

Abstract
The Rhyparochromidae, the largest family of Lygaeoidea, encompasses more than 1,850 described species, but no mitochondrial genome has been sequenced to date. Here we describe the first mitochondrial genome for Rhyparochromidae: a complete mitochondrial genome of Panaorus albomaculatus (Scott, 1874). This mitochondrial genome is comprised of 16,345 bp, and contains the expected 37 genes and control region. The majority of the control region is made up of a large tandem-repeat region, which has a novel pattern not previously observed in other insects. The tandem-repeats region of P. albomaculatus consists of 53 tandem duplications (including one partial repeat), which is the largest number of tandem repeats among all the known insect mitochondrial genomes. Slipped-strand mispairing during replication is likely to have generated this novel pattern of tandem repeats. Comparative analysis of tRNA gene families in sequenced Pentatomomorpha and Lygaeoidea species shows that the pattern of nucleotide conservation is markedly higher on the J-strand. Phylogenetic reconstruction based on mitochondrial genomes suggests that Rhyparochromidae is not the sister group to all the remaining Lygaeoidea, and supports the monophyly of Lygaeoidea.
The Rhyparochromidae, or dirt-colored seed bugs, are a relatively large group of Heteroptera and the largest family of Lygaeoidea (Hemiptera: Heteroptera)1. Rhyparochromidae includes two subfamilies (Plinthisinae and Rhyparochrominae), 372 genera, and more than 1,850 species2. The family was established by Amyot and Serville (1843) as Rhyparochromides, when it was regarded as a subfamily of the Lygaeidae3. Henry (1997) raised it to a family status and considered it as the basal group of Lygaeoidea based on morphological characters1. Dong and Zheng (1997), also using morphological characters, considered Rhyparochromidae as the sister group of Pyrrhocoroidea, which in turn forms the sister clade of the remaining Lygaeoidea4. In later molecular studies, analyses of 18S rDNA5, 18S + 28S rDNAs6, 16S rDNA7 and six Hox genes8 suggested that Rhyparochromidae is not the sister group to all the remaining Lygaeoidea. However, the phylogenetic relationships of Rhyparochromidae within Lygaeoidea have not been analyzed using mitochondrial genome (mt-genome) sequences to date, because no mt-genome data have been reported for the Rhyparochromidae.
Over the past decade, the mt-genome has become the most extensively used genomic resource in phylogenomic analyses of insects9, owing to its compact size (typically 15–18 kb), the availability of near-universal primers across insects, and the rapid development of sequencing technologies10,11,12. However, mt-genome representation at the family level is still limited, especially in most species-rich groups such as Heteroptera.
The size of true bug (Hemiptera: Heteroptera) mt-genomes range from 14,688 bp in Kleidocerys resedae resedae (Lygaeidae)13 to 17,544 bp in Nesidiocoris tenuis (Miridae)14. The main source of size variation is the variable number and length of tandem repeats in the control region15. Tandem repetition has been widely observed in the mitochondrial control region in animals16, and these duplications are thought to play a role in gene transcription or DNA methylation17,18. Moreover, tandem repeat units can be added or subtracted by slipped-strand mispairing during replication19, and this type of mutation happens at a rate at least 100,000 times higher compared to the rate of simple point mutations20. In true bugs, tandem repeats have been found in many groups, especially at the 3′-end of the control region (near the tRNA-I sequence), although the size and copy number of repeat units differ greatly among different species21,22,23. Length of the repeat unit can range from several base pairs to more than 200 bp24, and the copy number can range from at least 2 to 2023.
Here we describe the complete sequence of the mitochondrial genome of Panaorus albomaculatus, which represents the first sequenced mt-genome of the dirt-colored seed bugs. We evaluate compositional biases, codon usage, and nucleotide composition of this mt-genome. We then investigate the phylogenetic position of Rhyparochromidae within Heteroptera based on 13 protein-coding genes (PCGs) within the mt-genome. We compare the tRNA gene families (and their secondary structures) with other sequenced Pentatomomorpha and Lygaeoidea species. We also analyze two rRNA secondary structures across the sequenced Heteroptera and Lygaeoidea mt-genomes. Furthermore, we report novel patterns and large numbers of tandem repeats in the control region of the Panaorus albomaculatus mt-genome.
Results and Discussion
Genome organization and structure
The mitochondrial genome of P. albomaculatus contains 16,345 bp (GenBank Accession Number: KX216853; Fig. 1). The gene order of the 37 genes (including 13 PCGs, 22 tRNAs and two rRNAs) as well as the non-coding region (control region) is the same as observed in most other mt-genomes of true bugs25 (Table 1). The genome is relatively compact with gene overlaps at 16 gene junctions, involving a total of 75 bp. The longest overlap occurs between tRNA-Ser and ND1 and involves 20 bp. Two of the overlaps (between ATP8/ATP6 and ND4L/ND4) consist of the same seven nucleotides (ATGATAA). There are also 82 nucleotides in 4 intergenic spacers (in addition to the control region), which range in size from 1 to 77 bp.
Figure 1. Structure of the mitochondrial genome of Panaorus albomaculatus (GenBank accession number KX216853).
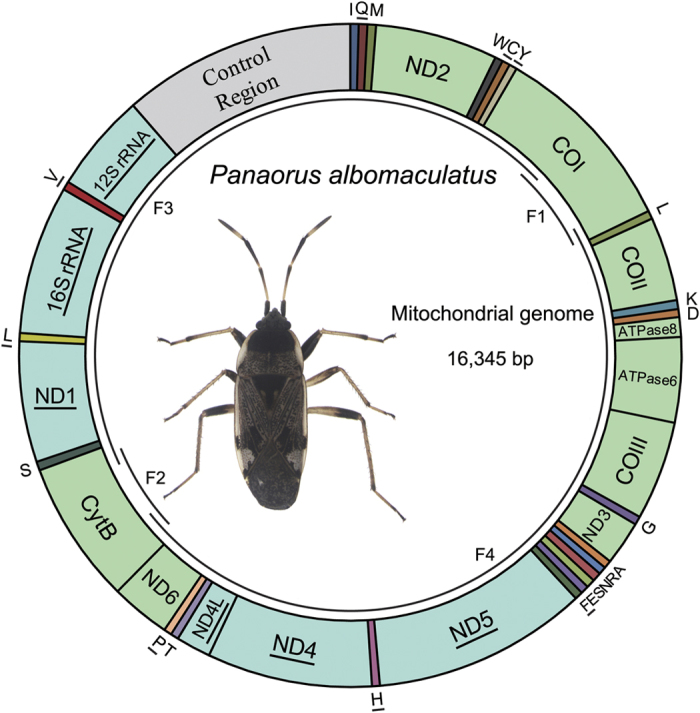
Gene names without an underline indicate genes transcribed from the majority strand (J-strand), and those with an underline indicate genes transcribed from the minority strand (N-strand). The tRNA genes are denoted by the color blocks and are named using single-letter amino acid abbreviations. Overlapping arcs (F1–F4) inside the circle indicate the PCR-amplified fragments.
Table 1. Organization of the Panaorus albomaculatus mitochondrial genome.
| Gene | Strand | Position | Anticodon | Size (bp) | Start codon | Stop codon | Intergenic Nucleotidesa |
|---|---|---|---|---|---|---|---|
| tRNA-Ile | J | 1–65 | GAT | 65 | |||
| tRNA-Gln | N | 63–131 | TTG | 69 | −3 | ||
| tRNA-Met | J | 131–198 | CAT | 68 | −1 | ||
| ND2 | J | 199–1170 | 972 | ATT | TAA | 0 | |
| tRNA-Trp | J | 1248–1310 | TCA | 63 | 77 | ||
| tRNA-Cys | N | 1311–1372 | GCA | 62 | 0 | ||
| tRNA-Tyr | N | 1373–1434 | GTA | 62 | 0 | ||
| CO1 | J | 1436–2974 | 1539 | TTG | TAA | 1 | |
| tRNA-Leu | J | 2970–3034 | TAA | 65 | −5 | ||
| CO2 | J | 3035–3701 | 667 | ATT | T- | 0 | |
| tRNA-Lys | J | 3702–3772 | CTT | 71 | 0 | ||
| tRNA-Asp | J | 3772–3834 | GTC | 63 | −1 | ||
| ATPase8 | J | 3835–3990 | 156 | ATT | TAA | 0 | |
| ATPase6 | J | 3984–4658 | 675 | ATG | TAA | −7 | |
| CO3 | J | 4645–5431 | 787 | ATG | T- | −14 | |
| tRNA-Gly | J | 5432–5498 | TCC | 67 | 0 | ||
| ND3 | J | 5499–5849 | 351 | ATA | TAG | 0 | |
| tRNA-Ala | J | 5848–5910 | TGC | 63 | −2 | ||
| tRNA-Arg | J | 5910–5971 | TCG | 62 | −1 | ||
| tRNA-Asn | J | 5972−6042 | GTT | 71 | 0 | ||
| tRNA-Ser | J | 6042−6110 | GCT | 69 | −1 | ||
| tRNA-Glu | J | 6110–6174 | TTC | 65 | −1 | ||
| tRNA-Phe | N | 6174–6237 | GAA | 64 | −1 | ||
| ND5 | N | 6237–7943 | 1707 | ATT | TAA | −1 | |
| tRNA-His | N | 7944–8005 | GTG | 62 | 0 | ||
| ND4 | N | 8006–9317 | 1312 | ATG | T- | 0 | |
| ND4L | N | 9311–9586 | 276 | GTG | TAA | −7 | |
| tRNA-Thr | J | 9589–9651 | TGT | 63 | 2 | ||
| tRNA-Pro | N | 9652–9714 | TGG | 63 | 0 | ||
| ND6 | J | 9717–10190 | 474 | ATC | TAA | 2 | |
| Cytb | J | 10183–11316 | 1134 | ATG | TAG | −8 | |
| tRNA-Ser | J | 11315–11384 | TGA | 70 | −2 | ||
| ND1 | N | 11365–12321 | 957 | ATT | TAA | −20 | |
| tRNA-Leu | N | 12322–12386 | TAG | 65 | 0 | ||
| 16S rRNA | N | 12387–13637 | 1251 | 0 | |||
| tRNA-Val | N | 13638–13705 | TAC | 68 | 0 | ||
| 12S rRNA | N | 13706–14492 | 787 | 0 | |||
| Control | 14493–16345 | 1853 | 0 |
aNumbers correspond to nucleotides separating a gene from an upstream one; negative numbers indicate that adjacent genes overlap.
Protein coding genes
In the mt-genome of P. albomaculatus, eleven of the thirteen protein-coding genes initiate with ATN as the start codon (five start with ATT, four start with ATG, one starts with ATA, and one starts with ATC) (Table 1). The COI gene initiates with a non-traditional start codon, TTG, as previously observed in other true bugs25,26. The ND4L gene has the unusual start codon GTG, as previously reported in assassin bugs27 (Hemiptera: Reduviidae). Most protein coding genes end with a complete termination codon. Eight genes (ND2, COI, ATP8, ATP6, ND5, ND4L, ND6 and ND1) terminate with TAA, and two genes (ND3 and CytB) terminate with TAG. The remaining three PCGs (COII, COIII and ND4) are terminated with an incomplete stop codon T.
In Pentatomomorpha mt-genomes, most PCGs use the standard start codon (ATN), but we also observed other initiation codons of ATCA (ND2), CTG (ND1), GTG (COII, ND1, ND4L and ND6), and TTG (ATP8, COI, COII, ND1, ND4L, ND5 and ND6; see Supplementary Table S1). Many PCGs use multiple start codons, but three PCGs always initiate with only one start codon (COI with TTG, COIII and ND4 with ATG). Furthermore, several PCGs initiate with a start codon only found within Lygaeoidea mt-genomes (e.g., ATP6 start with ATT only in Yemmalysus parallelus). Most PCGs stop with termination codons TAA/TAG, but truncated termination codons T/TA are also used in Pentatomomorpha mt-genomes. The phenomenon of incomplete stop codons is common in insect mt-genomes and it is likely that these truncated stop codons are completed by posttranscriptional polyadenylation28.
We compared the rate of nonsynonymous substitutions (Ka), the rate of synonymous substitutions (Ks), and the ratio of Ka/Ks for each PCG among the 13 PCGs in Pentatomomorpha and Lygaeoidea mt-genomes (see Supplementary Figure S1). The results showed that the evolutionary patterns of 13 PCGs were highly consistent between Pentatomomorpha and Lygaeoidea, and shared many common features: (1) ATP8 has the highest evolutionary rate and can be used to analyze intraspecific relationships29, while COI appears to have the lowest evolutionary rate and has been used for DNA barcoding markers30. (2) The Ka/Ks ratios for all PCGs are less than 1, indicating that these genes are likely evolving primarily under the purifying selection31. (3) The uniformly low evolutionary rates and low Ka/Ks ratios (Ka/Ks < 0.2) for four genes (COI, COII, COIII and CytB) indicate strong purifying selection and evolutionary constraints in cytochrome c oxidase and cytochrome b32. (4) Negative correlations have been detected between the Ka/Ks ratios and the G + C content of each PCG in Pentatomomorpha (R2 = 0.918) and Lygaeoidea (R2 = 0. 891) mt-genomes (see Supplementary Figure S2), suggesting that the variation of G + C content probably causes the different evolutionary patterns among genes22. Additionally, most PCGs of Lygaeoidea mt-genomes had lower evolutionary rates than Pentatomomorpha.
Nucleotide composition and codon usage
The nucleotide composition of the mt-genome of P. albomaculatus is significantly biased toward A/T. The overall A + T content of the J-strand is 76% (A = 44.4%, T = 31.6%, C = 14.5%, G = 9.5%; see Supplementary Table S2). This bias in A + T content falls within the known range for hemipteran mt-genomes (65.7% in Bemisia afer to 86.3% in Aleurodicus dugesii)29. The A + T content ranges from 75.1% for the PCGs, to 78.7% for the rRNAs. Among the PCGs, the lowest A + T content is 68.8% in COI, whereas the highest A + T content is 84% in ATP8. The nucleotide skew statistics33 for the mt-genome of P. albomaculatus reveal that the J-strand PCGs are AT-skewed and CG-skewed, whereas the N-strand PCGs are GC-skewed and TA-skewed. The observed strand bias is likely related to asymmetric mutation processes during replication34.
The overall A/T bias also influences codon usage. The A + T content of the third codon position (85.5%) is higher than either the first (71.3%) or second (68.2%) codon positions (see Supplementary Table S2). Moreover, the six most prevalent codons in P. albomaculatus (see Supplementary Table S3), Ile (ATT) (9.54%), Leu (TTA) (9.21%), Met (ATA) (7.87%), Phe (TTT) (7.41%), Asn (AAT) (4.92%), and Tyr (TAT) (4.48%) are all composed of A and/or T.
Comparison of tRNAs in Pentatomomorpha and Lygaeoidea mt-genomes
There are 22 tRNA genes in the mt-genome P. albomaculatus mt-genomes, as observed in other arthropod mt-geneomes. These genes range in length from 62 to 71 bp. Among these tRNAs, the tRNA-Ser (GCU) is the only one that does not fold into the typical cloverleaf secondary structure; the dihydrouridine (DHU) stem simply forms a loop (Fig. 2). Moreover, the predicted tRNA-Ser (GCU) anticodon stem is longer (9 bp vs. the normal 5 bp) and contains an unpaired nucleotide, as seen in many other insect mt-genomes35. A total of 20 unmatched G-U pairs exist in P. albomaculatus tRNA secondary structures. Of these, 16 mismatches are concentrated in 7 tRNAs that are encoded on the N-strand. These mismatches are common in arthropod mt-genomes; RNA editing processes are likely involved in correcting the mismatches36.
Figure 2. Putative secondary structure of the 22 tRNAs identified in the mitochondrial genome of Panaorus albomaculatus.

The tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashes indicate Watson-Crick base pairing and asterisks indicate G-U base pairing. Sites that are conserved 100% among sequenced Pentatomomorpha species are indicated with a red background. The 100% and > 80% conserved sites among sequenced Lygaeoidea species are indicated with green and orange backgrounds, respectively.
Most tRNA genes encoded on the J-strand are more conserved than those encoded on the N-strand, based on comparisons of aligned genes in Pentatomomorpha and Lygaeoidea (Fig. 2). Furthermore, some tRNAs (tRNA-Gln, tRNA-Gly, and tRNA-Met) within Lygaeoidea are much more conserved than they are within Pentatomomorpha, with changes mainly on the amino acid acceptor and TΨC stems. The anticodon stem and loop, and the DHU stem, are extremely conserved in most tRNAs within Pentatomomorpha, whereas most other tRNA regions have a high level of variation (the exceptions are the highly conserved TΨC stem of tRNA-Leu and acceptor stem of tRNA-Lys). The stems of each tRNA in Lygaeoidea mtDNA are more conserved than the remaining loops (TΨC, DHU and variable loops), except for several amino acid acceptor arms (tRNA-Ala, tRNA-Asp, tRNA-Cys, and tRNA-Val) and TΨC arms (tRNA-His, tRNA-Phe and tRNA-Pro). Most nucleotide substitutions are restricted to those loops (TΨC, DHU, and variable loops), which usually accompany insertion-deletion polymorphisms. The presence of indels at different taxonomic levels supports potential phylogenetic value of tRNA sequences in the study of insect phylogenetic relationships, especially when secondary structures are taken into account21,37.
We determined the percentage of identical nucleotides (INP%) for each tRNA family in Pentatomomorpha and Lygaeoidea mt-genomes, respectively (Fig. 3). The sequences and secondary structures of the tRNA-Leu (TAG), tRNA-Trp, and tRNA-Lys genes are highly conserved among most Pentatomomorpha mt-genomes (INP% > 55). In contrast, the most conserved tRNAs in Lygaeoidea mt-genomes (INP% > 75) are tRNA-Gln, tRNA-Thr, tRNA-Gly, tRNA-Ser (GCT), and tRNA-Leu (TAG). The highest level of nucleotide conservation in Lygaeoidea mt-genomes occurs in tRNA-Gln (INP% = 79.8), which is a much more variable gene in Pentatomomorpha (INP% = 28.9). Although the pattern of nucleotide conservation is markedly higher on the J-strand, the highest INP% values of tRNA-Leu (TAG) in Pentatomomorpha (INP% = 61.5) and tRNA-Gln (INP% = 79.8) in Lygaeoidea are both associated with genes located on the N-strand. None of the most frequently used codons (Ile, Leu-TTA, Met, Phe, Asn and Tyr) exhibit high INP% scores, whereas tRNA-Phe has the most variable tRNAs in both Pentatomomorpha and Lygaeoidea mtDNA.
Figure 3. Conservation of tRNA families in Pentatomomorpha and Lygaeoidea mitochondrial genomes.

The percentage of identical nucleotides for each tRNA family was inferred from a multiple alignment produced with CLUSTAL X61 and refined manually, taking into account the secondary structure.
Ribosomal RNAs
We assumed that the rRNA genes in the mt-genome of P. albomaculatus start and end at the boundaries of the flanking genes, as is the case in most other insect mt-genomes22,38. The large subunit (16S) rRNA gene is located between the tRNA-Val and tRNA-Leu (TAG) genes. The small subunit (12S) rRNA is located between the tRNA-Val gene and the control region. The 16S rRNA gene is 1251 bp with an A + T content of 79.9%, and the 12S rRNA gene is 787 bp with an A + T content of 76.8%. The secondary structure of P. albomaculatus 16S rRNA contains six structural domains (domain III is absent as in other arthropods) and 45 helices (Fig. 4), whereas the secondary structure of 12S rRNA contains three domains and 27 helices (Fig. 5).
Figure 4. Predicted secondary structure of the mitochondrial 16S rRNA of Panaorus albomaculatus.

Canonical Watson-Crick interactions are represented by a dash, non-canonical G-U interactions are represented by an asterisk, and all other non-canonical interactions are represented by a hollow circle. Roman numerals denote the conserved domain structure. The 100% conserved sites among sequenced true bugs are plotted with red background. The 100% and > 80% conserved sites among sequenced Lygaeoidea species are plotted with green and orange background, respectively.
Figure 5. Predicted secondary structure of the mitochondrial 12S rRNA of Panaorus albomaculatus.

The annotation is the same as for Fig. 4.
Based on the alignment of sequenced species between Heteroptera and Lygaeoidea, domains IV and V of the large subunit (16S) rRNA are more conserved than other three domains (I, II, and VI). Six helices (H563, H1775, H2064, H2507, H2547, and H2588) are highly conserved in both sequence (with 2–7 nucleotide substitutions) and secondary structure among sequenced heteropterans. In the variable domains II and VI, we found no conserved helices within heteropterans, but helices H579, H822, and the terminal loop of H2646 are highly conserved with 0–3 nucleotide substitutions in Lygaeoidea mtDNA. In domain IV, helices from H1830 to H1906 are highly conserved among Lygaeoidea. Our proposed secondary structure of helix H1835 is based on the model proposed by Buckley et al.39 and Cannone et al.40. In domain V, most helices are conserved among insect mtDNAs, except for helices H2077 and H2347, which are highly divergent in both length and secondary structure39. Helix H2077 has no apparent conserved motifs, but it contains an 18-paired-base stem and two loops in P. albomaculatus. Helix H2347 is composed of a short stem of the terminal 3 paired bases, which is similar to the structure proposed for Chauliops fallax (Hemiptera)10 and Zygaena sarpedon lusitanica (Lepidoptera)41.
Domain III of the 12S rRNA is among the most conserved regions among heteropterans in terms of sequence and secondary structure. Domains I and II are much less conserved than domain III, as most helices are highly variable among the studied taxa and are difficult to align. The primary exceptions are the distal sections of H511 and H769, which are highly conserved across insects42,43. In contrast to H511 and H769, there are considerable differences in the sequence and length of H47 across insects43,44, and alignments of this region across insects are ambiguous (partly because of its high AT content). We could not detect or predict a consistent secondary structure for H47 across the available insect 12S rRNA sequences. In P. albomaculatus, a possible secondary structure of H47 includes a long stem, two internal loops, and 10 bp terminal loop, which is similar to the model proposed by Niehuis et al.42. The nucleotide sequence between helices H577 and H673 suggests no obvious secondary folding, as in other heteropterans10,27. In domain III, there are several possible secondary structures for helices H1047, H1068, H1074, and H1113; this complex region likely contains several non-canonical base pairs43,44. Nonetheless, the sequence and predicted secondary structure of this region are highly conserved within Lygaeoidea. Helix H1399 is the most conserved helix of 12S rRNA among true bugs, except for its terminal loop.
Non-coding regions
There are only five non-coding regions in the mt-genome of P. albomaculatus, and most of them are shorter than 10 bp (Table 1). The largest non-coding region (1,853 bp) is located between the 12S rRNA and the tRNA-Ile (I)- tRNA-Gln (Q)- tRNA-Met (M) gene cluster (Fig. 1), as frequently found in other insects15,43. This region is considered to be the control region as it contains both an origin of transcription and replication9,45.
The control region is highly variable in length at all taxonomic scales within insects; most of the length variation is a result of variable numbers of tandem repeats13,16. Tandemly repeated sequences are commonly reported from the control region of most insect orders46, although both the size and copy number of tandem repeat units differ greatly across groups29. The repeat units of insect mt-genomes range from 8 bp to more than 700 bp, and the copy number of tandem repeats ranges from two to nearly 5016,47. However, the control region of P. albomaculatus has a novel feature not previously observed in other insect mt-genomes. The majority of the control region is made up of a 949 bp tandem-repeat region, which contains 53 tandem duplications including 52 repeat units (of 18 bp each) and a partial copy of the repeat (13 bp) (Fig. 6). This is the largest number of tandem repeats reported in insect mt-genomes to date. In addition, the nucleotide composition of this tandemly repeated region on the J-strand is strongly biased in composition toward A (A = 54.3%, T = 25.5%, C = 5.5%, G = 14.7%), which results in a higher AT-skew value for control region compared to the other genes of P. albomaculatus mt-genome (see Supplementary Table S2).
Figure 6. Control region of Panaorus albomaculatus mitochondrial genome.
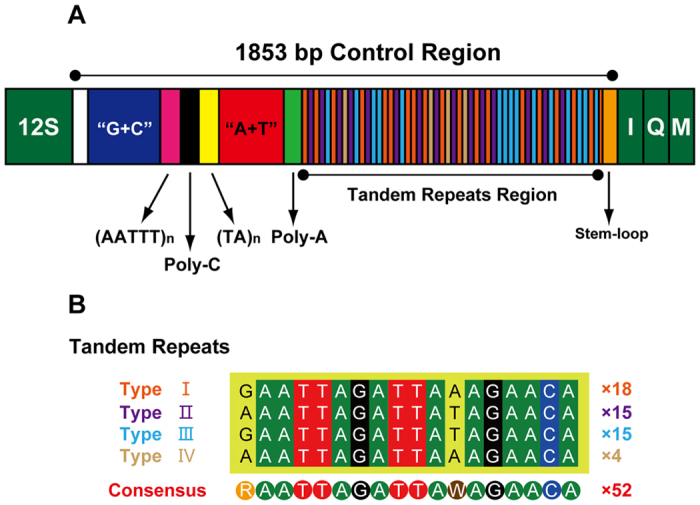
(A) Structure elements found in the control region of Panaorus albomaculatus. The control region flanking genes 12S rRNA, tRNA-I, tRNA-Q, and tRNA-M are represented in green boxes; “(AATTT)n” (magenta) and “(TA)n” (yellow) indicates the microsatellite-like repeat region; “A + T” (red) indicates high A + T content region; 53 boxes with four different colors indicate the tandem repeats region; the orange-red, purple, blue, and khaki boxes represent four different types of repeat units; orange boxes represent the stem-loop region. (B) The alignments of four different types of repeat units were generated by plotting the identities in different colors; numbers indicate the copy number.
The remainder of the control region can be divided into five sections (Fig. 6A): (1) the 5′-end of the control region contains a 321-bp region with 32.1% G + C content (higher than the average for the entire mt-genome); (2) a 8-bp poly-C structure is located between two microsatellite-like repeat regions ((AATTT)3 and (TA)5, respectively); (3) a 103-bp region with very high A + T composition (90.3%), as also found in the control region of other insects48,49; (4) a 8-bp poly-A structure is located 34-bp upstream of the tandem repeat region; and (5) a potential stem-loop secondary structure, with a ‘TATA’ element at the 5′ end (see Supplementary Figure S3).
Our comparative analyses of control regions among sequenced Pentatomomorpha mt-genomes revealed four distinct types of control region organization, which are summarized in Supplementary Figure S4. In some species of Pentatomomorpha, the control region contains four different motifs as summarized for arthropods50: tandem repeats, a poly-T stretch, a subregion of even higher A + T content, and stem-loop structures (e.g., Chauliops fallax in Supplementary Figure S4A). However, in most mt-genomes of Pentatomomorpha, tandem repeats occupy the majority of the control region, and two different types (see Supplementary Figure S4B) or identical tandem repeat units (Supplementary Figure S4C) are interrupted by a non-coding region23,51. A few control regions do not contain all four motifs or have only one; for example, Kleidocerys resedae resedae13 has just a stem-loop structure (see Supplementary Figure S4D). These various elements of the control region can provide valuable phylogenetic information within specific groups29,52,53.
Tandem repeats
The origin and persistence of tandem repeats is thought to involve slipped-strand mispairing during mtDNA replication15,19. Other mechanisms for repetitive sequences generation, such as recombination, are considered absent or rare in animal mt-genomes54. In P. albomaculatus, the repeat units consist of four different patterns with two variable sites (Fig. 6B): Type I (18 copies), Type II (15 copies), Type III (15 copies), and Type IV (4 copies). These four types are indicated by different colors in Fig. 6A.
These four types of repeat units occur throughout the tandem repeats region, with the following patterns (Fig. 6): (1) A partial copy Type I (GAATTAGATTAAA) is located at the 3′-end of the tandem repeats region, so Type I repeats occur at both the start and end of the repeat unit. (2) Most copies of Type II are located immediately downstream of Type I repeats, and all copies of Type IV are followed by Type II repeats. (3) Type III repeats are located between Type II and Type I repeats. (4) There are only four copies of Type IV repeats, but the other three types are all duplicated at least 15 times.
Considering these patterns of repeats, we suggest that the ancestral sequence consisted of a Type I-Type II-Type III-Type I cluster. The current distribution can then be generated from this ancestral sequence via replication slippage. Type IV repeats may be a product of slipped-strand mispairing from deletion of the unpaired section between Type II and Type I (Fig. 7). Duplication rates have been shown to be higher than deletion rates in several previous studies19,55, which likely accounts for the lower number of Type IV repeats compared to the other three types.
Figure 7. Proposed mechanism involved in the generation of Type IV induced by slipped-strand mispairing.
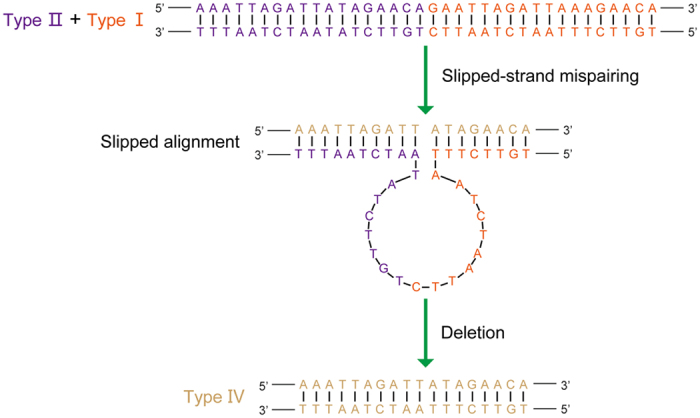
Slipped alignment produces a loop between the repeats of Type II and Type I, and results in a deletion after completion of replication.
There is a stem-loop structure, which is generally thought to be involved in the origin of replication15, located 34-bp downstream of the tandem repeats region (see Supplementary Figure S3). The significant number of tandem repeats adjacent to the origin of replication may indicate that they promote their own copy number amplification.
Phylogenetic analyses
We conducted phylogenetic analyses on the mt-genomes of 37 heteropteran species and 3 outgroup hemipteran insect species (Acyrthosiphon pisum, Sivaloka damnosa, and Lycorma delicatula). Maximum likelihood (ML) and Bayesian inference (BI) analyses yielded fully resolved trees with identical topologies (Fig. 8). The resulting phylogeny of Heteropteran infraordinal relationships is similar to that of Li et al.56. We found Enicocephalomorpha to be the sister group to all the remaining heteropterans, with high support values from both BI and ML analyses. In Fig. 8, all infraorders are monophyletic except Cimicomorpha. The non-monophyly of Cimicomorpha is incongruent with previous studies57,58; a more thorough taxon sampling of taxa within Cimicomorpha is needed to test these competing hypotheses. In Pentatomomorpha, our results support the groupings (Aradoidea + (Pentatomoidea + (Lygaeoidea + (Pyrrhocoroidea + Coreoidea)))), which have been widely supported in previous studies1,10,23. The monophyly of each superfamily within Pentatomomorpha is strongly supported in our analyses. The taxonomic status of Rhyparochromidae within Lygaeoidea is a highly contentious issue1,5,6,8. In our analyses, both BI and ML analyses provide strong support that Rhyparochromidae is not the sister group to the remaining Lygaeoidea, which is consistent with previous studies based on molecular data4,5,6. We also recovered the groups (Colobathristidae + (Berytidae + ((Lygaeidae + Malcidae) + (Geocoridae + Rhyparochromidae)))), but without strong support from either BI or ML analyses. We suggest that more thorough sampling of taxa will be needed to adequately resolve the family relationships within Lygaeoidea.
Figure 8. Phylogenetic tree inferred from the sequences of 13 protein coding genes of the mitochondrial genomes of 37 heteropterans and 3 outgroups.
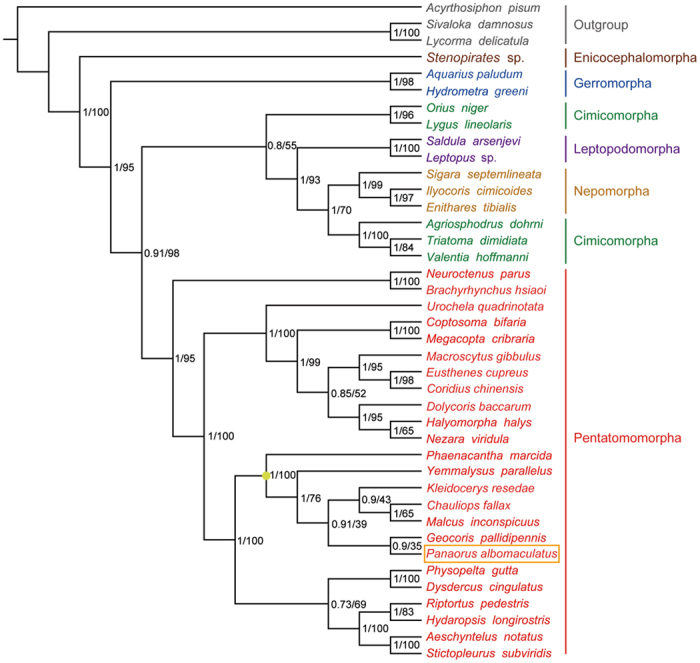
Numbers at the nodes are Bayesian posterior probabilities (left) and ML bootstrap values (right). The orange box indicates the position of Panaorus albomaculatus. The yellowish dot on the tree indicates the clade of Lygaeoidea.
Conclusions
We present the first description of a complete mitochondrial genome from a species of Rhyparochromidae, a large and diverse group within Heteroptera3. Comparative analysis of tRNA gene families showed that the level of conservation of many tRNAs was not consistent between sequenced Pentatomomorpha and Lygaeoidea mt-genomes, and most tRNA genes encoded on the J-strand are more conserved than those encoded on the N-strand. The control region of P. albomaculatus has novel patterns and large numbers of tandem repeats, including 53 tandem duplications, which can be explained by a replication slippage model. Phylogenomic analysis based on mt-genomes supported findings from nuclear genes that Rhyparochromidae is not the sister group to all the remaining Lygaeoidea, in contrast to the conclusions from morphological studies. This study should facilitate additional studies on the evolution and phylogeny of Rhyparochromidae, and on the evolution of tandem repeatswithin control regions of mt-genomes.
Materials and Methods
Specimen collection
We collected adult specimens of Panaorus albomaculatus from Nankai University (39°6.016N, 117°9.977E), Tianjin, China, on July 7th, 2008. We preserved specimens in 95% ethanol and stored them at −20 °C until tissues were used for DNA extraction. Voucher specimens are deposited in the Insect Molecular Systematic Lab, Institute of Entomology, College of Life Sciences, Nankai University, Tianjin, China.
PCR amplification and sequencing
We extracted genomic DNA from thoracic muscle tissue using the CTAB-based method59. We generated the entire mt-genome of Panaorus albomaculatus by amplification of four overlapping PCR fragments. We designed primers from the sequenced fragments, modifying primers from previous studies13 (see Supplementary Table S4). We performed PCR reactions using TaKaRa LA Taq under the following conditions: 1 min initial denaturation at 94 °C, followed by 30 cycles of 20 s at 94 °C, 1 min at 50 °C, and 2–8 min at 68 °C, and a final elongation for 10 min at 72 °C. We separated PCR products using electrophoresis in 1% agarose gels, purified the fragments, and then sequenced both strands of each fragments using a ABI 3730XL capillary sequencer with the BigDye Terminator Sequencing Kit (Applied Bio Systems).
Sequence analysis and annotation
We proofread the raw sequence files and then aligned them into contigs using BioEdit version 7.0.5.260. We identified PCGs using ORF Finder as implemented at the NCBI website (http://www.ncbi.nlm.nih.gov/gorf/gorf.html), using invertebrate mitochondrial genetic codes. We compared the boundaries of our predicted PCGs and rRNAs with published insect mt-genomes, and the alignments were produced with CLUSTAL X version 1.8361. We identified tRNA genes using tRNAscan-SE version 1.2162, with the invertebrate mitochondrial codon predictors, and a cove cut-off score of 5. A few of the tRNA genes that could not be detected using tRNAscan-SE were determined through comparing the sequences to other heteropterans. Our analyses of nucleotide composition and codon usage were conducted using MEGA 6.063. We calculated nucleotide skew statistics using the following formulas: AT skew = [A−T]/[A + T] and GC skew = [G−C]/[G + C]33. Tandem repeats were identified using the Tandem Repeats Finder server (http://tandem.bu.edu/trf/trf.html)64, and the stem-loop structure was inferred using the mfold web server (http://mfold.rna.albany.edu/)65.
Prediction of secondary structure of rRNAs
Our secondary structural predictions for 12S rRNA and 16S rRNA are based on previously published models of secondary structure in other insects, including Chauliops fallax (Hemiptera: Malcidae)10, Drosophila melanogaster (Diptera: Drosophilidae)40, Apis mellifera (Hymenoptera: Apidae)44, Manduca sexta (Lepidoptera: Sphingidae)43, and Stenopirates sp. (Hemiptera: Enicocephalidae)56. We followed Gillespie et al.44 and Cameron et al.43 in naming stem-loop structures. We inferred homology within regions of low sequence similarity using the mfold web server65.
Phylogenetic analyses
We conducted phylogenetic analyses on the 40 complete or nearly complete mt-genomes of true bugs that have sequences in GenBank, as well as three species from Sternorrhyncha and Auchenorrhyncha which we used as our outgroup (see Supplementary Table S5). We aligned PCGs based on amino acid sequence alignment using MUSCLE in MEGA version 6.063. Our alignments of individual genes excluded the stop codon and the third codon. We used GPU MrBayes66 for Bayesian inference and raxmlGUI 1.567 for ML phylogenetic analysis. All analyses were based on the GTR + I + Γ model of sequence evolution, as selected using on Modeltest version 3.768. We conducted two simultaneous runs of 10,000,000 generations each for the Bayesian analyses, with a burn-in of 2,500,000 generations and sampling every 100 generations. In ML analyses, we conducted 1000 bootstrap replicates with thorough ML searches.
Additional Information
How to cite this article: Li, T. et al. A Mitochondrial Genome of Rhyparochromidae (Hemiptera: Heteroptera) and a Comparative Analysis of Related Mitochondrial Genomes. Sci. Rep. 6, 35175; doi: 10.1038/srep35175 (2016).
Supplementary Material
Acknowledgments
We thank Dr. Zhen Ye (Nankai University) for help with editing the specimen photograph used in this study. We also thank Mr. Hongju Xia and Prof. Xiaoguang Liu (College of Information Technical Science, Nankai University) for help with parallel computation of the GPU MrBayes program. This research was supported by National Natural Science Foundation of China (No. 31372240, 31501879).
Footnotes
Author Contributions T.L. designed the experiments, carried out analyses, made all figures and drafted the manuscript. T.L. and Y.L. performed the experiments. J.Y. and Y.C. participated in the data analyses. J.Y. and Q.X. helped draft the manuscript. W.B. and D.M.H. directed this study, designed and reviewed analyses, and revised the manuscript. All authors read and approved the final manuscript.
References
- Henry T. J. Phylogenetic analysis of family groups within the infraorder Pentatomomorpha (Hemiptera: Heteroptera), with emphasis on the Lygaeoidea. Ann. Entomol. Soc. Am. 90, 275–301 (1997). [Google Scholar]
- Slater J. A. & O’Donnell J. E. In A catalogue of the Lygaeidae of the world (1960–1994) (New York Entomological Society, New York, 1995). [Google Scholar]
- Schuh R. T. & Slater J. A. In True bugs of the world (Hemiptera: Heteroptera): classification and natural history (Cornell University Press, Ithaca, NY, 1995). [Google Scholar]
- Dong J. Z. & Zheng L. Y. A preliminary study of the phylogenetic relastionships between the higher groups of Lygaeoid complex (Hemiptera: Heteroptera). Acta. Sci. Nat. Univ. Nankaiensis 30, 48–55 (1997). [Google Scholar]
- Xie Q., Bu W. J. & Zheng L. Y. The Bayesian phylogenetic analysis of the 18S rRNA sequences from the main lineages of Trichophora (Insecta: Heteroptera: Pentatomomorpha). Mol. Phylogenet. Evol. 34, 448–451 (2005). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Phylogenetic divergences of the true bugs (Insecta: Hemiptera: Heteroptera), with emphasis on the aquatic lineages: the last piece of the aquatic insect jigsaw originated in the Late Permian/Early Triassic. Cladistics 32, 390–405 (2016). [DOI] [PubMed] [Google Scholar]
- Li H. M. Phylogenetic research on the Lygaeoidea (Hemiptera: Heteroptera) based on mt16S rDNA sequences. J. Guangzhou Univ. (Nat. Sci. Ed.) 6, 30–34 (2007). [Google Scholar]
- Tian X. X. et al. Phylogeny of pentatomomorphan bugs (Hemiptera-Heteroptera: Pentatomomorpha) based on six Hox gene fragments. Zootaxa 2888, 57–68 (2011). [Google Scholar]
- Cameron S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014). [DOI] [PubMed] [Google Scholar]
- Li T., Gao C. Q., Cui Y., Xie Q. & Bu W. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera). PLoS ONE 8, e55381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M. et al. Multiplex sequencing of pooled mitochondrial genomes—a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic. Acids. Res. 42, e166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S. L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 39, 400–411 (2014). [Google Scholar]
- Li T., Yi W., Zhang H., Xie Q. & Bu W. Complete mitochondrial genome of the birch catkin bug Kleidocerys resedae resedae, as the first representative from the family Lygaeidae (Hemiptera: Heteroptera: Lygaeoidea). Mitochondrial DNA 27, 618–619 (2016). [DOI] [PubMed] [Google Scholar]
- Dai X. et al. The complete mitochondrial genome of the plant bug Nesidiocoris tenuis (Reuter) (Hemiptera: Miridae: Bryocorinae: Dicyphini). Zootaxa 3554, 30–44 (2012). [Google Scholar]
- Zhang D. X. & Hewitt G. M. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 25, 99–120 (1997). [Google Scholar]
- Lunt D. H., Whipple L. E. & Hyman B. C. Mitochondrial DNA variable number tandem repeats (VNTRs): utility and problems in molecular ecology. Mol. Ecol. 7, 1441–1455 (1998). [DOI] [PubMed] [Google Scholar]
- Vinces M. D., Legendre M., Caldara M., Hagihara M. & Verstrepen K. J. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324, 1213–1216 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Zheng J., He Y. & Luo C. Tandem repeat modification during double-strand break repair induced by an engineered TAL effector nuclease in zebrafish genome. PLoS ONE 8, e84176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson G. & Gutman G. A. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 (1987). [DOI] [PubMed] [Google Scholar]
- Fondon J. W. & Garner H. R. Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. USA 101, 18058–18063 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS ONE 7, e45925 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J. M. et al. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genomics 9, 610 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M. L., Zhang Q. L., Guo Z. L., Wang J. & Shen Y. Y. Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genomics 16, 460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li H., Wang P., Song F. & Cai W. Comparative mitogenomics of plant bugs (Hemiptera: Miridae): identifying the AGG codon reassignments between serine and lysine. PLoS ONE 9, e101375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J. M. et al. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): evidence from mitochondrial genomes. BMC Evol. Biol. 9, 134 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T. et al. Long-branch attraction and the phylogeny of true water bugs (Hemiptera: Nepomorpha) as estimated from mitochondrial genomes. BMC Evol. Biol. 14, 99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. Y. et al. Complete nucleotide sequence and organization of the mitochondrial genome of Sirthenea flavipes (Hemiptera: Reduviidae: Peiratinae) and comparison with other assassin bugs. Zootaxa 3669, 1–16 (2013). [DOI] [PubMed] [Google Scholar]
- Ojala D., Montoya J. & Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 (1981). [DOI] [PubMed] [Google Scholar]
- Wang Y., Chen J., Jiang L. Y. & Qiao G. X. Hemipteran mitochondrial genomes: features, structures and implications for phylogeny. Int. J. Mol. Sci. 16, 12382–12404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert P. D. N., Ratnasingham S. & deWaard J. R. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. B 270, S96–S99 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roques S., Fox C. J., Villasana M. I. & Rico C. The complete mitochondrial genome of the whiting, Merlangius merlangus and the haddock, Melanogrammus aeglefinus: a detailed genomic comparison among closely related species of the Gadidae family. Gene 383, 12–23 (2006). [DOI] [PubMed] [Google Scholar]
- Schmidt T. R., Wu W., Goodman M. & Grossman L. I. Evolution of nuclear- and mitochondrial-encoded subunit interaction in cytochrome c oxidase. Mol. Biol. Evol. 18, 563–569 (2001). [DOI] [PubMed] [Google Scholar]
- Perna N. T. & Kocher T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995). [DOI] [PubMed] [Google Scholar]
- Hassanin A., Leger N. & Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298 (2005). [DOI] [PubMed] [Google Scholar]
- Cui Y. et al. Phylogenomics of Hemiptera (Insecta: Paraneoptera) based on mitochondrial genomes. Syst. Entomol. 38, 233–245 (2013). [Google Scholar]
- Yokobori S. I. & Pääbo S. tRNA editing in metazoans. Nature 377, 490 (1995). [DOI] [PubMed] [Google Scholar]
- Nesnidal M. P., Helmkampf M., Bruchhaus I. & Hausdorf B. The complete mitochondrial genome of Flustra foliacea (Ectoprocta, Cheilostomata) - compositional bias affects phylogenetic analyses of lophotrochozoan relationships. BMC Genomics 12, 572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore J. L. The complete sequence of the mitochondrial genome of Nautilus macromphalus (Mollusca: Cephalopoda). BMC Genomics 7, 182 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley T. R., Simon C., Flook P. K. & Misof B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol. Biol. 9, 565–580 (2000). [DOI] [PubMed] [Google Scholar]
- Cannone J. J. et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3, 2 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehuis O., Yen S. H., Naumann C. M. & Misof B. Higher phylogeny of zygaenid moths (Insecta: Lepidoptera) inferred from nuclear and mitochondrial sequence data and the evolution of larval cuticular cavities for chemical defence. Mol. Phylogenet. Evol. 39, 812–829 (2006). [DOI] [PubMed] [Google Scholar]
- Niehuis O., Naumann C. M. & Misof B. Identification of evolutionary conserved structural elements in the mt SSU rRNA of Zygaenoidea (Lepidoptera): a comparative sequence analysis. Org. Divers. Evol. 6, 17–32 (2006). [Google Scholar]
- Cameron S. L. & Whiting M. F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408, 112–123 (2008). [DOI] [PubMed] [Google Scholar]
- Gillespie J. J., Johnston J. S., Cannone J. J. & Gutell R. R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol. Biol. 15, 657–686 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taanman J. W. The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta. 1410, 103–123 (1999). [DOI] [PubMed] [Google Scholar]
- Zhang D. X., Szymura J. M. & Hewitt G. M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 40, 382–391 (1995). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Extreme variation in patterns of tandem repeats in mitochondrial control region of yellow-browed tits (Sylviparus modestus, Paridae). Sci. Rep. 5, 13227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int. J. Biol. Sci. 8, 93–107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R. & Barker S. C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 20, 362–370 (2003). [DOI] [PubMed] [Google Scholar]
- Cook C. E. The complete mitochondrial genome of the stomatopod crustacean Squilla mantis. BMC Genomics 6, 105 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F. et al. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae). Sci. Rep. 6, 25725 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves R., Freitas A. I., Jesus J., De la Rua P. & Brehm A. Structure and genetic variation of the mitochondrial control region in the honey bee Apis mellifera. Apidologie 46, 515–526 (2015). [Google Scholar]
- Liu J., Bu C., Wipfler B. & Liang A. Comparative analysis of the mitochondrial genomes of Callitettixini Spittlebugs (Hemiptera: Cercopidae) confirms the overall high evolutionary speed of the AT-rich region but reveals the presence of short conservative elements at the tribal level. PLoS ONE 9, e109140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A., Ladoukakis E. & Zouros E. Animal mitochondrial DNA recombination revisited. Trends Ecol. Evol. 18, 411–417 (2003). [Google Scholar]
- Farabaugh P. J., Schmeissner U., Hofer M. & Miller J. H. Genetic studies of the lac repressor. VII. On the molecular nature of spontaneous hotspots in the lacI gene of Escherichia coli. J. Mol. Biol. 126, 847–857 (1978). [DOI] [PubMed] [Google Scholar]
- Li H. et al. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE 7, e29419 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh R. T., Weirauch C. & Wheeler W. C. Phylogenetic relationships within the Cimicomorpha (Hemiptera: Heteroptera): a total-evidence analysis. Syst. Entomol. 34, 15–48 (2009). [Google Scholar]
- Tian Y., Zhu W., Li M., Xie Q. & Bu W. Influence of data conflict and molecular phylogeny of major clades in Cimicomorphan true bugs (Insecta: Hemiptera: Heteroptera). Mol. Phylogenet. Evol. 47, 581–597 (2008). [DOI] [PubMed] [Google Scholar]
- Reineke A., Karlovsky P. & Zebitz C. P. Preparation and purification of DNA from insects for AFLP analysis. Insect Mol. Biol. 7, 95–99 (1998). [DOI] [PubMed] [Google Scholar]
- Hall T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic. Acids. Symp. Ser. 41, 95–98 (1999). [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F. & Higgins D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic. Acids. Res. 25, 4876–4882 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T. M. & Eddy S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A. & Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic. Acids. Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic. Acids. Res. 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Liu X., Stones D. S., Xie Q. & Wang G. MrBayes on a graphics processing unit. Bioinformatics 27, 1255–1261 (2011). [DOI] [PubMed] [Google Scholar]
- Silvestro D. & Michalak I. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2012). [Google Scholar]
- Posada D. & Crandall K. A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.