

Abstract
Atmospheric aerosols are the dominant source of Pb to the modern marine environment, and as a result, in most regions of the ocean the Pb isotopic composition of dissolved Pb in the surface ocean (and in corals) matches that of the regional aerosols. In the Singapore Strait, however, there is a large offset between seawater dissolved and coral Pb isotopes and that of the regional aerosols. We propose that this difference results from isotope exchange between dissolved Pb supplied by anthropogenic aerosol deposition and adsorbed natural crustal Pb on weathered particles delivered to the ocean by coastal rivers. To investigate this issue, Pb isotope exchange was assessed through a closed-system exchange experiment using estuarine waters collected at the Johor River mouth (which discharges to the Singapore Strait). During the experiment, a known amount of dissolved Pb with the isotopic composition of NBS-981 (206Pb/207Pb = 1.093) was spiked into the unfiltered Johor water (dissolved and particulate 206Pb/207Pb = 1.199) and the changing isotopic composition of the dissolved Pb was monitored. The mixing ratio of the estuarine and spike Pb should have produced a dissolved 206Pb/207Pb isotopic composition of 1.161, but within a week, the 206Pb/207Pb in the water increased to 1.190 and continued to increase to 1.197 during the next two months without significant changes of the dissolved Pb concentration. The kinetics of isotope exchange was assessed using a simple Kd model, which assumes multiple sub-reservoirs within the particulate matter with different exchange rate constants. The Kd model reproduced 56% of the observed Pb isotope variance. Both the closed-system experiment and field measurements imply that isotope exchange can be an important mechanism for controlling Pb and Pb isotopes in coastal waters. A similar process may occur for other trace elements.
This article is part of the themed issue ‘Biological and climatic impacts of ocean trace element chemistry’.
Keywords: isotope exchange, Pb, Pb isotopes, estuaries, particles, Singapore
1. Introduction
Fluvial transport has long been noted as an important source for metals into the ocean (e.g. [1,2] and references therein). The transport of metals involves the direct injection of dissolved metals and transport of particulate matter that may release or exchange elements when it contacts seawater [3–6]. Through their interaction with seawater, crustal-derived particulates can affect the concentration of some elements in estuarine waters (e.g. Ba, Cd, [6–10]); or can affect the isotopic composition of some elements without much change in concentration [11]. Such isotope exchange has been noted for Sr and Nd in both closed-system experiments [12,13] and in field observations [14–16]. There is no information on whether such isotope exchange influences Pb isotope data in coastal and estuarine waters.
Pb is widely used in tracing anthropogenic emissions into the earth surface environment, as its isotopic composition can distinguish sources from different reservoirs [17]. Understanding the processes that affect Pb isotopes in different environments is of critical importance in interpreting Pb data in a dynamic environment. In the modern open ocean, anthropogenically enhanced aeolian input is the major source of Pb [18]. And absent an abundant supply of crustal particulates, Pb isotopes found in surface oceans are generally the same as the aerosols depositing in the sea surface (e.g. [10,19,20]). In estuarine and coastal environments, the fluvial contribution of elemental Pb into the ocean and the potential exchanges in Pb isotopes by crustal particulates has not been clearly assessed, because of the dynamic nature of estuarine environments and the overwhelming supply of anthropogenic Pb into the modern environment.
As an alternative strategy, the fluvial and estuarine behaviour of Pb has been investigated extensively using radiogenic 210Pb. It has been found that the majority (greater than or equal to 79%) of 210Pb in an estuary was associated with particulate matter (e.g. [21–25]), although some exceptions have been found in rivers with high concentrations of dissolved organic matter (e.g. [26]). These studies concluded that 210Pb is scavenged within the estuaries, and, by analogy, it was suggested that stable Pb should also be scavenged. However, much of the elemental Pb is originally contained within mineral crystals, but these unstable phases break down and dissolve during weathering and release elemental Pb which largely adsorbs onto mineral surfaces (e.g. [27]). By contrast, 210Pb is produced within the 238U decay series by its precursor the inert gas 222Rn, which partially leaks into the atmosphere from which the daughter 210Pb is deposited. As a result, surface ocean water is relatively enriched in 210Pb and low in stable Pb, while the crustal particulates are relatively depleted in 210Pb but enriched in stable Pb. There might be significant differences between the pathways by which natural Pb and 210Pb move through the environment, although this subject has never been explored in the field.
Despite very few observations of stable Pb isotope ratios in estuarine and coastal water, a few studies did imply that isotope exchange may influence Pb in coastal and estuarine regions. One example comes from coral Pb studies in the Singapore Strait. Three corals were investigated on an east–west transect of the strait, and the Pb isotopes in these corals all shown 206Pb/207Pb = >1.190 [28,29] distinct from regional aerosols (206Pb/207Pb = ∼1.097–1.167 in Southeast Asia [30]). No high 206Pb/207Pb sources in the region have been found except regional natural soil. Hence, isotope exchange between seawater and natural crustal particulates is a possible explanation for why 206Pb/207Pb was higher in marine systems compared with the aerosols.
A second example is the Japanese GEOTRACES data on dissolved Pb and Pb isotopes in Indian Ocean seawater (KH09-5, November–December 2010; [31]). Thirteen stations were covered including the Arabian Sea and the Bay of Bengal. The 206Pb/207Pb in one profile obtained from the Bay of Bengal was 0.005–0.010 higher than in a profile obtained from the Arabian Sea, despite a concentration profile depth distribution that was almost identical. The isotopic offset could not be fully explained by different anthropogenic sources as the offset was observable throughout the water column, whereas the anthropogenic dissolved Pb contamination in the northern Indian Ocean was still confined to the upper ocean. From these observations, it was proposed that isotope exchange in the Arabian Sea and the Bay of Bengal may have created the isotopic offset between the two profiles.
These examples imply that isotope exchange might be an important mechanism modulating Pb isotopes in the ocean. To this end, the objective of this study is to experimentally illustrate the isotope exchange of Pb between the dissolved phase and coastal particulate matter, and investigate the kinetics of exchange using a simple Kd-type model.
2. Sampling and methodology
(a). Closed-system isotope exchange experiment
A closed-system isotope exchange experiment was carried out using estuarine water from the Johor River mouth (figure 1). The Johor River is located at the southern end of the Malaysian Peninsula and discharges a large quantity of freshwater from the Malaysian Peninsula to the Singapore Strait [32,33]. The mean annual discharge for the Johor River is approximately 1200 km3 yr−1 [34]. At the same time, a large quantity of crustal particulates is delivered to the Singapore Strait due to the mountainous terrain, intense chemical weathering and heavy rainfall in the region [35]. The upstream of the Johor River basin is mainly forest, while the downstream is agricultural land (oil palm and rubber, [36]). On 22 November 2013, 8 l water samples were collected from the estuarine waters near the Johor River mouth (1°25′22″ N 104°00′05″ E; figure 1). The samples were collected by lowering a plastic coated pole sampler near the front of the boat while the boat was slowly moving forward. A trace-metal-clean bottle was attached in the front of the pole to collect the water sample. The samples were transferred to the laboratory in a dark cooler box after collection.
Figure 1.

Map of the Singapore Strait and the Johor River with monsoonal wind and current directions. The sampling sites are illustrated in numbered boxes. The sampling sites include (1) Johor River mouth for isotope exchange water samples; (2) National University of Singapore for aerosol samples; (3) Kusu for seasonal seawater samples and (4) Hantu for seasonal seawater samples.
In the laboratory, 250 ml of unfiltered water and 250 ml of 0.4 µm filtered water were sub-sampled to provide the initial dissolved Pb and Pb isotope ratios in the water before the isotope exchange experiment. A subsample of the unfiltered water was acidified to pH ∼ 2 to determine ‘total dissolvable’ Pb and Pb isotope ratios (95% of which was derived from the particulate fraction). The remaining samples were divided into two groups (A and B): for group A, approximately 3.5 l of unfiltered sample was put into a 4 l trace-metal-clean narrow mouth bottle (bottle A). For group B, approximately 3.5 l of sample was filtered through 0.4 µm Nuclepore® polycarbonate filters, and kept in another identical narrow mouth bottle (bottle B) to serve as a control. The filters used in this study have been leached multiple times in ultrapure acid and then rinsed multiple times in 4× distilled deionized water, which should in theory contribute an immeasurable blank. The Pb concentration of the filtered sample at the beginning of the experiment was 18.3 pmol kg−1. At time t = 0, both bottles were spiked with 1.75 ml of pH ∼ 2, 21 nmol kg−1 NBS-981 (206Pb/207Pb = 1.093, [37]). The amount of spike added should result in 10.5 pmol kg−1 concentration increase in the sample (to 28.8 pmol kg−1) if no net adsorption occurs. The bottle was vigorously shaken during spike addition to minimize the possible effects of the acidic spike on the water chemistry. After spike addition, a 250 ml sample was separated and filtered from each 4 l bottle at different time points up to 60 days. A schematic about the isotope exchange experiment is shown in figure 2.
Figure 2.

Schematics of the isotope exchange experiments.
(b). Aerosol sampling
Aerosol samples for were collected on the roof of building S16 of National University of Singapore (NUS) from July 2011 through November 2012. From November 2012 to April 2013, the sampling station was moved to NUS CREATE building but the two buildings were within 1 km distance. The samples were collected by pumping air through a pre-cleaned 0.45 µm PTFE filter using a diaphragm pump (similar to that employed by Bollhöfer et al. [38]). Most aerosol samples were collected over about a week although some had longer collection periods (multiple weeks) due to the travel schedule of the sampling personnel.
(c). Seawater sampling
Seawater samples were collected seasonally in both Hantu and Kusu from September 2011 through November 2013. The samples were filtered (0.4 µm) and acidified to pH ∼ 2 with ultrapure 6N HCl. The time between sample collection and filtration varies from within 1 day to six months due to personnel limitations. Given the variable time between sample collection and filtration, only Pb isotopes are reported as Pb concentrations could have been affected by adsorption to the bottle walls. The filtered samples were stored acidified for at least two months before analysis to ensure the release of Pb adsorbed onto the surface of the container.
(d). Methods for analysing Pb and Pb isotopes
The filtered samples were analysed for both Pb concentration and Pb isotope ratios. The Pb concentrations were measured using isotope dilution after single batch nitrilotriacetate (NTA) resin extraction [31]. In brief, 1.3 ml of seawater samples was spiked with a known amount of 204Pb enriched spike (Oak Ridge National Laboratories) and then adjusted to pH = 5.3 by adding an ammonium acetate buffer. Approximately 2400 NTA superflow resin beads were added to each sample. After 4 days on a shaker table to allow the resin beads to bind Pb, the resin was rinsed several times with ultrapure water and then eluted using 0.1 M high-purity nitric acid. The eluted samples were then analysed on a Quadrupole ICP-MS (VG PlasmaQuad 2+). All samples were run in triplicate and accepted only if at least two out of three replicates agreed.
Pb isotope ratios in the seawater were measured using an IsoProbe multi-collector ICP-MS after Mg(OH)2 co-precipitation and HCl–HBr ion exchange chromatography as described in [39,41]. The seawater sample was spiked with a minimum dose of ammonia solution to induce Mg(OH)2 precipitation that scavenges Pb from the seawater. The precipitates were redissolved by a minimum amount of high-purity HCl and the Mg(OH)2 precipitation method was repeated for a second time to further concentrate the Pb. The final precipitates were redissolved in 200 µl of ultrapure 1.1 M HBr and loaded onto an Eichrom AG-1X8 (chloride form, 200–400 mesh) anion exchange resin column, and then eluted with 1 M and 6 M HCl to separate the Pb from the sample matrices. After ion exchange, the samples were dried in a class 100 clean environment and redissolved in ultrapure 0.2 M HNO3 for GV IsoProbe multi-collector sector ICP-MS analysis. Standardization and corrections were handled as discussed in Boyle et al. [40]. Although the expected precision and accuracy of the measurement depends on the concentration of the sample (limited by Johnson resistor noise), these 206Pb/207Pb data should be good to at least ±0.001 (2σ).
Pb isotope ratios in aerosols were measured using the MC-ICP-MS in the same way as seawater samples after leaching the filter in 6 M high-purity HCl, drying down, loading with 1.1 N HBr, and passing the leachates through the anion exchange columns.
3. Results
The Pb isotope ratios in aerosols (table 2) and seawater samples are shown in figure 3. The 206Pb/207Pb in Singapore seawater was consistently approximately 1.195 with no evident seasonal cycle. Additionally, no spatial difference for Pb isotopes was observed (figure 3). The 206Pb/207Pb in Singapore aerosols was approximately 1.147, with 206Pb/207Pb in the northeast monsoon season (November–March) being slightly higher (approx. 0.005) than in the southwest monsoon season (April–September). However, the Pb isotopes in Singapore aerosols are clearly distinct from Singapore Strait seawater at all times.
Table 2.
Singapore aerosol Pb isotope data. See Lee et al. [29] for data from July 2011 to April 2012.
| sampling period | 206/207 | 6/7 2 s.e. | 208/207 | 8/7 2 s.e. |
|---|---|---|---|---|
| 27–29 July 2011 | 1.1415 | 0.0001 | 2.4192 | 0.0001 |
| 3–7 Aug 2011 | 1.1508 | 0.0000 | 2.4267 | 0.0001 |
| 7–14 Aug 2011 | 1.1489 | 0.0001 | 2.4267 | 0.0001 |
| 14–23 Aug 2011 | 1.1496 | 0.0001 | 2.4268 | 0.0001 |
| 23 Aug–16 Sep 2011 | 1.1488 | 0.0001 | 2.4265 | 0.0001 |
| 16 Sep–1 Oct 2011 | 1.1481 | 0.0000 | 2.4245 | 0.0001 |
| 1–14 Oct 2011 | 1.1462 | 0.0001 | 2.4223 | 0.0002 |
| 14 Oct–1 Nov 2011 | 1.1467 | 0.0000 | 2.4213 | 0.0001 |
| 1–16 Nov 2011 | 1.1478 | 0.0001 | 2.4236 | 0.0001 |
| 16 Nov–16 Dec 2011 | 1.1467 | 0.0001 | 2.4221 | 0.0001 |
| 16 Dec 2011–2 Jan 2012 | 1.1539 | 0.0003 | 2.4319 | 0.0005 |
| 16–18 Jan 2012 | 1.1480 | 0.0003 | 2.4228 | 0.0004 |
| 2–7 Feb 2012 | 1.1496 | 0.0001 | 2.4276 | 0.0001 |
| 16–21 Feb 2012 | 1.1507 | 0.0001 | 2.4257 | 0.0001 |
| 4–9 Mar 2012 | 1.1474 | 0.0001 | 2.4216 | 0.0001 |
| 22–27 Mar 2012 | 1.1491 | 0.0000 | 2.4248 | 0.0002 |
| 5–10 Apr 2012 | 1.1469 | 0.0001 | 2.4214 | 0.0002 |
| 12–17 Apr 2012 | 1.1486 | 0.0001 | 2.4247 | 0.0002 |
| 19–24 Apr 2012 | 1.1445 | 0.0000 | 2.4217 | 0.0001 |
| 26 Apr–1 May 2012 | 1.1433 | 0.0001 | 2.4202 | 0.0001 |
| 3–8 May 2012 | 1.1478 | 0.0001 | 2.4236 | 0.0001 |
| 10–15 May 2012 | 1.1366 | 0.0000 | 2.4124 | 0.0001 |
| 17–24 May 2012 | 1.1464 | 0.0001 | 2.4247 | 0.0001 |
| 24–29 May 2012 | 1.1453 | 0.0001 | 2.4210 | 0.0001 |
| 1–6 June 2012 | 1.1478 | 0.0001 | 2.4246 | 0.0001 |
| 11–18 June 2012 | 1.1432 | 0.0000 | 2.4215 | 0.0001 |
| 18–22 June 2012 | 1.1468 | 0.0000 | 2.4223 | 0.0001 |
| 28 June–3 July 2012 | 1.1490 | 0.0003 | 2.4265 | 0.0006 |
| 5–17 July 2012 | 1.1438 | 0.0000 | 2.4215 | 0.0001 |
| 27 July–1 Aug 2012 | 1.1477 | 0.0001 | 2.4259 | 0.0001 |
| 2–7 Aug 2012 | 1.1464 | 0.0000 | 2.4243 | 0.0001 |
| 10–15 Aug 2012 | 1.1372 | 0.0001 | 2.4156 | 0.0001 |
| 17–23 Aug 2012 | 1.1377 | 0.0001 | 2.4179 | 0.0002 |
| 23–29 Aug 2012 | 1.1437 | 0.0001 | 2.4211 | 0.0001 |
| 31 Aug–5 Sep 2012 | 1.1459 | 0.0000 | 2.4247 | 0.0001 |
| 6–11 Sep 2012 | 1.1445 | 0.0000 | 2.4224 | 0.0001 |
| 13–18 Sep 2012 | 1.1407 | 0.0000 | 2.4185 | 0.0001 |
| 19–24 Sep 2012 | 1.1427 | 0.0000 | 2.4205 | 0.0001 |
| 7–14 Nov 2012 | 1.1550 | 0.0000 | 2.4345 | 0.0001 |
| 20–28 Nov 2012 | 1.1473 | 0.0000 | 2.4240 | 0.0001 |
| 29 Nov–4 Dec 2012 | 1.1518 | 0.0000 | 2.4272 | 0.0000 |
| 14–19 Dec 2012 | 1.1432 | 0.0000 | 2.4197 | 0.0001 |
| 20–26 Dec 2012 | 1.1486 | 0.0000 | 2.4246 | 0.0001 |
| 3–8 Jan 2013 | 1.1494 | 0.0000 | 2.4247 | 0.0001 |
| 16–21 Jan 2013 | 1.1569 | 0.0000 | 2.4378 | 0.0001 |
| 24–29 Jan 2013 | 1.1586 | 0.0001 | 2.4410 | 0.0002 |
| 12–18 Apr 2013 | 1.1439 | 0.0000 | 2.4218 | 0.0001 |
Figure 3.

The Pb isotope time series in Singapore aerosols (grey filled diamonds) and seawater (squares). The seawater samples were taken from the Singapore Strait near either Hantu (black filled squares) or Kusu (open squares). The dashed line illustrates an average 206Pb/207Pb ratio in Singapore Strait water. The aerosol samples were taken on top of the S16 building and CREATE building. The light band illustrates the regional aerosols [30] and the dark band illustrates the seawater feeding to the Singapore Strait [29,31].
The change in Pb concentration during the isotope exchange experiment is shown in figure 4. The dissolved Pb concentration in the collected water was 18.3 pmol kg−1 (before any manipulation); the unfiltered ‘total dissolvable’ Pb concentration was 371 pmol kg−1, indicating a large reservoir of particulate Pb (353 pmol kg−1). After spike addition, the Pb concentration in unfiltered bottle A increased to 25.4 pmol kg−1, and then fluctuated approximately between 20 and 30 pmol kg−1 within next 2 days. Afterwards, the Pb concentration increased approximately from 21 to 35 pmol kg−1 within 7 days and remained there until the end of two months. The detailed concentration variability can be found in table 1.
Figure 4.

The measured Pb concentration in seawater after spiking with NBS-981 standard reference material. Bottle A (grey filled triangles) is the unfiltered seawater; bottle B (open triangles) is the 0.4 µm filtered seawater; and the unspiked sample is shown in a black filled triangle. The main figure shows the detail change in Pb concentration in bottle A within first 200 h and the inset figure shows the two-month-long variation of Pb concentration in both bottles A and B.
Table 1.
Variability in Pb concentration and isotopes during closed-system isotope exchange experiment.
| name and timestep | 206/207 | 206/207 2 s.e.a | 208/207 | 208/207 2 s.e.a | [Pb] (pmol l−1) | SD for [Pb]b (pmol l−1) |
|---|---|---|---|---|---|---|
| Johor A unfiltered | 1.1987 | 0.0001 | 2.5438 | 0.0001 | 373.4 | 5.4 |
| Johor A T0 no spike | 1.1998 | 0.0001 | 2.5239 | 0.0003 | 18.3 | 2.0 |
| Johor A T0 | 1.1781 | 0.0001 | 2.5085 | 0.0003 | 25.4 | 1.4 |
| Johor A T 3 h | 1.1893 | 0.0002 | 2.5283 | 0.0004 | 32.7 | 0.5 |
| Johor A T 5 h | 1.1841 | 0.0005 | 2.5364 | 0.0013 | 27.1 | 2.4 |
| Johor A T 24 h | 1.1811 | 0.0003 | 2.5317 | 0.0004 | 21.1 | 1.5 |
| Johor A T 47.5 h | 1.1869 | 0.0002 | 2.5400 | 0.0006 | 20.9 | 1.0 |
| Johor A T 7 days | 1.1900 | 0.0002 | 2.5456 | 0.0004 | 35.2 | 0.5 |
| Johor A T 23 days | 1.1938 | 0.0005 | 2.4788 | 0.0009 | 33.9 | 2.0 |
| Johor A T two months | 1.1967 | 0.0001 | 2.5368 | 0.0003 | 33.0 | 3.7 |
| Johor B T 0 | 1.1668 | 0.0002 | 2.5210 | 0.0004 | 47.8 | 1.9 |
| Johor B T 2 h | 1.1671 | 0.0002 | 2.5253 | 0.0004 | 45.9 | 1.6 |
| Johor B T 21 h | 1.1675 | 0.0002 | 2.5255 | 0.0006 | 43.9 | 1.1 |
| Johor B T 44.5 h | 1.1678 | 0.0003 | 2.5206 | 0.0006 | 47.4 | 1.3 |
| Johor B T 7 days | 1.1667 | 0.0002 | 2.5234 | 0.0008 | c | |
| Johor B T 23 days | 1.1711 | 0.0003 | 2.4553 | 0.0008 | 36.0 | 1.8 |
| Johor B T two months | 1.1673 | 0.0004 | 2.5849 | 0.0005 | 36.8 | 2.3 |
aThe 2 s.e. was twice of the normalized standard deviation associated with the internal counting statistics of the ICP-MS during the run. The external reproducibility was monitored by measuring an in house standard (BAB3 deg, calibrated with NBS981 standard reference material) in the beginning and the end of the day and in between every five samples.
bEach seawater sample was measured at least three times and the s.d. was the standard deviation of the triplicates.
cRan out of sample.
The Pb concentration in the filtered bottle B was fairly stable at approximately 46.3 pmol kg−1 within 7 days and gradually decreased to approximately 36.4 pmol kg−1 during two months. We did not expect to see the Pb concentration increase by 28 pmol kg−1 (from 18.3 to 46.3 pmol kg−1) from the added spike, which should only have caused a 10.5 pmol kg−1 increase in the Pb concentration. We suspect that the higher Pb concentration in bottle B was introduced during the large-volume filtration process involved in filling-up bottle B (this filtration took up to 3 h) and some Pb from the particles might have been released into the water during the filtration process. The high Pb concentration could also have been caused by contamination during the filtration process. However, neither cause of the high Pb concentration affects the interpretation of Pb variability during the two months. It was encouraging to see the Pb concentration in bottle B decrease by only approximately 20% from 2 days to two months suggesting that the effect of bottle wall uptake should be minimal in the particle-buffered unfiltered experiment.
The isotopic variability during the isotope exchange experiment (table 1) is illustrated in figure 5. During the two months of the experiment, the 206Pb/207Pb of the filtered and spiked bottle ‘B’ was 1.168 ± 0.002 (2σ), indicating no isotope exchange happened in this bottle. Before the spike addition, the 206Pb/207Pb in both filtered and acidified unfiltered samples was 1.199, showing that the dissolved and acid-soluble particulate Pb have the same isotopic composition. Assuming that the filtered (dissolved) concentration of this water was 18.3 pmol kg−1, the spike addition (1.75 ml of 21 nmol kg−1) should have lowered the dissolved 206Pb/207Pb in the bottle to 1.161 (calculated T = 0). Filtering a subsample soon after the spike addition (within 5 min), the 206Pb/207Pb in bottle A was 1.178 and further increased to 1.189 by 3 h. From 3 h to 1 day, the 206Pb/207Pb in bottle A decreased from 1.189 to 1.181. After that brief drop, 206Pb/207Pb increased steadily from 1.181 to 1.197 at two months (figure 5 and table 2).
Figure 5.

The measured 206Pb/207Pb ratios of seawater after spiking with NBS-981 standard reference material (big open square). The main figure shows the detailed change in 206Pb/207Pb in the unfiltered bottle A (grey filled squares) after spike addition with the shaded area indicating the range of 206Pb/207Pb in Singapore Strait water. The inset figure shows the overview of the 206Pb/207Pb in unfiltered sample (big filled square), spike (big open square), the change in 206Pb/207Pb ratios in the unfiltered bottle A and filtered bottle B (filtered at t = 0, open squares). The two arrows in the inset figure illustrate the average 206Pb/207Pb in Singapore aerosol and natural regional soil.
4. Discussion
(a). Pb isotopes in Singapore seawater and aerosols
Pb isotope ratios in Singapore seawaters were generally stable during our 2-year sampling period, with 206Pb/207Pb = 1.195 ± 0.004 (2σ) and 208Pb/207Pb = 2.479 ± 0.006 (2σ). The steady Pb isotope ratios in Singapore seawater indicates that the strait Pb was dominated by a consistent Pb reservoir with high 206Pb/207Pb ratio. This consistency is at odds with the oceanographic setting of the Singapore Strait. The Singapore Strait is dominated by seasonal monsoon-driven currents [42]. It is flushed mainly by South China Sea water during the northeast monsoon (November–March) and mainly by the Malacca Strait and Java Sea water during southwest monsoon (April–September) [43]. There is almost no Pb isotope data in either of these regions, but neither the South China Sea water (one surface sample collected near Taiwan in 2000 had 206Pb/207Pb = 1.156 ± 0.015, 2σ, 208Pb/207Pb = 2.444 ± 0.017, 2σ, [29], table 3) nor the Indian Ocean water (eight samples of central Indian Ocean surface water had 206Pb/207Pb = 1.144 ± 0.006, 2σ, 208Pb/207Pb = 2.426 ± 0.009, 2σ [31]) can account for the isotope ratios of Pb in Singapore seawater.
Table 3.
The Pb isotope values in local or regional sources compare to the Singapore Strait water. Note the large difference in the Pb isotope of Singapore Strait water from all other sources in the region.
| sample | 206/207 | 208/207 | source |
|---|---|---|---|
| Singapore Strait water | 1.191–1.198 | 2.474–2.484 | this study |
| South China Sea water | 1.156 | 2.444 | Lee et al. [29] |
| Bay of Bengal water | 1.149 | 2.431 | Lee et al. [29] |
| Singapore aerosol | 1.137–1.159 | 2.412–2.441 | this study |
| Hong Kong aerosol | 1.149 | 2.440 | Bollhöfer & Rosman [30] |
| Vietnamese aerosol | 1.155–1.167 | 2.404–2.430 | Bollhöfer & Rosman [45] |
| Bangkok aerosol | 1.127 | 2.404 | Bollhöfer & Rosman [45] |
| Kuala Lumpur aerosol | 1.141 | 2.410 | Bollhöfer & Rosman [45] |
| Indonesian aerosol | 1.097–1.131 | 2.366–2.395 | Bollhöfer & Rosman [45] |
| Singapore Incineration ash | 1.141–1.153 | 2.412–2.427 | Chen et al. [28] |
| Singapore urban runoff | 1.169 | 2.451 | Carrasco et al. 2016, unpublished data |
The high 206Pb/207Pb ratio in Singapore seawater is also clearly distinct from Pb isotope ratios of aerosols from all of Southeast Asia. Singapore aerosol 206Pb/207Pb averaged 1.150 in the northeast monsoon season and 1.145 in the southwest monsoon season (figure 3). Atmospheric deposition appears to be the main source of Pb to the Singapore region as suggested by the sedimentary record from the central catchment reserve [44]. Besides Singapore aerosols, the regional aerosols (Kuala Lumpur, Bangkok, Vietnam, Indonesia, Hong Kong) 206Pb/207Pb range from 1.097 to 1.167 (table 3, [30,45]). In this case, regardless of local or distal sources, none of the 206Pb/207Pb in aerosols in this region comes close to the 1.195 ± 0.004 (2σ) ratio observed in Singapore Strait seawater (figure 3). Instead of reflecting the aerosol Pb isotope value, the high 206Pb/207Pb in Singapore water is similar to the 100-year-old natural soil we observe in this region (206Pb/207Pb ∼ 1.214, [44]), also in the same geological formation of the Johor River catchment [46]. In this case, the isotope differences between Singapore seawater and aerosols, together with the near-constancy of the Pb isotope ratio in strait seawater all suggest that the Pb isotope ratios in Singapore seawater have been converted from their primary aerosol values into more ‘crustal-like’ values by exchanging with crustal particulates.
(b). Mechanisms inferred from the closed-system isotope exchange experiment
Total dissolvable Pb in the Johor River mouth sample was 371 pmol kg−1, but only 18.3 pmol kg−1 was dissolved at the natural pH. These data indicate that less than 5% of Pb was in the dissolved form. The fraction of dissolved Pb is similar to that observed for 210Pb in many estuaries (e.g. [21–25]). Soon after spike addition, the Pb concentration in unfiltered bottle A increased to 25.4 ± 1.4 pmol kg−1, which was nearly that expected for the amount of Pb added. Within the first 3 h, the Pb concentration increased from 25.4 to 32.7 pmol kg−1, and then decreased steadily to 20.9 pmol kg−1 from 3 h to 2 days. It is difficult to be confident that the high Pb concentration at T = 3 h is a real signal as it was only a single sample that must be considered in view of Pb's contamination-prone character. The decreased Pb concentration from 3 h to 2 days was more prominent as the decreasing trend was visible at T = 5, 24 and 47.5 h. The decrease in Pb concentration implied that Pb was scavenged from the water from 3 h to 2 days. In the following 2 days to two months, the Pb concentration increased to 33.9 pmol kg−1 at T = 7 days and remained at approximately 33 pmol kg−1. The increase in Pb concentration from 2 days to two months suggested that processes in this experiment can release Pb from particulate matter into the water. The release of Pb from particles has been suggested in some estuaries (e.g. [47]). The dissolution and scavenging of Pb might function at different rates that result in the observed fluctuations of Pb concentration in bottle A, which has also been previously noted for thorium isotopes [48]. However, with limited sampling resolution, we cannot specify which processes account for these Pb removal/addition observations, but some possibilities are ion exchange with particle surfaces (+ or −), biological uptake (−), biological decomposition (+), scavenging onto mineral or biological surfaces (−), mineral dissolution (+) and mineral precipitation (−). The time dependence of these processes may differ substantially, i.e. some may occur quickly and others slowly. A limited role for colloids on isotope exchange has been observed in this experiment as the 0.4 µm filtered bottle B, excluding particulates but including commonly defined colloids (0.4–0.02 µm), showed limited change in Pb concentration and isotopes over the whole experiment, except adsorption onto the inner surface of the bottle wall after one month.
Pb isotope ratios in the unfiltered bottle A show large changes during the experiment. Soon after spike addition, the 206Pb/207Pb decreased from 1.199 to 1.178, reflecting the effect of the low 206Pb/207Pb from the NBS-981 spike (1.093). It should be noted that by simply adding 1.75 ml of 21 nmol kg−1 NBS-981 into the 18.3 pmol kg−1 of sample, the resulting 206Pb/207Pb should be 1.161 (calculated from mass balance assuming simple mixing of dissolved Pb), which is lower than the observed first sample 206Pb/207Pb ratio (1.178). The higher observed 206Pb/207Pb from simple mixing indicates that some isotope exchange (with Pb adsorbed onto particulates) happened within minutes so the 206Pb/207Pb in the bottle has already been altered to a higher value within a few minutes (our filtration would generally take approx. 5 min). The 206Pb/207Pb increased to 1.190 within 7 days, while the Pb concentration only increased by 9.8 pmol kg−1. If the increase in 206Pb/207Pb was purely due to release from particulate Pb (206Pb/207Pb = 1.199), the resulting 206Pb/207Pb at T = 7 days should be 1.184 by mass balance, which was again lower than the observed 1.190. The disproportional increase in 206Pb/207Pb also indicates that the particulates exchanged Pb isotopes and altered the 206Pb/207Pb to higher values. From 7 days to two months, the 206Pb/207Pb further increased to 1.197 without obvious change in Pb concentration, further supporting an isotope exchange mechanism. It is difficult to explain the 206Pb/207Pb data for T = 3 h and T = 1 day (figure 5) even though on a broader perspective, adsorption and desorption can operate at different rates (e.g. [48]). These speculations remain elusive given the coarse temporal resolution we have in this study. However, despite these uncertainties our closed-system experiment clearly demonstrates a rapid Pb isotope exchange process (within one week for the majority of isotope change; figure 5).
(c). A model for Pb isotope exchange
It has been previously suggested that isotope exchange without net element transfer occurs for Sr and Nd isotopes. The mechanism for this isotope exchange was proposed as contemporaneous dissolution and precipitation into secondary minerals [4,12–14]. That suggested mechanism for Sr isotopes might not be plausible for Pb as the concentration for Pb in seawater is generally in the pico-molar region (e.g. [49]), in which the formation of secondary Pb minerals by precipitation is unlikely.
We suggest that Pb isotope exchange happens between multiple distinct exchangeable sub-reservoirs. We make this assumption following the modifying premises proposed by Li et al. [50, p. 2012] and Nyffeler et al. [51]: ‘the radioisotopes and their natural stable counterparts may not be exchanged rapidly especially with those not in the surface sorption sites. Sorption of certain cations may not follow the reaction, but may involve oxidation and precipitation, ion exchange inside crystal lattices through diffusion, and adsorption of radiotracers on colloids <followed by> coagulation of colloids on larger particles etc.’ Hence we assume that there are multiple Pb reservoirs with different exchange rate time constants. As discussed in §4b, there must be at least one rapid exchange reservoir operating within a few minutes and one slow exchange reservoir that operates over days to months.
In modelling this process, we assume that Pb on the surface of suspended particulate matter approaches distribution coefficient (Kd) equilibrium with dissolved Pb with a time constant that is characteristic of sub-reservoirs within the particulate matter:
| 4.1 |
We estimate Kd = 2 × 106 from the total (acid) dissolvable Pb in the water sample compared to the 0.4 µm filtered dissolved Pb at the initiation of the experiment with the assumption that the total particulate concentration is 10 mg kg−1 (average particulate concentration measured in the Singapore Strait, [52]). It is possible that not all of the acid-soluble Pb participates in exchange equilibrium at the higher natural pH, but the total exchangeable Pb must be close to this number in order to be consistent with the high 206Pb/207Pb ratio observed at the end of the experiment. It should also be noted that a large variability exists for Kd in natural systems [21]; therefore, this estimated Kd represents the distribution coefficient in the bottle experiment, not throughout all of the actual estuarine system.
In the model, we assume Pb ions move in and out from each reservoir to the dissolved pool at the same rate for all of its isotopes. We treat each Pb isotope separately assuming the same Kd for each, and then calculate the resulting isotope ratios (it is known that small stable isotope ratio fractionations can occur during ion exchange, e.g. [53], but these fractionations are generally a few tenths of a per cent compared with the 11% radiogenic Pb isotope signature differences in nature and in this experiment). Taking 206Pb as an example, the instantaneous concentration of 206Pb in the reservoir Ri at time point t can be calculated from the following equation:
| 4.2 |
where Ti is the time constant for reservoir i; and Δt is the time step. The equilibrium concentration for reservoir i can be calculated from the instantaneous concentration in the dissolved seawater as equation (4.3):
 |
4.3 |
where the PM is the concentration of particulate matter (assumed 10 mg kg−1). The same equation can be applied for the other isotopes, and then the isotope ratios can be calculated by taking ratios of the concentrations of each Pb isotope in each reservoir.
The model was initiated based on the initial 18.3 pmol kg−1 concentration 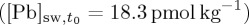 and measured isotopic composition before spike. And then the dissolved concentration is augmented by a 10.5 pmol kg−1 NBS-981 addition. This increases the concentrations of all the dissolved Pb isotopes, which then approach Kd equilibrium given assumed reservoir sizes and kinetic time constants. The size of each reservoir and its kinetic time constant was established by trial and error; but at least two reservoirs and time constants are necessary to explain our results. The model proceeds by time steps with rate constant-based transfer of Pb to the particulate phases (which is determined by the difference between the equilibrium adsorbed Pb for each isotope based on the dissolved concentration from the previous time step, equations (4.2) and (4.3)) and the calculated adsorbed Pb from the previous time step.
and measured isotopic composition before spike. And then the dissolved concentration is augmented by a 10.5 pmol kg−1 NBS-981 addition. This increases the concentrations of all the dissolved Pb isotopes, which then approach Kd equilibrium given assumed reservoir sizes and kinetic time constants. The size of each reservoir and its kinetic time constant was established by trial and error; but at least two reservoirs and time constants are necessary to explain our results. The model proceeds by time steps with rate constant-based transfer of Pb to the particulate phases (which is determined by the difference between the equilibrium adsorbed Pb for each isotope based on the dissolved concentration from the previous time step, equations (4.2) and (4.3)) and the calculated adsorbed Pb from the previous time step.
We began with a single reservoir that accounts for the long-term time evolution of the isotope ratios in the experiment, adjusting the time constant (69 days) to fit the later stages of the experiment (23 days to two months). Then we added a second reservoir that exchanges more rapidly (1.7 days), and used trial and error variations of the time constant and reservoir size to fit the intermediate stages of the experiment. Finally, we added a third reservoir with very rapid exchange constant (5 h) and approximated the earlier portion of the experiment. With the three exchangeable reservoirs, our simple model could reproduce the fast increase in 206Pb/207Pb as we observed in the close-system experiment with R2 = 0.56 (excluding the T = 3 and 24 h ratios, R2 rises to 0.89).
The change in 206Pb/207Pb during the experiment (figures 6 and 7) provides a direct evidence for isotope exchange between dissolved and particulate Pb and can explain the field observations for Singapore Strait seawater. Investigations in Singapore seawater indicated the 206Pb/207Pb in the seawater was more than 1.190 (figure 3), different from either regional aerosols (206Pb/207Pb ∼ 1.140) or the open-ocean seawater feeding the Singapore Strait (206Pb/207Pb =1.144–1.156 [29,31]). In the isotope exchange experiment, 206Pb/207Pb increased from 1.178 to greater than 1.190. The magnitude of the increase in 206Pb/207Pb during the experiment was comparable to the difference between Singapore seawater and aerosols.
Figure 6.

The modelled 206Pb/207Pb ratio (dashed grey line) compared to measured values from the close-system experiment (filled squares). The parameters used in the model are also provided.
Figure 7.

The modelled dissolved Pb concentration (dashed grey line) compared to measured values from the close-system experiment (filled squares). The parameters in the model are the same as illustrated in figure 6.
Besides the magnitude of increase, the timescale of the isotope exchange in the closed-system experiment also supports the consistent isotopic departure between Singapore seawater and aerosols. In the experiment, the major change in Pb isotopes occurred within a week. If the timescale of isotope exchange is much shorter than the residence time of Pb in Singapore water, the isotope exchange could maintain a consistent isotopic difference between Singapore seawater and aerosol (figure 3), which agrees well with what has been observed in Singapore. The residence time of Pb in the Singapore Strait has not been directly investigated, but there are a few clues: the 210Pb-derived residence time in the surface oceans feeding water to the Singapore Strait is 2–4 years (1.8 year in South China Sea [54], 4 years in the eastern Indian Ocean, [55]); although in estuary and coastal regions, a much shorter 210Pb-derived residence time (days–months) has been inferred [21]. We think that the former residence time is more likely for the Singapore Strait, because the variation of Pb/Ca in Singapore corals over more than a 50-year period consistently correlated with the local rainfall with a lag of 2–3 years [28]. The latter estimate should be regarded with caution as 210Pb and stable Pb might follow different mechanisms in estuarine areas. Therefore, we think that 2–3 years should be taken as a preliminary estimate of the residence time of Pb in Singapore water. In this case, the timescale of isotope exchange (hours–days) should be significantly shorter than the residence time of Pb in Singapore water and the isotope exchange could maintain the observed isotopic difference between anthropogenically sourced Pb and Singapore Strait seawater.
(d). The potential role of Pb isotope exchange in ocean chemistry
Crustal particulates transported by rivers have been proposed as an important factor regulating the chemistry of marine trace elements [4]. The global suspended riverine flux has been estimated as 15–20 Gt yr−1 [56,57], among which rivers in South Asia and maritime continent transport disproportionally high amounts of particulates to the Pacific and Indian Ocean (approx. 2/3 of the global suspended particle flux, [35]). These fluvial particulates could provide abundant materials for isotope exchange potentially happening in these regions, changing the Pb isotopes in the water to more ‘crustal-like’ ratios. The influence of isotope exchange on the geochemistry of Pb, and the extent of this influence is still poorly understood because of sparse seawater Pb isotope data in estuaries, coastal areas and near continental margins. The composition of particulates and their role in exchange process, the role of colloids and the dynamic nature of the natural aqueous systems are still poorly constrained. Despite this paucity of observational data, the batch isotope exchange experiment imply that isotope exchange can significantly influence the isotopic composition of Pb in coastal environments and illuminate the fate of particulate material upon its arrival to oceans. Further experimental investigations, including sterilized systems that cannot have biological uptake or decay, targeted exchange experiments using waters from other rivers (especially in regions where the geological Pb source is extreme); some simple single well-characterized phase experiments to illuminate the exchange process in a simple system, and better evidence on the spatial distributions of Pb isotopes in estuaries and coastal environments could provide important insights for the processes affecting Pb in the ocean.
This mechanism of isotope exchange also calls to attention the redistribution of dissolved and particulate Pb in the estuaries and has implications for the anthropogenic Pb impact as well as the natural oceanic Pb cycle. Pb isotopes have been widely used as a fingerprint for tracing anthropogenic sources [17]. The interpretation of Pb isotopes from coastal and estuarine environments is worth re-evaluating as the Pb isotopes in these environments could have been altered to different values from their sources via isotope exchange (figure 8a). It also has implications for the possible mechanisms contributing natural fluvial Pb to the ocean. Before extensive Pb contamination of the atmosphere, Pb would have weathered from unstable natural crustal materials and then adsorb back onto the residual stable weathered phases. When rivers carried this particulate matter into the ocean, particles that had experienced a relatively high-Pb environment would have encountered very low-Pb ocean waters (resulting from a short residence time created by efficient internal ocean scavenging). Based on this partitioning concept, some Pb adsorbed on the particulates would be released in a dissolved form in the estuary that would then mix out into the open ocean. This fluvial source (supplemented by the same process occurring on wind-blown dust deposited into the ocean) would have been the source of lead to the ancient ocean as represented by ferromanganese nodule records (e.g. [58]).
Figure 8.

An illustration of isotope exchange schematics in: (a) the Singapore Strait water. When high 206Pb/207Pb particulates meet low 206Pb/207Pb water, the 206Pb is preferentially released from particulate to water with other isotopes adsorbing from water to particulates. (b) general estuarine areas. When low 210Pb/Pb particulates meet high 210Pb/Pb water, the 210Pb is preferentially adsorbed from water to particulates with stable Pb releasing from particulates to water.
5. Implications for the use of 210Pb as a proxy for elemental Pb (and similar implications for other radioisotope systems)
We have shown that Pb with a different isotope composition added to the coastal marine environment takes on the natural crustal ratio of the regional continental crust, re-equilibrating the isotope ratios in the dissolved and particulate phases. It is clear that the same process would also occur for the uranium series radioisotope 210Pb, i.e. the 210Pb/Pb ratio of the crustal particles and the impinging open-ocean waters should be re-equilibrated in estuarine areas. Crustal materials should have a very low 210Pb/Pb ratio, while open-ocean water has a much higher 210Pb/Pb ratio. Based on the concept of isotope exchange, once low 210Pb/Pb particulates encounter high 210Pb/Pb open-ocean water in the estuarine region, the redistribution would favour the transfer of 210Pb from water onto particulates and stable Pb from particulates into water (figure 8b).
Taylor & McLennan [59] estimate that the average continental crust has 2.8 ppm U and 20 ppm Pb; left undisturbed until radiochemical equilibrium was attained that would result in a 210Pb/Pb ratio of 4.05 × 10+5 Bq mol−1 of Pb. The actual ratio will be lower for materials involved in the active weathering process because a lot of the 222Rn produced in the 238U series will be lost to the atmosphere, and a significant fraction of the daughter 210Pb is deposited in the ocean (e.g. [60]). Observations near Bermuda during the 1980s (even when anthropogenic Pb contamination was high) suggest that the 210Pb/Pb ratio of ocean waters was high (2.6 × 10+7 Bq mol−1 of Pb, [61]) compared with crustal materials. The ratio must have been much higher before anthropogenic elemental Pb contamination. Considering the spatial variability of 210Pb depositional flux, Bermuda is close to the average value of the published 210Pb depositional flux across the globe (varying a factor of approx. 6 [62]). Therefore, even adding spatial variability into consideration, the 210Pb/Pb ratio of ocean waters would still be at least an order of magnitude higher than the 210Pb/Pb ratio in crustal particulates. Hence, in the natural state, adsorbed Pb on weathered crustal material would have a low 210Pb/Pb ratio that would have encountered dissolved marine lead with a much higher 210Pb/Pb ratio (figure 8b). Isotope equilibration would have ensued, and because on an atom per atom basis there is much more stable elemental lead on the crustal material than 210Pb dissolved in the seawater, most of the marine 210Pb would be taken up onto the particles, releasing an immeasurably small amount of stable Pb in its place. Therefore, in this situation, it would appear that 210Pb was scavenged and lost from solution whereas elemental Pb could have been at least slightly released into the low-Pb dissolved phase, just by the operation of the Kd adsorption equilibration. The contrasting difference between the cycling of 210Pb and stable Pb results in a large difference in the 210Pb/Pb ratio between ocean waters and crustal particulates, which eventually results in their different behaviour in the estuarine environment. In other words, as we noted in our introduction ‘there may be some differences between the 210Pb budget compared to the elemental Pb budget’.
Benninger [22] quantified the 210Pb budget for Long Island Sound (USA) and concluded that 210Pb was scavenged in estuaries, and by extension argued that elemental Pb should also be scavenged in estuaries; therefore, rivers would not be a source of Pb into the ocean. As we have noted above, we show that 210Pb should be removed from solution by isotope re-equilibration even when there is a net release of elemental Pb from crustal particles. We suggest that some of the conclusions based on 210Pb to stable Pb should be is re-thought in the light of this process, and we suggest that the process of isotope equilibration should be quantified in other coastal marine environments to determine its generality.
Finally, we note that a similar process may occur for other radionuclide systems. For example, most of the thorium in crustal materials is long-lived 232Th with a minor contribution of 230Th from the 238U series, whereas the ocean contains a higher 230Th/232Th ratio because dissolved U is enriched in seawater and produces a steady supply of 230Th into the ocean. Therefore, there may be situations when net scavenging of Th is inferred from 230Th data, when instead exchange with crustal 232Th is the cause of the lost 230Th.
These considerations should remind us that Kd-style equilibration also may be an important factor in the elemental cycles of elements that do not have the strong isotope ratio contrasts that allowed us to deduce this process for Pb.
6. Conclusion
Isotope exchange of Pb in coastal waters has been investigated through a closed-system exchange experiment using estuarine waters sampled from the Johor River mouth. During the experiment, the 206Pb/207Pb in the sample changed from 1.178 to more than 1.190 within 7 days with disproportional changes in Pb concentration. The change in Pb isotope ratios in the batch experiment was significantly larger than the measurement error. With a timescale of isotope exchange much shorter than the residence time of Pb in a water environment, isotope exchange could maintain a consistent isotope departure from the Pb source feeding the water, which has been observed in the Singapore Strait. In addition to our closed-system experiment and field observations, a simple model was employed to simulate the kinetics of isotope exchange. Using a Kd-type exchange model, more than 50% of the observed 206Pb/207Pb variance can be simulated. The observations in both closed-system experiment and field measurements provide a compelling evidence for rapid (days–months) Pb isotope exchange between seawater and suspended particulate materials. Further investigation of the distributions of Pb and Pb isotopes around estuaries and coastal areas could enhance our understanding on the role of isotope exchange in marine Pb geochemistry. Finally, we suggest in some situations, isotope equilibration might need to be considered in interpreting data from 210Pb and other radioisotope systems.
Data accessibility
The datasets supporting this article are given in the tables.
Authors' contributions
M.C. collected samples, processed them, made Pb and Pb isotope measurements, analysed the data and contributed a large share of the manuscript preparation. E.A.B. participated in the Pb isotope measurements, quality-controlled data, helped in the data analysis and wrote parts of the paper. J.-M.L. processed aerosol filters for Pb isotope analysis. I.N. collected the aerosol and seawater samples. C.Z. made some of the Pb concentration analyses. A.D.S. supervised M.C. as a student and edited the manuscript. G.C. filtered some of the water samples and prepared them for Pb isotope analysis.
Competing interests
We declare we have no competing interests.
Funding
The research described in this project was funded in whole by the Singapore National Research Foundation (NRF) through the Singapore-MIT Alliance for Research and Technology (SMART) Center for Environmental Sensing and Modelling (CENSAM).
References
- 1.Salomons W, Förstner U. 1984. Metals in the hydrocycle. Acta 41, 1139–1144. ( 10.1007/978-3-642-69325-0) [DOI] [Google Scholar]
- 2.Turekian KK. 1977. The fate of metals in the oceans. Geochim. Cosmochim. Acta 41, 1139–1144. ( 10.1016/0016-7037(77)90109-0) [DOI] [Google Scholar]
- 3.Dupré B, Dessert C, Oliva P, Goddéris Y, Viers J, François L, Millot R, Gaillardet J. 2003. Rivers, chemical weathering and Earth's climate. C. R. Geosci. 335, 1141–1160. ( 10.1016/j.crte.2003.09.015) [DOI] [Google Scholar]
- 4.Jeandel C, Oelkers EH. 2015. The influence of terrigenous particulate material dissolution on ocean chemistry and global element cycles. Chem. Geol. 395, 50–66. ( 10.1016/j.chemgeo.2014.12.001) [DOI] [Google Scholar]
- 5.Viers J, Dupré B, Gaillardet J. 2009. Chemical composition of suspended sediments in World Rivers: new insights from a new database. Sci. Total Environ. 407, 853–868. ( 10.1016/j.scitotenv.2008.09.053) [DOI] [PubMed] [Google Scholar]
- 6.Shiller AM, Boyle EA. 1991. The Macalpine Hills Lunar Meteorite Consortium. Trace elements in the Mississippi River Delta outflow region: behavior at high discharge. Geochim. Cosmochim. Acta 55, 3241–3251. ( 10.1016/0016-7037(91)90486-O) [DOI] [Google Scholar]
- 7.Hanor JS, Chan L-H. 1977. Non-conservative behavior of barium during mixing of Mississippi River and Gulf of Mexico waters. Earth Planet. Sci. Lett. 37, 242–250. ( 10.1016/0012-821X(77)90169-8) [DOI] [Google Scholar]
- 8.Edmond JM, Boyle ED, Drummond D, Grant B, Mislick T. 1978. Desorption of barium in the plume of the Zaire (Congo) river. Neth. J. Sea Res. 12, 324–328. ( 10.1016/0077-7579(78)90034-0) [DOI] [Google Scholar]
- 9.Comans RNJ, van Dijk CPJ. 1988. Role of complexation processes in cadmium mobilization during estuarine mixing. Nature 336, 151–154. ( 10.1038/336151a0) [DOI] [Google Scholar]
- 10.Duce RA, et al. 1991. The atmospheric input of trace species to the world ocean. Glob. Biogeochem. Cycles 5, 193–259. ( 10.1029/91gb01778) [DOI] [Google Scholar]
- 11.Jones MT, Pearce CR, Jeandel C, Gislason SR, Eiriksdottir ES, Mavromatis V, Oelkers EH. 2012. Riverine particulate material dissolution as a significant flux of strontium to the oceans. Earth Planet. Sci. Lett. 355–356, 51–59. ( 10.1016/j.epsl.2012.08.040) [DOI] [Google Scholar]
- 12.Jones MT, Pearce CR, Oelkers EH. 2012. An experimental study of the interaction of basaltic riverine particulate material and seawater. Geochim. Cosmochim. Acta 77, 108–120. ( 10.1016/j.gca.2011.10.044) [DOI] [Google Scholar]
- 13.Pearce CR, Jones MT, Oelkers EH, Pradoux C, Jeandel C. 2013. The effect of particulate dissolution on the neodymium (Nd) isotope and Rare Earth Element (REE) composition of seawater. Earth Planet. Sci. Lett. 369–370, 138–147. ( 10.1016/j.epsl.2013.03.023) [DOI] [Google Scholar]
- 14.Jones MT, Gislason SR, Burton KW, Pearce CR, Mavromatis V, Pogge von Strandmann PAE, Oelkers EH. 2014. Quantifying the impact of riverine particulate dissolution in seawater on ocean chemistry. Earth Planet. Sci. Lett. 395, 91–100. ( 10.1016/j.epsl.2014.03.039) [DOI] [Google Scholar]
- 15.Lacan F, Jeandel C. 2001. Tracing Papua New Guinea imprint on the central Equatorial Pacific Ocean using neodymium isotopic compositions and Rare Earth Element patterns. Earth Planet. Sci. Lett. 186, 497–512. ( 10.1016/S0012-821X(01)00263-1) [DOI] [Google Scholar]
- 16.Lacan F, Jeandel C. 2005. Neodymium isotopes as a new tool for quantifying exchange fluxes at the continent–ocean interface. Earth Planet. Sci. Lett. 232, 245–257. ( 10.1016/j.epsl.2005.01.004) [DOI] [Google Scholar]
- 17.Komárek M, Ettler V, Chrastný V, Mihaljevič M. 2008. Lead isotopes in environmental sciences: a review. Environ. Int. 34, 562–577. ( 10.1016/j.envint.2007.10.005) [DOI] [PubMed] [Google Scholar]
- 18.Patterson CC, Settle DM. 1987. Review of data on eolian fluxes of industrial and natural lead to the lands and seas in remote regions on a global scale. Mar. Chem. 22, 137–162. ( 10.1016/0304-4203(87)90005-3) [DOI] [Google Scholar]
- 19.Church TM, Veron A, Patterson CC, Settle D, Erel Y, Maring HR, Flegal AR. 1990. Trace elements in the North Atlantic troposphere: shipboard results of precipitation and aerosols. Glob. Biogeochem. Cycles 4, 431–443. ( 10.1029/GB004i004p00431) [DOI] [Google Scholar]
- 20.Flegal AR. 1986. Lead in tropical marine systems: a review. Sci. Total Environ. 58, 1–8. ( 10.1016/0048-9697(86)90071-9)3810152 [DOI] [Google Scholar]
- 21.Baskaran M, Santschi PH. 1993. The role of particles and colloids in the transport of radionuclides in coastal environments of Texas. Mar. Chem. 43, 95–114. ( 10.1016/0304-4203(93)90218-D) [DOI] [Google Scholar]
- 22.Benninger LK. 1978. 210Pb balance in Long Island Sound. Geochim. Cosmochim. Acta 42, 1165–1174. ( 10.1016/0016-7037(78)90111-4) [DOI] [Google Scholar]
- 23.Benninger LK, Lewis DM, Turekian KK. 1975. The use of natural Pb-210 as a heavy metal tracer in the river—estuarine system. In Marine chemistry in the coastal environment (ed. TM Church), pp. 202–210. Washington, DC: American Chemical Society. [Google Scholar]
- 24.Rama, Koide M, Goldberg ED. 1961. Lead-210 in natural waters. Science 134, 98–99. ( 10.1126/science.134.3472.98) [DOI] [PubMed] [Google Scholar]
- 25.Windom H, Smith R, Rawlinson C, Hungspreugs M, Dharmvanij S, Wattayakorn G. 1988. Trace metal transport in a tropical estuary. Mar. Chem. 24, 293–305. ( 10.1016/0304-4203(88)90037-0) [DOI] [Google Scholar]
- 26.Baskaran M, Ravichandran M, Bianchi TS. 1997. Cycling of 7Be and 210Pb in a high DOC, shallow, turbid estuary of south-east Texas. Estuar. Coast. Shelf Sci. 45, 165–176. ( 10.1006/ecss.1996.0181) [DOI] [Google Scholar]
- 27.Erel Y, Morgan JJ, Patterson CC. 1991. Natural levels of lead and cadmium in a remote mountain stream. Geochim. Cosmochim. Acta 55, 707–719. ( 10.1016/0016-7037(91)90335-3) [DOI] [Google Scholar]
- 28.Chen M, Lee J-M, Nurhati IS, Switzer AD, Boyle EA. 2015. Isotopic record of lead in Singapore Straits during the last 50 years: spatial and temporal variations. Mar. Chem. 168, 49–59. ( 10.1016/j.marchem.2014.10.007) [DOI] [Google Scholar]
- 29.Lee J-M, Boyle EA, Suci Nurhati I, Pfeiffer M, Meltzner AJ, Suwargadi B. 2014. Coral-based history of lead and lead isotopes of the surface Indian Ocean since the mid-20th century. Earth Planet. Sci. Lett. 398, 37–47. ( 10.1016/j.epsl.2014.04.030) [DOI] [Google Scholar]
- 30.Bollhöfer A, Rosman KJR. 2000. Isotopic source signatures for atmospheric lead: the Southern Hemisphere. Geochim. Cosmochim. Acta 64, 3251–3262. ( 10.1016/S0016-7037(00)00436-1) [DOI] [Google Scholar]
- 31.Lee J-M, Boyle EA, Gamo T, Obata H, Norisuye K, Echegoyen Y. 2015. Impact of anthropogenic Pb and ocean circulation on the recent distribution of Pb isotopes in the Indian Ocean. Geochim. Cosmochim. Acta 170, 126–144. ( 10.1016/j.gca.2015.08.013) [DOI] [Google Scholar]
- 32.Thia-Eng C, Gorre IRL, Adrian Ross S, Bernad SR, Gervacio B, Corazon Ebarvia M. 2000. The Malacca Straits. Mar. Pollut. Bull. 41, 160–178. ( 10.1016/S0025-326X(00)00108-9) [DOI] [Google Scholar]
- 33.Hj. Wood AK, Ahmad Z, Md. Shazili NA, Yaakob R, Carpenter ROY. 1997. Geochemistry of sediments in Johor Strait between Malaysia and Singapore. Cont. Shelf Res. 17, 1207–1228. ( 10.1016/S0278-4343(97)00011-3) [DOI] [Google Scholar]
- 34.Kia MB, Pirasteh S, Pradhan B, Mahmud AR, Sulaiman WNA, Moradi A. 2012. An artificial neural network model for flood simulation using GIS: Johor River Basin, Malaysia. Environ. Earth Sci. 67, 251–264. ( 10.1007/s12665-011-1504-z) [DOI] [Google Scholar]
- 35.Milliman JD, Farnsworth KL. 2011. River discharge to the coastal ocean: a global synthesis. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 36.Tan ML, Ibrahim AL, Yusop Z, Duan Z, Ling L. 2015. Impacts of land-use and climate variability on hydrological components in the Johor River basin, Malaysia. Hydrol. Sci. J. 60, 873–889. ( 10.1080/02626667.2014.967246) [DOI] [Google Scholar]
- 37.Baker J, Peate D, Waight T, Meyzena C. 2004. Pb isotopic analysis of standards and samples using a 207Pb–204Pb double spike and thallium to correct for mass bias with a double-focusing MC-ICP-MS. Chem. Geol. 211, 275–303. ( 10.1016/j.chemgeo.2004.06.030) [DOI] [Google Scholar]
- 38.Bollhöfer A, Chisholm W, Rosman KJR. 1999. Sampling aerosols for lead isotopes on a global scale. Anal. Chim. Acta 390, 227–235. ( 10.1016/S0003-2670(99)00182-8) [DOI] [Google Scholar]
- 39.Lee J-M, Boyle EA, Echegoyen-Sanz Y, Fitzsimmons JN, Zhang R, Kayser RA. 2011. Analysis of trace metals (Cu, Cd, Pb, and Fe) in seawater using single batch nitrilotriacetate resin extraction and isotope dilution inductively coupled plasma mass spectrometry. Anal. Chim. Acta 686, 93–101. ( 10.1016/j.aca.2010.11.052) [DOI] [PubMed] [Google Scholar]
- 40.Boyle EA, et al. 2012. GEOTRACES IC1 (BATS) contamination-prone trace element isotopes Cd, Fe, Pb, Zn, Cu, and Mo intercalibration. Limnol. Oceanogr. Methods 10, 653–665. ( 10.4319/lom.2012.10.653) [DOI] [Google Scholar]
- 41.Reuer MK, Boyle EA, Grant BC. 2003. Lead isotope analysis of marine carbonates and seawater by multiple collector ICP-MS. Chem. Geol. 200, 137–153. ( 10.1016/S0009-2541(03)00186-4) [DOI] [Google Scholar]
- 42.Pang WC, Tkalich P. 2003. Modeling tidal and monsoon driven currents in the Singapore Strait. Singapore Marit. Port J. 2003, 151–162. [Google Scholar]
- 43.Chen M, Mural K, Khoo BC, Lou J, Kumar K. 2005. Circulation modelling in the strait of Singapore. J. Coast. Res. 21, 960–972. ( 10.2112/04-0412.1) [DOI] [Google Scholar]
- 44.Chen M, Boyle EA, Switzer AD, Gouramanis C. 2016. A century long sedimentary record of anthropogenic lead (Pb), Pb isotopes and other trace metals in Singapore. Environ. Pollut. 213, 446–459. ( 10.1016/j.envpol.2016.02.040) [DOI] [PubMed] [Google Scholar]
- 45.Bollhöfer A, Rosman KJR. 2001. Isotopic source signatures for atmospheric lead: the Northern Hemisphere. Geochim. Cosmochim. Acta 65, 1727–1740. ( 10.1016/S0016-7037(00)00630-X) [DOI] [Google Scholar]
- 46.Searle MP, et al. 2012. Tectonic evolution of the Sibumasu-Indochina terrane collision zone in Thailand and Malaysia: constraints from new U-Pb zircon chronology of SE Asian tin granitoids. J. Geol. Soc. 169, 489–500. ( 10.1144/0016-76492011-107) [DOI] [Google Scholar]
- 47.Guieu C, Martin JM, Tankéré SPC, Mousty F, Trincherini P, Bazot M, Dai MH. 1998. On trace metal geochemistry in the Danube River and western Black Sea. Estuar. Coast. Shelf Sci. 47, 471–485. ( 10.1006/ecss.1998.0377) [DOI] [Google Scholar]
- 48.Bacon MP, Anderson RF. 1982. Distribution of thorium isotopes between dissolved and particulate forms in the deep sea. J. Geophys. Res. Oceans 87, 2045–2056. ( 10.1029/JC087iC03p02045) [DOI] [Google Scholar]
- 49.Boyle EA, et al. 2014. Anthropogenic lead emissions in the ocean: the evolving global experiment. Oceanography 27, 69–75. ( 10.5670/oceanog.2014.10) [DOI] [Google Scholar]
- 50.Li Y-H, Burkhardt L, Teraoka H. 1984. Desorption and coagulation of trace elements during estuarine mixing. Geochim. Cosmochim. Acta 48, 1879–1884. ( 10.1016/0016-7037(84)90371-5) [DOI] [Google Scholar]
- 51.Nyffeler UP, Li Y-H, Santschi PH. 1984. A kinetic approach to describe trace-element distribution between particles and solution in natural aquatic systems. Geochim. Cosmochim. Acta 48, 1513–1522. ( 10.1016/0016-7037(84)90407-1) [DOI] [Google Scholar]
- 52.Dikou A, van Woesik R. 2006. Survival under chronic stress from sediment load: spatial patterns of hard coral communities in the southern islands of Singapore. Mar. Pollut. Bull. 52, 7–21. ( 10.1016/j.marpolbul.2005.07.021) [DOI] [PubMed] [Google Scholar]
- 53.Anbar AD, Roe JE, Barling J, Nealson KH. 2000. Nonbiological fractionation of iron isotopes. Science 288, 126–128. ( 10.1126/science.288.5463.126) [DOI] [PubMed] [Google Scholar]
- 54.Wei CL, Lin SY, Sheu DD, Chou WC, Yi MC, Santschi PH, Wen LS. 2011. Particle-reactive radionuclides (234Th, 210Pb, 210Po) as tracers for the estimation of export production in the South China Sea. Biogeosciences 8, 3793–3808. ( 10.5194/bg-8-3793-2011) [DOI] [Google Scholar]
- 55.Cochran JK, Bacon MP, Krishnaswami S, Turekian KK. 1983. 210Po and 210Pb distributions in the central and eastern Indian Ocean. Earth Planet. Sci. Lett. 65, 433–452. ( 10.1016/0012-821X(83)90180-2) [DOI] [Google Scholar]
- 56.Meybeck M, Laroche L, Dürr HH, Syvitski JPM. 2003. Global variability of daily total suspended solids and their fluxes in rivers. Glob. Planet. Change 39, 65–93. ( 10.1016/S0921-8181(03)00018-3) [DOI] [Google Scholar]
- 57.Walling DE. 2006. Human impact on land–ocean sediment transfer by the world's rivers. Geomorphology 79, 192–216. ( 10.1016/j.geomorph.2006.06.019) [DOI] [Google Scholar]
- 58.Frank M, Reynolds BC, Keith O'Nions R. 1999. Nd and Pb isotopes in Atlantic and Pacific water masses before and after closure of the Panama gateway. Geology 27, 1147–1150. ( 10.1130/0091-7613(1999)027%3C1147:napiia%3E2.3.co;2) [DOI] [Google Scholar]
- 59.Taylor SR, McLennan SM. 1995. The geochemical evolution of the continental crust. Rev. Geophys. 33, 241–265. ( 10.1029/95RG00262) [DOI] [Google Scholar]
- 60.Nozaki Y, Thomson J, Turekian KK. 1976. The distribution of 210Pb and 210Po in the surface waters of the Pacific Ocean. Earth Planet. Sci. Lett. 32, 304–312. ( 10.1016/0012-821X(76)90070-4) [DOI] [Google Scholar]
- 61.Boyle EA, Sherrell RA, Bacon MP. 1994. Lead variability in the western North Atlantic and Central Greenland: implications for the search for decadal trends in anthropogenic emissions. Geochim. Cosmochim. Acta 58, 3227–3238. ( 10.1016/0016-7037(94)90050-7) [DOI] [Google Scholar]
- 62.Baskaran M. 2011. Po-210 and Pb-210 as atmospheric tracers and global atmospheric Pb-210 fallout: a Review. J. Environ. Radioact. 102, 500–513. ( 10.1016/j.jenvrad.2010.10.007) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting this article are given in the tables.