L, et al. S-Nitrosylation of Sarcomeric Proteins Depresses Myofilament Ca2+)Sensitivity in Intact Cardiomyocytes. Antioxid Redox Signal. 2015;23(13):1017–1034. doi: 10.1089/ars.2015.6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463(7280):485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Vos MR, Schroder GF, Egelman EH. Near-atomic resolution for one state of F-actin. Structure. 2015;23(1):173–182. doi: 10.1016/j.str.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayarre J, Sanchez D, Sanchez-Gomez FJ, Terron MC, Llorca O, Perez-Sala D. Addition of electrophilic lipids to actin alters filament structure. Biochem Biophys Res Commun. 2006;349(4):1387–1393. doi: 10.1016/j.bbrc.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Gergely J. Contractile proteins. Annu Rev Biochem. 1966;35:691–722. doi: 10.1146/annurev.bi.35.070166.003355. [DOI] [PubMed] [Google Scholar]
- Ghesquiere B, Jonckheere V, Colaert N, Van Durme J, Timmerman E, Goethals M, Schymkowitz J, Rousseau F, Vandekerckhove J, Gevaert K. Redox proteomics of protein-bound methionine oxidation. Mol Cell Proteomics. 2011;10(5):M110 006866. doi: 10.1074/mcp.M110.006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridharan SS, Caplan S. MICAL-Family Proteins: Complex Regulators of the Actin Cytoskeleton. Antioxid Redox Signal. 2014;20(13):2059–2073. doi: 10.1089/ars.2013.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridharan SS, Rohn JL, Naslavsky N, Caplan S. Differential regulation of actin microfilaments by human MICAL proteins. J Cell Sci. 2012;125(Pt 3):614–624. doi: 10.1242/jcs.089367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170(11):5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- Go YM, Jones DP. The redox proteome. J Biol Chem. 2013;288(37):26512–26520. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Billault C, Munoz-Llancao P, Henriquez DR, Wojnacki J, Conde C, Caceres A. The role of small GTPases in neuronal morphogenesis and polarity. Cytoskeleton (Hoboken) 2012;69(7):464–485. doi: 10.1002/cm.21034. [DOI] [PubMed] [Google Scholar]
- Grigoriev I, Yu KL, Martinez-Sanchez E, Serra-Marques A, Smal I, Meijering E, Demmers J, Peranen J, Pasterkamp RJ, van der Sluijs P, et al. Rab6, Rab8, and MICAL3 Cooperate in Controlling Docking and Fusion of Exocytotic Carriers. Curr Biol. 2011;21(11):967–974. doi: 10.1016/j.cub.2011.04.030. [DOI] [PubMed] [Google Scholar]
- Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386(Pt 3):401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SM, Lehman SL. Accessibility of myofilament cysteines and effects on ATPase depend on the activation state during exposure to oxidants. PLoS One. 2013;8(7):e69110. doi: 10.1371/journal.pone.0069110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JQ, Almo SC, Reisler E, Chance MR. Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-actin solution structure is divalent cation dependent. Biochemistry. 2003;42(41):11992–12000. doi: 10.1021/bi034914k. [DOI] [PubMed] [Google Scholar]
- Guan JQ, Takamoto K, Almo SC, Reisler E, Chance MR. Structure and dynamics of the actin filament. Biochemistry. 2005;44(9):3166–3175. doi: 10.1021/bi048021j. [DOI] [PubMed] [Google Scholar]
- Hamnell-Pamment Y, Lind C, Palmberg C, Bergman T, Cotgreave IA. Determination of site-specificity of S-glutathionylated cellular proteins. Biochem Biophys Res Commun. 2005;332(2):362–369. doi: 10.1016/j.bbrc.2005.04.130. [DOI] [PubMed] [Google Scholar]
- Harper AM, Chaplin MF, Segal AW. Cytochrome b-245 from human neutrophils is a glycoprotein. Biochem J. 1985;227(3):783–788. doi: 10.1042/bj2270783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314(8):1765–1776. doi: 10.1016/j.yexcr.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Ho JR, Chapeaublanc E, Kirkwood L, Nicolle R, Benhamou S, Lebret T, Allory Y, Southgate J, Radvanyi F, Goud B. Deregulation of Rab and Rab effector genes in bladder cancer. PLoS One. 2012;7(6):e39469. doi: 10.1371/journal.pone.0039469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffjan S, Waldmuller S, Blankenfeldt W, Kotting J, Gehle P, Binner P, Epplen JT, Scheffold T. Three novel mutations in the ACTA2 gene in German patients with thoracic aortic aneurysms and dissections. Eur J Hum Genet. 2011;19(5):520–524. doi: 10.1038/ejhg.2010.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189(4):619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou ST, Nilchi L, Li X, Gangaraju S, Jiang SX, Aylsworth A, Monette R, Slinn J. Semaphorin3A elevates vascular permeability and contributes to cerebral ischemia-induced brain damage. Sci Rep. 2015;5:7890. doi: 10.1038/srep07890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R-J, Terman JR. Extracellular inhibitors, repellents, and Semaphorin/Plexin/MICAL-mediated actin filament disassembly. Cytoskeleton. 2011;68:415–433. doi: 10.1002/cm.20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, Pak CW, Terman JR. Direct redox regulation of F-actin assembly and disassembly by Mical. Science. 2011;334(6063):1710–1713. doi: 10.1126/science.1211956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, Spaeth CS, Yesilyurt HG, Terman JR. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat Cell Biol. 2013;15(12):1445–1454. doi: 10.1038/ncb2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, Yazdani U, Yoon J, Wu H, Yang T, Gupta N, Huang Z, van Berkel WJ, Terman JR. Mical links semaphorins to F-actin disassembly. Nature. 2010a;463(7282):823–827. doi: 10.1038/nature08724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RM, Yoon G, Hawkins CE, Halliday W, Biggar D, Vajsar J. Cap myopathy caused by a mutation of the skeletal alpha-actin gene ACTA1. Neuromuscul Disord. 2010b;20(4):238–240. doi: 10.1016/j.nmd.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Ishiwata S. Freezing of actin. Reversible oxidation of a sulfhydryl group and structural change. J Biochem. 1976;80(3):595–609. doi: 10.1093/oxfordjournals.jbchem.a131315. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3(2):193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Kang LT, Phillips TM, Vanderhoek JY. Novel membrane target proteins for lipoxygenase-derived mono(S)hydroxy fatty acids. Biochim Biophys Acta. 1999;1438(3):388–398. doi: 10.1016/s0167-4838(99)00100-4. [DOI] [PubMed] [Google Scholar]
- Kang LT, Vanderhoek JY. Mono (S) hydroxy fatty acids: novel ligands for cytosolic actin. J Lipid Res. 1998;39(7):1476–1482. [PubMed] [Google Scholar]
- Kanski J, Hong SJ, Schoneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem. 2005;280(25):24261–24266. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- Katsuyama M. NOX/NADPH oxidase, the superoxide-generating enzyme: its transcriptional regulation and physiological roles. J Pharmacol Sci. 2010;114(2):134–146. doi: 10.1254/jphs.10r01cr. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280(36):31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- Kevenaar JT, Hoogenraad CC. The axonal cytoskeleton: from organization to function. Front Mol Neurosci. 2015;8:44. doi: 10.3389/fnmol.2015.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielley WW. The Biochemistry of Muscle. Annu Rev Biochem. 1964;33:403–430. doi: 10.1146/annurev.bi.33.070164.002155. [DOI] [PubMed] [Google Scholar]
- Kim C, Dinauer MC. Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J Immunol. 2001;166(2):1223–1232. doi: 10.4049/jimmunol.166.2.1223. [DOI] [PubMed] [Google Scholar]
- Kim G, Weiss SJ, Levine RL. Methionine oxidation and reduction in proteins. Biochim Biophys Acta. 2014;1840(2):901–905. doi: 10.1016/j.bbagen.2013.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Gladyshev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem J. 2007;407(3):321–329. doi: 10.1042/BJ20070929. [DOI] [PubMed] [Google Scholar]
- Kim MA, Cho HJ, Bae SH, Lee B, Oh SK, Kwon TJ, Ryoo ZY, Kim HY, Cho JH, Kim UK, et al. Methionine Sulfoxide Reductase B3-Targeted In Utero Gene Therapy Rescues Hearing Function in a Mouse Model of Congenital Sensorineural Hearing Loss. Antioxid Redox Signal. 2016;24(11):590–602. doi: 10.1089/ars.2015.6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirilly D, Gu Y, Huang Y, Wu Z, Bashirullah A, Low BC, Kolodkin AL, Wang H, Yu F. A genetic pathway composed of Sox14 and Mical governs severing of dendrites during pruning. Nat Neurosci. 2009;12(12):1497–1505. doi: 10.1038/nn.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26(15):5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt F, Zdanov S, Levine RL, Pariser A, Zhang Y, Zhang B, Yu LR, Veenstra TD, Shacter E. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol. 2009;11(10):1241–1246. doi: 10.1038/ncb1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Tessier-Lavigne M. Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol. 2011;3(6):a001727. doi: 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno K, Morales MF. Exposure of actin thiols by the removal of tightly held calcium ions. Proc Natl Acad Sci U S A. 1985;82(23):7904–7908. doi: 10.1073/pnas.82.23.7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem. 1981;114(1):33–38. [PubMed] [Google Scholar]
- Kuhn TB. Oxygen radicals elicit paralysis and collapse of spinal cord neuron growth cones upon exposure to proinflammatory cytokines. Biomed Res Int. 2014;2014:191767. doi: 10.1155/2014/191767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing NG, Dye DE, Wallgren-Pettersson C, Richard G, Monnier N, Lillis S, Winder TL, Lochmuller H, Graziano C, Mitrani-Rosenbaum S, et al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1) Hum Mutat. 2009;30(9):1267–1277. doi: 10.1002/humu.21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb GD, Posterino GS. Effects of oxidation and reduction on contractile function in skeletal muscle fibres of the rat. J Physiol. 2003;546(Pt 1):149–163. doi: 10.1113/jphysiol.2002.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110(10):1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassing I, Schmitzberger F, Bjornstedt M, Holmgren A, Nordlund P, Schutt CE, Lindberg U. Molecular and structural basis for redox regulation of beta-actin. J Mol Biol. 2007;370(2):331–348. doi: 10.1016/j.jmb.2007.04.056. [DOI] [PubMed] [Google Scholar]
- Lee BC, Peterfi Z, Hoffmann FW, Moore RE, Kaya A, Avanesov A, Tarrago L, Zhou Y, Weerapana E, Fomenko DE, et al. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell. 2013;51(3):397–404. doi: 10.1016/j.molcel.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lepley RA, Fitzpatrick FA. 5-Lipoxygenase contains a functional Src homology 3-binding motif that interacts with the Src homology 3 domain of Grb2 and cytoskeletal proteins. J Biol Chem. 1994;269(39):24163–24168. [PubMed] [Google Scholar]
- Li WD, Jiao H, Wang K, Zhang CK, Glessner JT, Grant SF, Zhao H, Hakonarson H, Arlen Price R. A genome wide association study of plasma uric acid levels in obese cases and never-overweight controls. Obesity (Silver Spring) 2013;21(9):E490–E494. doi: 10.1002/oby.20303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TI, Kim M, Dowben RM. Accessibility of thiols in actin--a kinetic study with fluorescent maleimide probes. Prog Clin Biol Res. 1990;327:771–778. [PubMed] [Google Scholar]
- Liu DF, Wang D, Stracher A. The accessibility of the thiol groups on G- and F-actin of rabbit muscle. Biochem J. 1990;266(2):453–459. doi: 10.1042/bj2660453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria R, Bon G, Perotti V, Gallo E, Bersani I, Baldassari P, Porru M, Leonetti C, Di Carlo S, Visca P, et al. Sema6A and Mical1 control cell growth and survival of BRAFV600E human melanoma cells. Oncotarget. 2015;6(5):2779–2793. doi: 10.18632/oncotarget.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist MR, Storaska AJ, Liu TC, Larsen SD, Evans T, Neubig RR, Jaffrey SR. Redox Modification of Nuclear Actin by MICAL-2 Regulates SRF Signaling. Cell. 2014;156(3):563–576. doi: 10.1016/j.cell.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Xu Y, Zhu Q, Zhao F, Zhang Y, Peng X, Wang W, Wang X. Expression pattern of Mical-1 in the temporal neocortex of patients with intractable temporal epilepsy and Pilocarpine-induced rat model. Synapse. 2011 doi: 10.1002/syn.20961. [DOI] [PubMed] [Google Scholar]
- Luo Y, Raper JA. Inhibitory factors controlling growth cone motility and guidance. Curr Opin Neurobiol. 1994;4(5):648–654. doi: 10.1016/0959-4388(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Lusty CJ, Fasold H. Characterization of sulfhydryl groups of actin. Biochemistry. 1969;8(7):2933–2939. doi: 10.1021/bi00835a036. [DOI] [PubMed] [Google Scholar]
- Mannherz HG, Goody RS. Proteins of contractile systems. Annu Rev Biochem. 1976;45:427–465. doi: 10.1146/annurev.bi.45.070176.002235. [DOI] [PubMed] [Google Scholar]
- Marbiah MM, Harvey A, West BT, Louzolo A, Banerjee P, Alden J, Grigoriadis A, Hummerich H, Kan HM, Cai Y, et al. Identification of a gene regulatory network associated with prion replication. EMBO J. 2014;33(14):1527–1547. doi: 10.15252/embj.201387150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti S, Barravecchia I, Vindigni C, Pucci A, Balsamo M, Libro R, Senchenko V, Dmitriev A, Jacchetti E, Cecchini M, et al. MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget. 2015 doi: 10.18632/oncotarget.6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta M, Ruiz-Roig C, Sarria Y, Peiro JL, Nunez F, Ceron J, Munell F, Roig-Quilis M. Muscle genome-wide expression profiling during disease evolution in mdx mice. Physiol Genomics. 2009;37(2):119–132. doi: 10.1152/physiolgenomics.90370.2008. [DOI] [PubMed] [Google Scholar]
- McDonald CA, Liu YY, Palfey BA. Actin stimulates reduction of the MICAL-2 monooxygenase domain. Biochemistry. 2013;52(35):6076–6084. doi: 10.1021/bi4008462. [DOI] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508(7495):199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YI, Chang MK, Funk CD, Feramisco JR, Witztum JL. 12/15-lipoxygenase translocation enhances site-specific actin polymerization in macrophages phagocytosing apoptotic cells. J Biol Chem. 2001;276(22):19431–19439. doi: 10.1074/jbc.M011276200. [DOI] [PubMed] [Google Scholar]
- Milzani A, DalleDonne I, Colombo R. Prolonged oxidative stress on actin. Arch Biochem Biophys. 1997;339(2):267–274. doi: 10.1006/abbi.1996.9847. [DOI] [PubMed] [Google Scholar]
- Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R, DalleDonne I. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 2000;9(9):1774–1782. doi: 10.1110/ps.9.9.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamide LS, Maiti S, Boyle JA, Davis RC, Coppinger JA, Bao Y, Huang TY, Yates J, Bokoch GM, Bamburg JR. Isolation and characterization of cytoplasmic cofilin-actin rods. J Biol Chem. 2010;285(8):5450–5460. doi: 10.1074/jbc.M109.063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan L, Moldovan NI, Sohn RH, Parikh SA, Goldschmidt-Clermont PJ. Redox changes of cultured endothelial cells and actin dynamics. Circ Res. 2000;86(5):549–557. doi: 10.1161/01.res.86.5.549. [DOI] [PubMed] [Google Scholar]
- Morinaka A, Yamada M, Itofusa R, Funato Y, Yoshimura Y, Nakamura F, Yoshimura T, Kaibuchi K, Goshima Y, Hoshino M, et al. Thioredoxin Mediates Oxidation-Dependent Phosphorylation of CRMP2 and Growth Cone Collapse. Sci Signal. 2011;4(170):ra26. doi: 10.1126/scisignal.2001127. [DOI] [PubMed] [Google Scholar]
- Muhlrad A, Corsi A, Granata AL. Studies on the properties of chemically modified actin. I. Photooxidation, succinylation, nitration. Biochim Biophys Acta. 1968;162(3):435–443. doi: 10.1016/0005-2728(68)90129-1. [DOI] [PubMed] [Google Scholar]
- Muller T, Concannon CG, Ward MW, Walsh CM, Tirniceriu AL, Tribl F, Kogel D, Prehn JH, Egensperger R. Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD) Mol Biol Cell. 2007;18(1):201–210. doi: 10.1091/mbc.E06-04-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnamalai V, Suter DM. Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J Neurochem. 2009;108(3):644–661. doi: 10.1111/j.1471-4159.2008.05787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnamalai V, Weaver CJ, Weisheit CE, Venkatraman P, Agim ZS, Quinn MT, Suter DM. Bidirectional interactions between NOX2-type NADPH oxidase and the F-actin cytoskeleton in neuronal growth cones. J Neurochem. 2014;130(4):526–540. doi: 10.1111/jnc.12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadella M, Bianchet MA, Gabelli SB, Barrila J, Amzel LM. Structure and activity of the axon guidance protein MICAL. Proc Natl Acad Sci U S A. 2005;102(46):16830–16835. doi: 10.1073/pnas.0504838102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122(4):277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- Nayernia Z, Jaquet V, Krause KH. New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal. 2014;20(17):2815–2837. doi: 10.1089/ars.2013.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez MT, Urrutia P, Mena N, Aguirre P, Tapia V, Salazar J. Iron toxicity in neurodegeneration. Biometals. 2012;25(4):761–776. doi: 10.1007/s10534-012-9523-0. [DOI] [PubMed] [Google Scholar]
- Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11(6):1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Iwasa M, Aihara T, Maeda Y, Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457(7228):441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- Olguin-Albuerne M, Moran J. ROS produced by NOX2 control in vitro development of cerebellar granule neurons development. ASN Neuro. 2015;7(2) doi: 10.1177/1759091415578712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paclet MH, Henderson LM, Campion Y, Morel F, Dagher MC. Localization of Nox2 N-terminus using polyclonal antipeptide antibodies. Biochem J. 2004;382(Pt 3):981–986. doi: 10.1042/BJ20040954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao M, Wiggs EA, Anastacio MM, Hyun J, DeCarlo ES, Miller JT, Anderson VL, Malech HL, Gallin JI, Holland SM. Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics. 2004;45(3):230–234. doi: 10.1176/appi.psy.45.3.230. [DOI] [PubMed] [Google Scholar]
- Parkos CA, Dinauer MC, Walker LE, Allen RA, Jesaitis AJ, Orkin SH. Primary structure and unique expression of the 22-kilodalton light chain of human neutrophil cytochrome b. Proc Natl Acad Sci U S A. 1988;85(10):3319–3323. doi: 10.1073/pnas.85.10.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro GO, Ogut O. Impact of actin glutathionylation on the actomyosin-S1 ATPase. Biochemistry. 2009;48(31):7533–7538. doi: 10.1021/bi900669m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326(5957):1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost P, Doucet J, Hammarberg T, Gerisch G, Samuelsson B, Radmark O. 5-Lipoxygenase interacts with coactosin-like protein. J Biol Chem. 2001;276(19):16520–16527. doi: 10.1074/jbc.M011205200. [DOI] [PubMed] [Google Scholar]
- Regalado ES, Guo DC, Estrera AL, Buja LM, Milewicz DM. Acute aortic dissections with pregnancy in women with ACTA2 mutations. Am J Med Genet A. 2014;164A(1):106–112. doi: 10.1002/ajmg.a.36208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes RC, Brennan AM, Shen Y, Baldwin Y, Swanson RA. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J Neurosci. 2012;32(37):12973–12978. doi: 10.1523/JNEUROSCI.1597-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RL, Tang DG, Haddad M, Honn KV, Taylor JD. 12(S)-hydroxyeicosatetraenoic acid increases the actin microfilament content in B16a melanoma cells: a protein kinase-dependent process. Int J Cancer. 1998;77(2):271–278. doi: 10.1002/(sici)1097-0215(19980717)77:2<271::aid-ijc17>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, Spaetti A, Pollock JD, Borneo JB, Bradford GB, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10(2):183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- Rojas F, Gonzalez D, Cortes N, Ampuero E, Hernandez DE, Fritz E, Abarzua S, Martinez A, Elorza AA, Alvarez A, et al. Reactive oxygen species trigger motoneuron death in non-cell-autonomous models of ALS through activation of c-Abl signaling. Front Cell Neurosci. 2015;9:203. doi: 10.3389/fncel.2015.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Gonzalez A, Salido GM, Pariente JA. Effects of reactive oxygen species on actin filament polymerisation and amylase secretion in mouse pancreatic acinar cells. Cell Signal. 2002;14(6):547–556. doi: 10.1016/s0898-6568(01)00273-x. [DOI] [PubMed] [Google Scholar]
- Rotrosen D, Kleinberg ME, Nunoi H, Leto T, Gallin JI, Malech HL. Evidence for a functional cytoplasmic domain of phagocyte oxidase cytochrome b558. J Biol Chem. 1990;265(15):8745–8750. [PubMed] [Google Scholar]
- Rumpf S, Bagley JA, Thompson-Peer KL, Zhu S, Gorczyca D, Beckstead RB, Jan LY, Jan YN. Drosophila Valosin-Containing Protein is required for dendrite pruning through a regulatory role in mRNA metabolism. Proc Natl Acad Sci U S A. 2014;111(20):7331–7336. doi: 10.1073/pnas.1406898111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EF, Shim SO, Strittmatter SM. Release of MICAL autoinhibition by semaphorin-plexin signaling promotes interaction with collapsin response mediator protein. J Neurosci. 2008;28(9):2287–2297. doi: 10.1523/JNEUROSCI.5646-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988(1–2):193–198. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev. 2000;32(3–4):307–326. doi: 10.1081/dmr-100102336. [DOI] [PubMed] [Google Scholar]
- Shartava A, Monteiro CA, Bencsath FA, Schneider K, Chait BT, Gussio R, Casoria-Scott LA, Shah AK, Heuerman CA, Goodman SR. A posttranslational modification of beta-actin contributes to the slow dissociation of the spectrin-protein 4.1-actin complex of irreversibly sickled cells. J Cell Biol. 1995;128(5):805–818. doi: 10.1083/jcb.128.5.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebold C, Berrow N, Walter TS, Harlos K, Owens RJ, Stuart DI, Terman JR, Kolodkin AL, Pasterkamp RJ, Jones EY. High-resolution structure of the catalytic region of MICAL (molecule interacting with CasL), a multidomain flavoenzyme-signaling molecule. Proc Natl Acad Sci U S A. 2005;102(46):16836–16841. doi: 10.1073/pnas.0504997102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirokmany G, Donko A, Geiszt M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol Sci. 2016;37(4):318–327. doi: 10.1016/j.tips.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Sobierajska K, Skurzynski S, Stasiak M, Kryczka J, Cierniewski CS, Swiatkowska M. Protein disulfide isomerase directly interacts with beta-actin Cys374 and regulates cytoskeleton reorganization. J Biol Chem. 2014;289(9):5758–5773. doi: 10.1074/jbc.M113.479477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorce S, Krause KH, Jaquet V. Targeting NOX enzymes in the central nervous system: therapeutic opportunities. Cell Mol Life Sci. 2012;69(14):2387–2407. doi: 10.1007/s00018-012-1014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5(5):577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- Stanley A, Thompson K, Hynes A, Brakebusch C, Quondamatteo F. NADPH oxidase complex-derived reactive oxygen species, the actin cytoskeleton, and Rho GTPases in cell migration. Antioxid Redox Signal. 2014;20(13):2026–2042. doi: 10.1089/ars.2013.5713. [DOI] [PubMed] [Google Scholar]
- Stournaras C, Drewes G, Blackholm H, Merkler I, Faulstich H. Glutathionyl(cysteine-374) actin forms filaments of low mechanical stability. Biochim Biophys Acta. 1990;1037(1):86–91. doi: 10.1016/0167-4838(90)90105-o. [DOI] [PubMed] [Google Scholar]
- Su D, Shukla AK, Chen B, Kim JS, Nakayasu E, Qu Y, Aryal U, Weitz K, Clauss TR, Monroe ME, et al. Quantitative site-specific reactivity profiling of S-nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free Radic Biol Med. 2013;57:68–78. doi: 10.1016/j.freeradbiomed.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Kondrikov D, Block ER. Beta-actin: a regulator of NOS-3. Sci STKE. 2007;2007(404):pe52. doi: 10.1126/stke.4042007pe52. [DOI] [PubMed] [Google Scholar]
- Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338(1):677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- Tait JF, Frieden C. Chemical modification of actin. Acceleration of polymerization and reduction of network formation by reaction with N-ethylmaleimide, (iodoacetamido)tetramethylrhodamine, or 7-chloro-4-nitro-2,1,3-benzoxadiazole. Biochemistry. 1982;21(24):6046–6053. doi: 10.1021/bi00267a004. [DOI] [PubMed] [Google Scholar]
- Takamoto K, Kamal JK, Chance MR. Biochemical implications of a three-dimensional model of monomeric actin bound to magnesium-chelated ATP. Structure. 2007;15(1):39–51. doi: 10.1016/j.str.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Tang JX, Janmey PA, Stossel TP, Ito T. Thiol oxidation of actin produces dimers that enhance the elasticity of the F-actin network. Biophys J. 1999;76(4):2208–2215. doi: 10.1016/S0006-3495(99)77376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29(1):97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman JR, Kashina A. Post-translational modification and regulation of actin. Curr Opin Cell Biol. 2013;25(1):30–38. doi: 10.1016/j.ceb.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 2002;109(7):887–900. doi: 10.1016/s0092-8674(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Thom SR, Bhopale VM, Mancini DJ, Milovanova TN. Actin S-nitrosylation inhibits neutrophil beta2 integrin function. J Biol Chem. 2008;283(16):10822–10834. doi: 10.1074/jbc.M709200200. [DOI] [PubMed] [Google Scholar]
- Tsapara A, Kardassis D, Moustakas A, Gravanis A, Stournaras C. Expression and characterization of Cys374 mutated human beta-actin in two different mammalian cell lines: impaired microfilament organization and stability. FEBS Lett. 1999;455(1–2):117–122. doi: 10.1016/s0014-5793(99)00848-0. [DOI] [PubMed] [Google Scholar]
- Ullevig S, Zhao Q, Lee CF, Seok Kim H, Zamora D, Asmis R. NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler Thromb Vasc Biol. 2012;32(2):415–426. doi: 10.1161/ATVBAHA.111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia A, Duran C, San Martin A. The role of Nox-mediated oxidation in the regulation of cytoskeletal dynamics. Curr Pharm Des. 2015;21(41):6009–6022. doi: 10.2174/1381612821666151029112624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Battum EY, Gunput RA, Lemstra S, Groen EJ, Yu KL, Adolfs Y, Zhou Y, Hoogenraad CC, Yoshida Y, Schachner M, et al. The intracellular redox protein MICAL-1 regulates the development of hippocampal mossy fibre connections. Nat Commun. 2014;5:4317. doi: 10.1038/ncomms5317. [DOI] [PubMed] [Google Scholar]
- Villegas R, Martinez NW, Lillo J, Pihan P, Hernandez D, Twiss JL, Court FA. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J Neurosci. 2014;34(21):7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Ecken J, Muller M, Lehman W, Manstein DJ, Penczek PA, Raunser S. Structure of the F-actin-tropomyosin complex. Nature. 2015;519(7541):114–117. doi: 10.1038/nature14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT. Expanding nature's inventory. Englewood, CO: Roberts and Company Publishers; 2006. Posttranslational modifications of proteins. [Google Scholar]
- Walsh KP, Minamide LS, Kane SJ, Shaw AE, Brown DR, Pulford B, Zabel MD, Lambeth JD, Kuhn TB, Bamburg JR. Amyloid-β and Proinflammatory Cytokines Utilize a Prion Protein-Dependent Pathway to Activate NADPH Oxidase and Induce Cofilin-Actin Rods in Hippocampal Neurons. PLoS ONE. 2014;9(4):e95995. doi: 10.1371/journal.pone.0095995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276(51):47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- Weaver CJ, Fai Leung Y, Suter DM. Expression dynamics of NADPH oxidases during early zebrafish development. J Comp Neurol. 2015 doi: 10.1002/cne.23938. [DOI] [PubMed] [Google Scholar]
- Webb JL, Harvey MW, Holden DW, Evans TJ. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect Immun. 2001;69(10):6391–6400. doi: 10.1128/IAI.69.10.6391-6400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Gonzalez-Billault C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front Cell Neurosci. 2015;9:381. doi: 10.3389/fncel.2015.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Nunez MT, Gonzalez-Billault C. Contribution of NADPH oxidase to the establishment of hippocampal neuronal polarity in culture. J Cell Sci. 2015;128(16):2989–2995. doi: 10.1242/jcs.168567. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Tsang LY, Haworth SG. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L749–L760. doi: 10.1152/ajplung.00361.2004. [DOI] [PubMed] [Google Scholar]
- Xu J, Schwarz WH, Kas JA, Stossel TP, Janmey PA, Pollard TD. Mechanical properties of actin filament networks depend on preparation, polymerization conditions, and storage of actin monomers. Biophys J. 1998;74(5):2731–2740. doi: 10.1016/S0006-3495(98)77979-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Desiderio DM. The human pituitary nitroproteome: detection of nitrotyrosyl-proteins with two-dimensional Western blotting, and amino acid sequence determination with mass spectrometry. Biochem Biophys Res Commun. 2004;325(4):1180–1186. doi: 10.1016/j.bbrc.2004.10.169. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Adolfs Y, Pijnappel WW, Fuller SJ, Van der Schors RC, Li KW, Sugden PH, Smit AB, Hergovich A, Pasterkamp RJ. MICAL-1 is a Negative Regulator of MST-NDR Kinase Signaling and Apoptosis. Mol Cell Biol. 2011a;31(17):3603–3615. doi: 10.1128/MCB.01389-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Gunput RA, Adolfs Y, Pasterkamp RJ. MICALs in control of the cytoskeleton, exocytosis, and cell death. Cell Mol Life Sci. 2011b;68(24):4033–4044. doi: 10.1007/s00018-011-0787-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini D, Caprini G, Pasterkamp RJ, Tedeschi G, Vanoni MA. Kinetic and spectroscopic characterization of the putative monooxygenase domain of human MICAL-1. Arch Biochem Biophys. 2011;515(1–2):1–13. doi: 10.1016/j.abb.2011.08.004. [DOI] [PubMed] [Google Scholar]