

Abstract
Two novel oral drugs that target androgen signaling have recently become available for the treatment of metastatic castration-resistant prostate cancer (mCRPC). Abiraterone acetate inhibits the synthesis of the natural ligands of the androgen receptor, whereas enzalutamide directly inhibits the androgen receptor by several mechanisms. Abiraterone acetate and enzalutamide appear to be equally effective for patients with mCRPC pre- and postchemotherapy. Rational decision making for either one of these drugs is therefore potentially driven by individual patient characteristics. In this review, an overview of the pharmacokinetic characteristics is given for both drugs and potential and proven drug–drug interactions are presented. Additionally, the effect of patient-related factors on drug disposition are summarized and the limited data on the exposure–response relationships are described. The most important pharmacological feature of enzalutamide that needs to be recognized is its capacity to induce several key enzymes in drug metabolism. The potency to cause drug–drug interactions needs to be addressed in patients who are treated with multiple drugs simultaneously. Abiraterone has a much smaller drug–drug interaction potential; however, it is poorly absorbed, which is affected by food intake, and a large interpatient variability in drug exposure is observed. Dose reductions of abiraterone or, alternatively, the selection of enzalutamide, should be considered in patients with hepatic dysfunction. Understanding the pharmacological characteristics and challenges of both drugs could facilitate decision making for either one of the drugs.
Key Points
| Understanding the pharmacology of abiraterone and enzalutamide could facilitate rational therapeutic decision making for either one of the drugs based on patient-specific factors. |
| Abiraterone bioavailability is low and is majorly affected by food intake. |
| Enzalutamide affects the activity of multiple hepatic enzymes and is therefore prone to cause drug interactions. |
Introduction
Prostate cancer is the second most commonly diagnosed cancer in men and one of the most common causes of death related to cancer in men [1]. After primary treatment with chemical or surgical castration, most patients progress to a state of the disease termed metastatic castration-resistant prostate cancer (mCRPC), with a median survival of <2 years [2]. Until recently, only docetaxel chemotherapy provided prolongation of survival in this stage of the disease [3]. The understanding of the remaining dependency of prostate cancer on the androgen receptor pathway was an incentive to develop targeted therapies for extra-gonadal androgen signaling [4–6]. Two novel oral androgen receptor targeted drugs, abiraterone acetate and enzalutamide, have recently become available and can substantially prolong survival [5, 7–10].
Abiraterone acetate (Zytiga®), a prodrug of abiraterone, is a selective and irreversible blocker of the cytochrome P450 (CYP) C17 enzyme (CYP17), a crucial enzyme in testosterone and estrogen synthesis, resulting in virtually undetectable serum and intratumoral androgen levels [11, 12]. Abiraterone acetate is registered in combination with low-dose prednisolone because the inhibition of CYP17 decreases the production of endogenous glucocorticoids.
Enzalutamide (Xtandi®) is a potent androgen signaling receptor inhibitor and acts in multiple ways on the androgen pathway. Enzalutamide (1) competitively inhibits the binding of androgen to the androgen receptor; (2) inhibits nuclear translocation and recruitment of cofactors; and (3) inhibits the association of the activated androgen receptor with DNA [6]. The active metabolite N-desmethyl enzalutamide is equipotent to the parent compound in vitro, and is present in plasma at the same concentration level as enzalutamide, which may add to the antitumor activity of enzalutamide in patients treated with this drug [13].
Both of these novel drugs are administered orally once daily in a fixed dose, are well tolerated [14, 15], and appear to be equipotent in the pre- and postchemotherapy setting [7–10, 16]. Since these drugs show comparable efficacy, other characteristics might drive rational therapeutic decision making. A better understanding of the pharmacology of these drugs in relation to awareness of patient-specific conditions (e.g. comedication, comorbidities) could facilitate the choice for either one of these drugs.
The aim of this review is to give an overview of the pharmacokinetic characteristics of abiraterone acetate and enzalutamide. Moreover, results from additional studies on potential drug–drug interactions, the involvement of drug transporters, and data on the exposure–response relationship have been incorporated in this overview. Finally, the effect of specific patient characteristics on drug disposition will be discussed.
Search Strategy
PubMed and EMBASE searches were performed using the following search terms: ‘Abiraterone OR Zytiga OR abiraterone acetate OR CB7630’ OR ‘Enzalutamide OR MDV3100 OR Xtandi OR N-desmethylenzalutamide’ in combination with ‘Excretion’ OR ‘Distribution’ OR ‘Absorption’ OR ‘Bioavailability’ OR ‘availability’ OR ‘pharmacokinetics’ OR ‘metabolism’ OR ‘biotransformation’ OR ‘cytochrome p450 enzyme’ OR ‘drug–drug interaction’ OR ‘transporter’. In addition, citation snowballing was used to find other relevant studies. Registration information from the European Medicines Agency (EMA) and the US FDA was used, and results reported in ClinicalTrials.gov were screened. Results were limited to studies in humans and English language full-text articles published until 22 February 2016.
Our search revealed 491 hits: 142 articles did not contain pharmacokinetic data, 170 articles were reviews that did not obtain original data, 18 articles were not accessible through our library, 35 articles were not in English, 40 articles described data not in humans, 39 articles represented expert opinions, letters, and editorials, and 25 articles did not contain data on abiraterone acetate and enzalutamide. The remaining 22 original articles regarding the pharmacokinetics of enzalutamide and abiraterone were included in this study. For both drugs, the most recent drug label of the EMA and FDA was included.
An overview of the pharmacokinetic properties of abiraterone, enzalutamide and N-desmethyl enzalutamide is presented in Table 1, while Table 2 describes the effect of renal and hepatic impairment. Table 3 shows an overview of hepatic enzymes that are involved in the metabolism of abiraterone and enzalutamide, as well as hepatic enzymes that are inhibited or induced by these drugs. Tables 4 and 5 summarize drug–drug interactions, and Table 6 shows examples of frequently used drugs that are potentially affected by enzalutamide and abiraterone. Figures 1 and 2 show the metabolism of abiraterone acetate and enzalutamide, respectively.
Table 1.
Pharmacokinetic parameters
| F [%] | Protein binding [%] |
t max [h] | t ½ [h] | AUC [ng h/mL] (CV %) | Vd/F (L) | CL/F [L/h] (CV %) | C trough [ng/mL] | C max [ng/mL] | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| Abiraterone | NA | ~99.8 | 2 | 24 | 993a (64) | 5620 | 1198 (65) | 11.1 | 226 | [4, 17, 20] |
| Enzalutamide | NA | ~98 | 1–2 | 5.8 days | 322,000a (26.6) | 110 | 0.55 (18) | 11,400 | 16,600 | [13, 15, 20, 31] |
| N-desmethyl enzalutamide | NA | ~95 | 4 | 7.8 days | 278,000a (30.7) | NA | NA | 13,000 | 12,700 | [13, 15, 20, 31] |
F absolute bioavailability, t max median time to peak concentration, t ½ mean elimination half-life, AUC mean area under the concentration–time curve from time zero to 24 h, Vd/F mean apparent volume of distribution, CL/F apparent oral clearance, C trough mean minimum concentration, C max mean maximum concentration, NA not available, CV % percentage coefficient of variation
aAUC24 for abiraterone and AUCtau for enzalutamide
Table 2.
Effect of renal and hepatic impairment on the pharmacokinetics of abiraterone acetate and enzalutamide
| Hepatic impairment | Renal impairment | References | |||||
|---|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Mild | Moderate | Severe | ||
| Abiraterone | AUCinf increased (111)a | AUCinf increased (357)a | DN AUCInf increased (614)*,a | NAb | NAb | AUCinf no changec | [14, 21] |
| Enzalutamide and N-desmethyl enzalutamide | AUCinf increased (14 %) | AUCinf increased (14 %) | AUCinf increased (34 %) | No changed | No changed | NA | [15, 20, 31] |
AUC inf area under the concentration–time curve from time zero to infinity, DN dose normalized, NA not available
* Dose normalized; in this study, patients with severe liver impairment received 1/16th of the dose of healthy volunteers: 125 vs. 2000 mg of a suspension
aEstimated geomatric mean ratios
bNot studied since patients with severe renal impairment did not show exposure more than twice the normal exposure
cEnd-stage renal disease patients who required hemodialysis
dCalculated in a post hoc population pharmacokinetic analysis
Table 3.
Enzymes and transporters involved in pharmacokinetics
| Substrate of: | Effector of: | References | |||
|---|---|---|---|---|---|
| Enzyme phase I | Enzyme phase II | Inhibitor | Inducer | ||
| Abiraterone | CYP3A4 (minor) | SULT2A1 (major) | CYP2D6 (strong) CYP2C8 (moderate) CYP1A2 (weak) |
[14, 18, 22] | |
| Enzalutamide | CYP2C8 CYP3A4/5 |
CYP2C8 (weak) P-gp (in vitro) |
CYP3A4 (strong) CYP2C19(moderate) CYP2C9 (moderate) CYP2D6 (moderate) CYP1A2 (weak) |
[30, 32] | |
| N-desmethyl enzalutamide | CYP2C8 CYP3A4 |
Unknown | Unknown | Unknown | [32] |
CYP cytochrome P450, SULT2A1 sulfotransferase 2A1, P-gp P-glycoprotein
Table 4.
Drugs that interact with abiraterone or enzalutamide
| Inducing drug | Inhibitory drug | Effects observed | Authors’ recommendations | References | |
|---|---|---|---|---|---|
| Abiraterone | Rifampicin | AUCinf decreased (45 %) | Avoid combination if possible If not possible, the dose of abiraterone acetate should be increased to 1000 mg bid |
[14, 22, 25] | |
| Ketoconazole | AUCinf increased (15 %) | Not clinically relevant | [14, 22, 25] | ||
| Enzalutamide + N-desmethyl enzalutamide | Rifampicin | AUCinf decreased (37 %) | Switch to alternatives that are not inducers of CYP3A4 If not possible, the dose of enzalutamide should be increased to 240 mg once daily |
[15, 20] | |
| Itraconazole | AUCinf increased (1.3-fold) | Not clinically relevant | [15, 20, 32] | ||
| Gemfibrozil | AUCinf increased (2.2-fold) | Switch to alternatives that are not inhibitors of CYP2C8 If not possible, the dose of enzalutamide should be reduced to 80 mg once daily |
[15, 20, 32] |
AUC inf area under the concentration–time curve from time zero to infinity, bid twice daily
Table 5.
Effect of abiraterone and enzalutamide on coadministrated drugs
| Drugs | Enzyme | Effects observed | Authors’ recommendations | References | ||
|---|---|---|---|---|---|---|
| Abiraterone Acetate | Dextromethorphan | CYP2D6 | AUC24 increased (2.9-fold) | If possible, switch to alternatives that are not metabolized through CYP2D6 Be aware that substrates of CYP2D6 are presumably more active. Be aware of side effects and toxicity. Consider dose reduction of concomitant CYP2D6 substrate with at least twofold Avoid use of CYP2D6 substrates with a narrow therapeutic index |
[14, 22, 26] | |
| Theophylline | CYP1A2 | No effect | Not clinically relevant: no change in medication is required | [14, 22, 26] | ||
| Pioglitazone | CYP2C8 | AUC24 increased (46 %) | If possible, switch to alternatives that are not metabolized through CYP2C8 Be aware that substrates of CYP2C8 are presumably more active. Be aware of side effects and toxicity Avoid use of CYP2C8 substrates with a narrow therapeutic index |
[14, 22] | ||
| Enzalutamide | Pioglitazone | CYP2C8 | AUCinf increased (20 %) | Not clinically relevant: no change in medication is required | [15, 20, 32] | |
| S-Warfarin | CYP2C9 | AUCinf decreased (56 %) | If possible, switch to alternatives that are not metabolized through CYP2C9 Be aware that substrates of CYP2C9 are presumably less active and elevate the dose based on efficacy Avoid use of CYP2C9 substrates with a narrow therapeutic index |
[15, 20, 32] | ||
| Omeprazole | CYP2C19 | AUCinf decreased (70 %) | If possible, switch to alternatives that are not metabolized through CYP2C19 Be aware that substrates of CYP2C19 are less active and elevate the dose based on efficacy Avoid use of CYP2C19 substrates with a narrow therapeutic index |
[15, 20, 32] | ||
| Midazolam | CYP3A4 | AUCinf decreased (86 %) | If possible, switch to alternatives that are not metabolized through CYP3A4 Be aware that substrates of CYP3A4 are substantially less active. The dose of CYP3A4 substrates should be adjusted to achieve comparable efficacy Avoid use of CYP3A4 substrates |
[15, 20, 32] | ||
| Dextromethorphan | CYP2D6 | AUCinf decreased (31 %) | Be aware that substrates of CYP2D6 are moderately less active Avoid concomitant use of CYP2D6 substrates with a narrow therapeutic index |
[15, 20] | ||
| Caffeine | CYP1A2 | AUCinf decreased (11 %) | Not clinically relevant: no change in medication is required | [15, 20] | ||
CYP cytochrome P450, AUC 24 area under the concentration–time curve from time zero to 24 h
Table 6.
Examples of drugs subject to drug–drug interactions with enzalutamide and abiraterone
| Substrates | Examples of potentially affected drugs | Expected effect of enzalutamide | Expected effect of abiraterone | References |
|---|---|---|---|---|
| CYP1A2 substrates | Duloxetine, clozapine | No relevant effect | No relevant effect | [24] |
| CYP2C8 substrate | Repaglinide | No relevant effect | AUC increase | [24] |
| CYP2C9 substrates | Warfarin, acenocoumarol, losartan, diclofenac, tolbutamide | AUC decrease | NA | [24] |
| CYP2C19 substrates | Omeprazole, esomeprazole, clopidogrel, citalopram, diazepam | AUC decrease | NA | [24] |
| CYP2D6 substrates | Oxycodone (into highly active metabolite), metoprolol, haloperidol, flecainide, paroxetine | AUC decrease | AUC increase | [24] |
| CYP3A4 substrates | Oxycodone (into less active metabolite), methadone, ticagrelor, simvastatin, nifedipine, fentanyl, St John’s Wort | AUC decrease | NA | [24] |
The presented list of examples is not inexhaustive, and the extent of the effect on the probe substrate is described in Table 4
CYP cytochrome P450, AUC area under the concentration–time curve, NA not available
Fig. 1.
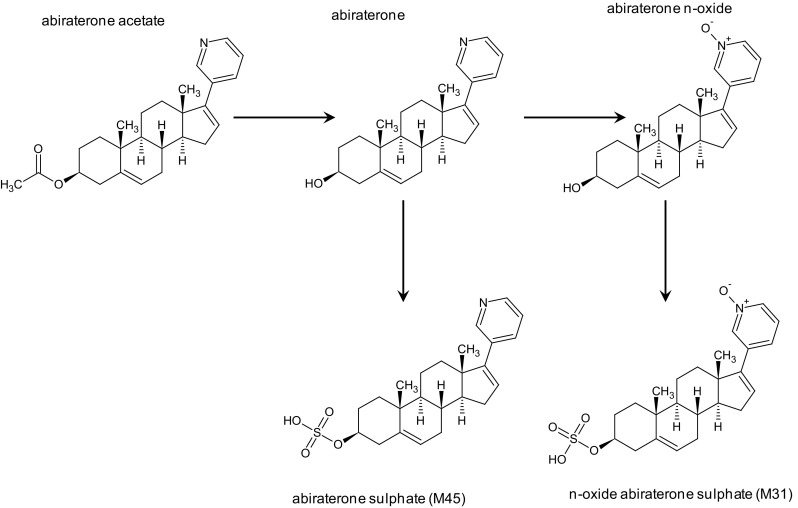
Abiraterone acetate metabolism. The prodrug abiraterone acetate is readily converted to abiraterone through hydrolysis. Thereafter, abiraterone is extensively metabolized through several pathways, primarily by SULT2A1 to abiraterone sulphate, and by SULT2A1 and CYP3A4 to n-oxide abiraterone sulphate. SULT2A1 sulfotransferase 2A1, CYP cytochrome P450
Fig. 2.
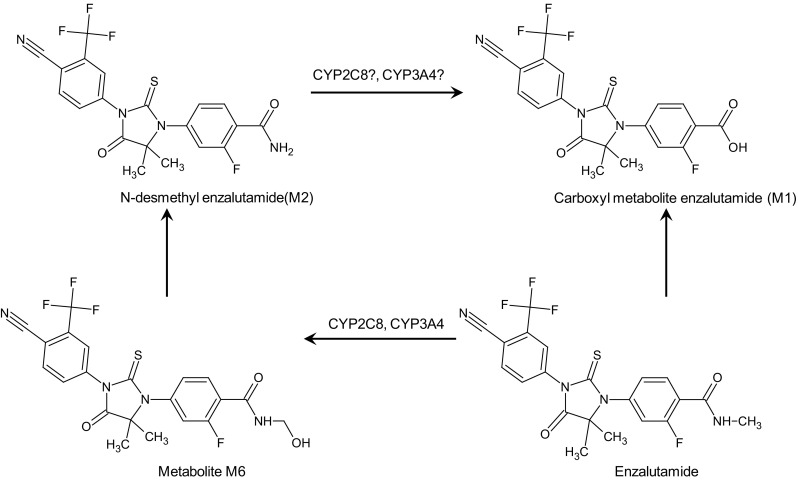
Enzalutamide metabolism. The proposed pathway to form N-desmethyl enzalutamide is via M6 and M1 through CYP2C8 and CYP3A4 metabolism. N-desmethyl enzalutamide is metabolized by carboxylesterase 1 to the carboxyl metabolite; no CYP enzymes involvement in further metabolism were identified. CYP cytochrome P450
Abiraterone Acetate
Absorption
The prodrug abiraterone acetate is rapidly hydrolyzed into abiraterone in vivo. No abiraterone acetate could be detected in plasma in patients treated with abiraterone acetate [12]; therefore, only abiraterone will be responsible for the clinical activity. After a median of 2 h, the maximum plasma concentration (C max) of abiraterone was achieved (Table 1) [17]. Abiraterone acetate is classified as a Biopharmaceutics Classification System (BCS) class IV drug due to its low solubility and low permeability characteristics. The solubility of abiraterone acetate rapidly declines at pH >1 [17]. The absolute bioavailability of this agent has not been studied; however, results from the mass balance study after intake of abiraterone acetate on an empty stomach suggest that bioavailability can be 50 % at its maximum since unchanged abiraterone acetate accounted for 50 % of the drug recovered in feces [18].
In a relative bioavailability study, a formulation of abiraterone acetate in olive oil increased exposure 4.5-fold compared with the tablet formulation, indicating that a fatty environment significantly improves bioavailability [17]. These findings support the major food effect on abiraterone exposure. In a single-dose study of abiraterone in healthy volunteers, the area under the plasma concentration–time curve (AUC) and C max increased 10- and 17-fold after a high-fat meal, respectively, and sevenfold and fivefold after a low-fat meal compared with intake without food, respectively [17]. In cancer patients, a 4.4-fold increase in AUC was observed after intake of abiraterone acetate with a high-fat meal compared with fasted conditions; in this study, a capsule was used instead of the currently registered tablets, which may have affected the outcome [4]. However, in a more recent study in cancer patients, only a twofold increase in exposure was observed with a high-fat meal compared with modified fasting [19]. The origin of this difference in food effect between patients and healthy volunteers is as yet unclear.
Some of the observed large increases in abiraterone exposure with food versus intake on an empty stomach seem to contradict data from the mass balance study. Differences in patient populations, analytical techniques, drug formulation, and types of meals may all contribute to these paradoxical observations.
It is recommended that abiraterone acetate be taken in fasting conditions [14]. A plausible reason for this recommendation is that the composition of meals (especially the fat content) can vary, and this will substantially affect abiraterone exposure.
Distribution
Abiraterone is highly protein bound to both albumin (95.6–99.9 %) and α1-glycoprotein (89.4–95.6 %). It is extensively distributed in tissue, with an apparent central volume of distribution (Vd) of 5630 L [14]. The ratio of whole blood to plasma concentrations of abiraterone was 0.523, which indicates no substantial binding to red blood cells. This observation is supported by in vitro data showing that radioactivity of abiraterone and metabolites was limited to the plasma component of the blood [18]. Animal studies showed that abiraterone passes the blood–brain barrier, however this has not been explored in humans [20].
Metabolism
The prodrug abiraterone acetate is readily converted to abiraterone through hydrolysis; the esterases involved herein are not yet identified [18]. Thereafter, abiraterone is extensively metabolized through several pathways, primarily by sulfotransferase 2A1 (SULT2A1) to abiraterone sulphate (M45), and by SULT2A1 and CYP3A4 to N-oxide abiraterone sulphate (M31) [Fig. 1] [17, 18]. M45 and M31 are the major inactive metabolites and both are responsible for >40 % of drug present in the plasma [18].
Excretion
Abiraterone acetate is primarily excreted in feces. No unchanged abiraterone acetate or abiraterone could be detected in urine, and only approximately 5 % of abiraterone metabolites were recovered in urine, of which 4 % were N-oxide abiraterone sulphate (M45) [18]. In patients undergoing hemodialysis, the exposure of abiraterone was comparable to that of patients with normal renal function, underscoring the minor involvement of the renal elimination pathway [21]; therefore, no dose adjustments are indicated, which is in accordance with the label (Table 2) [14, 22].
Since abiraterone is extensively metabolized by the liver, an effect of hepatic impairment on abiraterone pharmacokinetics is expected. In patients with moderate hepatic impairment (Child–Pugh B), the exposure increased fourfold. In severe hepatic impairment (Child–Pugh C) a 16-fold reduced dose resulted in a decreased exposure of only 56 % compared with healthy controls. The dose-normalized AUC was increased approximately sevenfold in severe hepatic impaired patients compared with healthy controls [21].
Hepatic impairment decreases the elimination of abiraterone, resulting in an increase in elimination half-life of 4.6–5.5 h for patients with mild/moderate liver impairment (Child–Pugh A/B) (Table 2) [21]. Remarkably, in patients with severe hepatic impairment (Child–Pugh C), the half-life only increased by 3.2 h [21].
Abiraterone is contraindicated in patients with severe hepatic impairment [21]. For patients with moderate hepatic impairment, for whom alternative treatments are not an option, a starting dose of 250 mg could be considered. Further dose individualization in this group of patients could be driven by either the side effects or therapeutic drug monitoring of abiraterone.
Something to be aware of is that abiraterone itself can cause elevated liver function tests (ALT, AST, bilirubin) [22].
Drug–Drug Interactions
Abiraterone is a substrate of SULT2A1 and CYP3A4 (Table 3). Concomitant administration with rifampicin, a strong inducer of CYP3A4 enzymes as well as sulfation enzymes, decreased exposure to abiraterone by 55 % [23–25], while ketoconazole, a strong CYP3A4 inhibitor, increased exposure to abiraterone by only 15 %, which was not considered clinically relevant (Table 4) [25]. This discrepancy could be explained by the major role of SULT2A1 in the metabolism of abiraterone. The EMA and FDA accepted different labeling recommendations regarding coadministration of abiraterone with substrates of CYP3A4. Both agencies advise avoiding coadministration of abiraterone with strong CYP3A4 inducers. In case avoidance is not possible, the US label also advises increasing the dosing frequency to twice daily when coadministered with a CYP3A4 inducer. Although this is not based on clinical data (Table 4) [14, 22], in our opinion the dose should be elevated when abiraterone acetate is used together with strong CYP3A4 inducers. Measurement of drug concentrations of abiraterone could support the clinician in these situations.
In vitro studies indicated that abiraterone is a strong inhibitor of CYP2D6 and CYP1A2, and a moderate inhibitor of CYP3A4, CYP2C9, and CYP2C19 [17]; no further in vivo drug interaction studies were conducted for the CYP3A4, CYP2C9, and CYP2C19 substrates. A study in patients treated with abiraterone acetate showed a 2.9-fold higher exposure to the CYP2D6 substrate dextromethorphan, confirming that abiraterone is a strong CYP2D6 inhibitor; however, coadministration of the CYP1A2 substrate theophylline with abiraterone did not alter theophylline exposure [26]. Finally coadministration with the CYP2C8 substrate pioglitazone increased pioglitazone exposure by 46 % [14, 22]. Based on these data, coadministration of abiraterone with CYP2D6 substrates should be avoided when possible. If treatment with the CYP2D6 substrate cannot be interrupted, the dose should be reduced on forehand for substrates with a narrow therapeutic index. Careful monitoring of side effects is advised for drugs with a broader therapeutic index [14, 22]. Since the effect of abiraterone on CYP2C8 is more modest, only coadministered CYP2C8 substrates with a narrow therapeutic index should be monitored more closely and dose adjusted accordingly (Table 5) [14, 22]. Examples of drugs that are potentially subject to drug–drug interactions with abiraterone are described in Table 6.
Acid-reducing agents may affect abiraterone exposure since the solubility of abiraterone acetate decreased rapidly as pH increases >1 [17]. No formal interaction studies have been conducted to investigate the effect of acid-reducing agents on abiraterone exposure.
Transporters
In vitro studies show that abiraterone acetate and abiraterone are not substrates for the efflux transporter P-glycoprotein (P-gp) [17]. Additionally, abiraterone showed little inhibitory effect on P-gp [17]; therefore no clinically relevant interactions with P-gp substrates are expected. The two major metabolites abiraterone sulphate and N-oxide abiraterone sulphate showed in vitro inhibition of the hepatic uptake transporter organic anion transporting polypeptide B1 (OATP1B1) [27], which could imply potential interactions with drugs that are OATP substrates (Table 6); however, no information is available to confirm drug–drug interactions attributable to the inhibition or induction of transporters [14].
Patient Characteristics
No formal studies have been conducted to assess the influence of age, weight, height, or genetic polymorphisms on drug exposure and treatment response [17]; however, in a post hoc population pharmacokinetic analysis, concomitant food intake, and mild and moderate liver impairment were identified as relevant covariates that affect abiraterone exposure [28]. Other covariates (age, testosterone, body mass index, and total plasma protein) showed no significant effect on abiraterone exposure [28]. Ethnicity (Asian vs. Caucasian) does not appear to affect the pharmacokinetics of abiraterone [29].
Exposure–Effect Relationships
An association was shown between trough abiraterone concentrations and decreased Prostate Specific Antigen (PSA) and increased survival [14]. Large interpatient variability in abiraterone exposure was observed (40.5–140.6 %) [14]; therefore, hypothetically, a subgroup of patients will be undertreated at the normal dose of 1000 mg once daily, and monitoring of C trough levels of abiraterone may be a tool for identifying this subgroup of patients. A better understanding of abiraterone pharmacokinetics might help to optimize the dose of the individual patient. For example, knowledge of the food effect in patients, the impact of dose increases in patients with suboptimal exposure, as well as the effect of patient characteristics on the pharmacokinetics of abiraterone should be studied since patients in clinical trials are rather homogeneous due to strict exclusion criteria.
Enzalutamide
Absorption
Enzalutamide is rapidly absorbed, with a C max reached after 1–2 h (Table 1) [30]. N-desmethyl enzalutamide (M2) and the carboxylic enzalutamide (M1) are formed slowly, with a median C max in plasma reached after 132 and 96 h, respectively [30]. For enzalutamide, the absolute bioavailability is unknown since no intravenous formulation of this agent is available; however, based on data from the mass balance study, approximately 84 % of enzalutamide appears to be absorbed after oral administration [30]. In order to improve the solubility of this BCS class II substance (indicating high permeability and low solubility), the formulation of a soft capsule with enzalutamide dissolved in caprylocaproyl macrogolglycerides (CCMG) was developed [20]. A moderate food effect was observed on the rate of enzalutamide absorption but not on the extent of absorption [30]. Dose-proportional pharmacokinetics were observed from 40 to 360 mg [15].
Distribution
Enzalutamide is extensively distributed into tissues and is highly protein bound (approximately 97–98 %), which results in an apparent oral distribution volume (Vd/F) of approximately 110 L [20]. The active metabolite N-desmethyl enzalutamide is equally protein bound (95 %) [20]. In the mass balance and biotransformation study, the overall whole blood to plasma ratio was 0.55, which indicates little binding or distribution to red blood cells [30]. Partitioning to the brain was evaluated in rats [31], and penetration in human brain tissue still needs to be explored.
Metabolism
As confirmed in a drug interaction study, in vitro studies indicated that enzalutamide is mainly metabolized by CYP2C8, with minor CYP3A4/5 involvement [32]. In a 14C-enzalutamide mass balance study, a total of seven metabolites were identified [13]. The two main metabolites in circulation are the active N-desmethyl enzalutamide (M2), which in vitro is equally active as enzalutamide, and the inactive carboxylic acid metabolite (M1) (Fig. 2) [15, 20]. The mean steady-state C trough concentrations are similar for enzalutamide and its active metabolite N-desmethyl enzalutamide, and therefore both substances contribute to pharmacological activity. The inactive carboxylic metabolite accounts for approximately 75 % of the exposure [15, 20]. The proposed pathway to form M2 is via M6 and M1, through CYP2C8 and CYP3A4 metabolism (Fig. 2) [32]. N-desmethyl enzalutamide (M2) is metabolized by carboxylesterase 1 to the carboxyl metabolite (M1). No CYP enzymes involved in further metabolism were identified [13].
Excretion
Renal elimination is the major route of excretion. Seventy-one percent of the total dose was recovered in urine, primarily as the carboxyl metabolite (approximately 63 %), and only a trace amount of unchanged parent and metabolites were found in urine and feces [13]. In a post hoc population pharmacokinetic analysis based on pre-existing renal function, no difference in clearance for mild and moderate renal impairment was observed [13] (Table 2). No data are available on pharmacokinetic changes in patients with severe renal impairment.
The influence of mild, moderate, and severe hepatic impairment was evaluated. Enzalutamide and N-desmethyl enzalutamide exposure increased approximately 14 % in patients with mild (Child–Pugh A) and moderate (Child–Pugh B) liver impairment [30], while in severe hepatic impairment (Child–Pugh C), exposure increased 34 %. Nevertheless, in patients with severe hepatic impairment, the elimination half-life of enzalutamide was increased twofold [30]. Considering the marginal effect on exposure, an adjusted starting dose is not required in patients with mild/moderate hepatic impairment (Table 2) [15, 20, 30].
Drug–Drug Interactions
As mentioned previously, enzalutamide is primarily metabolized by CYP2C8 and also, to a lesser extent, CYP3A4 (Table 3). Therefore, an interaction study was conducted to investigate the effect of gemfibrozil, a strong CYP2C8 inhibitor, on the sum exposure of enzalutamide and N-desmethyl enzalutamide, which increased 2.2-fold [32]. The strong CYP3A4 inhibitor itraconazole increased the sum exposure 1.3-fold [32]. An interaction study with the inducer rifampin showed that exposure to enzalutamide decreased by 66 %, and exposure to N-desmethyl enzalutamide increased 15 %. Consequently, the sum exposure decreased only 37 %, which could be explained by the major effect of rifampicin on enzalutamide metabolism (CYP3A4 and CYP2C8), with only limited effect on N-desmethyl enzalutamide, which is mainly metabolized by carboxylesterase [15, 20] (Fig. 1) [ClinicalTrials.gov identifier NCT02138799). Table 4 provides a summary of drug–drug interactions that affect enzalutamide exposure.
Enzalutamide strongly induces CYP3A4 and moderately induces CYP2C19 and CYP2C9. A decrease in exposure of 86, 70, and 56 %, respectively, was observed for the CYP3A4 substrate midazolam, the CYP2C19 substrate omeprazole, and the CYP2C9 substrate S-warfarin when coadministered with enzalutamide [32]. The drug label warns for coadministration of drugs that are substrates of these enzymes, with a narrow therapeutic index [20]. In contrary to the in vitro observation, the induction of CYP2C8 was deemed not clinically significant when the effect of enzalutamide on pioglitazone exposure, a CYP2C8 substrate, was evaluated [32]. Gibbons et al. reported that the pregnane X receptor (PXR) might also be involved as exposure to the hydroxyl metabolites of the tested substrates decreased while elevated exposures were expected. Induction of the UDP glucuronosyltransferase 1 polypeptide A1 (UGT1A1) via PXR might explain this phenomenon since many metabolites are glucuronidated via UGT1A1 [32].
Finally, in vitro studies suggested that enzalutamide is an inhibitor of CYP2B6, CYP1A2, and CYP2D6 [20]. Unexpectedly, in a clinical study with CYP1A2 and CYP2D6 probes, the exposure of the CYP1A2 substrate caffeine was reduced by 11 %. Exposure of dextromethorphan, a CYP2D6 substrate, was also reduced by 31 % due to enzalutamide coadministration. In this study, the metabolites of dextromethorphan were elevated. It was suggested that the induction of CYP3A4 and UGT might contribute to the reduced exposure of dextromethorphan (ClinicalTrials.gov identifier NCT02225093). An additional explanation is that enzalutamide is not a CYP2D6 (or CYP1A2) inhibitor in vivo, but a weak CYP2D6 inducer. Lastly, no data are available on the effect of enzalutamide on CYP2B6 substrates in vivo. On the other hand, this might be of less relevance considering the limited number of CYP2B6 substrates used in clinical practice.
In conclusion, strong CYP2C8 inhibitors and strong CYP3A4 inducers have a clinically relevant effect on exposure of enzalutamide, and enzalutamide dose adjustments are indicated when they are combined. The dosage of enzalutamide should be reduced to 80 mg once daily in combination with strong CYP2C8 inhibitors [15, 20]; in combination with strong CYP3A4 inducers, the FDA and EMA approved conflicting labeling. Both regulatory authorities advise against the use of enzalutamide with strong CYP3A inducers. In contrast to the EMA drug label, the drug label approved by the FDA suggests dose elevation to 240 mg once daily when avoidance is undesirable, although this is not based on clinical data [15, 20]. In our opinion, it could be possible to combine enzalutamide with strong CYP3A inducers when the dose of enzalutamide is elevated to 240 mg once daily and drug levels can be measured. In our opinion, drugs that are a substrate of CYP3A4 should be avoided in combination with enzalutamide. Concomitant use of drugs that are substrate to CYP2C19, CYP2C9, or CYP2D6 might also require dose adjustments due to the gradual loss of efficacy in combination with enzalutamide. In our opinion, coadministered substrates of CYP2C19 and CYP2C9 should be avoided, especially when they have a narrow therapeutic index. If dosing is necessary, awareness of this phenomenon is warranted and doses of substrates should be elevated based on efficacy. CYP2D6 substrates will be moderately less active, and substrates with a narrow therapeutic index should be avoided when possible (Table 5). Examples of drugs that are potentially subject to drug–drug interactions with enzalutamide are provided in Table 6.
Transporters
Enzalutamide is not a substrate of P-gp or the breast cancer resistance protein (BCRP). In in vitro experiments, enzalutamide and N-desmethyl enzalutamide were shown to be inhibitors of P-gp, and the carboxyl metabolite was not an inhibitor or a substrate of P-gp [20]. In the drug–drug interaction study, Gibbons et al. hypothesized that the P-gp transporter may be induced via induction of PXR, while in vitro studies do not support this hypothesis [32]. Furthermore, inhibition of multidrug resistance-associated protein 2 (MRP2), BCRP and OATP1B1 could not be excluded based on in vitro work [20]. The emulsifier CCMG that is used to improve the bioavailability of enzalutamide is known to inhibit P-gp in vitro [20]. In the drug interaction trial of Gibbons et al., a placebo with this emulsifier was used as a comparator, therefore the effect of CCMG could not be determined [20]. Thus far, no clinical studies have been conducted to confirm transporter-mediated drug–drug interactions. Nevertheless, the drug label approved by the EMA states that drugs that are substrates of P-gp should be used with caution [20].
Patient Characteristics
No formal study has yet been conducted to investigate the influence of patient characteristics on exposure of enzalutamide and N-desmethyl enzalutamide. For registration purposes, a post hoc population pharmacokinetic analysis (healthy volunteers and patients) was conducted in which no significant influence of covariates was identified [13]. The effect of severe renal impairment on the pharmacokinetics of enzalutamide has not been studied. Caution regarding the effects of the enzyme induction of enzalutamide is especially warranted when coadministered with antiepileptic drugs as this may result in lower pharmacokinetic exposure and loss of seizure control [13]. Ethnicity (Asian vs. Caucasian) does not appear to affect the pharmacokinetics of enzalutamide [33].
Exposure–Response Relationships
Steady-state concentrations of enzalutamide are reached after approximately 1 month. For enzalutamide, no exposure–response relationship was identified for overall survival when administered at 160 mg. This was expected since interpatient variability in enzalutamide and N-desmethyl enzalutamide exposure (AUC, C min and C max) is low (≤30 %). In a retrospective analysis, efficacy was similar in the four different exposure quartiles based on steady-state C trough [13], which may imply that patients in the lower quartile are treated equally effectively.
In a phase I study, more patients had seizures from dose levels higher than 240 mg/day [5] Due to this finding and the high incidence of grade 3 fatigue, the dose was lowered before phase III trials started. In the phase III trials, patients with a risk or history of seizures were excluded; however, 0.9 % of patients experienced seizures [16]. An analysis of the association between exposure and new-onset seizures was precluded by the low incidence of seizures in this selected subgroup and the limited variability in the pharmacokinetics of the drug. [13]. A postmarketing safety trial was requested in order to assess the risk of seizure in patients who were excluded from the randomized clinical trial [13].
Conclusions
Both abiraterone and enzalutamide have pharmacokinetic characteristics that need to be recognized in order to adequately treat patients with mCRPC. Abiraterone is moderately absorbed, and its bioavailability is majorly increased by food, while enzalutamide is well absorbed and has no food interference. Abiraterone is registered in combination with low-dose prednisolone in order to overcome side effects due to CYP17 blockade. Patients who experience nausea around drug intake, or for whom fasting around drug intake is difficult, might prefer treatment with enzalutamide. Both drugs are primarily metabolized in the liver, and the influence of mild, moderate, and severe liver impairment has been described for the pharmacokinetics of both drugs. Abiraterone requires dose reduction in patients with impaired liver function, whereas no dose adjustment is required for enzalutamide. Abiraterone is mainly excreted in feces, while enzalutamide is predominantly excreted in urine as inactive metabolite. Enzalutamide causes drug–drug interactions since it induces several CYP enzymes for which many potential coadministered drugs are substrates. Caution should be taken when coadministering enzalutamide with antiepileptic drugs as the pharmacokinetic exposure of these drugs can be lowered by induction of hepatic enzymes. In addition, in the phase III trial, seizures (0.9 %) were reported as an adverse event in a cohort of patients without a known seizure disorder. Abiraterone has a much smaller drug–drug interaction potential, and this aspect should be considered in patients who are using comedication. Furthermore, exposure–effect relationships have been studied for both drugs. For abiraterone, an exposure–efficacy relationship has been described.
A better understanding of the pharmacokinetics of abiraterone may facilitate dose optimization for the individual patient which may beneficially affect treatment outcome. For enzalutamide, no difference in efficacy was shown across the concentration/exposure range when administered at a single fixed dose of 160 mg daily, which may imply that enzalutamide is administered at the higher end of the exposure–response curve. These results warrant further research in order to apply this knowledge for personalized therapy in the future.
Both drugs appear to be equipotent in the treatment of patients with mCRPC pre- and postchemotherapy. The therapeutic choice might therefore be driven by patient-specific features (e.g. comorbidities, comedication). This review describes the presently available pharmacokinetic data of abiraterone acetate and enzalutamide, and provides an overview of the pharmacological aspects and challenges involved in adequate treatment with these drugs.
Compliance with Ethical Standards
Funding
No sources of funding were used to assist in the preparation of this review.
Conflict of interest
Guillemette E. Benoist, Rianne J. Hendriks, Peter F. A. Mulders, Winald R. Gerritsen, Diederik M. Somford, Jack A. Schalken, Inge M. van Oort, David M. Burger, and Nielka P. van Erp have no conflicts of interest that are directly relevant to the content of this review.
References
- 1.GLOBOCAN . Estimated cancer incidence, mortality and prevalence worldwide in 2012. Geneva: World Health Organization; 2012. [Google Scholar]
- 2.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 3.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 4.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13:983–992. doi: 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- 8.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90:2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–1488. doi: 10.1200/JCO.2009.24.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Us FDA. Clinical pharmacology and biopharmaceutics review: Xtandi (enzalutamide) Silver Spring (MD): US FDA; 2012. pp. 1–83. [Google Scholar]
- 14.European Medicines Agency . European Public Assessment Report (EPAR): Zytiga (abiraterone acetate) London: European Medicines Agency; 2016. [Google Scholar]
- 15.Us FDA. Prescribing information: Xtandi (enzalutamide) Silver Spring: US FDA; 2015. [Google Scholar]
- 16.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 17.Us FDA. Clinical pharmacology and biopharmaceutics review: Zytiga (abiraterone acetate) Silver Spring: US FDA; 2010. pp. 1–86. [Google Scholar]
- 18.Acharya M, Gonzalez M, Mannens G, De Vries R, Lopez C, Griffin T, et al. A phase I, open-label, single-dose, mass balance study of 14C-labeled abiraterone acetate in healthy male subjects. Xenobiotica. 2013;43:379–389. doi: 10.3109/00498254.2012.721022. [DOI] [PubMed] [Google Scholar]
- 19.Chi KN, Spratlin J, Kollmannsberger C, North S, Pankras C, Gonzalez M, et al. Food effects on abiraterone pharmacokinetics in healthy subjects and patients with metastatic castration-resistant prostate cancer. J Clin Pharmacol. 2015;55(12):1406–1414. doi: 10.1002/jcph.564. [DOI] [PubMed] [Google Scholar]
- 20.European Medicines Agency . European Public Assessment Report (EPAR): Xtandi (enzalutamide) London: European Medicines Agency; 2015. [Google Scholar]
- 21.Marbury T, Lawitz E, Stonerock R, Gonzalez M, Jiao J, Breeding J, et al. Single-dose pharmacokinetic studies of abiraterone acetate in men with hepatic or renal impairment. J Clin Pharmacol. 2014;54:732–741. doi: 10.1002/jcph.253. [DOI] [PubMed] [Google Scholar]
- 22.US FDA . Prescribing information: Zytiga (abiraterone acetate) tablets. Silver Spring: US FDA; 2015. [Google Scholar]
- 23.Nijland HM, Ruslami R, Suroto AJ, Burger DM, Alisjahbana B, van Crevel R, et al. Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin Infect Dis. 2007;45:1001–1007. doi: 10.1086/521894. [DOI] [PubMed] [Google Scholar]
- 24.Flockhart DA. Drug interactions: P450 drug interaction table. Indianapolis: Indiana University School of Medicine; 2007. [Google Scholar]
- 25.Bernard A. Impact on abiraterone pharmacokinetics and safety: open-label drug–drug interaction studies with ketoconazole and rifampicin. Clin Pharmacol Drug Dev. 2014;4:63–73. doi: 10.1002/cpdd.132. [DOI] [PubMed] [Google Scholar]
- 26.Chi KN, Tolcher A, Lee P, Rosen PJ, Kollmannsberger CK, Papadopoulos KP, et al. Effect of abiraterone acetate plus prednisone on the pharmacokinetics of dextromethorphan and theophylline in patients with metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013;71:237–244. doi: 10.1007/s00280-012-2001-0. [DOI] [PubMed] [Google Scholar]
- 27.European Medicines Agency. Zytiga-H-C-2321-PSUV-19. European public assessment report: scientific conclusions and grounds recommending the variation to the terms of the marketing authorisation. London: European Medicines Agency; 2014. p. 1–11.
- 28.Stuyckens K, Saad F, Xu XS, Ryan CJ, Smith MR, Griffin TW, et al. Population pharmacokinetic analysis of abiraterone in chemotherapy-naive and docetaxel-treated patients with metastatic castration-resistant prostate cancer. Clin Pharmacokinet. 2014;53:1149–1160. doi: 10.1007/s40262-014-0178-6. [DOI] [PubMed] [Google Scholar]
- 29.Inoue K, Shishido A, Vaccaro N, Jiao J, Stieltjes H, Bernard A, et al. Pharmacokinetics of abiraterone in healthy Japanese men: dose-proportionality and effect of food timing. Cancer Chemother Pharmacol. 2015;75:49–58. doi: 10.1007/s00280-014-2616-4. [DOI] [PubMed] [Google Scholar]
- 30.Gibbons JA, Ouatas T, Krauwinkel W, Ohtsu Y, van der Walt JS, Beddo V, et al. Clinical pharmacokinetic studies of enzalutamide. Clin Pharmacokinet. 2015;54:1043–1055. doi: 10.1007/s40262-015-0271-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Us FDA. Pharmacology review: Xtandi (enzalutamide) Silver Spring: US FDA; 2012. pp. 1–154. [Google Scholar]
- 32.Gibbons JA, de Vries M, Krauwinkel W, Ohtsu Y, Noukens J, van der Walt JS, et al. Pharmacokinetic drug interaction studies with enzalutamide. Clin Pharmacokinet. 2015;54:1057–1069. doi: 10.1007/s40262-015-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akaza H, Uemura H, Tsukamoto T, Ozono S, Ogawa O, Sakai H, et al. A multicenter phase I/II study of enzalutamide in Japanese patients with castration-resistant prostate cancer. Int J Clin Oncol. 2016. doi:10.1007/s10147-016-0952-6. [DOI] [PMC free article] [PubMed]