

Abstract
Seaweed is receiving an increasing amount of attention as a “sea vegetable”. The microbiota of coastal populations may acquire seaweed associated enzymes through marine food. Several agarases have been found in non-marine environments; however, their origin is unknown. In this study, a hypothetical protein, Aga1, was identified as an agarase from an inland soil agar-degrading bacterium, Paenibacillus sp. SSG-1.Having low similarity to known glycoside hydrolases, Aga1 may be a distant member of the glycoside hydrolase family 86. Aga1 has good pH stability (pH 3–11) and is stable in the presence of various metal ions. Aga1 is an exo-type β-agarase that produces NA 4 (neoagarotetraose) and NA 6 (neoagarohexaose) as its main products. In addition, Aga1 may be a cell-surface-binding protein. The bioinformatic analysis showed aga1 may have been transfered together with its surrounding genes, from marine bacteria to soil bacteria via human microbiota. The use of seaweed as food and the disposal of human faeces or saliva were the most likely reasons for this gene transfer pathway. Notably, the results also indicated that microbes from inland humans may degrade agar and that these microbes may have acquired seaweed associated genes because of increased seaweed in diets.
In 2012, the world’s annual production of seaweed reached 23.8 million tons, which is 3.5 times more than that produced in the 1990s1. In recent decades, seaweed has become increasingly popular as food, not only in Asian countries, but also in western countries2,3,4,5. Compared to traditional crops, seaweed has many superior characteristics, such as being fertilizer-free and irrigation-free and having no land conflicts with traditional agricultural crops6,7. Considering its nutrient composition, including amino acids and fatty acids, seaweed is a promising food source.
Red seaweed, as an important marine plant, is widely used as food. Agar is the main component of red seaweed, and it consists of 3-O-linked β-D-galactopyranose and 4-O-linked α-3,6-anhydro-L-galactopyranose linked to sulfate groups, methyl groups or pyruvic acid acetal8. Agarase is the enzyme that degrades agar into oligosaccharides or monosaccharides8,9. Based on cleavage patterns, agarases may be classified as α-agarases, β-agarases and β-porphyranases10. According to the CAZy (carbohydrate-active enzymes) database, approximately only 40 agarases have been characterized11. Some agarases belong to existing glycoside hydrolase (GH) families, such as GH1612,13 and GH5014,15,16. Others have defined new families, such as GH8617,18, GH9619,20, GH11721,22 and GH11823,24. Compared with other well-studied glycoside hydrolases, such as cellulases or xylanases25, the number of agarases is small. Identifying and studying new agarases is essential.
Until now, most reported agarases have come from marine environments13,14,15,16,17,19,23. Agar-degrading bacteria have also been found in different non-marine environments, such as soil26,27,28, plant endogenous environments29, rivers30 and even the human gut31,32. It is interesting that agarase exists in environments containing nearly no seaweed. It has been reported that coastal human microbiota may acquire glycoside hydrolases from the marine environment through food connections33. It is not known where non-marine source agarases come from, although marine environments or soil environments are possibilities. An outstanding question is how non-marine agarases came into existence.
In our previous work, we purified and characterized a natural agarase from the agar-degrading soil bacteria Paenibacillus sp. SSG-127. This natural agarase is a hypothetical protein in the genome of Paenibacillus sp. SSG-1. Furthermore, this hypothetical protein (Aga1) was synthesized, purified and characterized as agarase. Aga1 had low similarity to known agarases and was a distant member of GH86. A detailed biochemical characterization was conducted to determine the specific properties of Aga1. Bioinformatic analysis revealed that aga1 may be the result of horizontal gene transfer from a marine environment to a soil environment via human microbiota.
Results
Cloning and identification of agarase aga1
Using the matched peptide sequences of SSG1a, a BLAST search against the Paenibacillus sp. SSG-1 genome identified one hypothetical protein, which was subsequently designated Aga1.
As demonstrated in Fig. 1A, Aga1 contains one signal peptide (1–55), a conserved region (330–613), and three S-layer homology (SLH) domains (1495–1536, 1554–1597 and 1624–1668).The signal peptide and SLH domains indicate that Aga1 is probably a secreted protein and may be located on the cell wall surface, which was supported by the subcellular location prediction results.
Figure 1.

(A) Structure of Aga1 from Paenibacillus sp. SSG-1. SLH regions represent the S-layer homology regions. (B) SDS-PAGE and Coomassie staining of purified Aga1. (C) Aga1’s enzymatic activity against different substrates. (D) Phylogenetic tree between aga1 and the characterized agarases. Numbers at nodes are levels of bootstrap support (%).
The recombinant protein Aga1, excluding the signal peptide and SLH domains, was inserted into the pET28a vector (His-tag fusion) and expressed in E. coli (DE3) as a soluble protein. SDS-PAGE showed the Aga1 protein had an apparent molecular mass of 165 kDa (Fig. 1B), which matched the calculated molecular mass of 166.3 kDa. Tandem mass analysis of the purified Aga1 confirmed that it was correctly expressed and purified (Supplemental Table S1).
As shown in Fig. 1C, Aga1 is an agarase that is active only in the presence of agarose. Aga1 had very low similarity (lower than 30%) to other glycoside hydrolases. The phylogenetic tree consisting of Aga1 and known agarases showed that Aga1 may be a distant member of GH86 (Fig. 1D).
Biochemical analysis of agarase
Aga1 maintained over 40% of its activity across a wide range of temperatures (0–70 °C), and 50 °C was the optimal temperature for Aga1 (Fig. 2A). In addition, as shown in Fig. 2B, Aga1 showed strong stability over a wide pH range (pH 1–12). The decrease in Aga1 activity at pH 3.0 may be related to the pI of Aga1. Because the predicted pI of Aga1 was 4.5, it is possible that the real pI was close to pH 3.0, which caused this decrease. Meanwhile, most metal ions (1 mM) did not affect the activity of Aga1 (Supplemental Table S2). However, Cu2+ strongly inhibited its activity (37% activity).
Figure 2.

(A) Effects of temperature on stability and activity of Aga1. (B) Effects of pH on stability and activity of Aga1. Data are mean ± SD of three independent experiments. (C) TLC analysis of the catalytic property against the oligosaccharide. (D) The TLC analysis of the end products at different time. NA2, NA4, NA6, NA8 represent the neoagarooctaose, neoagarotetraose, neoagarohexaose and neoagarooctaose, respectively.
As shown in Fig. 2C, Aga1 could not hydrolyse neoagarobiose (NA 2), neoagarotetraose (NA 4) and neoagarohexaose (NA 6). Neoagarooctaose (NA 8) was the smallest oligosaccharide that Aga1 could hydrolyse.
TLC analysis of the end product showed that Aga1 hydrolysed agarose into two main products, which should be NA 4 and NA 6 according to the standards (Fig. 2D). These two products were also subjected to HPLC, and the results showed that they had the same retention times as NA 4 and NA 6 (Fig. 3A). Moreover, as shown in Fig. 3B, the MALDI-TOF mass spectrometry results of the end products showed two main peaks, i.e., 653.2 m/z and 959 m/z, which corresponded to [M + Na]+. These two peaks were attributed to NA 4 and NA 6, respectively. Taking these three results into consideration, the main hydrolysis products were NA 4 and NA 6.
Figure 3.

(A) The HPLC result of the hydrolysis product produced by Aga1. (B) MALDI-TOF mass result of the end products. (C) The 13C NMR of the end products. G β-D-galactopyranose, A 3,6-anhydro-a-L-aglactopyranose, r reducing end, nr non-reducing end, α α anomer, β β anomer.
To analyse the cleavage pattern of Aga1, 13C NMR was conducted. As shown in Fig. 3C, resonances at approximately 97.04 ppm and 93.05 ppm corresponded to the β and α anomeric carbons, respectively, of the galactose residues. Resonance at 90.72 ppm, the typical signal of an α-agarase, was not observed. Thus Aga1 is a β-agarase.
TLC (Fig. 2D) and HPLC analysis (Supplemental Fig. S1) showed that the amounts of NA 4 and NA 6 increased as the hydrolysis time increased, and no other oligosaccharides were observed during hydrolysis. Endo-agarase decomposed agarose in a random way and produced oligosaccharides with different degrees of polymerization during hydrolysis. As only two products were observed in the hydrolysis procedure, Aga1 was determined to be an exo-type agarase.
Inland aga1 may be from inland human symbionts
To investigate the distribution of aga1, we searched for homologues of aga1 in NCBI none-redundant database. Interestingly, almost all of aga1’s similar genes came from other class or phylum. Among the 208 genomes of Paenibacillus sp., aga1 like gene only appeared in Paenibacillus sp. D14. This rare distribution of aga1’s similar gene in its own genus indicated that aga1 may be a foreign gene. Meanwhile, the GC content, GC3s content, and Codon Bias Index of aga1 were all significantly different from those of the genome (Table 1 and Supplemental Table S3). Additionally, the Relative Synonymous Codon Usage (RSCU) value of aga1 was clearly distinct from the genome’s value (Supplemental Table S4). Thus, these evidences strongly indicated that aga1 had been transfered horizontally from other microbes.
Table 1. Correspondence analysis of codon usage of aga1, Paenibacillus sp. SSG-1 and control genes.
| CAI | CBI | Fop | Nc | GC | GC3s | |
|---|---|---|---|---|---|---|
| Genome | 0.267 | 0.127 | 0.489 | 47.43 | 0.536 | 0.633 |
| aga1 | 0.251 | -0.029 | 0.414 | 52.18 | 0.457 | 0.445 |
| PA2609 | 0.235 | 0.092 | 0.468 | 49.23 | 0.561 | 0.634 |
| PA1054 | 0.217 | 0.083 | 0.464 | 52.60 | 0.575 | 0.632 |
| PA4965 | 0.246 | 0.013 | 0.425 | 57.25 | 0.511 | 0.630 |
| PA4486 | 0.265 | 0.188 | 0.508 | 43.03 | 0.507 | 0.631 |
Control genes were protein-coding genes in Paenibacillus sp. SSG-1 and had same GC3s value with the genome. CAI: Codon Adaptation Index. CBI: codon bias index. Nc: effective number of codons. Fop: frequency of optimal codons. GC3s: GC of silent 3rd codon posit.GC: GC content of gene.
Given that aga1 was from an inland soil environment that was far from the sea and contained almost no seaweed, it was difficult for the enzymes to evolve into agarases without a substrate, and the distant geographic position of the enzyme made gene transfer nearly impossible. As showed in Fig. 4A, aga1 had a closer relationship with genes from symbiotic environments, other than those from marine environments. Meanwhile, we noticed that four aga1 like genes came from inland people’s microbiota; i.e., from the human gut or human mouth. Human faeces are usually used as fertilizer, and discarding saliva is also common. Given that aga1 was also from an inland location, it is reasonable to infer that aga1 may come from inland human microbiota.
Figure 4.

(A) Maximum likelihood tree of Aga1 and similar proteins. Numbers at nodes are levels of bootstrap support calculated from 100 bootstrap replicates (%). (B) The GC content change around the aga1. The red arrow indicates the position of aga1. The blue line represents the GC content of the whole genome of Paenibacillus sp. SSG-1. (C) Schematics of clusters containing aga1 like genes in different species. Sequence related genes (higher than 30% identity) are linked. NABH: α-neoagarobiose hydrolase; cycloisomerase: 3,6-anhydro-L-galactonate cycloisomerase; reductase: 2,5-diketo-3-deoxy-L-galactonate 5-reductase; dehydrogenase-1: 3,6-anhydro-L-galactose dehydrogenase; dehydrogenase-2: 2-keto-3-deoxy-L-galactonate 5-dehydrogenase.
Horizontal gene transfer linkage
Analysis of Paenibacillus sp. SSG-1’s genome showed that aga1 was surrounded by other genes coding for agar, including α-neoagarobiose hydrolase (NABH), galactosidase. 3,6-anhydro-L-galactose (L-AnG) metabolic enzymes and sulfatase. All these genes were located in a region which had an atypical GC content value with the genome (Fig. 4B). Moreover, NABH, and galactosidase were also uncommon in Paenibacillus sp. Their closest homologues (>70% identify) were found in other microbes, such as Clostridium sp. D5, Paenibacillus sp. D14. As showed in Fig. 4A, aga1 had a closer relationship with genes from human symbiotic environments. Thus, aga1 and its surrounding genes in Paenibacillus sp. SSG-1 may have been horizontally transfered from other microbes, such as human oral or gut symbionts. As discussed above, the most possible mode for this transfer was the disposal of human faeces or saliva.
When the distribution of aga1 like gene, NABH and galactosidase genes from human symbionts, Paenibacillus sp. D14 and Clostridium sp. D5, were investigated, we also found that these genes were rare in their corresponding genus. Meanwhile, the homologue of these genes could be found in the marine bacterium Rhodopirellula sallentina SM41. Conserved gene pair was also the indicator of horizontal gene transfer34. As showed in Fig. 4C, conserved gene pairs could be observed among the clusters. Gene pair, encoding for dehydrogenase-2 and reductase, was conserved between Paenibacillus sp. D14 (genes 2759 and 2758) and Rhodopirellula sallentina SM41 (genes 1698 and 1699). Gene pair, encoding for cycloisomerase and dehydrogenase-2, was conserved between Clostridium sp. D5 (genes 2446 and gene 2447) and Rhodopirellula sallentina SM41 (genes 1696 and 1697). Moreover, transposase genes were found around the gene cluster in Paenibacillus sp. D14 and integrase gene was also found in the downstream of cluster in Clostridium sp. D5 (data not shown). Both of them were associated with horizontal gene transfer35,36. Combining these evidences, aga1 like gene and surrounding genes in human symbiotic bacteria may have been horizontally transfered from marine bacteria. According to previous work of Hehemann et al.33, the most possible reason for gene transfer from marine to human microbiota may be seafood diet.
To further confirm this inferred pathway, phylogenetic trees of two other soil agarases were constructed. As shown in Fig. 5A,B, the same trend could be observed. Soil agarase showed a closer relationship to the agarases of symbiotic environments, such as the human gut or human mouth, than the agarases from marine environments. This evidence also indicated the same mode of gene dissemination; i.e., from marine to symbiotic environments to soil.
Figure 5.

(A,B) Maximum likelihood tree of two soil agarases. Numbers at nodes are levels of bootstrap support calculated from 1000 bootstrap replicates. (C) The sketch map of the predicted gene transfer pathway.
Based on these results, we developed the hypothesis that soil agarases may be the result of horizontal gene transfer from a marine environment to a soil environment via human microbiota, and human symbiotic microbiota and human faeces and saliva serving as the link between human microbiota and the soil environment (Fig. 5C).
Inland human microbiota may use agar
Horizontal gene transfer linkage of marine-symbiont-soil was inferred. Agarases are abundant in marine environments and are found in inland soil environments. However, agarases have not been found in other inland populations, the missing link in the above chain.
Thus, we used 37 characterized agarases as queries to investigate their distribution in human symbiotic microbes. Several possible agarases could be found in the microbes of inland people (Table 2). Twenty faecal samples from inland people were used to test for the capacity to degrade agarose. Interestingly, 8 of 20 samples were found to degrade agarose partially (Fig. 6). The screening of agar-degrading bacteria on an agar plate did not achieve positive results, possibly because of unsuitable cultural conditions. Given these results, the microbiota of inland people may also utilize agar.
Table 2. Distribution of the potential agarase genes in human microbiome.
| Protein ID | Strain | Identity | Source |
|---|---|---|---|
| BACPLE_01670 | Bacteroide splebeius DSM 17135 | 135/286 (47%) | NS |
| BACPLE_01689 | Bacteroides plebeius DSM_17135 | 321/321 (100%) | NS |
| HMPREF0240_02413 | Clostridium sp. D5 | 212/343 (61%) | Inland |
| HMPREF0240_02442 | Clostridium sp. D5 | 155/348 (44%) | Inland |
| VE20221DRAFT_03240 | Clostridiales bacteriumVE202-21 | 150/345 (43%) | NS |
| VE20221DRAFT_03208 | Clostridiales bacterium VE202-21 | 198/338 (58%) | NS |
| VE20221DRAFT_03199 | Clostridiales bacterium VE202-21 | 183/321 (57%) | NS |
| RSAG_01951 | Ruminococcus sp. 5_1_39BFAA | 205/348 (58%) | NS |
| RSAG_01951 | Ruminococcus sp. 5_1_39BFAA | 205/348 (58%) | NS |
| RSAG_01951 | Ruminococcus sp. 5_1_39BFAA | 194/347 (55%) | NS |
| POTG_02958 | Paenibacillus sp. oral taxon 786 | 220/352 (62%) | Inland |
| POTG_02762 | Paenibacillus sp. oral taxon 786 | 211/355 (59%) | Inland |
Protein IDs are from IMG database. NS: source is not specified.
Figure 6. The TLC analysis of the hydrolysis product by inland human fecal sample. NA2, NA4, NA6:
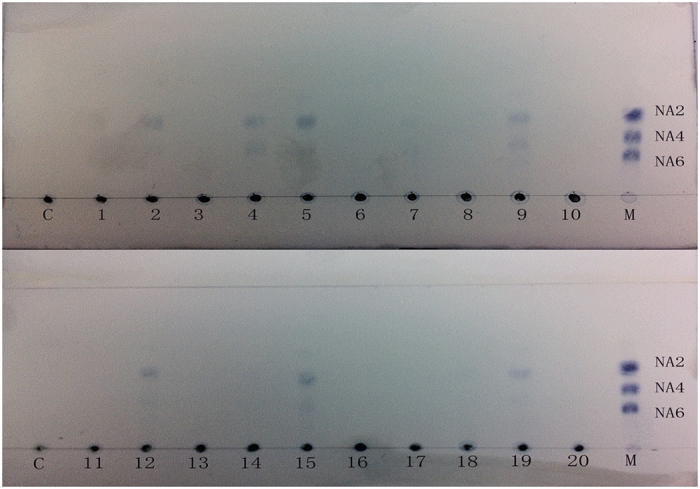
neoagarobiose, neoagarotetraose, neoagarohexaose.
Discussion
Until now, most studied agarases have come from marine environments, and very few studies have focused on agarases from terrestrial environments, such as soil. In our previous study, an agarase was purified from the soil agar-degrading bacterium Paenibacillus sp. SSG-127, whose genome was subsequently sequenced (data not published). Using the identified peptides (tandem mass result), we found that natural agarase matched a hypothetical protein in the genome. This hypothetical protein showed specific activity against agarose and was thus designated as Aga1. Aga1 showed very low similarity (lower than 30%) to characterized proteins and was thought to be a distant member of GH86.
Aga1 is an exo-type β-agarase, which hydrolyses agarose into NA 4 and NA 6 as end products. In contrast, most exo-agarases produce only one type of oligosaccharide instead of a mixture; for example, Aga50D37, Aga2138, and AgWH50A15 produce NA2 as an end product, and AgWH50C14 produces NA4 as an end product. The structure of agarase greatly affects its end products39. Thus, the catalytic pattern of Aga1 may be different from those of known agarases.
Aga1 has S-layer homology (SLH) domains that are associated with anchoring to the cell wall surface, and the subcellular prediction results showed that Aga1 was located on the cell wall. Previously, most agarases were intracellular or extracellular40. The SLH domain was not found in other reported agarases, nor was a domain with similar function. The cell-wall-binding enzymes were thought to be an efficient way to decompose polysaccharides into smaller components that were suitable for cells to absorb41. This was also found in the starch utilization system, which showed multiple functional proteins displayed on the surfaces of bacteria42. Thus, Aga1 was possibly located on the cell surface to degrade the agar, which has seldom been reported in agarases.
Given the scarcity of agar in inland environments and the distance between these environments and geographic locations with a sea environment, aga1 is unlikely to be the result of self-evolution or direct gene transfer from the sea. With the appearance of several agarases in inland symbiotic microbes, inland human microbiota are the most likely source. It has been reported that using human faeces as manure may cause antibiotic resistance gene transfer43. Additionally, saliva has also been shown to spread microbes in the environment. The “contamination” of soil with human faeces or saliva maybe the precondition for the gene transfer of agarase in an inland soil environment.
An anomalous nucleotide composition indicated that aga1 was the result of horizontal gene transfer. In Paenibacillus sp. SSG-1, aga1 and other agar utilization genes, which encoding for NABH, galactosidase and L-AnG metabolic enzymes, were clustered. Meanwhile, gene clusters from different species were compared. The scarcity of agar utilization genes in their corresponding genus, the closest homologues between different microbes, the conserved gene pairs between different taxa and the appearance of transposase and integrase indicated aga1 may have been transfered together with other genes from marine environments to human microbiota, and to soil environments. This gene cluster transfer was also found in other human gut bacterium32. This gene cluster transfer is reasonable, because gene cluster transfer may enable microbes to utilize agar, while transfer of one agarase is not sufficient.
All in all, soil agarase aga1 may be a result of horizontal gene transfer, from marine environment to soil environment via human microbiota. This gene transfer was also observed with two other soil agarases. In particular, Sco3481, the soil neoagarobiose hydrolase, was founded in microbes from terrestrial plants. This was consistent with the appearance of agar-degrading bacteria in plant associated environments29. Additionally, this evidence confirmed that human faeces and saliva affect not only soil but also plants in soil environments. Acquiring agarase may enable soil or plant associated microbes to use agar or agar-like polysaccharides. However, it is still unknown if this gene transfer provides an advantage.
In this study, aga1 was from an inland bacterium. It is known that microbiota in coastal humans may acquire agarase genes through seafood, which has not been reported in inland populations. If this gene transfer linkage exists, it is reasonable to infer that inland human microbiota have agarase genes.
Using the 37 known agarases as queries, several possible agarases were found in human reference genomes, and some of them were from inland human microbes. In addition, a previous study indicated that agarases from GH86 and GH117 specifically appeared in the human digestive system. Moreover, these agarases were distributed in a North American population, as well as a Japanese population44. The lack of studies on agarases has made bioinformatic analysis difficult. As many agarases or seaweed associated genes may be annotated as hypothetical proteins or simple glycoside hydrolases, bioinformatic analysis are restricted. These data suggest that agarases may be distributed in human microbiota, including microbiota from inland people.
In our study, 8 of 20 faecal samples from inland people were shown to have agar-degrading capacity. Given the difficulty in screening for agar-degrading bacteria due to unfavourable culture conditions, these findings are still encouraging. Additionally, a previous study showed that inland people’s microbiota could degrade agar-oligosaccharides and that an agar-degrading bacterium, B. uniformis L8, was isolated from inland human faecal samples31. These results suggest that microbiota from an inland population can degrade agar.
A previous study of Hehemann et al.33 showed that seaweed associated genes were horizontally transfered into Bacteroides plebeius of the Japanese population, which traditionally eats non-cooked seaweed. Seaweed food, known as “sea vegetables”, is a popular food, not only in coastal regions but also in inland areas2,3,4,45. Diet changes have always altered human microbiota46, and microbes living inside the body have employed gene transfer to gain functions to adapt to changes44. Taking seaweed as food may explain how microbiota from inland people have acquired the agarases from marine bacteria. Meanwhile, agar-degrading bacteria have been reported to produce agar-oligosaccharides with biological functions, which may influence human microbiota47. Thus, using seafood as food may influence the balance of human microbiota, which has been shown to be important for human health. Further study is needed to determine whether eating seaweed can affect human microbiota.
Conclusion
In conclusion, we first cloned and characterized an exo-type β-agarase; i.e., Aga1, from Paenibacillus sp. SSG-1. aga1 showed low similarity to known glycoside hydrolases and may be a distant member of the GH 86 family. aga1 gene may be the result of horizontal gene transfer from marine environments to humans to soil. Using seaweed as food and human faeces or saliva are the most likely linkages for this gene transfer pathway. Our results indicate that inland human microbiota also have the opportunity to acquire seaweed-associated genes from microbes that attach to the surface of seaweed foods.
Methods
Bacterial strains and culture medium
E. coli DH5α was used as the general gene-cloning host, and E. coli BL21 (DE3) was used as the host for protein expression. Unless otherwise noted, E. coli trains were cultured in Luria-Bertani (LB) medium with 100 μg/mL kanamycin. Paenibacillus sp. SSG-1 was cultured at 37 °C in LB medium. Strain SSG-1 had been deposited in the China Center Type Culture Collection (CCTCC) with the accession number CCTCC CB 2015001.
Gene cloning
After overnight culturing, the cell pellet of Paenibacillus sp.SSG-1 was harvested, and the genomic DNA was extracted. The aga1 gene was amplified using high-fidelity PrimeSTAR Max DNA Polymerase (Takara, Japan). The PCR product was digested with XholI/Not I and then ligated into the pET28a vector, which was also digested with Xhol I /Not I. After transformation into E. coli BL21, the recombinant plasmid was sequenced to confirm the accuracy of PCR. The sequences of the primers are listed in Supplemental Table S5.
Domain analysis of Aga1
Conserved domains of Aga1 were analysed using InterPro, and secondary structure analysis was conducted at the PSIPRED site (http://bioinf.cs.ucl.ac.uk/psipred/). Cell-PLos 2.0 was used to predict the subcellular location of Aga1, and signal peptides were predicted using SignalP 3.0.
Protein production and purification
Recombinant Aga1 was produced with an auto-induction method. E. coli BL21 harbouring the recombinant plasmid was cultured in LB medium containing 100 μg/mL kanamycin. After overnight culturing, the E. coli cells were inoculated into 2 L auto-induction-medium and then cultured at 28 °C for 48 h. After centrifugation, the cell pellet was collected and then suspended in 50 mL of 20 mM PB buffer with sonication. The supernatant was harvested. The protein was further purified using Ni-column (0.7 × 2.5 cm; GE Healthcare). The elution fractions with agarase activity were collected and further analysed by SDS-PAGE. Unless otherwise noted, the protein purification procedure was conducted at 4 °C.
Biochemical characterization of Aga1
To analyse the substrate specificity of Aga1, CMC-Na, pectin, carrageenan (mixture of κ, λ and ι), sodium alginate, arabic gum, neoagarooctaose, neoagarotetraose, neoagarohexaose and neoagarooctaose were tested.
To confirm whether Aga1 was correctly produced and purified, purified protein was obtained to conduct tandem mass spectrometry (MS) analysis. A local database was created using the protein data of Paenibacillus sp. SSG-1 and the Mascot search engine were used to identify the matched protein. The DNS method was used to assay agarase activity, with D-galactose as the standard. The assay procedure was conducted as previously described. Enzyme activity (U) was defined as the amount of enzyme that liberated 1 μmol D-galactose per minute. The optimal pH and temperature and the stability at different pH values and temperatures were tested as previously described. Various metal ions and chemical reagents (1 mM) were added to the reaction solution to investigate their effects on agarase activity. All experiments were conducted in triplicate.
Analysis of the degrading pattern of Aga1
To investigate the hydrolysis pattern of Aga1, 100 μg of purified enzyme was added to 50 mL of 0.5% substrate solution (0.5% agarose in deionized water). The reaction solution was incubated at 40 °C. Different samples were collected at fixed intervals. The collected samples were applied to silica G plates (Qingdao Haiyang Chemical Co., Ltd) using n-butyl alcohol:water:acetic acid = 2:1:1 as the developing solvent and then visualized using phenylamine/diphenylamine solution. High performance liquid chromatography (HPLC) was also used to detect the reaction (column HPX87-H Biorad 300 × 7.8 mm).
Bioinformatic analysis of Aga1 and related agarases
aga1 similar sequences were obtained from NCBI, Integrated Microbial Genomes (IMG) and the NIH Human Microbiome Project (HMP). Sequences were aligned with the Clustal W program and modified using Gblocks. The phylogenetic tree was constructed using a maximum likelihood method in PhyML. The nucleotide composition and codon usage analyses were conducted using the CodonW online service (http://mobyle.pasteur.fr/cgi-bin/portal.py#forms::CodonW). The species tree was constructed in MEGA 6.0 using a neighbour-joining method. Analysis of other two soil agarases was conducted using the same procedure.
Agar degrading experiments using microbiomes from an inland population
This study was approved by the Ethics Committee of Sichuan University. 20 persons, who lived in Chengdu, were enrolled to collect stool specimens. Informed consent was obtained from all participants. All experimental procedures were carried out in accordance with the Committee’s approved guidelines. The faecal samples were diluted (0.1 g samples were added to 10 mL of deionized water) and 50 μl of the diluted sample was added into 3 mL of the medium that contained agarose as a sole carbon source. The medium contained 0.1% NaCl, 0.1%K2HPO4, 0.1% (NH4)2SO4, 0.05% MgSO4, 0.01% CaCl2, 0.2% yeast extract and 0.2% agarose. After cultivation at 37 °C for 5 days, 50 μl of supernatant was collected and subjected to TLC analysis.
Investigation of the distribution of agarase in human symbiotic microbes
The protein sequences of 37 characterized agarases were obtained from the CAZy database and were used as search queries. IMG online Blast service (https://img.jgi.doe.gov/cgi-bin/mer/main.cgi?section=FindGenesBlast&page=geneSearchBlast) was used to search for proteins with high similarity (over 40%). Similar proteins from human-related microbes were chosen,and information on geographic locations was also collected.
Identification of agar utilization proteins
The agar’s utilization enzymes were studied in recent years, proteins in the database may be not annotated. Thus, experimental confirmed enzymes were used as the search queries and proteins with confident identity (higher than 30%) were deduced to have same function. The accession numbers of the search sequences were listed in the Supplemental Table S6.
Sequence accession number
The nucleotide and protein sequences of the aga1 gene were submitted to the DDBJ under the accession numbers LC094956 and BAT46645.1, respectively. The accession numbers of aga1’s surrounding genes were listed in Supplemental Table S7.
Additional Information
How to cite this article: Song, T. et al. Horizontal Transfer of a Novel Soil Agarase Gene from Marine Bacteria to Soil Bacteria via Human Microbiota. Sci. Rep. 6, 34103; doi: 10.1038/srep34103 (2016).
Supplementary Material
Acknowledgments
We thank Dr. Luo Dan for providing advice on gene cloning and appreciate Dr. Jingqiang Tang’s help with HPLC. We thank Dr. Jan-Hendrik Hehemann for advice on the classification of aga1. This work was supported by the National Twelfth Five-year Science and Technology support program (2014BAD02B02), the National Natural Science Foundation of China (31272659), the National Infrastructure of Natural Resources for Science and Technology Program of China (NIMR-2014-8) and the Sichuan Science and Technology Bureau (2014GXZ0005, 2017GXZ0005, 2012GZ0008, and 2015JPT0032).
Footnotes
Author Contributions T.S. and H.X. conducted most of the experiments, analysed the results and wrote most of the manuscript. C.W., T.J. and S.Q. conducted the bioinformatic analysis. W.Z., Y.C., C.H. and F.Z. purified proteins and analysed the data. D.Q. and Y.C. conceived the idea for this project and wrote the paper with T.S. and H.X.
References
- FAO, R. FAO yearbook: fishery and aquaculture statistics 2012. Rome: Food and Agriculture Organization of the United Nations (2014).
- Mouritsen O. G. et al. On the human consumption of the red seaweed dulse (Palmaria palmata (L.) Weber & Mohr). J Appl Phycol 25, 1777–1791, doi: 10.1007/s10811-013-0014-7 (2013). [DOI] [Google Scholar]
- Fleurence J. et al. What are the prospects for using seaweed in human nutrition and for marine animals raised through aquaculture? Trends Food Sci Technol 27, 57–61, doi: 10.1016/j.tifs.2012.03.004 (2012). [DOI] [Google Scholar]
- Edwards M. D., Holdt S. L. & Hynes S. Algal eating habits of phycologists attending the ISAP Halifax Conference and members of the general public. J Appl Phycol 24, 627-633, doi: 10.1007/s10811-011-9760-6 (2011).
- Dawczynski C., Schubert R. & Jahreis G. Amino acids, fatty acids, and dietary fibre in edible seaweed products. Food Chem 103, 891–899, doi: 10.1016/j.foodchem.2006.09.041 (2007). [DOI] [Google Scholar]
- Wargacki A. J. et al. An engineered microbial platform for direct biofuel production from brown macroalgae. Science 335, 308–313, doi: 10.1126/science.1214547 (2012). [DOI] [PubMed] [Google Scholar]
- Enquist-Newman M. et al. Efficient ethanol production from brown macroalgae sugars by a synthetic yeast platform. Nature 505, 239–243, doi: 10.1038/nature12771 (2013). [DOI] [PubMed] [Google Scholar]
- Lahaye M., Yaphe W., Viet M. T. P. & Rochas C. 13 Cn. mr spectroscopic investigation of methylated and charged agarose oligosaccharides and polysaccharides. Carbohydr Res 190, 249–265 (1989). [Google Scholar]
- Usov A. Structural analysis of red seaweed galactans of agar and carrageenan groups. Food Hydrocoll 12, 301–308 (1998). [Google Scholar]
- Chi W.-J., Chang Y.-K. & Hong S.-K. Agar degradation by microorganisms and agar-degrading enzymes. Appl Microbiol Biotechnol 94, 917–930, doi: 10.1007/s00253-012-4023-2 (2012). [DOI] [PubMed] [Google Scholar]
- Lombard V., Ramulu H. G., Drula E., Coutinho P. M. & Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42, D490–D495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M., Yu Z. & Xu X. A novel beta-agarase with high pH stability from marine Agarivorans sp. LQ48. Mar Biotechnol (NY) 12, 62–69, doi: 10.1007/s10126-009-9200-7 (2010). [DOI] [PubMed] [Google Scholar]
- Fu X. T. et al. Gene cloning, expression, and characterization of a beta-agarase, agaB34, from Agarivorans albus YKW-34. J Microbiol Biotechnol 19, 257–264 (2009). [PubMed] [Google Scholar]
- Liu N., Mao X., Yang M., Mu B. & Wei D. Gene cloning, expression and characterisation of a new beta-agarase, AgWH50C, producing neoagarobiose from Agarivorans gilvus WH0801. World J Microbiol Biotechnol 30, 1691–1698, doi: 10.1007/s11274-013-1591-y (2014). [DOI] [PubMed] [Google Scholar]
- Liu N., Mao X., Du Z., Mu B. & Wei D. Cloning and characterisation of a novel neoagarotetraose-forming-beta-agarase, AgWH50A from Agarivorans gilvus WH0801. Carbohydr Res 388, 147–151, doi: 10.1016/j.carres.2014.02.019 (2014). [DOI] [PubMed] [Google Scholar]
- Lee D. G. et al. Over-production of a glycoside hydrolase family 50 beta-agarase from Agarivorans sp. JA-1 in Bacillus subtilis and the whitening effect of its product. Biotechnol Lett 30, 911–918, doi: 10.1007/s10529-008-9634-4 (2008). [DOI] [PubMed] [Google Scholar]
- Ohta Y. et al. Cloning, expression, and characterization of a glycoside hydrolase family 86 beta-agarase from a deep-sea Microbulbifer-like isolate. Appl Microbiol Biotechnol 66, 266–275, doi: 10.1007/s00253-004-1757-5 (2004). [DOI] [PubMed] [Google Scholar]
- Ekborg N. A. et al. Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl Environ Microbiol 72, 3396–3405, doi: 10.1128/aem.72.5.3396-3405.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y. et al. Purification and characterization of a novel alpha-agarase from a Thalassomonas sp. Curr Microbiol 50, 212–216, doi: 10.1007/s00284-004-4435-z (2005). [DOI] [PubMed] [Google Scholar]
- Potin P., Richard C., Rochas C. & Kloareg B. Purification and characterization of the α‐agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur J Biochem 214, 599–607 (1993). [DOI] [PubMed] [Google Scholar]
- Rebuffet E. et al. Discovery and structural characterization of a novel glycosidase family of marine origin. Environ Microbiol 13, 1253–1270 (2011). [DOI] [PubMed] [Google Scholar]
- Ha S. C. et al. Crystal structure of a key enzyme in the agarolytic pathway, alpha-neoagarobiose hydrolase from Saccharophagus degradans 2-40. Biochem Biophys Res Commun 412, 238–244, doi: 10.1016/j.bbrc.2011.07.073 (2011). [DOI] [PubMed] [Google Scholar]
- Ma C. et al. Molecular Cloning and Characterization of a Novel β-Agarase, AgaB, from Marine Pseudoalteromonas sp. CY24. J Biol Chem 282, 3747–3754, doi: 10.1074/jbc.M607888200 (2007). [DOI] [PubMed] [Google Scholar]
- Dong J., Hashikawa S., Konishi T., Tamaru Y. & Araki T. Cloning of the novel gene encoding beta-agarase C from a marine bacterium, Vibrio sp. strain PO-303, and characterization of the gene product. Appl Environ Microbiol 72, 6399–6401, doi: 72/9/6399 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert H. J. & Hazlewood G. P. Bacterial cellulases and xylanases. Microbiology 139, 187–194 (1993). [Google Scholar]
- Lakshmikanth M., Manohar S., Souche Y. & Lalitha J. Extracellular beta-agarase LSL-1 producing neoagarobiose from a newly isolated agar-liquefying soil bacterium, Acinetobacter sp., AG LSL-1. World J Microbiol Biotechnol 22, 1087–1094, doi: 10.1007/s11274-006-9147-z (2006). [DOI] [Google Scholar]
- Song T. et al. Purification and characterization of a novel beta-agarase of Paenibacillus sp. SSG-1 isolated from soil. J Biosci Bioeng 118, 125–129, doi: 10.1016/j.jbiosc.2014.02.008 (2014). [DOI] [PubMed] [Google Scholar]
- Li X., Dong X., Zhao C., Zhen C. & Feng C. Isolation and some properties of cellulose-degrading Vibrio sp. LX-3 with agar-liquefying ability from soil. World J Microbiol Biotechnol 129, 375–379 (2003). [Google Scholar]
- Song T. et al. Isolation and characterization of agar-degrading endophytic bacteria from plants. Curr Microbiol 70, 275–281, doi: 10.1007/s00284-014-0713-6 (2015). [DOI] [PubMed] [Google Scholar]
- Agbo J. A. & Moss M. O. The isolation and characterization of agarolytic bacteria from a lowland river. J Gen Microbiol 115, 355–368 (1979). [Google Scholar]
- Li M. et al. Isolation and characterization of an agaro-oligosaccharide (AO)-hydrolyzing bacterium from the gut microflora of Chinese individuals. PLoS ONE 9, e91106, doi: 10.1371/journal.pone.0091106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann J. H. & Boraston A. B. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. P Natl Acad Sci USA 109, 19786–19791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann J. H. et al. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912, doi: 10.1038/nature08937 (2010). [DOI] [PubMed] [Google Scholar]
- Koonin E. V., Makarova K. S. & Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification1. Annu Rev Microbiol 55, 709–742 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchaya C., Fournous G., Chibani-Chennoufi S., Dillmann M.-L. & Brüssow H. Phage as agents of lateral gene transfer. Curr Opin Microbiol 6, 417–424 (2003). [DOI] [PubMed] [Google Scholar]
- Salyers A. A., Shoemaker N. B., Stevens A. M. & Li L.-Y. Conjugative transposons: an unusual and diverse set of integrated gene transfer elements. Microbiol Rev 59, 579–590 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. T. et al. Overexpression and molecular characterization of Aga50D from Saccharophagus degradans 2-40: an exo-type beta-agarase producing neoagarobiose. Appl Microbiol Biotechnol 86, 227–234, doi: 10.1007/s00253-009-2256-5 (2010). [DOI] [PubMed] [Google Scholar]
- Li J., Hu Q., Li Y. & Xu Y. Purification and characterization of cold-adapted beta-agarase from an Antarctic psychrophilic strain. Braz J Microbiol 46, 683–690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W. J. et al. An extra peptide within the catalytic module of a beta-agarase affects the agarose degradation pattern. J Biol Chem 288, 9519–9531, doi: 10.1074/jbc.M112.412247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X. T. & Kim S. M. Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar Drugs 8, 200–218, doi: 10.3390/md8010200 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabisch A. et al. Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J 8, 1492–1502, doi: 10.1038/ismej.2014.4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn D. W. et al. Molecular details of a starch utilization pathway in the human gut symbiont Eubacterium rectale. Mol Microbiol 95, 209–230, doi: 10.1111/mmi.12859 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer H., Schmitt H. & Smalla K. Antibiotic resistance gene spread due to manure application on agricultural fields. Curr Opin Microbiol 14, 236–243 (2011). [DOI] [PubMed] [Google Scholar]
- Cantarel B. L., Vincent L. & Bernard H. Complex carbohydrate utilization by the healthy human microbiome. PLoS ONE 7, e28742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchugh D. J. A guide to the seaweed industry. FAO Fish Tech Pap (2003). [Google Scholar]
- David L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdt S. L. & Kraan S. Bioactive compounds in seaweed: functional food applications and legislation. J Appl Phycol 23, 543–597, doi: 10.1007/s10811-010-9632-5 (2011). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.