

Abstract
Nikkomycins and polyoxins are antifungal peptidylnucleoside (PN) antibiotics active against human and plant pathogens. Here, we report that during PN biosynthesis in Streptomyces cacaoi and Streptomyces tendae, the C5′-extension of the nucleoside essential for downstream structural diversification is catalyzed by a conserved radical S-adenosyl-L-methionine (SAM) enzyme, PolH or NikJ. This is distinct from the nucleophilic mechanism reported for antibacterial nucleosides and represents a novel mechanism of nucleoside natural product biosynthesis.
Natural products with modified nucleosides exhibit diverse biological activities including anti-cancer, antibacterial, antiviral, and antifungal activities.1 One of the most characteristic and prevalent modifications is C-C bond extension at the C-5′ of ribose (Supplementary Results, Supplementary Fig. 1),1 which allows for the connection of the nucleoside to structurally diverse functional groups. Among the C5′-extended nucleosides, polyoxins and nikkomycins (Fig. 1a) constitute an important class of antifungal PNs2 used in agriculture to treat plant fungal diseases3 and have been clinically investigated to treat invasive fungal infections in humans.4
Figure 1.
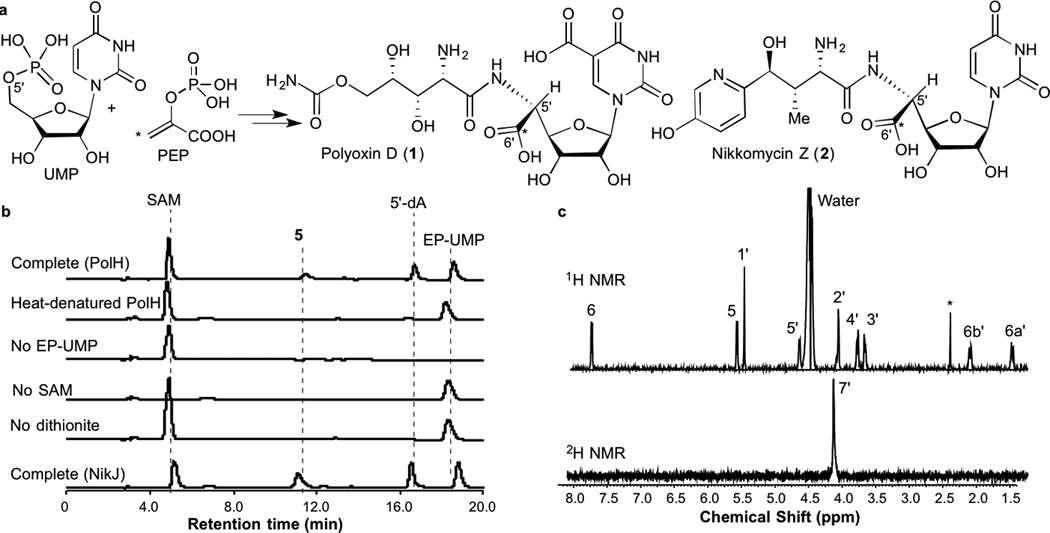
C5′-Extension in antifungal PN biosynthesis. a. Proposed biosynthetic pathway for polyoxins and nikkomycins. The asterisks show the fate of C-3 of PEP. UMP is the only experimentally demonstrated precursor5,9 and the timing of nucleobase modifications is unknown. b. HPLC analysis of PolH assays and negative controls without EP-UMP, SAM, or dithionite, or with heat-inactivated PolH. Also shown is a result of NikJ assay (bottom trace). c. 1H and 2H NMR spectra of U-OAP formed in the PolH assay in deuterium oxide. The H-7′ signal was absent in the 1H NMR spectrum and, instead, observed in the 2H NMR spectrum (compare with Supplementary Fig. 7). The 1H NMR signal with an asterisk at 2.5 ppm is an unidentified impurity.
The common structure among antifungal PNs is aminohexuronic acid (AHA, 3). During the biosynthesis, AHA is formed from UMP and phosphoenol pyruvate (PEP, Fig. 1a)5 via enolpyruvyl UMP (EP-UMP, 4) and a C5′-extended high-carbon nucleoside, octosyl acid or its derivative (see Supplementary Figure 2 for detail).5–7 While EP-UMP is formed by the transfer of enolpyruvyl group of PEP to 3′-OH of UMP by an UMP enolpyruvyltransferase (PolA in the polyoxin pathway),8,9 the mechanism of C5′-extension from EP-UMP to octosyl acid remains unknown. Previously, the C5′-extension was proposed to proceed via a hypothetical 5′-aldehyde intermediate (Supplementary Fig. 2b and 2c) by an action of a conserved α-ketoglutarate (α-KG) dependent dioxygenase (polD or polK in polyoxin).8,10 However, considering the poor nucleophilicity of enolethers, this model does not explain the mechanism of C5′-C6′ bond formation. Therefore, we considered an alternative mechanism in which a conserved putative radical SAM enzyme (PolH; Supplementary Table 1) uses a transient 5′-deoxyadenosyl radical (5′-dA•)11,12 to oxidize C-5′ and/or initiate C5′-C6′ bond formation.
To investigate the mechanism of C5′ extension, the candidate enzymes, PolD, PolK and PolH were individually expressed in E. coli, purified and reconstituted for their metallo-cofactors under strict anaerobic conditions. The resulting PolH exhibited broad UV-vis absorption at 420 nm, and harbored 3.1 ± 0.3 eq. Fe2+ and S2− per polypeptide, consistent with the presence of one 4Fe-4S cluster per monomer (Supplementary Fig. 3, Supplementary Table 2). PolD and PolK were characterized to have a non-heme mono-iron center capable of binding and oxidizing α-KG (Supplementary Fig. 4). Their catalytic functions were investigated using nucleotide and nucleoside substrate candidates (UMP, EP-UMP, uridine, or EP-uridine). Among all the combinations tested, only the assay with PolH and EP-UMP resulted in formation of a new HPLC peak at retention time 11.5 min (Fig. 1b). This peak was not observed in the assays with other substrate candidates or in the absence of PolH. Subsequently, the PolH product was isolated and characterized by HR-MS and 1D and 2D NMR (Supplementary Fig. 7 – 13), which were most consistent with uracil OAP (U-OAP, 5, see Supplementary Fig. 13 for details of structural characterization). These observations all together suggested that PolH alone is sufficient to establish the C5′-C6′ bond.
To investigate the functional conservation, NikJ, a homolog of PolH in the nikkomycin pathway, was also characterized. Purified NikJ (Supplementary Fig. 3, Supplementary Table 2) catalyzed the conversion of EP-UMP into U-OAP (Fig. 1b). PolH and NikJ exhibited similar steady state kinetic parameters: Km = 17 – 19 µM for EP-UMP; and kcat/Km = 0.016 – 0.023 min−1 µM−1 (Supplementary Table 4, Supplementary Fig. 14). These values are comparable to those reported for the conversion of UMP to EP-UMP by NikO (kcat/Km = 0.10 – 0.45 min−1 µM−1).13 The low Km values support high specificity of NikJ and PolH towards EP-UMP. Together with the previously established relevance of octosyl acid to PN biosynthesis,5–7 these observations suggest that the observed OAP synthase activity is likely the physiologically relevant function of NikJ and PolH.
PolH does not exhibit significant sequence homology to other radical SAM enzymes, and hence constitutes a unique subfamily. Therefore, we characterized the catalytic mechanism of PolH. In general, radical SAM enzymes use SAM in either catalytic or stoichiometric amounts.12 PolH produced stoichiometric amounts of U-OAP and 5′-dA (Supplementary Fig. 15a), suggesting that PolH consumes SAM as a co-substrate. When the PolH assay was performed in D2O, a deuterium was incorporated at the 7′-position (Fig. 1c). These observations suggest that the PolH catalysis involves a C-7′ centered free radical intermediate (C7′•), which is stereospecifically reduced by a reductant with a solvent exchangeable proton.
Since PolH does not harbor any redox active cofactors other than the 4Fe-4S cluster essential for SAM cleavage, the C7′• reduction is likely catalyzed by a redox active amino acid. To test this hypothesis, we characterized PolH variants with Ala or Phe mutations in one of the conserved Cys or Tyr residues (Supplementary Fig. 16). Nine such variants were expressed as soluble proteins with Fe-S cluster loading comparable to wt-PolH (2.9 – 3.2 Fe/monomer, Supplementary Table 2). While all of these variants were capable of producing U-OAP (Supplementary Fig. 17), C209A-PolH uniquely produced a second product that migrated faster than U-OAP on HPLC (retention time 7.0 min, Fig. 2a), and exhibited an UV-vis absorption spectrum characteristic for uridine nucleotides. This compound was not observed when EP-UMP or SAM was omitted from the assay, or in the assays of other variants (Supplementary Fig. 17b). The sum of the amounts of the two products was stoichiometric to the amount of 5′-dA (Supplementary Fig. 15b). The C209A-PolH-specific product was then isolated and characterized by HR-MS, and 1D and 2D NMR (Supplementary Fig. 18 – 24), which were most consistent with a stereoisomer of U-OAP with 7′-R configuration (epi-U-OAP; see Supplementary Fig. 19 for details of structural characterization). The formation of epi-U-OAP was also observed in assays of C209S-PolH (Supplementary Fig. 17), suggesting that the formation of epi-U-OAP is caused by the absence of the redox capability at the 209 residue.
Figure 2.
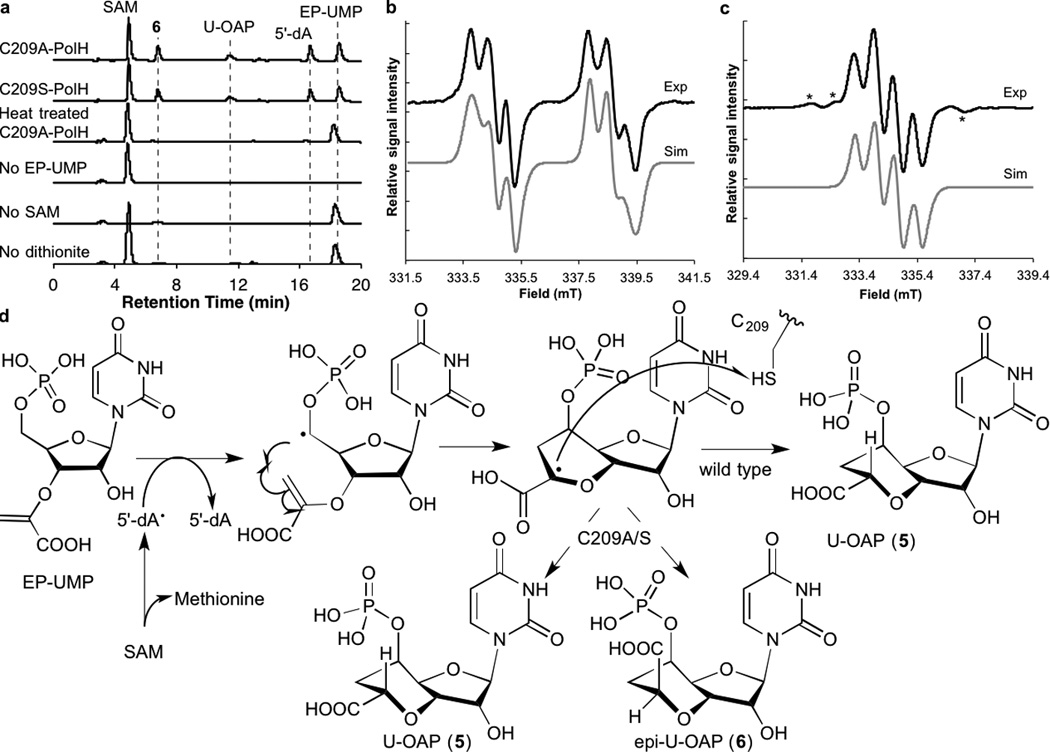
Characterization of PolH C209 variants. a. HPLC analysis of assays for C209A-PolH and C209S-PolH and negative controls performed for C209A-PolH assays. b and c. X-band continuous wave (CW) EPR spectra of C7′• observed in the C209S-PolH reaction with EP-UMP (b) or with [3′′-2H2]EP-UMP (95% 2H) (c). Top traces are experimental spectra and bottom traces are simulation with parameters in Supplementary Table 6. Features shown in asterisks in c are associated with residual non-deuterated EP-UMP, and constitutes 2 – 5% of the total signal intensity. d. Proposed mechanism of the reactions catalyzed by WT- and C209A/S-PolH.
To further investigate the redox function of C209, C209S-PolH was characterized by electron paramagnetic resonance (EPR) spectroscopy. In the absence of EP-UMP, C209S-PolH exhibited a broad EPR signal at 15 K characteristic for [4Fe-4S]+ clusters (39 ± 6% of C209S-PolH, Supplementary Fig. 28a), but no signal was observed at 77 K, where organic radicals would be detectable. In contrast, under turnover conditions, the [4Fe-4S]+ cluster signal was significantly diminished (<10% of C209S-PolH, Supplementary Fig. 28d), and a narrow EPR signal at g ~2.0 was observed at 77 K, consistent with a formation of an organic radical (15 ± 2% of C209S-PolH, Fig. 2b). Simulation of this EPR spectrum suggested hyperfine interactions with two nuclei with I = 1/2 (Fig. 2b, Supplementary Table 6). Furthermore, when the C209S-PolH reaction was performed with [3′′-2H2]EP-UMP, the EPR signal at 77 K exhibited narrower hyperfine splitting (Fig. 2c). Simulation of this signal suggested hyperfine interactions with one I = 1 and one I = 1/2 nucleus (Fig. 2c, Supplementary Table 6), consistent with a substitution of 1H with 2H. Further analysis of the hyperfine coupling constants and dihedral angles combined with molecular simulation (Supplementary Fig. 29) suggested that the observed radical is located β to H-6′, and most consistent with C7′-centered radical intermediate (C7′•). C209A-PolH also accumulated the same organic radical (Supplementary Fig. 28c), whereas wt-PolH or any other PolH variants did not, suggesting the specific accumulation of C7′• in PolH variants without the redox capability at the 209 residue.
Based on these observations, we propose a catalytic mechanism of PolH (Fig. 2d). In this model, PolH catalyzes the H-5′ abstraction from EP-UMP using 5′-dA• to generate C-5′ radical, which then attacks C3′′ to form the bicyclic structure of U-OAP. This radical cyclization reaction yields C7′•, which is stereospecifically reduced by C209 to yield U-OAP. The redox function of C209 is consistent with the accumulation of C7′• in the C209A- and C209S-PolH. In WT-PolH, the catalytic cycle completes upon reduction of the C209 thiyl radical (C209•) likely by another redox active amino acid, such as Tyr, followed by a radical propagation to protein surface and reduction by exogenous reductant. The reductive environment of the active site is consistent with the observation that in C209A- and C209S-PolH, C7′• is slowly reduced to U-OAP and epi-U-OAP.
Over the past five years, radical SAM enzymes that catalyze C-C bond formations have been reported in medically significant pathways.14–17 All of these enzymes require quenching of radical intermediates to complete catalysis, but their mechanisms are unknown. The mechanistic finding in PolH is, to our knowledge, the first demonstration of a radical quenching mechanism in C-C-bond forming radical SAM enzymes. The Cys-mediated radical reduction could be a common mechanism for radical SAM enzymes that require stereospecific reduction of a radical intermediate.18,19
The functional characterization of PolH also revealed the novel free-radical mediated mechanism of C5′-extention in nucleoside natural product biosynthesis. The only other experimentally demonstrated mechanism of C5′ extension was for the biosynthesis of caprazamycin class of antibacterial nucleosides, in which C5′ of a nucleotide precursor is oxidized to an aldehyde by an α-KG dependent dioxygenase, followed by nucleophilic condensation (Supplementary Fig. 30).20,21 Therefore, the free radical mediated C5′ extension found in the current study is mechanistically novel. Considering that octosyl acid structures were found in other antifungal nucleosides, such as ezomycins and malayamycins (Supplementary Fig. 1), the free radical mediated C5′ extension could be operating in other nucleoside natural product biosynthesis pathways. In fact, when PolH homologs were searched in the genome sequence database, more than 25 unique amino acid sequences with the overall sequence identity of > 40% were found. In all of these homologs, the H-atom donor Cys residue (C209 in PolH) is conserved. Importantly, more than 10 of these genes are in operons that also encode homologs of PolA/NikO (Supplementary Fig. 31). Therefore, these operons are potentially involved in biosynthesis of OAP-related nucleoside natural products. Overall, our analysis suggests the potential abundance of OAP-derived antifungal nucleoside natural products, and the potential to use genome mining to aid in their discovery.
Online Methods
Gereral
QAE A25 Sephadex resin, UMP (> 99% purity), SAM p-toluenesulfonate salt (> 80%), 5′-dA, deuterium oxide (99.9 atom% enriched) and sodium dithionite (85%) were purchased from Sigma-Aldrich. PEP (99%) was purchased from Alfa Aesar. Dithiothreitol (DTT, 99.4%) was purchased from Amresco. EP-UMP (> 95%) and EP-uridine (88%) were enzymatically prepared from UMP and PEP as described in the Supplementary Note. G-25 sephadex and DEAE sepharose FF resins were from GE Healthcare. Chemically competent E. coli DH5α and BL21(DE3) cells, pCR2.1-TOPO plasmid and all PCR primers were from Invitrogen. pET28 plasmid was from Novagen. Calf-intestine alkaline phosphatase (CIAP, 20 U/µL) was from NEB. UV-vis absorption spectra were determined using a U-3900 UV-VIS ratio recording double-beam spectrometer (HITACHI) or Nanodrop 1000 (Thermo Scientific). Non-linear least square fitting of kinetic data was carried out using KaleidaGraph software (Synergy Software, Reading, PA). Anaerobic experiments were carried out in a UNIlab workstation glove box (MBaun) maintained at 10 ± 2 °C with O2 concentration < 0.1 ppm. All anaerobic solutions were degassed on a Schlenk line, and equilibrated in the glove box atmosphere for > 12 h. All plastic devices used for anaerobic use were evacuated for > 12 h in the glove box antechamber before bringing into the glove box. All DNA sequences were confirmed by Eton Bioscience Inc. PCR was carried out using PfuUltraII polymerase (Stratagene) according to the manufacturer’s protocol. All HPLC experiments were performed on a Hitachi L-2130 Pump equipped with an L-2455 diode array detector, an L-2485 fluorescence detector, an L-2200 autosampler and an L-2300 column oven maintained at 40 °C. All MS analyses were performed using an Agilent 6224 ESI-TOF MS operated in positive ion mode. The typical mass accuracy of the instrument was 5 ppm. All NMR spectra were recorded on 500 or 800 MHz Varian Innova NMR spectrometers operated with VNMRJ 3.1 software and analyzed by the ACD/NMR processor (ACD/Laboratories). S. tendae and S. cacaoi were purchased from ATCC. EPR spectra were collected using an EMXplus 9.5/2.7 EPR spectrometer system equipped with an In-Cavity Cryo-Free VT system (Bruker Biospin Corporation).
Cloning of NikO, NikJ, PolH, PolD and PolK
nikO, nikJ, polD, polK, and polH genes were cloned by PCR from isolated genomic DNA of S. tendae or S. cacaoi using primers described in Supplementary Table 7. PCR was carried out for 25 cycles with an annealing temperature of 65 °C. Adenine was then added to the 3′-terminus of the PCR product for TA ligation by incubation with Taq Hot start polymerase (Promega) in the presence of 0.2 mM dATP at 72°C for 30 min. The product was cloned into pCR2.1-TOPO (Invitrogen) by following the manufacturer’s protocol. After confirmation of the DNA sequence, the resulting plasmids were digested with NdeI and HindIII and the PolD, PolK, PolH, NikO and NikJ genes were individually subcloned into the corresponding site of pET28b, yielding pET-HisPolD, pET-HisPolK, pET-HisPolH, pET-HisNikO, and pET-HisNikJ, respectively. PolH variants with single-point mutations were prepared by following the Stratagene’s QuikChange site-directed mutagenesis protocol using the primers shown in Supplementary Table 7 and pET-HisPolH as a template.
Expression of WT-PolH, WT-NikJ and PolH variants
E. coli BL21 (DE3) cells were co-transformed with pET28b with the nikJ, polH or polH variant gene and a plasmid containing the Suf proteins (SufA, SufB, SufC, SufD, SufS, and SufE)22. Transformants were grown overnight at 37 °C on LB-Agar plates containing chloramphenicol (35 µg/mL) and kanamycin (50 µg/mL). All subsequent growths contain the same selection markers. A single colony was picked and grown in LB media (5 mL) until saturation at 37 °C. The saturated culture was diluted into LB media (250 mL) and grown until saturation. A portion (35 mL) of this saturated culture was used to inoculate 3.5 L of LB media supplemented with 35 mg of ferric chloride and 85 mg of L-cysteine in a 4.0 L Erlenmyer flask. Cells were grown aerobically at 37 °C with rigorous shaking (200 rpm). When the OD600 reached 0.8–1.0, the culture was cooled to 15 °C, and protein expression was induced with 0.25 mM IPTG. Then, the flasks were flushed with Ar gas, and sealed with a rubber plug. The culture was continued anaerobically at 15 °C with slow shaking (100 rpm) for 20 hours. Cells were harvested by centrifugation, frozen in liquid N2, and stored at −80 °C. Typically, 1.5–2.0 g of wet cell paste/L was obtained.
Purification and reconstitution of WT-PolH, WT-NikJ and PolH variants
WT-PolH was anaerobically purified typically from 10–30 g of a cell paste of E. coli BL21(DE3) harboring pET-HisPolH. The cell pellet was brought into an mBraun anaerobic glove box ([O2] < 0.1 ppm). Then, each gram of cell pellet was suspended and homogenized in 4 mL of anaerobic buffer A (50 mM Tris-HCl pH 7.6, 300 mM NaCl, and 10% (v/v) glycerol) supplemented with 3 mM β-mercaptoethanol (β-ME). The cell suspension was brought out of the glovebox, and lysed by 2 passages through a French pressure cell operating at 14,000 psi under constant Ar flow. The resulting lysate was cleared by centrifugation (20,000 g, 20 min, 4 °C) using centrifugal tubes filled with Ar gas, and brought back to the glovebox. All subsequent purification steps were carried out under strict anaerobic conditions in the glove box ([O2] < 0.1 ppm) maintained at 10 °C. The dark brown-colored supernatant was incubated with Ni-NTA agarose resin (Qiagen, 20 mL equilibrated in buffer A containing 40 mM imidazole and 3 mM β-ME) for 1 hour. The resin was washed with 10 volume of buffer A with 20 mM imidazole and 3 mM β-ME, and the protein eluted with buffer A with 400 mM imidazole and 3 mM β-ME. Fractions containing dark brown colored PolH were exchanged into buffer A containing 5 mM DTT using a Sephadex G-25 column. The concentration of PolH was determined on the basis of UV absorption at 280 nm using an extinction coefficient (ε280nm = 64.5 mM−1 cm−1) determined by the Edelhoch’s method23. The amounts of Fe2+/3+ and S2− were quantified by following published protocols24,25. Typically, the preparation yielded PolH with 1.5 ± 0.2 Fe per monomer. The resulting purified PolH was immediately used for anaerobic reconstitution carried out at 10 °C by adding FeII(NH4)2(SO4)2 and Na2S slowly over the course of 10 min. The amounts of FeII(NH4)2(SO4)2 and Na2S were adjusted based on the amounts of Fe2+/3+ and S2− associated with the as-isolated PolH, so that the final concentrations of Fe2+/3+ and S2− becomes 6 eq. per PolH monomer. The resulting mixture was incubated for 60 min at 10 °C. The protein was then desalted using a Sephadex G-25 column equilibrated with buffer A. The amounts of Fe and sulfide were determined by the ferrozine assay24 and sulfide assay25, respectively. The resultant protein solution (~150 µM) was frozen in liquid N2, and stored at −80 °C. WT-NikJ and PolH variants were purified in identical manner.
Anaerobic Size Exclusion Chromatography
NikJ and PolH (200 µL of 60 µM) were anaerobically analyzed at 4 °C by an FPLC system (AKTA purifier, GE Healthcare Life Science) equipped with a Superdex 200 10/300 GL column (GE Healthcare Life Science, Part Number # 17-5175-01). The column was washed with 10 CV of anaerobic buffer A supplemented with 5 mM DTT and 5 mM sodium dithionite, and then equilibrated in anaerobic buffer A supplemented with 5 mM DTT and 5 µM sodium dithionite. To avoid oxygen contamination, these buffers were kept under a constant flow of Ar throughout the experiments. The chromatography was performed in buffer A supplemented with 5 mM DTT and 5 µM sodium dithionite with a flow rate of 0.4 mL/min, and the elution was monitored by absorbance at 280 nm. Elution containing NikJ or PolH were collected under the flow of Ar gas and analyzed by SDS-PAGE. The resulting proteins were characterized for the amount of Fe by the ferrozine assay,24 and for their catalytic functions by the PolH/NikJ activity assay (see below). Molecular weight of NikJ and PolH were determined using gel filtration standards (Bio-Rad, Part Number 151-1901).
Activity Assay of PolH and NikJ
PolH or NikJ (2.5 µM) was anaerobically incubated with nucleoside/nucleotide substrate candidates (EP-UMP, EP-uridine, UMP or uridine; 100 µM) in the presence of SAM (100 µM) and sodium dithionite (5 mM) in 300 µL buffer A supplemented with 5 mM DTT for 60 min at 25 °C. The reaction was initiated by adding PolH. After 10, 20, 30, 40, and 60 min of incubation, an aliquot (45 µL) was removed and mixed with 5 µL of 25% TCA to quench the reaction. After removal of precipitation by centrifugation, an aliquot (40 µL) of the supernatant was injected to HPLC equipped with an Alltech Apollo C18 column (4.6 × 250 mm, 2.7 µm, Part No #36511) equilibrated in 0.1 M KH2PO4, pH 6.0, 8 mM tetrabutylammonium hydrogen sulfate (Solvent A). The elution was made with a flow rate at 1.0 mL/min using solvent A and solvent B (0.1 M KH2PO4, pH 6.0, 8 mM tetrabutylammonium hydrogen sulfate, 30% (v/v) ACN): 0% B for 3 min, 0–100% B for 3–18 min. Chromatography was monitored by the L-2455 diode array detector.
Steady state kinetic characterization of PolH and NikJ
For steady-state kinetic analysis, substrate and enzyme concentrations described in the legend of Supplementary Fig. 14 were used. Briefly, PolH or NikJ (1 µM) was incubated with a fixed concentration (250 µM) of one of the substrates (SAM or EP-UMP) and varied concentrations (5 – 100 µM) of the other. Under these conditions, the effective concentration of NikJ/PolH was 0.39 µM, based on the EPR observation that 39% of PolH has catalytically competent [4Fe-4S]+. Other conditions were identical to above described NikJ/PolH assays. The amount of U-OAP was quantified by injecting an aliquot (40 µL) of the supernatant of the quenched solution into an HPLC system equipped with a Hypersil GOLD SAX column (4.6 × 150 mm, 3.0 µm) equilibrated in water (Solvent A). The elution was made with a flow rate at 1.5 mL/min using solvent A and solvent B (1 M ammonium phosphate pH 3.1): 2% B for 3 min, 2–15% B for 3–20 min. Chromatography was monitored by the L-2455 diode array detector. The lower limit of detection of U-OAP was 20 pmol (0.5 µM in 40 µL injection). In each assay, we monitored formation of at least 0.5 µM U-OAP, which is more than a single turnover, and determined the initial velocity based on the linear correlation between the amount of U-OAP and incubation time. The substrate consumption was less than 20% in all conditions, which allowed us to apply the initial velocity assumption. The initial velocity was plotted against the substrate concentration, and analyzed by non-linear curve fitting to the Michaelis-Menten equation (Supplementary Fig. 14).
Expression of PolD and PolK
PolD and PolK were expressed using E. coli BL21(DE3) transformed with pET28b-HisPolD or with pET28b-HisPolK. Transformants were grown overnight at 37 °C on LB-Agar plates containing kanamycin (50 µg/mL). All subsequent growths contain kanamycin (50 µg/mL). A single colony was picked and grown in LB media (5 mL) until saturation at 37 °C. The saturated growth solution was diluted into LB media (250 mL) and grown until saturation. A portion (15 mL) of this saturated culture was used to inoculate 1.5 L of LB media supplemented with 15 mg of ferric chloride. Cells were grown aerobically at 37 °C with rigorous shaking (180 rpm). When the OD600 reached 0.6–0.8, the culture was cooled to 15 °C, the protein expression was induced with 0.25 mM IPTG. The culture was continued at 15 °C for 20 hours. Cells were harvested by centrifugation, frozen in liquid N2, and stored at −80 °C. Typically, 3–4 g of wet cell paste/L was obtained.
Purification and Reconstitution of PolD and PolK
PolD and PolK were expressed and purified using essentially identical procedures. The cell pellet of E. coli BL21(DE3) expressing PolD or PolK was brought into the glovebox. Each gram of cell pellet was suspended and homogenized in 4 mL of buffer A supplemented with 3 mM β-ME. The cell suspension was brought out of the glovebox, and lysed by two passages through a French pressure cell operating at 14,000 psi under continuous Ar gas flow. The resulting lysate was cleared by centrifugation (20,000 g, 20 min, 4 °C) using centrifugal tubes filled with Ar gas, and brought back into the glovebox. All subsequent purification steps were carried out under strict anaerobic conditions in the glove box ([O2] < 0.1 ppm) maintained at 10 °C. The supernatant was incubated with Ni-NTA agarose resin (Qiagen, 20 mL equilibrated in buffer A containing 40 mM imidazole and 3 mM β-ME) for 1 hr. The column was then washed with 10 volumes of buffer A containing 40 mM imidazole and 3 mM β-ME, and the bound protein was subsequently eluted with 400 mM imidazole in buffer A with 3 mM β-ME. Fractions containing PolD (or PolK) were identified by SDS-PAGE, combined, and exchanged into buffer A containing 5 mM DTT and 5 mM α-ketoglutarate (α-KG) on a Sephadex G-25 column. The concentration of PolD and PolK was determined on the basis of UV absorption at 280 nm using an extinction coefficient (PolD: ε280nm = 37.9 mM−1 cm−1; PolK: ε280nm = 36.6 mM−1 cm−1). The resulting purified PolD and PolK were immediately used for anaerobic reconstitution carried out at 10 °C by adding 2 eq. of FeII(NH4)2(SO4)2 slowly over the course of 10 min followed by incubation for an additional 60 min. The enzymes were then exchanged into buffer A containing 5 mM α-KG, by Sephadex G-25 column chromatography. The resulting protein solutions (~300 µM PolD; ~50 µM PolK) were aliquoted, frozen in liquid N2, and stored at −80 °C.
Activity Assay for PolD and PolK
PolD or PolK (10 µM) was anaerobically incubated with 100 µM of UMP, Uridine, EP-UMP, or EP-Uridine in 100 µL buffer A supplemented with α-KG (5 mM), sodium ascorbate (5 mM) and FeII(NH4)2(SO4)2 (2 eq. of PolD or PolK) for 15 min at 25 °C. The mixtures were taken out of the glovebox and the reactions initiated by adding 200 µL of oxygen saturated buffer A with 5 mM α-KG. After 10, 20, 30, 40, and 80 min of incubation at 25 °C, aliquots (45 µL each) were taken out from the assay mixture, and mixed with 5 µL of 25% TCA. After centrifugal removal of precipitation, an aliquot (40 µL) of the supernatant was injected onto and analyzed by HPLC. The assays with nucleotide substrates were analyzed using a Hypersil GOLD SAX column (4.6 × 150 mm, 3.0 µm) equilibrated in water (Solvent A). The elution was made with a flow rate at 1.5 mL/min using solvent A and solvent B (1 M ammonium phosphate pH 3.1): 0% B for 3 min, 0–20% B for 3–20 min. The assays with nucleoside substrates were analyzed using the ODS-Hypersil C-18 column (4.6 × 150 mm, 3.0 µm) equilibrated in 0.1% TFA (Solvent A). The elution was made with a flow rate at 1.0 mL/min using solvent A and solvent B (Methanol): 0% B for 3 min, 0–100% B for 3–17.5 min. In all cases, chromatography was monitored by the L-2455 diode array detector.
To detect succinate formation, the assays were performed with PolD or PolK (75 µM) in the presence of α-KG (5 mM), FeII(NH4)2(SO4)2 (150 µM) and sodium ascorbate (5 mM) in the oxygen saturated buffer A at 25 °C for 40 min. An aliquot (45 µL) of the reaction was then quenched with 5 µL of 25% TCA, and centrifuged to remove protein precipitation. The resulting supernatant (40 µL) was neutralized by adding 360 µL of 100 mM Tris base, and treated with SP sepharose cation exchange resin (NH4+ form) to remove Tris base. The resulting samples were analyzed by HR-ESI-TOF-MS equipped with a Poroshell HILIC column (4.6 × 100 mm, 2.7 µm) equilibrated in 95% ACN (Solvent A) and 5% Ammonium Acetate pH 6.8 (Solvent B). The elution was made with a flow rate at 0.4 mL/min using solvent A and solvent B: 5–70% B min from 0–12 min. All mass spectroscopy experiments were performed in positive ion mode. Calculated [M+H]+ 119.0339, [M+NH4]+ 136.0604; observed [M+H]+ 119.0351, [M+NH4]+ 136.0617.
Stoichiometry of reactions catalyzed by WT- and C209A-PolH
Enzyme reactions were performed and quenched as described for the WT PolH activity assay. After the TCA quenching, the solution was clarified by centrifugation, and an aliquot (40 µL) of the supernatant was injected to HPLC. Chromatography was performed on an Alltech Apollo C18 column under conditions described for the WT PolH activity assay. Chromatography was monitored by the L-2455 diode array detector.
EPR Measurements
WT-, C209S-, and C209A-PolH (100 µM) were anaerobically pre-reduced with sodium dithionite (5 mM) at 10 °C for 60 min in buffer A. To the resulting solution, EP-UMP or [3′′-2H]-EP-UMP (500 µM) and SAM (500 µM) were added and incubated for 30, 45, 60, and 120 s at 25 °C. At each time point, aliquots (300 µL) of the reaction mixture were transferred to EPR tubes and flash frozen by hand in liquid nitrogen. The EPR spectra for [4Fe-4S]+ and C7′• were determined at 15 K and 77 K, respectively. EPR characterization of all the other PolH variants were carried out under identical conditions. EPR parameters were a microwave frequency of 9.38 – 9.44 GHz; a power of 5.0 µW and 6.3 mW for C7′• and [4Fe-4S]+, respectively; modulation amplitudes of 0.5 and 10 G for C7′• and [4Fe-4S]+, respectively; modulation frequencies of 60 and 100 kHz for C7′• and [4Fe-4S]+, respectively; time constants of 0.04 and 0.01 ms for C7′• and [4Fe-4S]+, respectively; a scan time of 30 s, and conversion time of 15 and 25 ms for C7′• and [4Fe-4S]+, respectively. EPR spin quantitation was carried out using Cu(II)SO4 as a standard.26 All EPR spectral simulations were performed using EasySpin software.27
Supplementary Material
Acknowledgments
This work was supported by the Duke University Medical Center and National Institute of General Medical Sciences R01 GM115729 (to K. Y.). We thank George R. Dubay (Duke University, Department of Chemistry) for assistance on the MS measurements. We thank Dr. Anthony A. Ribeiro at the Duke NMR center for the assistance in collecting NMR data. EPR spectrometer was supported by the Institutional Development Grant (ID 37940) from North Carolina Biotechnology Center.
Footnotes
Author contributions
K.Y. conceived the project. K.Y and E.A.L. designed the experiments. E.A.L. performed the experiments. K.Y. and E.A.L. analyzed the data and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Isono K. J Antibiot (Tokyo) 1988;41:1711–1739. doi: 10.7164/antibiotics.41.1711. [DOI] [PubMed] [Google Scholar]
- 2.Munro CA. Adv Appl Microbiol. 2013;83:145–172. doi: 10.1016/B978-0-12-407678-5.00004-0. [DOI] [PubMed] [Google Scholar]
- 3.Reuveni M, Cohen H, Zahavi T, Venezian A. Crop Prot. 2000;19:393–399. [Google Scholar]
- 4.Shubitz LF, et al. J Infect Dis. 2014;209:1949–1954. doi: 10.1093/infdis/jiu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isono K, Sato T, Hirasawa K, Funayama S, Suzuki S. J Am Chem Soc. 1978;100:3937–3939. [Google Scholar]
- 6.Schuz TC, Fiedler HP, Zahner H, Rieck M, Konig WA. J Antibiot. 1992;45:199–206. doi: 10.7164/antibiotics.45.199. [DOI] [PubMed] [Google Scholar]
- 7.Isono K, Crain PF, Mccloskey JA. J Am Chem Soc. 1975;97:943–945. [Google Scholar]
- 8.Chen W, et al. J Biol Chem. 2009;284:10627–10638. doi: 10.1074/jbc.M807534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginj C, Ruegger H, Amrhein N, Macheroux P. Chembiochem. 2005;6:1974–1976. doi: 10.1002/cbic.200500208. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, et al. J Ind Microbiol Biotechnol. 2015;43:401–417. doi: 10.1007/s10295-015-1636-3. [DOI] [PubMed] [Google Scholar]
- 11.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frey PA. Acc Chem Res. 2014;47:540–549. doi: 10.1021/ar400194k. [DOI] [PubMed] [Google Scholar]
- 13.Oberdorfer G, Binter A, Ginj C, Macheroux P, Gruber K. J Biol Chem. 2012;287:31427–31436. doi: 10.1074/jbc.M112.352096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. Nature. 2010;465:891–896. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hover BM, Loksztejn A, Ribeiro AA, Yokoyama K. J Am Chem Soc. 2013;135:7019–7032. doi: 10.1021/ja401781t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahanta N, Fedoseyenko D, Dairi T, Begley TP. J Am Chem Soc. 2013;135:15318–15321. doi: 10.1021/ja408594p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kudo F, Hoshi S, Kawashima T, Kamachi T, Eguchi T. J Am Chem Soc. 2014;136:13909–13915. doi: 10.1021/ja507759f. [DOI] [PubMed] [Google Scholar]
- 19.Wyszynski FJ, et al. Nat Chem. 2012;4:539–546. doi: 10.1038/nchem.1351. [DOI] [PubMed] [Google Scholar]
- 20.Yang ZY, et al. J. Biol. Chem. 2011;286:7885–7892. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnard-Britson S, et al. J Am Chem Soc. 2012;134:18514–18517. doi: 10.1021/ja308185q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods Only
- 23.Edelhoch H. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 24.Fish WW. Method Enzymol. 1988;158:357–364. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 25.Beinert H. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 26.Palmer G. Method Enzymol. 1967;10:594–609. [Google Scholar]
- 27.Stoll S, Schweiger A. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.