

Abstract
The incidence and prevalence of inflammatory bowel disease (IBD) continues to rise with time, signifying its emergence as a global disease. Clinical onset of IBD, comprising Crohn’s disease and ulcerative colitis, typically occurs prior to or at peak reproductive age. Although active disease in female patients is associated with reduced fertility and adverse obstetric outcomes in pregnancy, the molecular mechanisms underlying this altered reproductive course, as well as its impact on IBD transmission to offspring, remain poorly understood. Clinical and experimental studies have now begun to elucidate the hormonal, environmental and microbial factors that modulate immune-reproductive crosstalk in IBD, and define their impact on maternal health, fetal development and heritability of disease risk. Evolving insight into maternal-fetal imprinting in IBD has important implications for patient counseling and disease management during pregnancy, and may help predict clinical outcomes for both mother and child.
Keywords: inflammation, epithelium, mucosa, pregnancy, reproduction, environment, microbiota
Introduction
Inflammatory bowel disease (IBD) defines a spectrum of chronic and debilitating gastrointestinal disorders, with Crohn’s disease (CD) and ulcerative colitis (UC) comprising the predominant entities. While IBD currently affects approximately 2–5 per 1000 people in developed countries, rising prevalence worldwide and increased diagnoses in newly industrialized nations signify its emergence as a truly global disease.1 Disease onset typically occurs at adolescence or early adulthood, thus affecting most patients at their peak reproductive age.
The influence of IBD diagnosis, disease activity, and medical therapy on fertility as well as pregnancy and neonatal outcomes has been the subject of extensive clinical investigation in recent years2. IBD disease activity has consistently been identified as a significant risk factor for adverse outcomes in pregnancy.3, 4 Women with IBD in remission appear to have fertility rates and pregnancy outcomes comparable with the general population. While disease activity does not appear to impact fertility rates in UC, patients with prior restorative proctocolectomy with ileal pouch anal anastomosis (IPAA) have a nearly 4-fold increased rate of infertility5 related to post-surgical pelvic adhesions and associated tubal obstruction. Conversely, population-based studies do indicate slightly increased rates of infertility (5–14%) in the setting of active CD irrespective of surgery,6 supported by evidence of decreased anti-Müllerian hormone (AMH) levels in female patients with active disease.7 Likewise, active IBD at conception or during pregnancy has repeatedly been identified as the most significant risk factor for adverse pregnancy outcomes including spontaneous abortion, preterm and low-weight birth, ischemic placental disease, stillbirth and cesarean delivery.8 As such, patients are counseled to conceive during quiescent stages of disease and to maintain quiescence throughout pregnancy. In addition to the benefits of continued IBD medications during pregnancy, pregnancy itself may have a favorable impact on the clinical course of IBD. Clinical evidence indicates improved IBD symptoms during gestation in a number of patient cohorts,9 with hormonal surges associated with various reproductive stages proposed to play a contributory role.10 Despite the associations of disease activity with infertility and adverse pregnancy outcomes, the molecular mechanisms underlying this association and how these may be influenced by gestational hormones, specific inflammatory pathways, IBD therapies, or other factors remain largely unknown.
Beyond immediate pregnancy outcomes, there is even less known about how maternal disease during gestation may affect the child’s risk for developing IBD. While the precise etiology of IBD is incompletely understood, growing evidence indicates a dysregulated immune response to enteric microbiota in genetically susceptible individuals.11 The familial nature of IBD was first described in the early twentieth century, and genome-wide association studies (GWAS) have to date identified 200 IBD risk loci.12 Having one or more first degree relatives with IBD remains the most significant identified risk factor for IBD, with the lifetime risk to offspring of two affected parents exceeding 30%.13 Despite this, the absolute risk imparted by these variants is uniformly low (<1.3-fold), and do not show complete concordance in monozygotic twin studies. This suggests a profound environmental influence on IBD initiation and progression and such influences are likely to begin as early as in utero or the perinatal period. Interestingly, maternal diet, mode of delivery and breastfeeding can impact initial infant gut microbial colonization and epigenetics, and as such are thought to modify offspring susceptibility to autoimmune disease.14 Likewise, clinical studies have linked IBD susceptibility to active maternal IBD during pregnancy and preterm birth, although underlying mechanisms remain elusive.15
Significant insight into IBD pathogenesis has come from gene-targeted and chemically-induced animal models of intestinal inflammation, which have enabled investigation of the interplay between host immunity, genetics and the associated microbiome. As outlined in a number of recent articles, these in vivo models have also facilitated detailed investigations of immune-reproductive crosstalk in the context of intestinal disease, and defined novel pathways for maternal transmission of IBD risk. This review outlines recent advances in our understanding of the reproductive impacts of IBD, including gender bias and hormonal effects on disease, influence of inflammatory cytokines on fetal inflammation and development, and the impact of maternal nutrition and microbiota on disease in both mother and offspring.
Estrogen signaling and pregnancy factors in IBD
Gender disparity is a defining feature of multiple autoimmune diseases, and IBD is no exception. Both the incidence and severity of CD are reportedly increased in female patients compared to males,16 and female gender is a risk factor for relapse in UC.17 Moreover, female patients experience dichotomous changes in disease symptoms during puberty, pregnancy and menopause. Genome linkage studies have revealed high frequency haplotypes in chromosome X associated with IBD in females, although the specific genetic loci that confer this bias remain elusive.18
Gonadal sex hormones, in particular estrogen, have been proposed as candidate immunomodulatory factors in the gastrointestinal tract. Although estrogen signaling through nuclear estrogen receptors (ERs) is generally considered to promote immunity, correlating with enhanced immune responses against infectious agents in female patients,19 divergent estrogen effects have been reported in other disease settings. Several genetic and bacterial models of IBD in mice display a similar female bias for severe intestinal inflammation20, 21 and studies of estrogen signaling in these systems have revealed cell-specific mechanisms of estrogen-mediated immunomodulation. Exogenous estrogen treatment in Th1-linked experimental colitis has been shown to reduce tissue inflammatory and injury indices, and suppress proinflammatory cytokine expression and mast cell protease levels in an ER-dependent manner.22 Estrogen-mediated blockade of mast cell activation may in part contribute to the reduced neutrophilic infiltrate and tissue damage in these models.22 Helicobacter hepaticus inoculation of ER knockout mice further revealed that functional ERβ signaling in the absence of ERα is sufficient to dampen intestinal inflammation and proinflammatory cytokine expression in bacterially-induced IBD.20 Estrogen signaling through intestinal epithelial cell ERβ supports tight junction development and decreases intestinal permeability, thereby reinforcing the primary cellular mucosal barrier to omnipresent gut microbes.23 Furthermore, ERβ mRNA levels are reduced in colonic biopsies from IBD patients and IL-10 knockout (IL-10−/−) mice, with increased colonic permeability preceding the onset of colitis in this genetic mouse model.24 More recently, studies in the gene-targeted SAMP1/YitFc (SAMP) model of CD-like ileitis revealed a deficit in T-regulatory cell (Treg) frequency and suppressive function specifically in females as compared to males.21 Within the mucosal immune system, Tregs orchestrate tolerance to self-antigens and dampen deleterious immune responses, thereby maintaining normal gut homeostasis.25 Estrogen treatment expanded gut-associated lymphoid tissue Treg frequency and attenuated ileitis exclusively in male mice, suggesting both that estrogen signaling differentially influences T-cell proliferation and conversion in male versus female mice, and that a functional impairment of mucosal Tregs in females contributes to accelerated, more severe intestinal disease.21
Pregnancy elicits beneficial effects in a number of autoimmune disorders such as rheumatoid arthritis and multiple sclerosis, and clinical symptoms of IBD are reported to improve during gestation. In addition to gonadal hormones, other candidate immunomodulating factors have recently emerged that likely contribute to attenuated intestinal inflammation during gestation. Pregnancy-specific glycoproteins (PSG) are a family of Ig-like soluble proteins that are secreted by placental syncytiotrophoblasts and are present in the maternal circulation at high concentrations throughout pregnancy.26 PSGs play a multifaceted immunoregulatory role in promoting fetal tolerance, and low serum PSG1 levels in the first trimester correlate with adverse pregnancy outcomes, including preterm delivery and small birth weight.27 Recent work showed that PSG1 administration is profoundly protective in DSS-induced experimental colitis, as evidenced by reduced weight loss, abrogated colonic leukocyte infiltrate and absence of bloody stool in PSG1-treated mice compared to controls. PSG1 was found to repress colonic expression of Th1 proinflammatory mediators, and to increase the frequency of LAP- and FoxP3-expressing CD4+ Tregs in colonic lamina propria. PGS1-mediated protection is elicited in large part through activation of TGF-β, an important modulator of intestinal tolerance, as these effects were inhibited in the presence of neutralizing anti-TGF-β antibody.28 TGF-β plays a prominent immunomodulatory role during pregnancy, suppressing CD8+ T-cell and NK cell cytotoxic functions and driving differentiation of extrathymic Tregs to promote maternal tolerance to paternal antigens expressed by the developing fetus.29 Interestingly, clinical studies conducted in women with primary ovarian insufficiency associated with the chromosome X monosomy Turner syndrome revealed a significantly increased prevalence of IBD and autoimmune disease compared with control populations, commensurate with lower anti-inflammatory TGF-β2 levels and lower lifetime estrogen exposure.30 This work further implies that haploinsufficiency for chromosome X-linked genes likely plays a prominent role in immune dysfunction and the pathogenesis of IBD.
Maternal supplementation in IBD
Vitamin D
The role of vitamin D in human immunologic and reproductive health has garnered significant interest in recent years. Low serum 25-hydroxy vitamin D [25(OH)D] levels have been described in myriad disorders and global trends indicate an increasing prevalence of vitamin D insufficiency (serum 25(OH)D of 21–29ng/ml) or deficiency (serum 25(OH)D less than 20ng/ml) in both pediatric and adult populations.31 In patients with CD, vitamin D deficiency may precede diagnosis and increase disease risk, while low serum levels in established disease are associated with a more aggressive clinical course and increased risk of surgery.32 GWAS have identified several genetic variants that influence circulating vitamin D levels, and recent work confirmed an association of these loci with vitamin D deficiency in IBD.33 Given the reported prevalence of vitamin D deficiency in IBD, an interesting correlate is the impact of vitamin D on reproductive health. High levels of vitamin D and its metabolites in the decidua during early pregnancy indicate an important role in implantation, and clinical pregnancy and live-birth rates were lower among vitamin D–deficient recipients than among vitamin D–replete recipients in an egg donation program, suggesting an influence on pregnancy, mediated via the endometrium.34 Vitamin D deficiency is highly prevalent during pregnancy35 and has been associated with increased risk of obstetrical complications such as low birth weight, preterm delivery and recurrent pregnancy loss.36
Much of our understanding of the role of vitamin D in intestinal health and inflammation has derived from studies of supplementation and transgenic receptor knockout in mouse IBD models. Early work employing the IL-10−/− mouse model of spontaneous enterocolitis demonstrated that vitamin D supplementation markedly ameliorated clinical symptoms and disease progression in mice with established disease, while vitamin D deficiency induced accelerated weight loss and mortality.37 Similarly, genetic deletion of the vitamin D receptor (VDR) exacerbated intestinal inflammation in both IL-10−/− and CD45RB T-cell transfer murine models of experimental colitis, coincident with thymic atrophy in VDR/IL-10 double knockout animals.38 Profiling of VDR/IL-10 knockout mice revealed marked alterations in lymphoid organs, reduced proliferative capacity of CD4+ T-cells and increased local levels of proinflammatory cytokines such as IL1-β, IFNγ and TNFα. As such, vitamin D signaling promotes thymocyte survival and function by blunting systemic inflammatory responses in this experimental model.39 Further interrogation of T-cell function in VDR deficiency alone demonstrated that VDR expression on T-cells mediates their homing to the gut40 and is critical for induction of Tregs, suppression of Th17 cells41 and development of invariant natural killer (NK) T-cells.40 Given the independent associations of vitamin D signaling with both IBD and reproductive outcomes, as well as the prevalence of vitamin D deficiency in these patient populations, it stands to reason that vitamin D deficiency likely carries important clinical consequences for IBD patients who are pregnant or trying to conceive.
Intriguingly, endometriosis provides another correlation between IBD, vitamin D signaling, and reproductive health. Endometriosis is characterized by the growth of ectopic endometrial tissue outside the uterine cavity, and has long been associated with significantly increased rates of infertility42. A defect in immune-mediated clearance of refluxed menstrual debris has been proposed to contribute to the pathogenesis of endometriosis, and recent work revealed that local uterine NK cell maturation is significantly impaired in female patients with endometriosis-associated infertility.43 Eutopic endometrial tissue from subjects with endometriosis also demonstrates enhanced proliferative capacity and increased angiogenesis, a phenotype that likely promotes ectopic endometrial cell invasion and persistence in the peritoneal cavity.44 Interestingly, a large European cohort of 37,600 women with a prior diagnosis of endometriosis revealed a significantly increased likelihood of a subsequent diagnosis of either Crohn’s disease or ulcerative colitis.45 Likewise, among Crohn’s patients, a confirmed diagnosis of endometriosis at surgery has been associated with an increased risk of a stricturing phenotype.46 Lastly, several studies utilizing murine surgical models of endometriosis have shown that vitamin D and the VDR agonist elocalcitol can repress the growth of endometriotic lesions by inhibiting neovascularization and attenuating levels of inflammatory cytokines and matrix metalloproteinase.47, 48 Dysregulation of some vitamin D enzymes and receptors has also been shown in the endometrium of women with endometriosis.49 More recently, a large prospective clinical study demonstrated an inverse correlation between serum 25(OH)D levels and endometriosis, supporting the hypothesis that vitamin D insufficiency or deficiency is associated with increased risk of disease50.
Folic acid and other methyl donor micronutrients
Growing epidemiological and clinical evidence indicates that both genetic factors and diverse environmental modulators such as geographical location and diet underlie the evolving IBD burden worldwide.51 Dynamic responses to such extrinsic stimuli can be elicited through epigenetic changes, meiotically and mitotically heritable molecular modifications that modulate gene expression without altering DNA sequence. Epigenetic alterations include chromatin re-arrangement, histone modifications and DNA methylation of cytosine residues. Methylation patterns established during embryogenesis can be modified by shifts in gut microbiota or oxidative stress during intestinal inflammation. Of note, cellular methylation has been associated with inflammatory metabolic signatures in mouse models of IBD.52 Epigenetic changes are now also appreciated to occur in utero and via effects on the paternal epigenome. The DNA methylation pathway is influenced by dietary micronutrients that serve as cofactors or methyl donors such as betaine, vitamin B12, choline and folate, many of which are included in periconceptional/prenatal supplements. As such, there is growing interest in the impact of maternal nutrition on developmental epigenomic programming in utero and disease risk in later life. Patients with IBD exhibit impaired uptake of folic acid, and therapeutic intervention with 5-aminosalicyclic acid compounds can further attenuate folate uptake and synthesis.53 A recent study in mice showed that dietary folic acid mediates methylation of the carcinoembryonic antigen-related cell-adhesion molecule 6 (CEACAM6) gene promoter.54 Low levels of dietary folate were found to enhance CEACAM6 expression on gut epithelia, thus favoring the colonization of IBD-associated adherent-invasive Escherichia coli (AIEC).
Limited studies in mouse IBD models have begun to investigate the epigenetic and phenotypic changes elicited by maternal methyl donor supplementation. Maternal dietary exposure to methyl donor micronutrients throughout gestation and lactation was found to induce persistent changes in offspring colonic mucosal DNA methylation status compared to non-supplemented controls.55 Select genomic regions were found in this study to be epigenetically responsive, including the IBD-linked gene locus Ptpn22,56 with over 50% of hypomethylated regions located on chromosome X. Maternal supplementation similarly induced persistent changes in colonic mucosal gene expression. Microarray profiling revealed specific enrichment of genes involved in immune responses in methyl donor-exposed offspring compared to control groups. Strikingly, this epigenomic reprogramming was associated with a markedly increased susceptibility to DSS-induced colitis that persisted through pediatric development to young adulthood. Extension of these studies revealed that methyl donor supplementation during the prenatal period alone was sufficient to augment IBD risk, highlighting gestation as the critical developmental period for epigenetic changes that imprint sensitivity to mucosal injury, at least in this DSS model.57 Interestingly, prenatal methyl donor exposure was also shown to induce changes in gut microbial composition independent of postnatal maternal colonization, resulting in a colitogenic microbiome. This microbial dysbiosis is attributed, at least in part, to methyl donor-derived epigenetic changes in the host that modulate mucosal immune development and function.57 While this work has intriguing implications for the developmental origins of nutritional imprinting and IBD risk, further studies are needed to unravel how host genetics, environment and the microbiota interact to shape the mucosal epigenetic landscape over time.
Tumor Necrosis Factor (TNF)
While the contribution of TNF to an altered reproductive course has recently been posited, the critical role of this proinflammatory cytokine in IBD pathogenesis is well established, comprising effects on mucosal barrier integrity, immune cell recruitment and granuloma formation. Secretion of TNF from both immune and epithelial compartments58 and subsequent receptor (TNFR) binding activates proinflammatory signaling cascades that impact T-cell, neutrophil and macrophage responses, tight junction remodeling, endothelial adhesion molecule expression and intestinal epithelial apoptosis.59 Levels of this mediator are significantly elevated in the serum, stool and mucosal tissue of IBD patients60. The TNF gene is located within the IBD3 locus on chromosome 6p21.1-23, a region that has been consistently implicated in linkage studies for both Crohn’s disease and ulcerative colitis. Studies in a zebrafish model of intestinal inflammation implicate hypomethylation in the TNF gene promoter and resultant high TNF levels as a risk factor for IBD.61 Polymorphisms within the TNF promoter have also been reported to alter cytokine expression62 and have been independently linked to increased risk for IBD63 and preterm birth64 in certain patient populations. Similarly, elevated serum TNF-α levels persisting through the second trimester of pregnancy were associated with preterm delivery in a small cohort of IBD patients.65 Anti-TNF monoclonal antibodies are increasingly used in clinical practice to treat IBD, and their use in the third trimester of pregnancy contraindicates the use of live vaccines in immunosuppressed neonates until levels are undetectable in infant circulation (up to 7 months postpartum).66 As such, critical questions arise regarding the impact of TNF and anti-TNF biologics on fetal and neonatal immune development.
Animal models of IBD have highlighted a multifaceted role of TNF in intestinal pathology and homeostasis, reproductive health and IBD susceptibility in offspring. TNF neutralization effectively ameliorates intestinal inflammation in many models, but has been shown to exacerbate acute mucosal injury in DSS colitis,59 indicating that TNF may function as a critical cue for epithelial barrier repair following acute damage or inflammation. Studies with gene-targeted TNF knockout mice have further demonstrated a more severe disease phenotype in DSS or IL-10−/− colitis models in the absence of TNF.67, 68 Interestingly, fertility and reproductive problems were also reported in female mice deficient for both TNF and IL-10 (Tnf−/−:Il-10−/−), but not in mice harboring single deletions.67 The pathogenic role of TNF overproduction in IBD has been successfully modelled using the TNFΔAU-rich element (TNFΔARE) mouse, which displays chronic elevated TNF levels and spontaneously develops a progressive, CD8+ T-cell dependent Crohn’s-like ileitis.69 Recent work adopted this TNFΔARE model to assess the impact of chronic, TNF-driven maternal inflammation in utero on postnatal IBD development and severity in normal and genetically-susceptible offspring.70 While ileal pathology of adult healthy and TNFΔARE offspring was found to be unaffected by maternal inflammation, gene expression profiling revealed a significant influence of maternal disease on fetal epithelial gene signatures. Strikingly, similar patterns of gene regulation were observed in normal and TNFΔARE progeny exposed in utero to maternal disease, indicating that TNF-driven intestinal inflammation during pregnancy strongly influences the fetal epithelial transcriptional profile, and that fetal genotype does not significantly impact gut programing in early development. In contrast, the adult epithelial transcriptome was substantially modulated by disease phenotype, suggesting that fetal gene signatures can be overwritten by signals derived from the postnatal environment.70
Extensions of this work have examined maternal diet as a candidate environmental modulator of disease initiation in the TNFΔARE model.71 The impact of Westernized diet and maternal obesity on offspring metabolic and inflammatory status has been intensely investigated in mice72 and a number of studies have now demonstrated a link between diet-induced obesity and perturbed intestinal immunity and barrier function.73, 74 Maternal exposure to high-fat diet in the nonobese diabetic (NOD) mouse model correlates with altered offspring gut function, including enhanced intestinal permeability, increased oxidative stress and reduced goblet cell density.75 In adult TNFΔARE mice, a high-fat diet was found to accelerate onset of Crohn’s disease-like ileal inflammation,76 with increased endotoxin translocation into portal vein circulation and decreased expression of tight junction marker occludin in distal ileal epithelium. Dendritic cell recruitment and Th17-biased immune responses were also augmented in response to high-fat diet.76 Exposure to maternal high-fat diet during gestation was similarly found to promote early ileitis onset and increase disease severity in TNFARE progeny as compared to control diet-exposed cohorts.71 Enhanced inflammation and tissue damage in early adulthood was commensurate with increased mucosal neutrophil infiltration and proinflammatory cytokine expression, while TNF plasma levels were significantly correlated with portal vein endotoxemia and tissue pathology. Whether increased mucosal barrier permeability in this TNF-driven model results from aggravated intestinal injury or from a direct influence of maternal nutrition remains to be determined. In addition, future cross-fostering and cohousing studies are needed to determine the potential impact of maternally-derived, high-fat diet associated microbiota on disease acceleration in genetically susceptible offspring. However, the implications of this work for IBD and reproduction are significant, given the recent association of obesity with a more severe IBD clinical course and as a potential risk factor for development of CD.77
Microbiota in IBD and reproduction
The mammalian intestinal tract is host to a diverse microbial ecosystem. Commensal gut bacteria produce metabolites that influence barrier function and host energy, and play a central role in shaping immune tolerance and homeostasis78. Three distinct and predominant microbiota enterotypes have been described in diverse human adult populations79 and community structure is strongly influenced by environmental factors such as diet,80 intestinal oxygenation81 and pregnancy.82 Perturbations in commensal diversity and defects in host containment mechanisms and tolerance have been shown to stimulate pathogenic immune responses in IBD.78 In particular, studies of IBD-associated microbiota have identified reduced taxonomic diversity, a decreased abundance of butyrate-producing Firmicutes microrganisms such as Faecalibacterium praunitzii and increased Proteabacteria such as Escherichia coli.83 Our understanding of host-microbial interactions and their role in IBD pathogenesis has been greatly advanced by studies in rodent IBD models and studies of gut microbial community transfer into germ-free mice. Using these tools, recent seminal work has begun to unravel the gut microbial contribution to fetal metabolic and immune programming, defining novel mechanisms for maternal transmission of disease.
Microbiota and IBD: Evidence from mouse models
Compelling evidence for a critical link between commensal microbes and IBD is provided by the absence of intestinal inflammation in germ-free genetically-susceptible mice such as the IL-10−/− model, and the rapid development of disease after bacterial colonization of the gut. Moreover, antibiotic treatment has been shown to prevent disease onset and alleviate established intestinal inflammation in multiple experimental IBD models.78 Transfer of dysbiotic, disease-associated microbiota was recently shown to initiate ileal inflammation and attenuate Paneth cell antimicrobial function in germ-free TNFΔARE mice, while antibiotic treatment reduced disease severity and relapse of established ileitis.84 Gut microbiome profiling studies revealed that prenatal methyl donor exposure induces significant shifts in microbial diversity. Furthermore, fecal microbial transfer from methyl donor-exposed offspring potentiates susceptibility to DSS-induced colitis in germ-free recipients.57 Several gene-targeted mouse models also demonstrate indigenous microbial dysbiosis characteristic of human IBD. Intestinal epithelial VDR knockout mice were found to harbor increased levels of Bacteroides and Escherichia coli and reduced abundance of butyrate-producing microorganisms.85 Cohousing and cross-fostering experiments in mice have further demonstrated that genotype-dependent disease is communicable from knockout animals to healthy hosts via maternally transmitted microbiota.86 These studies reveal a complex interplay between genetics and maternal microbial imprinting, and suggest that both pre- and postnatal factors play a critical role in shaping early gut colonization patterns and host physiology.
Gut microbial influences at the fetal-maternal interface
During pregnancy, the maternal immune system is modulated to transiently tolerate highly immunogenic paternal alloantigens in order to sustain fetal integrity, without compromising systemic immunity that defends both mother and fetus from invasive pathogens.87 A landmark study revealed that the human maternal gut microbiota is also markedly altered between the first and third trimesters of pregnancy, independent of maternal diet and health status.82 First trimester gut microbial composition was reported to resemble that of healthy non-pregnant women, whereas third trimester microbial profiling showed reduced taxonomic diversity and increased abundance of Proteobacteria and Actinobacteria. Interestingly, this microbial shift included depletion of the IBD-linked Firmicutes bacteria Faecalibacterium prausnitzii.83 Transfer of third trimester microbiota to germ-free mice produced metabolic changes similar to aspects of metabolic syndrome in recipient animals, including insulin insensitivity, inflammation and weight gain. In contrast to the long-term health detriments associated with metabolic syndrome and obesity, such pregnancy-elicited metabolic changes are posited to support fetal development and energetic demands of lactation. Third trimester stool samples also exhibited increased markers of inflammation, suggesting that temporal immune and/or hormonal changes during pregnancy may promote a low-grade inflammation at intestinal mucosal surfaces, which reciprocally drives microbial dysbiosis and alters host metabolism.82 An important open question is whether similar modifications of the maternal immune milieu and gut microbial ecosystem occur in pregnant IBD patients, and whether these alterations may underlie reported changes in clinical symptoms or the adverse obstetric outcomes associated with active disease.
The developing fetus and associated placenta has conventionally been considered a sterile unit, with earliest microbial exposure and colonization proposed to occur at birth. Experimental and clinical studies have begun to challenge this paradigm, and indicate that feto-placental immune programming may be modulated indirectly through maternal bacterial exposure or directly via intrauterine contact with microbes or microbial components.88 Maternal mucosal contact with Actinetobacter lwoffi F78 modulates placental toll-like receptor (TLR) expression in an experimental mouse model and attenuates asthma development offspring.89 In the clinical setting, direct microbial colonization of the placenta has been found after infection or premature birth as well as in normal pregnancy and term placentas90 and recent profiling studies of placental microorganisms identified a low-abundance but metabolically rich placental microbiome.91 Live bacteria originating from the skin and intestinal tract were discovered in approximately 50% of 1365 placental samples obtained upon second-trimester, preterm delivery at 23–28 weeks gestation.92 DNA from intestinal commensal microbes Lactobacillus spp. and Bifidobacterium spp. has also been identified in term placentas after vaginal and caesarean delivery.93 Similarly, bacterial DNA is found at term in fetal membranes without concomitant inflammation94 and the presence of microbial DNA in infant meconium has also been reported, implicating a prenatal origin.95 Additional studies confirmed the presence of nonpathogenic species Lactobacillus and Bifidobacterium in term placental and amniotic fluid samples without signs of infection, membrane rupture or onset of labor. Moreover, detection of bacterial DNA in placental samples was associated with alterations in TLR-related innate immune gene expression in the fetal gut.96 Although the source of microbial entry to the amniotic cavity is currently unknown, potential routes include ascension from the birth canal, retrograde spread though the fallopian tubes or hematogenous transmission from distal maternal sites via umbilical cord blood.97 Hematogenous transfer of oral microbiota has been modelled in mice, and indicates that a diverse group of oral bacteria are capable of colonizing the murine placenta.98 Overall, these findings imply that fetal microbial exposure may occur much earlier than previously appreciated, and that maternally-derived microbiota can fundamentally shape immunological and metabolic programming in utero. Understanding the potential impact of IBD-associated dysbiosis on prenatal microbial colonization may help to determine maternal and fetal outcomes, and provide a foundation for novel clinical management strategies.
Conclusions
Symptomatic IBD is associated with adverse effects on fertility and pregnancy outcomes, and the safety of medical intervention throughout conception, pregnancy and lactation is a significant clinical consideration. Although the number of prospective clinical studies focused on the impact of IBD medications during pregnancy is increasing, there is still a marked deficit in our understanding of how active disease elicits immediate pregnancy risks and how maternal disease throughout gestation may impact fetal immune development and lifelong IBD susceptibility. As such, this highlights the need for additional basic mechanistic studies of IBD-associated pathways in immune-reproductive crosstalk. In vivo models of IBD provide a relevant and useful platform to systematically dissect the impact of pregnancy on disease, and the influence of maternal inflammation, nutrition and microbial colonization on fertility, fetal development and neonatal outcomes (Figure 1). Understanding the molecular cues, transcriptional programs and metabolic milieu that regulate maternal-fetal imprinting during intestinal disease will provide substantial insight for patient management during pregnancy and projected clinical outcomes for both mother and child.
Figure 1.
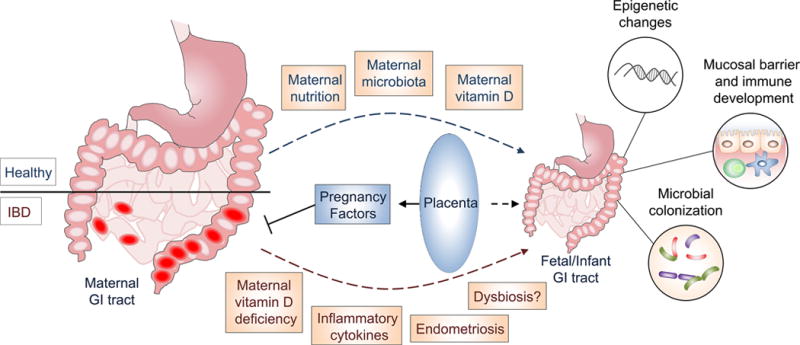
IBD, pregnancy and maternal disease transmission. Active IBD during pregnancy increases the risk of adverse obstetric outcomes. Maternal IBD-associated factors that are thought to contribute to this risk include vitamin D deficiency and overproduction of proinflammatory mediators such as TNF. Evidence from mouse models indicates that maternal environment markedly impacts fetal immune and metabolic development, fetal epigenomic programming and gut colonization, and fetal exposure to maternal microbes or microbial components likely shapes mucosal immune development and tolerance. Symptomatic maternal disease during pregnancy may therefore also influence the life-long IBD susceptibility of offspring. Conversely, pregnancy-elicited hormonal surges and placental-derived immunomodulatory pregnancy glycoproteins can modulate disease though altered immune and barrier cell function.
Acknowledgments
This work was supported by National Institutes of Health Grant DK103712.
Footnotes
Conflict of interest statement: The authors declare no financial interests in any of the work submitted here.
References
- 1.Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720–7. doi: 10.1038/nrgastro.2015.150. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen OH, Maxwell C, Hendel J. IBD medications during pregnancy and lactation. Nat Rev Gastroenterol Hepatol. 2014;11:116–27. doi: 10.1038/nrgastro.2013.135. [DOI] [PubMed] [Google Scholar]
- 3.Broms G, Granath F, Linder M, et al. Birth outcomes in women with inflammatory bowel disease: effects of disease activity and drug exposure. Inflamm Bowel Dis. 2014;20:1091–8. doi: 10.1097/MIB.0000000000000060. [DOI] [PubMed] [Google Scholar]
- 4.Norgard B, Hundborg HH, Jacobsen BA, et al. Disease activity in pregnant women with Crohn’s disease and birth outcomes: a regional Danish cohort study. Am J Gastroenterol. 2007;102:1947–54. doi: 10.1111/j.1572-0241.2007.01355.x. [DOI] [PubMed] [Google Scholar]
- 5.Rajaratnam SG, Eglinton TW, Hider P, et al. Impact of ileal pouch-anal anastomosis on female fertility: meta-analysis and systematic review. Int J Colorectal Dis. 2011;26:1365–74. doi: 10.1007/s00384-011-1274-9. [DOI] [PubMed] [Google Scholar]
- 6.van der Woude CJ, Ardizzone S, Bengtson MB, et al. The second European evidenced-based consensus on reproduction and pregnancy in inflammatory bowel disease. J Crohns Colitis. 2015;9:107–24. doi: 10.1093/ecco-jcc/jju006. [DOI] [PubMed] [Google Scholar]
- 7.Senates E, Colak Y, Erdem ED, et al. Serum anti-Mullerian hormone levels are lower in reproductive-age women with Crohn’s disease compared to healthy control women. J Crohns Colitis. 2013;7:e29–34. doi: 10.1016/j.crohns.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut. 2007;56:830–7. doi: 10.1136/gut.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riis L, Vind I, Politi P, et al. Does pregnancy change the disease course? A study in a European cohort of patients with inflammatory bowel disease. Am J Gastroenterol. 2006;101:1539–45. doi: 10.1111/j.1572-0241.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- 10.Mogadam M, Korelitz BI, Ahmed SW, et al. The course of inflammatory bowel disease during pregnancy and postpartum. Am J Gastroenterol. 1981;75:265–9. [PubMed] [Google Scholar]
- 11.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–86. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon H, Trier Moller F, Andersen V, et al. Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflamm Bowel Dis. 2015;21:1428–34. doi: 10.1097/MIB.0000000000000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawrylowicz C, Ryanna K. Asthma and allergy: the early beginnings. Nat Med. 2010;16:274–5. doi: 10.1038/nm0310-274. [DOI] [PubMed] [Google Scholar]
- 15.Sonntag B, Stolze B, Heinecke A, et al. Preterm birth but not mode of delivery is associated with an increased risk of developing inflammatory bowel disease later in life. Inflamm Bowel Dis. 2007;13:1385–90. doi: 10.1002/ibd.20206. [DOI] [PubMed] [Google Scholar]
- 16.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(Suppl 1):S3–9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 17.Bitton A, Peppercorn MA, Antonioli DA, et al. Clinical, biological, and histologic parameters as predictors of relapse in ulcerative colitis. Gastroenterology. 2001;120:13–20. doi: 10.1053/gast.2001.20912. [DOI] [PubMed] [Google Scholar]
- 18.Saruta M, Targan SR, Mei L, et al. High-frequency haplotypes in the X chromosome locus TLR8 are associated with both CD and UC in females. Inflamm Bowel Dis. 2009;15:321–7. doi: 10.1002/ibd.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol. 2008;8:737–44. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cook LC, Hillhouse AE, Myles MH, et al. The role of estrogen signaling in a mouse model of inflammatory bowel disease: a Helicobacter hepaticus model. PLoS One. 2014;9:e94209. doi: 10.1371/journal.pone.0094209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman WA, Garg RR, Reuter BK, et al. Loss of estrogen-mediated immunoprotection underlies female gender bias in experimental Crohn’s-like ileitis. Mucosal Immunol. 2014;7:1255–65. doi: 10.1038/mi.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harnish DC, Albert LM, Leathurby Y, et al. Beneficial effects of estrogen treatment in the HLA-B27 transgenic rat model of inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2004;286:G118–25. doi: 10.1152/ajpgi.00024.2003. [DOI] [PubMed] [Google Scholar]
- 23.Wada-Hiraike O, Imamov O, Hiraike H, et al. Role of estrogen receptor beta in colonic epithelium. Proc Natl Acad Sci U S A. 2006;103:2959–64. doi: 10.1073/pnas.0511271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Looijer-van Langen M, Hotte N, Dieleman LA, et al. Estrogen receptor-beta signaling modulates epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. 2011;300:G621–6. doi: 10.1152/ajpgi.00274.2010. [DOI] [PubMed] [Google Scholar]
- 25.Himmel ME, Hardenberg G, Piccirillo CA, et al. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunology. 2008;125:145–53. doi: 10.1111/j.1365-2567.2008.02939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou GQ, Baranov V, Zimmermann W, et al. Highly specific monoclonal antibody demonstrates that pregnancy-specific glycoprotein (PSG) is limited to syncytiotrophoblast in human early and term placenta. Placenta. 1997;18:491–501. doi: 10.1016/0143-4004(77)90002-9. [DOI] [PubMed] [Google Scholar]
- 27.Pihl K, Larsen T, Laursen I, et al. First trimester maternal serum pregnancy-specific beta-1-glycoprotein (SP1) as a marker of adverse pregnancy outcome. Prenat Diagn. 2009;29:1256–61. doi: 10.1002/pd.2408. [DOI] [PubMed] [Google Scholar]
- 28.Blois SM, Sulkowski G, Tirado-Gonzalez I, et al. Pregnancy-specific glycoprotein 1 (PSG1) activates TGF-beta and prevents dextran sodium sulfate (DSS)-induced colitis in mice. Mucosal Immunol. 2014;7:348–58. doi: 10.1038/mi.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. 2004;5:266–71. doi: 10.1038/ni1037. [DOI] [PubMed] [Google Scholar]
- 30.Bakalov VK, Gutin L, Cheng CM, et al. Autoimmune disorders in women with turner syndrome and women with karyotypically normal primary ovarian insufficiency. J Autoimmun. 2012;38:315–21. doi: 10.1016/j.jaut.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–81. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 32.Ananthakrishnan AN, Cagan A, Gainer VS, et al. Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn’s disease. Inflamm Bowel Dis. 2013;19:1921–7. doi: 10.1097/MIB.0b013e3182902ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ananthakrishnan AN, Cagan A, Cai T, et al. Common Genetic Variants Influence Circulating Vitamin D Levels in Inflammatory Bowel Diseases. Inflamm Bowel Dis. 2015;21:2507–14. doi: 10.1097/MIB.0000000000000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudick BJ, Ingles SA, Chung K, et al. Influence of vitamin D levels on in vitro fertilization outcomes in donor-recipient cycles. Fertil Steril. 2014;101:447–52. doi: 10.1016/j.fertnstert.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Vandevijvere S, Amsalkhir S, Van Oyen H, et al. High prevalence of vitamin D deficiency in pregnant women: a national cross-sectional survey. PLoS One. 2012;7:e43868. doi: 10.1371/journal.pone.0043868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ota K, Dambaeva S, Han AR, et al. Vitamin D deficiency may be a risk factor for recurrent pregnancy losses by increasing cellular immunity and autoimmunity. Hum Reprod. 2014;29:208–19. doi: 10.1093/humrep/det424. [DOI] [PubMed] [Google Scholar]
- 37.Cantorna MT, Munsick C, Bemiss C, et al. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–52. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- 38.Froicu M, Weaver V, Wynn TA, et al. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol. 2003;17:2386–92. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 39.Froicu M, Zhu Y, Cantorna MT. Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology. 2006;117:310–8. doi: 10.1111/j.1365-2567.2005.02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu S, Bruce D, Froicu M, et al. Failure of T cell homing, reduced CD4/CD8alphaalpha intraepithelial lymphocytes, and inflammation in the gut of vitamin D receptor KO mice. Proc Natl Acad Sci U S A. 2008;105:20834–9. doi: 10.1073/pnas.0808700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruce D, Yu S, Ooi JH, et al. Converging pathways lead to overproduction of IL-17 in the absence of vitamin D signaling. Int Immunol. 2011;23:519–28. doi: 10.1093/intimm/dxr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buyalos RP, Agarwal SK. Endometriosis-associated infertility. Curr Opin Obstet Gynecol. 2000;12:377–81. doi: 10.1097/00001703-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Thiruchelvam U, Wingfield M, O’Farrelly C. Increased uNK Progenitor Cells in Women With Endometriosis and Infertility are Associated With Low Levels of Endometrial Stem Cell Factor. Am J Reprod Immunol. 2016;75:493–502. doi: 10.1111/aji.12486. [DOI] [PubMed] [Google Scholar]
- 44.Healy DL, Rogers PA, Hii L, et al. Angiogenesis: a new theory for endometriosis. Hum Reprod Update. 1998;4:736–40. doi: 10.1093/humupd/4.5.736. [DOI] [PubMed] [Google Scholar]
- 45.Jess T, Frisch M, Jorgensen KT, et al. Increased risk of inflammatory bowel disease in women with endometriosis: a nationwide Danish cohort study. Gut. 2012;61:1279–83. doi: 10.1136/gutjnl-2011-301095. [DOI] [PubMed] [Google Scholar]
- 46.Lee KK, Jharap B, Maser EA, et al. Impact of Concomitant Endometriosis on Phenotype and Natural History of Inflammatory Bowel Disease. Inflamm Bowel Dis. 2016;22:159–63. doi: 10.1097/MIB.0000000000000577. [DOI] [PubMed] [Google Scholar]
- 47.Mariani M, Vigano P, Gentilini D, et al. The selective vitamin D receptor agonist, elocalcitol, reduces endometriosis development in a mouse model by inhibiting peritoneal inflammation. Hum Reprod. 2012;27:2010–9. doi: 10.1093/humrep/des150. [DOI] [PubMed] [Google Scholar]
- 48.Yildirim B, Guler T, Akbulut M, et al. 1-alpha,25-dihydroxyvitamin D3 regresses endometriotic implants in rats by inhibiting neovascularization and altering regulation of matrix metalloproteinase. Postgrad Med. 2014;126:104–10. doi: 10.3810/pgm.2014.01.2730. [DOI] [PubMed] [Google Scholar]
- 49.Sayegh L, Fuleihan Gel H, Nassar AH. Vitamin D in endometriosis: a causative or confounding factor? Metabolism. 2014;63:32–41. doi: 10.1016/j.metabol.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Harris HR, Chavarro JE, Malspeis S, et al. Dairy-food, calcium, magnesium, and vitamin D intake and endometriosis: a prospective cohort study. Am J Epidemiol. 2013;177:420–30. doi: 10.1093/aje/kws247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng SC, Bernstein CN, Vatn MH, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62:630–49. doi: 10.1136/gutjnl-2012-303661. [DOI] [PubMed] [Google Scholar]
- 52.Kominsky DJ, Keely S, MacManus CF, et al. An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol. 2011;186:6505–14. doi: 10.4049/jimmunol.1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baggott JE, Morgan SL, Ha T, et al. Inhibition of folate-dependent enzymes by non-steroidal anti-inflammatory drugs. Biochem J. 1992;282(Pt 1):197–202. doi: 10.1042/bj2820197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denizot J, Desrichard A, Agus A, et al. Diet-induced hypoxia responsive element demethylation increases CEACAM6 expression, favouring Crohn’s disease-associated Escherichia coli colonisation. Gut. 2015;64:428–37. doi: 10.1136/gutjnl-2014-306944. [DOI] [PubMed] [Google Scholar]
- 55.Schaible TD, Harris RA, Dowd SE, et al. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum Mol Genet. 2011;20:1687–96. doi: 10.1093/hmg/ddr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diaz-Gallo LM, Espino-Paisan L, Fransen K, et al. Differential association of two PTPN22 coding variants with Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2287–94. doi: 10.1002/ibd.21630. [DOI] [PubMed] [Google Scholar]
- 57.Mir SA, Nagy-Szakal D, Dowd SE, et al. Prenatal methyl-donor supplementation augments colitis in young adult mice. PLoS One. 2013;8:e73162. doi: 10.1371/journal.pone.0073162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roulis M, Armaka M, Manoloukos M, et al. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc Natl Acad Sci U S A. 2011;108:5396–401. doi: 10.1073/pnas.1007811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dube PE, Punit S, Polk DB. Redeeming an old foe: protective as well as pathophysiological roles for tumor necrosis factor in inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2015;308:G161–70. doi: 10.1152/ajpgi.00142.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komatsu M, Kobayashi D, Saito K, et al. Tumor necrosis factor-alpha in serum of patients with inflammatory bowel disease as measured by a highly sensitive immuno-PCR. Clin Chem. 2001;47:1297–301. [PubMed] [Google Scholar]
- 61.Marjoram L, Alvers A, Deerhake ME, et al. Epigenetic control of intestinal barrier function and inflammation in zebrafish. Proc Natl Acad Sci U S A. 2015;112:2770–5. doi: 10.1073/pnas.1424089112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson AG, Symons JA, McDowell TL, et al. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci U S A. 1997;94:3195–9. doi: 10.1073/pnas.94.7.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fan W, Maoqing W, Wangyang C, et al. Relationship between the polymorphism of tumor necrosis factor-alpha-308 G>A and susceptibility to inflammatory bowel diseases and colorectal cancer: a meta-analysis. Eur J Hum Genet. 2011;19:432–7. doi: 10.1038/ejhg.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harper M, Zheng SL, Thom E, et al. Cytokine gene polymorphisms and length of gestation. Obstet Gynecol. 2011;117:125–30. doi: 10.1097/AOG.0b013e318202b2ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Katsumi T, Tomita K, Leung PS, et al. Animal models of primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48:142–53. doi: 10.1007/s12016-015-8482-y. [DOI] [PubMed] [Google Scholar]
- 66.Nielsen OH, Loftus EV, Jr, Jess T. Safety of TNF-alpha inhibitors during IBD pregnancy: a systematic review. BMC Med. 2013;11:174. doi: 10.1186/1741-7015-11-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hale LP, Greer PK. A novel murine model of inflammatory bowel disease and inflammation-associated colon cancer with ulcerative colitis-like features. PLoS One. 2012;7:e41797. doi: 10.1371/journal.pone.0041797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noti M, Corazza N, Mueller C, et al. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med. 2010;207:1057–66. doi: 10.1084/jem.20090849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kontoyiannis D, Pasparakis M, Pizarro TT, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–98. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 70.Hemmerling J, Heller K, Hormannsperger G, et al. Fetal exposure to maternal inflammation does not affect postnatal development of genetically-driven ileitis and colitis. PLoS One. 2014;9:e98237. doi: 10.1371/journal.pone.0098237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gruber L, Hemmerling J, Schuppel V, et al. Maternal High-fat Diet Accelerates Development of Crohn’s Disease-like Ileitis in TNFDeltaARE/WT Offspring. Inflamm Bowel Dis. 2015;21:2016–25. doi: 10.1097/MIB.0000000000000465. [DOI] [PubMed] [Google Scholar]
- 72.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 73.Garidou L, Pomie C, Klopp P, et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORgammat and Controls Metabolic Disease. Cell Metab. 2015;22:100–12. doi: 10.1016/j.cmet.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 74.Lam YY, Ha CW, Campbell CR, et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS One. 2012;7:e34233. doi: 10.1371/journal.pone.0034233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xue Y, Wang H, Du M, et al. Maternal obesity induces gut inflammation and impairs gut epithelial barrier function in nonobese diabetic mice. J Nutr Biochem. 2014;25:758–64. doi: 10.1016/j.jnutbio.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gruber L, Kisling S, Lichti P, et al. High fat diet accelerates pathogenesis of murine Crohn’s disease-like ileitis independently of obesity. PLoS One. 2013;8:e71661. doi: 10.1371/journal.pone.0071661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khalili H, Ananthakrishnan AN, Konijeti GG, et al. Measures of obesity and risk of Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2015;21:361–8. doi: 10.1097/MIB.0000000000000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–94. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 79.Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Albenberg L, Esipova TV, Judge CP, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;147:1055–63 e8. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koren O, Goodrich JK, Cullender TC, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–80. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–83. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 84.Schaubeck M, Clavel T, Calasan J, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65:225–37. doi: 10.1136/gutjnl-2015-309333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64:1082–94. doi: 10.1136/gutjnl-2014-307436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garrett WS, Gallini CA, Yatsunenko T, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Munoz-Suano A, Hamilton AB, Betz AG. Gimme shelter: the immune system during pregnancy. Immunol Rev. 2011;241:20–38. doi: 10.1111/j.1600-065X.2011.01002.x. [DOI] [PubMed] [Google Scholar]
- 88.Rautava S, Luoto R, Salminen S, et al. Microbial contact during pregnancy, intestinal colonization and human disease. Nat Rev Gastroenterol Hepatol. 2012;9:565–76. doi: 10.1038/nrgastro.2012.144. [DOI] [PubMed] [Google Scholar]
- 89.Conrad ML, Ferstl R, Teich R, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med. 2009;206:2869–77. doi: 10.1084/jem.20090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stout MJ, Conlon B, Landeau M, et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol. 2013;208:226 e1–7. doi: 10.1016/j.ajog.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aagaard K, Ma J, Antony KM, et al. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6:237ra65. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Onderdonk AB, Delaney ML, DuBois AM, et al. Detection of bacteria in placental tissues obtained from extremely low gestational age neonates. Am J Obstet Gynecol. 2008;198:110 e1–7. doi: 10.1016/j.ajog.2007.05.044. [DOI] [PubMed] [Google Scholar]
- 93.Satokari R, Gronroos T, Laitinen K, et al. Bifidobacterium and Lactobacillus DNA in the human placenta. Lett Appl Microbiol. 2009;48:8–12. doi: 10.1111/j.1472-765X.2008.02475.x. [DOI] [PubMed] [Google Scholar]
- 94.Steel JH, Malatos S, Kennea N, et al. Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr Res. 2005;57:404–11. doi: 10.1203/01.PDR.0000153869.96337.90. [DOI] [PubMed] [Google Scholar]
- 95.Mshvildadze M, Neu J, Shuster J, et al. Intestinal microbial ecology in premature infants assessed with non-culture-based techniques. J Pediatr. 2010;156:20–5. doi: 10.1016/j.jpeds.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rautava S, Collado MC, Salminen S, et al. Probiotics modulate host-microbe interaction in the placenta and fetal gut: a randomized, double-blind, placebo-controlled trial. Neonatology. 2012;102:178–84. doi: 10.1159/000339182. [DOI] [PubMed] [Google Scholar]
- 97.Jimenez E, Fernandez L, Marin ML, et al. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol. 2005;51:270–4. doi: 10.1007/s00284-005-0020-3. [DOI] [PubMed] [Google Scholar]
- 98.Fardini Y, Chung P, Dumm R, et al. Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect Immun. 2010;78:1789–96. doi: 10.1128/IAI.01395-09. [DOI] [PMC free article] [PubMed] [Google Scholar]