

Abstract
Krabbe’s disease is a leukodystrophy due to the deficiency of galactosyl-ceramidase and the accumulation of galactosyl-sphingosine (psychosine) in the nervous system. Psychosine is believed to cause central demyelination by killing oligodendrocytes. Quantitative analysis of this process is lacking. To address this, we generated a new transgenic reporter twitcher line where myelinating oligodendrocytes are genetically marked by the expression of LacZ under the control of the myelin basic protein (MBP) promoter. MBP-LacZ-twitcher transgenic mice were used for unbiased stereological quantification of β-galactosidase(+) oligodendrocytes in the spinal cord. As expected, we found decreased numbers of these cells in mutant cords, paralleling the severity of clinical disease. Interestingly, the decrease of oligodendrocytes does not correlate well with the increase of psychosine. The new MBP-LacZ-twitcher line will be a useful genetic tool for measuring changes in oligodendrocyte numbers in different regions of the mutant central nervous system (CNS) and in pre-clinical trials of therapies to prevent demyelination.
Keywords: oligodendrocytes, demyelination, Krabbe’s disease, psychosine
Graphical Abstract
Staining for the expression of β-galactosidase revealed a noticeable reduction of β-galactosidase+ oligodendrocytes in coronal sections from P15 MBP-LacZ-Twitcher spinal cord.
Introduction
Krabbe’s disease is an inherited leukodystrophy caused by the deficient activity of the lysosomal enzyme galactosylceramidase (GALC) and the progressive accumulation of the lipid-raft associated sphingolipid galactosylsphingosine (psychosine), (Igisu and Suzuki 1984; Wenger et al. 2000; White et al. 2009). First described by Knud Krabbe in 1916 (Krabbe 1916), demyelination is diffuse, involving most myelinated circuits in the brain and spinal cord. The twitcher mouse is the short lived authentic murine model for Krabbe’s disease (Duchen et al. 1980), with minimal or no demyelination during the first two weeks of life (Nagara et al. 1982). However, as twitcher mice age and accumulate psychosine (Shinoda et al. 1987), diffuse central demyelination becomes evident after the third week of postnatal life (Nagara et al. 1982; Takahashi and Suzuki 1984; Tanaka et al. 1989).
Demyelination has been interpreted as the consequence of massive oligodendrocyte death (Jatana et al. 2002; Taniike et al. 1999; Zaka and Wenger 2004). The study of Taniike and collaborators is specially important because it clearly shows how oligodendrocytes die of apoptosis during disease [taniike 1989]. However, the underpinning mechanism of demyelination remains incompletely characterized. This is particularly intriguing since reports depict segmental demyelination affecting the nervous system of infantile cases vs thinning (a sign of hypomyelination) in adult onset forms of the disease (Bischoff and Ulrich 1969; Lake 1968; Matsumoto et al. 1996; Sabatelli et al. 2002). In vitro, twitcher oligodendrocytes develop abnormal cellular processes and atrophic myelin-like membranes (Costantino-Ceccarini et al. 1999; Ida et al. 1990; LeVine and Torres 1992), defects attributed to the increased accumulation of psychosine in the membrane (Ida et al. 1990).
How psychosine affects oligodendrocytes is not completely clear but it appears to involve the deregulation of phospholipase A2 (Giri et al. 2006), AP-1 (Giri et al. 2002; Haq et al. 2003), and peroxisomes (Khan et al. 2005). We have shown that psychosine exerts most of its pathogenic defects via its preferential enrichment in lipid rafts (White et al. 2009). This effect may have important consequences on local membrane deformation and downstream signaling (Simons and Gruenberg 2000; White et al. 2011; White et al. 2009). Noteworthy, physicochemical raft alterations may also exert side effects perturbing the surrounding non-raft membrane physiology (Fessler and Parks 2011; Williamson and Sutherland 2011). Thus, the presence of local increases of psychosine may be key to initiate a cascade of effects leading to myelin disassembly and oligodendrocyte death.
To assist in future mechanistic studies of oligodendrocyte loss and demyelination, we have generated various reporter twitcher lines to genetically identify oligodendrocytes by means of histochemical (β-galactosidase) or fluorescent (green-fluorescent protein) marking. This study presents the first of these transgenic lines where mutant oligodendrocytes are identified by the expression of β-galactosidase under the transcriptional control of the MBP promoter transgenic line 18.2 (Wrabetz et al. 1998). The human 750bp MBP promoter was selected originally to explore what was the minimal length of human MBP promoter to maintain properly regulated expression of a reporter gene in amplitude and location in developing brain. The 18-2 line was most advantageous line for the twitcher study because 1) its expression is extinguished well after the full lifespan of twitcher mice, 2) the level of β-galactosidase activity produces mainly cell body staining and not process staining, and 3) immunohistochemistry showed a one-to-one correspondence between immunohistochemistry for β-galactosidase and the olidogendrocyte markers MBP and PLP (Wrabetz et al. 1998). Our experiments focused on the spinal cord, for simplicity of anatomical structure and therefore, stereological counting. We show that this new reporter line correctly identifies myelinating oligodendrocytes, allowing for unbiased stereological quantification of changes in cell numbers during disease.
MATERIALS AND METHODS
Animals
twitcher heterozygous breeders were housed under standard vivarium conditions and their homozygous progeny identified by PCR (Sakai et al. 1996). twitcher heterozygous mice were crossed with mice carrying the MBP-LacZ transgene (Wrabetz et al. 1998) to obtain double heterozygotes (MBP-LacZ+/−/twitcher+/−). Homozygous MBP-LacZ-twitcher (MBP-LacZ+/−/ twitcher+/+) pups were identified by PCR (Sakai et al. 1996; Wrabetz et al. 1998). Animal work was performed as described in protocols approved by the appropriate Institutional Animal Care and Use Committees.
Clinical Score
Mice were scored by calculating an arbitrary score system ranging from 0=normal to 8=moribund. Scores were calculated three times per week by an operator blind to the genotype. Body weight, twitching, body tremor, and ataxic gait were measured. Individual scores assigned as follows: net loss of weight 0.1–0.5g=+0.5, 0.6–1.0g=+1, 1.1–2g=+1.5; >2g=2; visible tremor (twitching)=+1, and ataxic gait= waddling=+1; dragging one leg=+1.5; unable to ambulate=+2.
Glia Cell Cultures
Oligodendrocyte cells were cultured as described (Bongarzone et al. 1996). Briefly, brain cortices from newborn (0 to 3 day-old) pups were dissociated in DMEM/F12 with 10% fetal calf serum and passed through a nylon mesh. Cells were collected and poured sequentially through two collector tissue sieves (230 μm and 140 μm pore size, respectively). Cells were resuspended in cell medium at 5×105 cells/ml and plated on poly-L-lysine coated coverslips. Culture medium was replenished every 4 days. Cultures were maintained for up to 21 days.
Tissue collection, and β-galactosidase (β-gal) histochemistry
Mice were anesthetized with isofluorane and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA)-PBS. Spinal cords were postfixed with 4% PFA for 2 h, cryoprotected in 30% sucrose overnight, embedded in OCT (Tissue-Tek, Sakura Finetek USA, inc., Torrance, CA, USA) and sectioned on a cryotome at 50 μm thickness. Coronal sections from cervical, thoracic and lumbar segments were air-dried, rinsed in PBS and stained in 0.8mg/ml X-Gal in 35 mM K3Fe(CN)6 35 mM K4Fe(CN)6 2 mM MgCl2 PBS at 37°C for 2 hours. Sections were dehydrated through serial graded ethanol, cleared in xylenes, and coverslipped with Permount. In some experiments, sections of spinal cord were postfixed in 4% PFA 1% glutaraldehyde-PBS and embedded in araldite. Plastic ultrathin (90 nm) sections were prepared and stained with toluidine blue.
Stereology
For unbiased stereological studies, 50-μm-thick spinal cord cross-sections were selected at intervals of 10 sections and stained. Quantification of positive cell markers was performed with design-based stereology system (StereoInvestigator version 8, MBF Bioscience, Williston, VT, USA). Spinal cord ventral or dorsal white matter was traced under 5X objective and all cell markers were counted under 63X objective (Zeiss AX10 microscope, Carl Zeiss Ltd., Hertfordshire, England). The sampling parameters were set up according to the software guide to achieve a coefficient of error ranging from 0.09 to 0.12 (Gundersen test), using a counting frame size of 100×100 μm, optical dissector height 20 μm, and an average of 10 sampling sites per section.
Immunocytochemistry
Cell cultures were maintained for 7, 14 and 21 days before fixation. β-gal activity was detected as described above. Cultures were washed with PBS, then stained using monoclonal antibodies A2B5, O4, anti-O1 (kind gifts from Dr Anthony Gard, University of Alabama), and fluorescently labeled using Alexa-488-secondary antibodies. Samples were photographed using a Leica upright microscope.
β-gal Enzyme Assay
Brainstem and spinal cord tissues were homogenized in 250 mM Tris buffer (pH 7.8) and centrifuged at 15,000 rpm for 15 min at 4 ºC. β-gal assay was performed using a modified o-nitrophenyl-β-D-galactopyranoside substrate technique as described (Wrabetz et al. 1998). Fifty μl protein supernatants were incubated with a 1.75 ml reaction solution (3.5 mg/ml ONPG, 100 mM Na2HPO3, 2 mM MgCl2, 50 mM β-mercaptoethanol) for 1–3 hours at 37 ºC. The reaction was terminated by 1M Na2CO3 and absorbance was measured at 420 nm. Standard curves were prepared from commercially synthesized β-galactosidase (Sigma). Units of β-gal activity were expressed as nmol of ONPG hydrolyzed per minute per milligram protein.
Reverse-Transcriptase PCR Analysis
RNA was isolated from tissues using CsCl2 gradient centrifugation as described (Chirgwin et al. 1979). cDNA was synthesized using Superscript cDNA synthesis kit (Invitrogen). cDNA product was treated with RNAse to remove remaining RNA and amplified using Taqman one step real-time PCR master mix (Invitrogen). The following primers were used for MBP: 5′-CACAGAAGAGACCCTCACAGCGACA-3′ and 5′-CCGCTAAAGAAGCGCCCG ATGGA-3′; GAPDH: 5′-GTATGACTCTACCCACGG-3′ and 5′-GTTCAGCTCTG GGATGAC-3′; 94 ºC for 30 s, 50 ºC for 60 s, and 72 ºC for 60 s.
Mass Spectrometry
Spinal cord samples were weighed, homogenized, and psychosine was extracted, and analyzed using liquid chromatography-tandem mass spectrometry, as described (Galbiati et al. 2007). In brief, samples were extracted with chloroform and methanol and then partially purified on a strong cation exchanger column. Dissolved residue was analyzed using liquid chromatography tandem mass spectrometry (LC-MS/MS). Psychosine was measured using a Waters XTerra 3.5 μm, MS C18, 2.1 × 100 mm analytical column. Positive ion electrospray tandem mass spectrometry was performed using an Applied Biosystems (Foster City, CA) API 4000 triple quadrupole mass spectrometer.
Statistical analysis
Results are the mean ± statistical error from three to five different experiments. Data were analyzed by the Student’s t test or ANOVA where appropriate. p values <0.05 were considered statistically significant.
RESULTS
Twitcher mice were crossed with the 18.2 MBP-LacZ line (Wrabetz et al. 1998) to generate a GALC deficient (MBP-LacZ-twitcher) line where oligodendrocytes could be identified by the expression of a reporter gene (β-gal), and used for unbiased analysis of oligodendrocyte abundance.
One benefit of using the 18.2MBP-LacZ transgenic is that it allows marking of only myelinating oligodendrocytes but not immature oligodendrocyte progenitors (OPCs). Figure 1A–1C shows that OPCs (identified by labeling with monoclonal antibody A2B5) isolated from MBP-LacZ-twitcher pups did not express detectable levels of β-gal (identified by β-gal histochemical staining). In contrast, intermediate O4+ and mature O1+ oligodendrocytes were readily identified by β-gal staining (Figure 1D–1I). β-gal staining showed that β-gal protein was restricted to the cell body of oligodendrocytes and minimally present in cellular processes or myelin-like membranes. This genetic marking simplifies the labeling and identification of the cell body of myelinating oligodendrocytes for stereological quantification.
Figure 1. LacZ expression in oligodendrocytes from the MBP-LacZ-twitcher mouse.
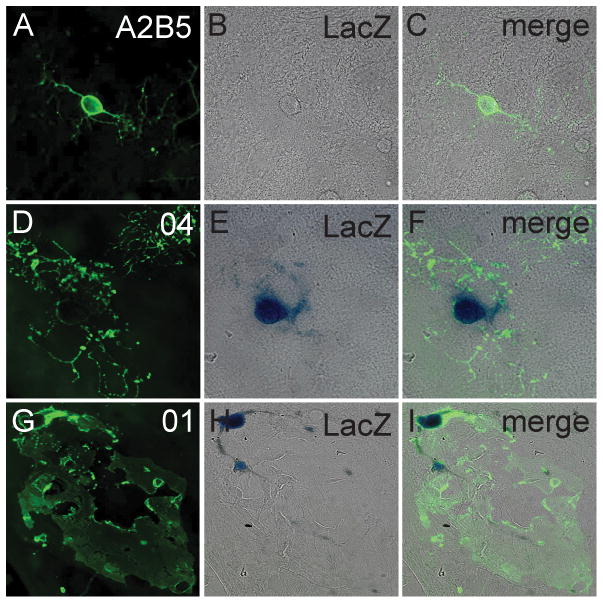
A–I) Mouse glial primary cultures were prepared from MBP-LacZ-twitcher cortices and β-gal staining was correlated with oligodendrocyte maturation by co-labeling with monoclonal antibodies A2B5 (A–C), O4 (D–F) and O1 (G–I). Cultures were analyzed at 7 (A–C), 14 (D–F) and 21 (G–I) days of incubation.
The expression of the 18.2 MBP-LacZ transgene is regulated by axonal contact (Wrabetz et al. 1998). Because the twitcher CNS and PNS develops axonal damage towards the endstage of the disease (Cantuti Castelvetri et al. 2013; Cantuti-Castelvetri et al. 2012; Castelvetri et al. 2011), we aimed to examine how this situation influences the expression of the transgene. For this, we performed β-gal histochemical staining on longitudinal sections of postnatal (P) 40 mutant and control brains. We selected this postnatal age because it is close to the time of death in twitcher mice (Duchen et al. 1980) when neurodegeneration is highest (Cantuti Castelvetri et al. 2013; Cantuti-Castelvetri et al. 2012; Castelvetri et al. 2011). Figure 2A (magnified area shown in Fig. 2B) shows a visible reduction of β-gal staining in myelinated regions of P40 mutant brains (compare with Fig. 2C, 2D), which could indicate either a decrease in the expression level of LacZ and/or a decrease in the number of β-gal(+) oligodendrocytes in MBP-LacZ-twitcher brains.
Figure 2. LacZ expression in the brain of MBP-LacZ-twitcher mice.
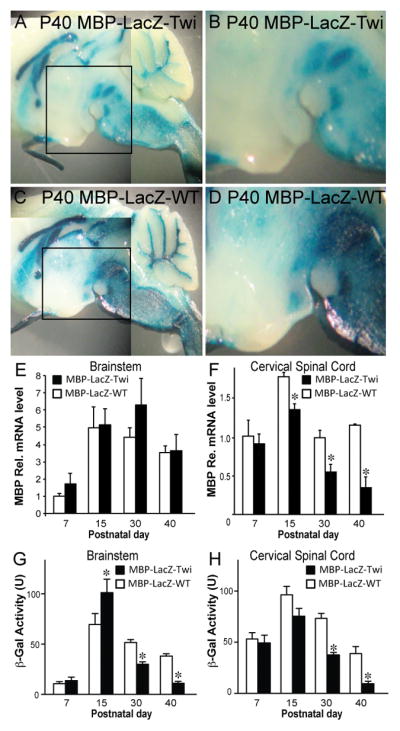
A–D) P40 brains from MBP-LacZ-twitcher and wild-type mice were stained for β-gal. A noticeable reduction of LacZ expression was observed in the mutant brain (A and its magnified boxed area in B). E, F) Expression levels of MBP transcripts in the brainstem (E) and cervical spinal cord (F) were quantified by Taqman PCR. G, H) Lysates from brainstem (G) and spinal cord (H) from MBP-LacZ-twitcher (TWI) and MBP-LacZ-WT were assayed for β-gal activity (U, unit). *p<0.05. ANOVA. n=4–6 animals per genotype.
To examine this question in more detail, we measured the levels of MBP mRNA and of transgenic β-gal activity in the mutant brainstem and cervical spinal cord between P7 to P40. Transcription by the MBP promoter (thus of the transgene) and β-gal activity correlated well between P7 to P15 in the brainstem and cord independently of the genotype (Fig. 2E–2H). At P30 and P40, although general levels of MBP mRNA were not significantly affected in the mutant brainstem, β-gal activity was lower. MBP mRNA levels were more affected in the mutant cord after 30 days of age, leading to ~50% and ~75% reductions of β-gal activity in P30 and P40 cords, respectively. The sharp reduction of β-gal activity makes stereological counting unreliable in P40 cords. Instead, β-gal positive oligodendrocytes were relieably detected by β-gal staining in endstage disease in the hindbrain, and up to P30 in the cervical spinal cord, despite loss of axons, and consequentially reduced levels of MBP promoter activation in the transgene (Wrabetz et al. 1998) (Fig. 2G and H) and MBP mRNA levels at P30 and after (Fig. 2F).
To determine that the new transgenic MBP-LacZ-twitcher mutant retains all neurological signs of disease, mutants and age-matched wild-type reporter mice were scored for disease severity (clinical score) taking into consideration changes in body weight, body tremor, and ataxic gait. MBP-LacZ-twitcher mutants were without neurological signs (clinical score 0–1, Fig. 3A) and without significant loss of body weight (Fig. 3B) until about P25. As expected for this mutation and seen in the parental twitcher background, MBP-LacZ-twitcher mutants rapidly declined (clinical score >2) afterwards. All MBP-LacZ-twitcher mutants died by P45 with clinical scores >5, in accordance with disease progression described for original twitcher mouse (Duchen et al. 1980).
Figure 3. Clinical scoring of MBP-LacZ-twitcher mice.
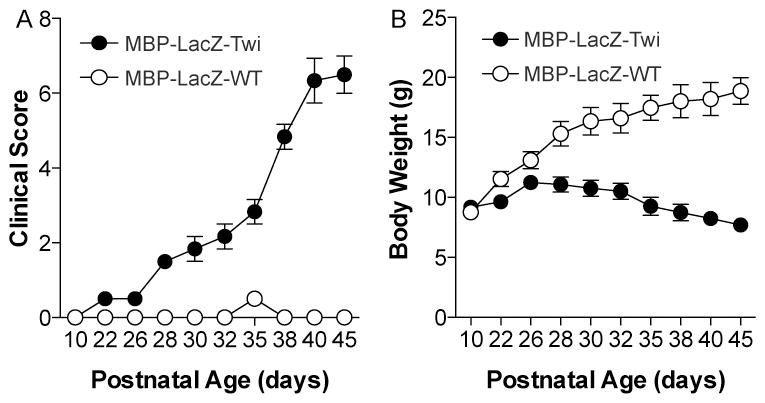
The new MBP-LacZ-Twitcher mice were examined for disease severity and their clinical score (A) and body weight (in grams, g) (B) was measured. Differences in clinical scores and weights were significant (p<0.05) after postnatal (P) day 26. N=6 mice per group. T-test.
Further studies were focused on the spinal cord, for simplicity of anatomical structure and therefore, stereological counting. Plastic-embedded cross sections of MBP-LacZ-twitcher cords clearly showed degeneration of myelinated tracks increasing with age (Fig. 4A–4F). This was evident in ventral white matter columns at P30 (inset in Fig. 4B) and P40 (inset in Fig. 4C) as well as in dorsal white matter columns (not shown). Quantitative analyses of demyelination were performed by counting the proportion of large calibre axons (>1.5 μm) showing clear signs of demyelination in the ventral column of the lumbar spinal cord (Fig. 4G, 4H). As expected, demyelination in MBP-LacZ-twitcher mice paralleled the progression of disease, with a significant increase of demyelinated axons in mutant cords (Fig. 4H), paralleling significant decreases of myelinated axons (Fig. 4G), particularly from P30 and P40 mutants.
Figure 4. Demyelination of the spinal cord in MBP-LacZ-twitcher mice.
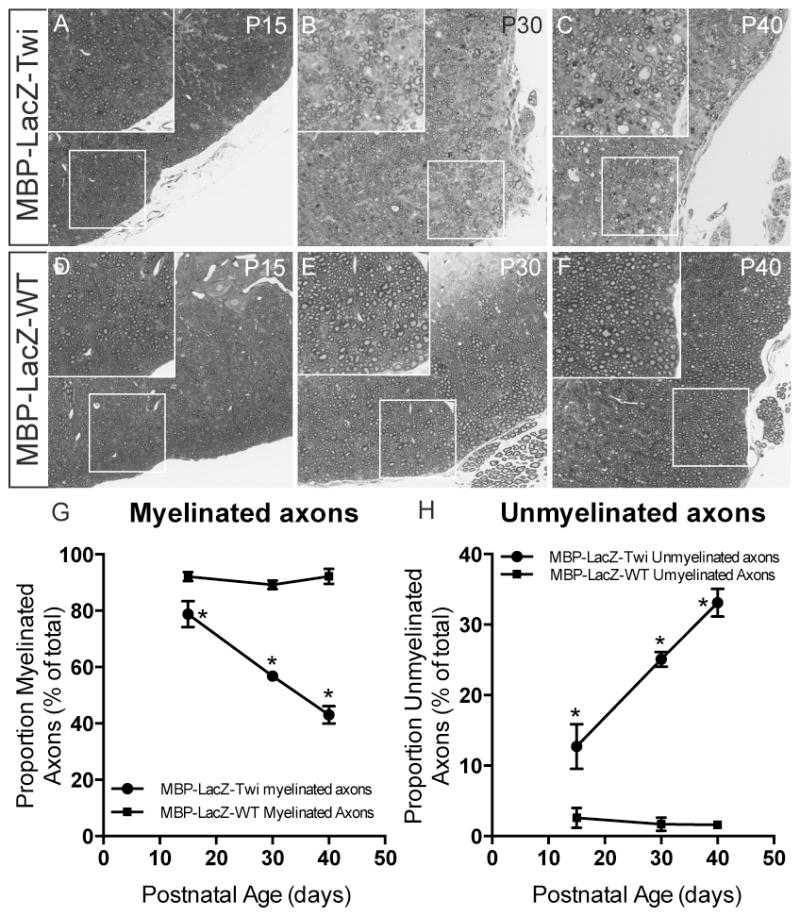
A–F) Toluidine blue staining of thin plastic sections of the lumbar spinal cord of mutant mice at P15, P30 and P40 (A–C) show progressive signs of demyelination (magnified fields in insets). G–H) The proportion of myelinated (G) and unmyelinated (H) axons as percent from the total was determined. *, p<0.05; t-test.
To further characterize the new MBP-LacZ-twitcher line, we measured the levels of psychosine by LC-MS-MS of extracts from mutant cords. For this, we dissected the dorsal and ventral hemihalves from cervical, thoracic, and lumbar segments from mutants spanning from 7 to 40 days of postnatal age. As expected, psychosine levels significantly increased along the spinal cord during disease progression. Psychosine increased from ~8 pmol/mg protein to ~220 pmol/mg protein in dorsal regions along the entire spinal cord from P7 to P40 MBP-LacZ-twitcher mice, respectively (Fig. 5A, 5D, with normal levels ranging between 2 to 4 pmol/mg protein). Interestingly, ventral regions of the cord contained significantly more psychosine than the dorsal counter-regions in each segment and at every time point of this analysis, with values increasing from ~12 pmol/mg protein to ~375 pmol/mg protein in P7 and P40 cords, respectively (Fig. 5B, 5D, with normal levels ranging between 3 to 6 pmol/mg protein). The increase in psychosine correlated with the general development of neurological signs (Fig. 5C).
Figure 5. Accumulation of psychosine in the MBP-LacZ-twitcher spinal cord.
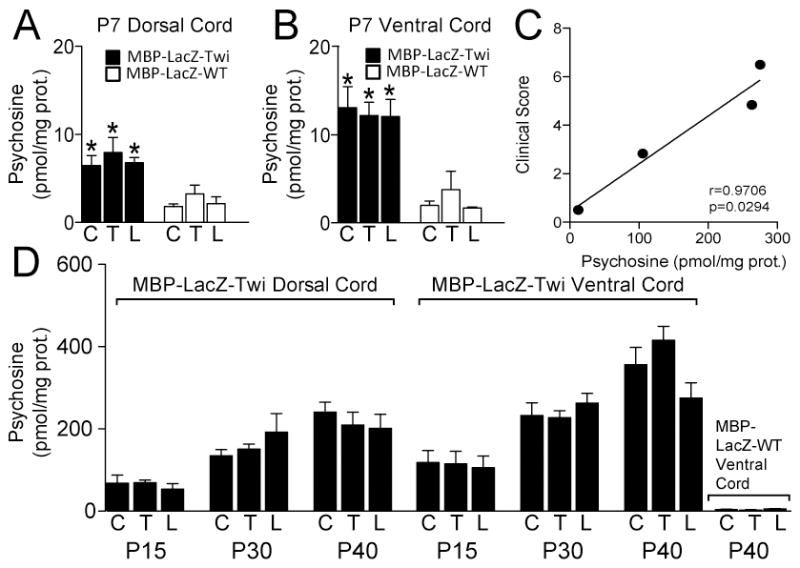
Psychosine was measured by LC-MS-MS from lipid extracts prepared from dorsal and ventral hemisections of cervical (C), thoracic (T), and lumbar (L) from MBP-LacZ-twitcher and MBP-LacZ-WT littermates at P7 (A, B) and P15, 30 and 40 (D). Differences between mutant and control samples were significant (p<0.05) at all time points. ANOVA. n=3–5 samples per condition. Correlation analysis between clinical scoring and total psychosine levels is shown in C.
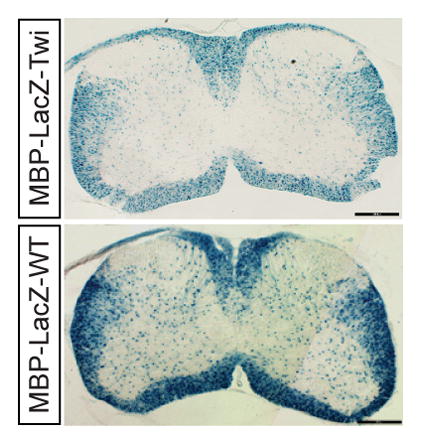
To examine the power of stereological counting to detect changes in β-gal(+) oligodendrocyte numbers using the new MBP-LacZ-twitcher mice, we stained serially prepared cross-sections from cervical, thoracic, and lumbar segments from spinal cords between P7 to P30 and used the Stereoinvestigator software for unbiased calculations. Histological images from representative cervical (Fig. 6A, 6D), thoracic (Fig. 6B, 6E) and lumbar (Fig. 6C, 6F) sections from P15 cords and stained for β-gal are presented. Mutant sections readily showed reduced numbers of β-gal(+) oligodendrocytes in all white matter regions (Fig. 6D–6F). Stereological counting revealed significant reductions of β-gal(+) oligodendrocytes in the dorsal and ventral regions throughout the entire mutant spinal cord (Fig. 6G, 6H shows the density of β-gal(+) oligodendrocytes in the P15 mutant thoracic segment).
Figure 6. Reduction of β-gal(+) oligodendrocytes in the MBP-LacZ-twitcher spinal cord.
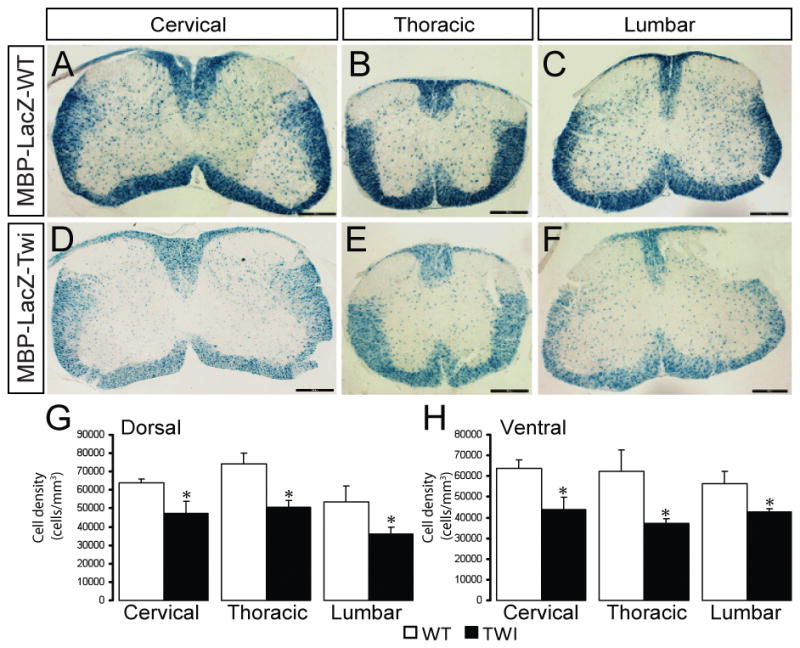
Coronal sections from P15 MBP-LacZ-WT (A–C) and MBP-LacZ-Twitcher (D–F) spinal cords were stained for the expression of the reporter gene. Reduced staining was noticeable in cervical, thoracic and lumbar segments. Unbiased stereology was used to quantitate the changes in the density of β-gal(+) oligodendrocytes in the dorsal and ventral white matter at P7 (not shown), P15 (G,H), and P30 (not shown). Decreases in the dorsal (G) and ventral (H) for the P15 cord are shown. p<0.05, ANOVA, n=3–6 per condition.
Next we calculated the proportion of β-gal(+) oligodendrocyte reduction (or loss) during disease as the percent of normal density values of β-gal(+) oligodendrocytes in age-matched wild type littermates. Interestingly, we observed that the reduction of β-gal(+) oligodendrocytes in mutant cords tended to plateau between 15 and 30 days of age (Fig. 7A, 7B). Demyelination has been historically considered the consequence of massive psychosine-mediated apoptotic death of myelinating oligodendrocytes (Costantino-Ceccarini et al. 1999; Ida et al. 1990; Jatana et al. 2002; LeVine and Torres 1992; Taniike et al. 1999; Zaka and Wenger 2004). In addition, Figure 7 shows that the reduction of β-gal(+) oligodendrocytes in the mutant cord did not parallel the accumulation of psychosine, which otherwise increased at a constant positive rate in dorsal (Fig. 7A) and ventral (Fig. 7B) regions of the mutant cord between 7 and 30 days of age. These results underline the possibility that mutant oligodendrocytes may be more resistant to increasing psychosine levels than expected.
Figure 7. Analysis of β-gal(+) oligodendrocytes and psychosine changes in the postnatal MBP-LacZ-twitcher cord.
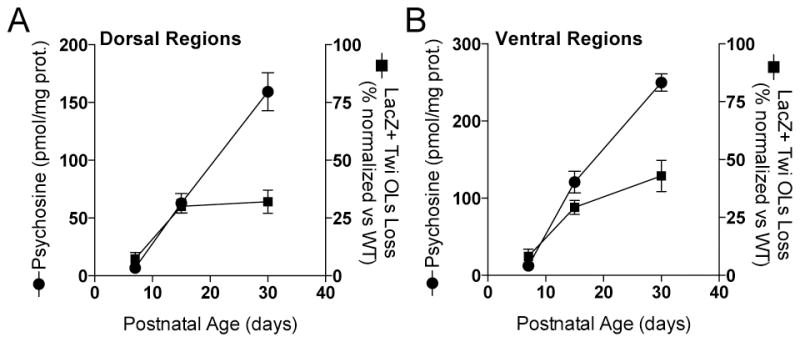
Unbiased stereology was used to determine the reduction (as loss of LacZ staining) of β-gal(+) oligodendrocytes as a function of the increment of psychosine in dorsal (A) and ventral (B) regions of the spinal cord at P7, P15, and P30.
Together these results provide evidence of the fidelity of the new MBP-LacZ-twitcher line as a model of the parental twitcher line in terms of demyelination, development of clinical signs, and psychosine content.
DISCUSSION
Demyelination of the twitcher spinal cord, which starts around 25 days of postnatal life and increases rapidly thereafter (Nagara et al. 1982; Takahashi and Suzuki 1984; Tanaka et al. 1989), is thought to be caused by the apoptotic depletion of mutant oligodendrocytes (Jatana et al. 2002; Taniike et al. 1999; Zaka and Wenger 2004). However, stereological quantitative studies addressing this assumption have been hampered due to the lack of appropriate genetic reporters in Krabbe animal models. As a first step, we have generated various new reporter lines in which myelinating oligodendrocytes can be identified by genetic marking. This study characterizes the first of these new reporter twitcher lines. The MBP-LacZ-twitcher line allows the identification of myelinating oligodendrocytes by the expression of the LacZ reporter gene under the control of the 750 bp proximal MBP promoter.
The MBP-LacZ-twitcher mouse developed all expected signs of disease with loss of body weight, twitching, ataxic movements, muscle wasting, deficient paw grip, and short survival as seen in the parental twitcher line. Metabolic deficiency of GALC (evidenced by the accumulation of psychosine) in the MBP-LacZ-twitcher nervous system was also present, and led to accumulation of total psychosine levels identical to those measured previously in twitcher mice. Genetic marking of myelinating oligodendrocytes was stable across multiple generations, and reliably identified myelinating oligodendrocytes in the brain and spinal cord of affected homozygous twitcher mice. As expected, only mature myelinating oligodendrocytes manifested active expression of the reporter transgene. We found no evidence of LacZ expression in immature oligodendrocytes. In addition, the LacZ staining is restricted to the cell body of oligodendrocytes, facilitating unbiased steriological analysis of oligodendrocyte numbers. Therefore, the new MBP-LacZ-twitcher mouse provides an authentic genetic tool for the correlative study of degenerative mechanisms of oligodendrocytes and myelin in the context of galactosylceramidase deficiency.
Previous studies employing the original twitcher strain determined total levels of psychosine in the mutant cord (Kobayashi et al. 1987; Mitsuo et al. 1989; Shinoda et al. 1987). Tanaka and collaborators showed an analysis of psychosine levels in micropunches taken from dorsal and ventral columns (Tanaka et al. 1988), but differentiation between levels in the cervical, thoracic and lumbar regions was not reported. By accurate microdissection of the spinal cords, our study measured psychosine levels in dorsal and ventral hemihalves along the entire mutant spinal cord. Interestingly, we found that the psychosine content in the ventral regions of the mutant cord was 1.5–2 fold higher than in the respective dorsal regions for most cord segments at all studied time points. This difference may reflect intrinsic differences in the rate of psychosine accumulation, which is largely driven by its synthetic rate (i.e. higher synthesis of psychosine in the ventral vs the dorsal). Psychosine is synthesized via the Cleland and Kennedy cycle (Cleland and Kennedy 1960; Mitsuo et al. 1989). Differences in local accumulation likely depend on multiple variables including the rates of synthesis and of catabolism, its transport within cellular compartments, the cell type where it is synthesized, the differentiation state of that cell, and likely cellular responses to the environment. Unfortunately, most of the metabolic and dynamic aspects of the process of psychosine accumulation in membranes are unclear (Mitsuo et al. 1989). For example, it is unknown whether the rate of psychosine synthesis is the same in immature and mature oligodendrocytes, or whether psychosine synthesis varies in brain vs spinal cord oligodendrocytes or in gray matter vs white matter oligodendrocytes or among the different neuronal types. Equivalent questions could be raised for the catabolism of psychosine. Because psychosine is thought to be the leading cause for oligodendrocyte death in Krabbe’s disease, investigating these questions may reveal the basis for regional differences in vulnerability of the nervous system, and how to better adjust therapeutic approaches.
Our analyses showed that the number of β-gal(+) oligodendrocytes decreased in parallel with the progression of clinical signs in mutant mice. However, unbiased stereology showed that this decrease of β-gal(+) oligodendrocytes was not linear, and showed poor correlation with the progressive and constant accumulation of psychosine in the mutant cord (Fig. 7). This discrepancy underlines the lack of solid quantitative data demonstrating a direct relationship between accumulation of psychosine, oligodendrocyte death, and demyelination in the twitcher spinal cord. In other words, the stoichiometry by which psychosine kills oligodendrocyte and/or induces demyelination remains undetermined. Psychosine is capable of inducing significant killing of oligodendrocytes in vitro (Giri et al. 2008; Giri et al. 2006; Haq et al. 2003; Jatana et al. 2002; Won et al. 2013; Zaka et al. 2005; Zaka and Wenger 2004). However, a direct role for psychosine induction of oligodendrocyte death in vivo (and hence, inducing demyelination by oligodendrocyte death) is less clear. Our study using genetic marking of myelinating oligodendrocytes revealed a lower-than expected reduction of myelinating oligodendrocytes despite the steady accumulation of psychosine. In the context of the progressive demyelination reported in the mutant cord, we speculate that demyelination occurs faster than oligodendrocyte death. Various animal models of oligodendrocyte cell death and demyelination have in fact revealed discrepancies between the time of cell death and the damage to myelin (Buch et al. 2005; Caprariello et al. 2012; Oluich et al. 2012; Pohl et al. 2011; Traka et al. 2010). Psychosine, which is highly toxic to a large range of cell types, and in high concentrations is capable of killing cultured oligodendrocytes (Jatana et al. 2002) could be inducing a rapid “myelin-dying back” mechanism. Being a lipid raft associated sphingolipid (White et al. 2009), psychosine may induce local changes in myelin fluidity, leading to instability and disassembly of myelin (Hawkins-Salsbury et al. 2013; White et al. 2011) (D’auria and Bongarzone, 2016, in press).
An additional possibility is that a compensatory response to demyelination triggers the generation of newly born oligodendrocyte progenitors to replace dying oligodendrocytes. An earlier study by Taniike and Suzuki (Taniike and Suzuki 1995) indeed reported that a fraction of oligodendrocytes undergo mitosis in the cord of sick twitcher mice. Undoubtedly, cell death of myelinating oligodendrocytes is a key pathological process in the Krabbe CNS, underpinning the overall process of demyelination. Apoptosis of oligodendrocytes has been proven to be sufficient to cause rapid and focal areas of demyelination (Caprariello et al. 2012). However, focal myelin lesions may trigger the recruitment of resident oligodendrocyte progenitor cells to engage in remyelination. This is also an area where solid unbiased quantitative analyses are lacking. Investigating glial responses during the progression of the disease is important not only to obtain a clear understanding of the dynamics of demyelination and glial loss but also to improve the timing of therapeutic interventions.
In summary, this study is a first step in the quantitative and unbiased analysis of oligodendrocyte responses during disease progression in the spinal cord of the twitcher mouse. Further studies will examine the mechanism of demyelination and oligodendrocyte death and how remyelination is regulated in the twitcher CNS.
Significance.
Krabbe’s disease is a leukodystrophy due to the deficiency of galactosyl-ceramidase and the accumulation of psychosine. Psychosine is believed to cause central demyelination by killing oligodendrocytes. In this study, we generated a new transgenic reporter twitcher line where myelinating oligodendrocytes are genetically marked by the expression of LacZ to perform quantitative and unbiased analysis of oligodendrocyte responses during disease progression in the spinal cord of the twitcher mouse.
Acknowledgments
We thank Paola Saveri for excellent technical assistance. Support for this study was provided in part by grants from NIH (R01NS065808) and the Legacy for Angels Foundation to ERB.
Footnotes
DISCLOSURE OF CONFLICT OF INTEREST
ERB is a consultant with Lysosomal Therapeutics, Inc.
References
- Bischoff A, Ulrich J. PERIPHERAL NEUROPATHY IN GLOBOID CELL LEUKODYSTROPHY (KRABBE’S DISEASE). ULTRASTRUCTURAL AND HISTOCHEMICAL FINDINGS. Brain : a journal of neurology. 1969;92(4):861–870. doi: 10.1093/brain/92.4.861. [DOI] [PubMed] [Google Scholar]
- Bongarzone ER, Foster LM, Byravan S, Verity AN, Landry CF, Schonmann VV, Amur-Umarjee S, Campagnoni AT. Conditionally Immortalized Neural Cell Lines: Potential Models for the Study of Neural Cell Function. Methods. 1996;10(3):489–500. doi: 10.1006/meth.1996.0126. [DOI] [PubMed] [Google Scholar]
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nature methods. 2005;2(6):419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- Cantuti Castelvetri L, Givogri MI, Hebert A, Smith B, Song Y, Kaminska A, Lopez-Rosas A, Morfini G, Pigino G, Sands M, Brady ST, Bongarzone ER. The Sphingolipid Psychosine Inhibits Fast Axonal Transport in Krabbe Disease by Activation of GSK3beta and Deregulation of Molecular Motors. J Neurosci. 2013;33(24):10048–10056. doi: 10.1523/JNEUROSCI.0217-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Zhu H, Givogri MI, Chidavaenzi RL, Lopez-Rosas A, Bongarzone ER. Psychosine induces the dephosphorylation of neurofilaments by deregulation of PP1 and PP2A phosphatases. Neurobiol Dis. 2012;46(2):325–335. doi: 10.1016/j.nbd.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprariello AV, Mangla S, Miller RH, Selkirk SM. Apoptosis of oligodendrocytes in the central nervous system results in rapid focal demyelination. Ann Neurol. 2012;72(3):395–405. doi: 10.1002/ana.23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelvetri LC, Givogri MI, Zhu H, Smith B, Lopez-Rosas A, Qiu X, van Breemen R, Bongarzone ER. Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta neuropathologica. 2011;122(1):35–48. doi: 10.1007/s00401-011-0814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Cleland WW, Kennedy EP. The enzymatic synthesis of psychosine. J Biol Chem. 1960;235:45–51. [PubMed] [Google Scholar]
- Costantino-Ceccarini E, Luddi A, Volterrani M, Strazza M, Rafi MA, Wenger DA. Transduction of cultured oligodendrocytes from normal and twitcher mice by a retroviral vector containing human galactocerebrosidase (GALC) cDNA. Neurochem Res. 1999;24(2):287–293. doi: 10.1023/a:1022574323784. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F. Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain : a journal of neurology. 1980;103(3):695–710. doi: 10.1093/brain/103.3.695. [DOI] [PubMed] [Google Scholar]
- Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187(4):1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Basso V, Cantuti L, Givogri MI, Lopez-Rosas A, Perez N, Vasu C, Cao H, van Breemen R, Mondino A, Bongarzone ER. Autonomic denervation of lymphoid organs leads to epigenetic immune atrophy in a mouse model of Krabbe disease. J Neurosci. 2007;27(50):13730–13738. doi: 10.1523/JNEUROSCI.3379-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Jatana M, Rattan R, Won JS, Singh I, Singh AK. Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: implications for Krabbe disease. Faseb J. 2002;16(7):661–672. doi: 10.1096/fj.01-0798com. [DOI] [PubMed] [Google Scholar]
- Giri S, Khan M, Nath N, Singh I, Singh AK. The role of AMPK in psychosine mediated effects on oligodendrocytes and astrocytes: Implication for Krabbe Disease. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Khan M, Rattan R, Singh I, Singh AK. Krabbe disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res. 2006;47(7):1478–1492. doi: 10.1194/jlr.M600084-JLR200. [DOI] [PubMed] [Google Scholar]
- Haq E, Giri S, Singh I, Singh AK. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86(6):1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x. [DOI] [PubMed] [Google Scholar]
- Hawkins-Salsbury JA, Parameswar AR, Jiang XT, Schlesinger PH, Bongarzone E, Ory DS, Demchenko AV, Sands MS. Psychosine, the cytotoxic sphingolipid that accumulates in globoid cell leukodystrophy, alters membrane architecture. Journal of Lipid Research. 2013;54(12):3303–3311. doi: 10.1194/jlr.M039610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida H, Kawame F, Kim SU, Eto Y. Abnormality in cultured oligodendrocytes and Schwann cells isolated from the twitcher mouse. Mol Chem Neuropathol. 1990;13(3):195–204. doi: 10.1007/BF03159922. [DOI] [PubMed] [Google Scholar]
- Igisu H, Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224(4650):753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Singh AK. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci Lett. 2002;330(2):183–187. doi: 10.1016/s0304-3940(02)00655-9. [DOI] [PubMed] [Google Scholar]
- Khan M, Haq E, Giri S, Singh I, Singh AK. Peroxisomal participation in psychosine-mediated toxicity: implications for Krabbe’s disease. J Neurosci Res. 2005;80(6):845–854. doi: 10.1002/jnr.20529. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Shinoda H, Goto I, Yamanaka T, Suzuki Y. Globoid cell leukodystrophy is a generalized galactosylsphingosine (psychosine) storage disease. Biochem Biophys Res Commun. 1987;144(1):41–46. doi: 10.1016/s0006-291x(87)80472-2. [DOI] [PubMed] [Google Scholar]
- Krabbe K. A new familial, infantile form of diffuse brain. sclerosis. Brain : a journal of neurology. 1916;39:74–114. doi: 10.1093/brain/awt232. [DOI] [PubMed] [Google Scholar]
- Lake BD. Segmental demyelination of peripheral nerves in Krabbe’s disease. Nature. 1968;217(5124):171–172. doi: 10.1038/217171a0. [DOI] [PubMed] [Google Scholar]
- LeVine SM, Torres MV. Morphological features of degenerating oligodendrocytes in twitcher mice. Brain Res. 1992;587(2):348–352. doi: 10.1016/0006-8993(92)91018-a. [DOI] [PubMed] [Google Scholar]
- Matsumoto R, Oka N, Nagahama Y, Akiguchi I, Kimura J. Peripheral neuropathy in late-onset Krabbe’s disease: histochemical and ultrastructural findings. Acta neuropathologica. 1996;92(6):635–639. doi: 10.1007/s004010050573. [DOI] [PubMed] [Google Scholar]
- Mitsuo K, Kobayashi T, Shinnoh N, Goto I. Biosynthesis of galactosylsphingosine (psychosine) in the twitcher mouse. Neurochem Res. 1989;14(9):899–903. doi: 10.1007/BF00964821. [DOI] [PubMed] [Google Scholar]
- Nagara H, Kobayashi T, Suzuki K, Suzuki K. The twitcher mouse: normal pattern of early myelination in the spinal cord. Brain Res. 1982;244(2):289–294. doi: 10.1016/0006-8993(82)90087-7. [DOI] [PubMed] [Google Scholar]
- Oluich LJ, Stratton JA, Xing YL, Ng SW, Cate HS, Sah P, Windels F, Kilpatrick TJ, Merson TD. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci. 2012;32(24):8317–8330. doi: 10.1523/JNEUROSCI.1053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl HB, Porcheri C, Mueggler T, Bachmann LC, Martino G, Riethmacher D, Franklin RJ, Rudin M, Suter U. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci. 2011;31(3):1069–1080. doi: 10.1523/JNEUROSCI.5035-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatelli M, Quaranta L, Madia F, Lippi G, Conte A, Lo Monaco M, Di Trapani G, Rafi MA, Wenger DA, Vaccaro AM, Tonali P. Peripheral neuropathy with hypomyelinating features in adult-onset Krabbe’s disease. Neuromuscular disorders : NMD. 2002;12(4):386–391. doi: 10.1016/s0960-8966(01)00285-1. [DOI] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe’s disease. J Neurochem. 1996;66(3):1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- Shinoda H, Kobayashi T, Katayama M, Goto I, Nagara H. Accumulation of galactosylsphingosine (psychosine) in the twitcher mouse: determination by HPLC. J Neurochem. 1987;49(1):92–99. doi: 10.1111/j.1471-4159.1987.tb03399.x. [DOI] [PubMed] [Google Scholar]
- Simons K, Gruenberg J. Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol. 2000;10(11):459–462. doi: 10.1016/s0962-8924(00)01847-x. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Suzuki K. Demyelination in the spinal cord of murine globoid cell leukodystrophy (the twitcher mouse) Acta neuropathologica. 1984;62(4):298–308. doi: 10.1007/BF00687612. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nagara H, Kobayashi T, Goto I. The twitcher mouse: accumulation of galactosylsphingosine and pathology of the sciatic nerve. Brain Res. 1988;454(1–2):340–346. doi: 10.1016/0006-8993(88)90835-9. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nagara H, Kobayashi T, Goto I. The twitcher mouse: accumulation of galactosylsphingosine and pathology of the central nervous system. Brain Res. 1989;482(2):347–350. doi: 10.1016/0006-8993(89)91198-0. [DOI] [PubMed] [Google Scholar]
- Taniike M, Mohri I, Eguchi N, Irikura D, Urade Y, Okada S, Suzuki K. An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J Neuropathol Exp Neurol. 1999;58(6):644–653. doi: 10.1097/00005072-199906000-00009. [DOI] [PubMed] [Google Scholar]
- Taniike M, Suzuki K. Proliferative capacity of oligodendrocytes in the demyelinating twitcher spinal cord. J Neurosci Res. 1995;40(3):325–332. doi: 10.1002/jnr.490400306. [DOI] [PubMed] [Google Scholar]
- Traka M, Arasi K, Avila RL, Podojil JR, Christakos A, Miller SD, Soliven B, Popko B. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain. 2010;133(10):3017–3029. doi: 10.1093/brain/awq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Molecular genetics and metabolism. 2000;70(1):1–9. doi: 10.1006/mgme.2000.2990. [DOI] [PubMed] [Google Scholar]
- White AB, Galbiati F, Givogri MI, Lopez Rosas A, Qiu X, van Breemen R, Bongarzone ER. Persistence of psychosine in brain lipid rafts is a limiting factor in the therapeutic recovery of a mouse model for Krabbe disease. J Neurosci Res. 2011;89(3):352–364. doi: 10.1002/jnr.22564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AB, Givogri MI, Lopez-Rosas A, Cao H, van Breemen R, Thinakaran G, Bongarzone ER. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(19):6068–6077. doi: 10.1523/JNEUROSCI.5597-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R, Sutherland C. Neuronal membranes are key to the pathogenesis of Alzheimer’s disease: the role of both raft and non-raft membrane domains. Curr Alzheimer Res. 2011;8(2):213–221. doi: 10.2174/156720511795256008. [DOI] [PubMed] [Google Scholar]
- Won JS, Kim J, Paintlia MK, Singh I, Singh AK. Role of endogenous psychosine accumulation in oligodendrocyte differentiation and survival: implication for Krabbe disease. Brain Res. 2013;1508:44–52. doi: 10.1016/j.brainres.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, Taveggia C, Feltri ML, Quattrini A, Awatramani R, Scherer SS, Messing A, Kamholz J. A minimal human MBP promoter-lacZ transgene is appropriately regulated in developing brain and after optic enucleation, but not in shiverer mutant mice. J Neurobiol. 1998;34(1):10–26. doi: 10.1002/(sici)1097-4695(199801)34:1<10::aid-neu2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Zaka M, Rafi MA, Rao HZ, Luzi P, Wenger DA. Insulin-like growth factor-1 provides protection against psychosine-induced apoptosis in cultured mouse oligodendrocyte progenitor cells using primarily the PI3K/Akt pathway. Mol Cell Neurosci. 2005;30(3):398–407. doi: 10.1016/j.mcn.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Zaka M, Wenger DA. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci Lett. 2004;358(3):205–209. doi: 10.1016/j.neulet.2003.12.126. [DOI] [PubMed] [Google Scholar]