

Abstract
The kinetochore is an essential structure that mediates accurate chromosome segregation in mitosis and meiosis. While many of the kinetochore components have been identified, the mechanisms of kinetochore assembly remain elusive. Here, we identify a novel role for Snap29, an unconventional SNARE, in promoting kinetochore assembly during mitosis in Drosophila and human cells. Snap29 localizes to the outer kinetochore and prevents chromosome mis‐segregation and the formation of cells with fragmented nuclei. Snap29 promotes accurate chromosome segregation by mediating the recruitment of Knl1 at the kinetochore and ensuring stable microtubule attachments. Correct Knl1 localization to kinetochore requires human or Drosophila Snap29, and is prevented by a Snap29 point mutant that blocks Snap29 release from SNARE fusion complexes. Such mutant causes ectopic Knl1 recruitment to trafficking compartments. We propose that part of the outer kinetochore is functionally similar to membrane fusion interfaces.
Keywords: kinetochore, KMN network, membrane, microtubules, mitosis, Snap29, SNARE
Subject Categories: Cell Cycle, Membrane & Intracellular Transport
Introduction
Cell division relies on organization of a microtubule (MT) spindle to which replicated chromosomes become attached for equal segregation. Defective MT attachment to kinetochores (KTs) leads to chromosome miss‐segregation and formation of fragmented nuclei after cell division. These events have been proposed to contribute to the genome instability observed in many cancers, indicating that control of MT attachment is a major tumor suppressing process (for review, see Santaguida & Musacchio, 2009).
The molecular nature of the outer KT, the structure that mediates MT attachment, has been studied extensively. MTs are engaged and stabilized by the Knl1, Mis12, and Ndc80 complexes, together referred to as the KMN network. The KMN network also holds in place the Rod‐Zw10‐Zwilch (RZZ) complex and spindle assembly checkpoint (SAC) proteins, which are important for signaling incomplete attachment. In mammalian cells, MT attachment is further assisted by the KNL1‐interactor ZWINT and by the SKA complex, which associates with curved MT ends at KTs (for review, see DeLuca & Musacchio, 2012).
In sheer contrast with the extensive molecular knowledge of the outer KT, much less is known about the steps that regulate its assembly. In Drosophila, a widely used metazoan model system for KT studies, part of the Mis12 complex resides at the KT throughout the cell cycle, while the rest of the outer KT is created de novo in early prophase, by stepwise addition of components. The earliest components added to the outer KT in early prophase appears to be Knl1, followed by the Ndc80 complex (Venkei et al, 2012), both of which are recruited from unknown cellular locales, and SAC components, such as Mad1 and Mad2, which are recruited from nuclear pores (Buffin et al, 2005; Katsani et al, 2008).
Unexpectedly, we have recently found that the Drosophila SNARE protein Snap29 can be isolated from cell extracts together with multiple components of the KMN network. These are the Drosophila Knl1 ortholog Spc105R, three out of four components of the Ndc80 complex (Nuf2 and the Drosophila Spc24 ortholog Kmn2) as well as three of the four subunits of the Mis12 complex (Mis12, Nnf1b, and the Drosophila Nsl1 ortholog Kmn1) (Morelli et al, 2014). SNARE (Soluble NSF Attachment REceptor) proteins (SNAREs) are part of the conserved coiled‐coil machinery that brings membranes in close proximity during trafficking, a prerequisite for most membrane fusion events (Holt et al, 2006). The Synaptosomal‐Associated Protein (SNAP) family of SNAREs in metazoans includes Snap25, Snap23, and Snap29, which are composed by two SNARE domains, separated by a linker region. The first two proteins are membrane‐associated and control synaptic transmission and a wide range of non‐neuronal membrane fusion processes, respectively. In contrast, Snap29 only transiently associates with membranes and contains an acidic NPF motif that mediates its association with endocytic factors (Steegmaier et al, 1998; Rotem‐Yehudar et al, 2001; Su et al, 2001). Such unconventional features, which are exclusive of Snap29 among the SNARE proteins, predict involvement in a versatile set of membrane trafficking processes, in line with reports in the literature (Steegmaier et al, 1998; Wong et al, 1999; Hohenstein & Roche, 2001; Rotem‐Yehudar et al, 2001; Su et al, 2001; Schardt et al, 2009; Rapaport et al, 2010; Kang et al, 2011; Sato et al, 2011; Wesolowski et al, 2012; Willett et al, 2013). Consistent with this, we and others recently have shown that Snap29 also controls fusion of autophagosomes with endo‐lysosomes in Drosophila and human cells, together with the SNAREs syntaxin17 (Syx17) and Vamp7 (VAMP8 in human cells) (Itakura et al, 2012; Takats et al, 2013; Morelli et al, 2014).
Despite involvement of Snap29 in multiple trafficking pathways in interphase, a possible function during cell division has not been explored. Here we investigated whether Snap29 localizes and acts at the KT in cells and tissues. Our data identify a novel step of KT formation that is conserved and supports tissue formation.
Results
Drosophila Snap29 localizes to the outer KTs during mitosis
To test whether Snap29 is present at KTs, we immunoprecipitated the outer KT protein Ndc80 in Drosophila Schneider‐2 (S2) cell extracts. We found Snap29 among Ndc80 coimmunoprecipitants (Fig 1A). In agreement with this, Ndc80 coimmunoprecipitates with Snap29 (Fig 1A), as we have previously reported (Morelli et al, 2014). Thus, Snap29 appears to be associated with the KMN network in S2 cells.
Figure 1. Drosophila Snap29 localizes to the mitotic KT .
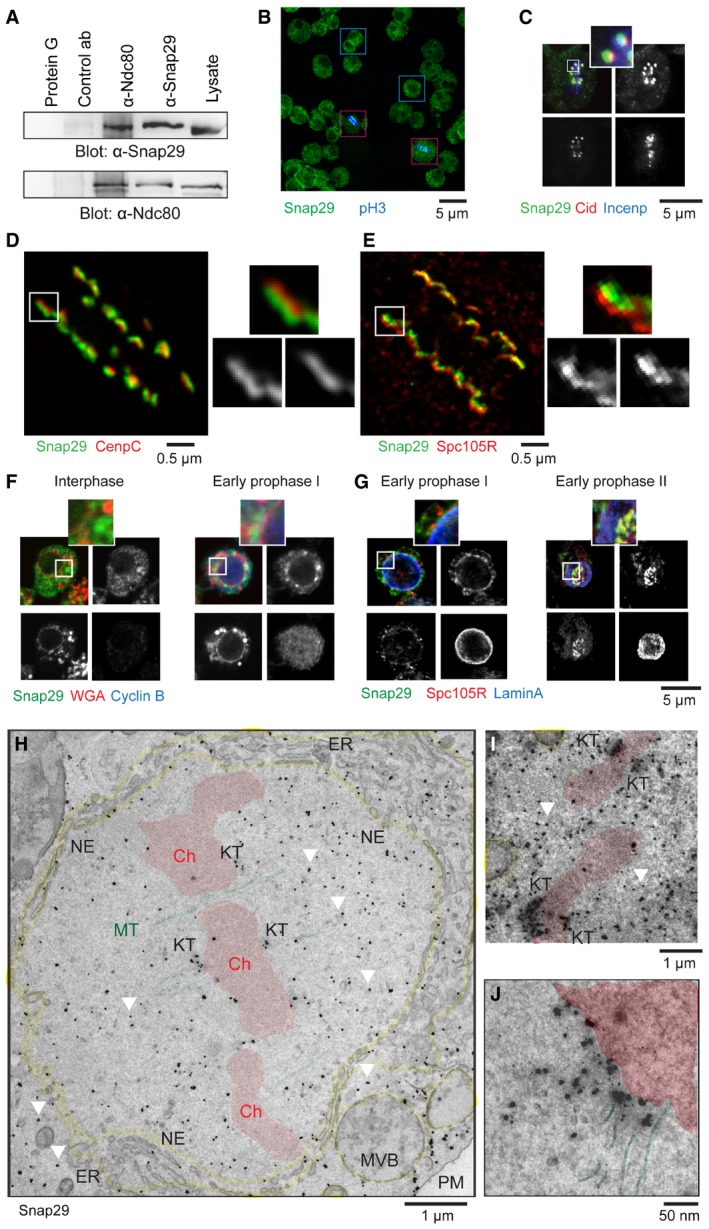
-
AImmunoprecipitations of extracts from asynchronous S2 cells using 1 μg of the indicated antibodies at 4°C overnight. The control antibody (ab) is a rabbit anti‐yeast Mad2.
-
BSingle confocal section of a population of S2 cells. Examples of interphase cells are boxed in blue. Mitotic cells positive for the anti‐phospho‐histone 3 (pH3) are boxed in red.
-
CSingle confocal section of a S2 cell. Anti‐Incenp and anti‐Cid mark the centromeric chromatin and the inner KT, respectively.
-
D, EMaximal projections of S2 cells imaged by STED microscopy. Seven z‐sections for a total of 1.5 μm (D) and 4 z‐sections for a total of 0.5 μm (E) are shown. Anti‐CenpC and anti‐Spc105R mark the proximal and the distal regions of different portions of the outer KT, respectively.
-
F, GSingle confocal sections of S2 cells at the indicated mitotic stages stained to detect Snap29, nuclear pores (WGA), and cyclin B (F), or Snap29, Spc105R, and the nuclear lamina (lamin A) (G). Note the transient perinuclear enrichment of Snap29 in early prophase. WGA stains also cytoplasmic speckles in hemocyte‐derived S2 cells (Rizki & Rizki, 1983).
-
H–JImmunogold localization of Snap29 in cryosections of S2 cells in pro‐metaphase. Pseudo‐coloring in red identifies chromosomes (Ch), in yellow membrane organelles, and in green MTs. The white arrows indicate examples of signal in the nucleoplasm that is not associated with membranes, but rather with electron‐dense material. MVB: multivesicular body; NE: nuclear envelope; PM: plasma membrane.
We next studied the subcellular localization of endogenous Snap29 in S2 cells by immunofluorescence. While the protein is not present in the nucleus of cells in interphase, we discovered a pool of Snap29 puncta associated with chromosomes of cells in mitosis, which are positive for phospho‐histone 3 (pH3), a marker of the centromeric chromatin of dividing cells (Fig 1B). At metaphase, such pool colocalizes with Cid (Drosophila CENPA), a inner KT component, but not with Incenp, a centromeric chromatin protein (Fig 1C), indicating that Snap29 is present at KTs during mitosis. By super‐resolution confocal microscopy, Snap29 appears to localize distal to CenpC, with connects the inner and other part of the KT (Fig 1D, inset). In contrast, Snap29 localizes proximal to Spc105R (Fig 1E, inset). These data reveal that Snap29 resides in the proximal part of the outer KT in mitotic cells.
Formation of the outer KT in Drosophila is concomitant with nuclear envelope fenestration, nuclear lamina dissolution, and nuclear pore disassembly (Kiseleva et al, 2001). One of the earliest landmarks of outer KT formation in prophase is the nuclear localization of Spc105R (Venkei et al, 2012). To test when Snap29 is recruited to KTs relative to these events, we analyzed Snap29 localization in S2 cells relative to Spc105R, cyclin B, which is upregulated from prophase to metaphase (Knoblich & Lehner, 1993), lamin A, a marker of the nuclear lamina, and WGA (wheat germ agglutinin), a lectin that associates with O‐linked N‐acetyl‐D‐glucosamine‐modified proteins, including nucleoporins (Holt et al, 1987). In interphase, Snap29 is distributed in cytoplasmic puncta, as previously reported (Morelli et al, 2014), and very low levels of Spc105R are detectable (Fig 1F and G, interphase; Appendix Fig S1A and B). In sheer contrast, in early prophase cells with high levels of the mitotic cyclin B, Snap29 is found enriched in a perinuclear compartment (Fig 1F, early prophase I; Appendix Fig S1A), where it partly colocalizes with low amounts of Spc105R (Fig 1G, early prophase I; Appendix Fig S1B). Note that in these cells, the nuclear lamina and nuclear pores are still mostly intact (Fig 1F and G, early prophase I; Appendix Fig S1A and B). Before full disassembly of the nuclear lamina, Snap29 and Spc105R are corecruited to KTs (Fig 1G, early prophase II; Appendix Fig S1B). Finally, from prophase until chromosome de‐condensation in telophase, Snap29 is found at the KT at similar levels (Appendix Fig S1A and B). However, both Snap29 and Spc105R are never found at nuclear pores or at the nuclear lamina, as revealed by lack of colocalization with WGA or lamin A (Fig 1F and G, early prophase I, insets; Appendix Fig S1A and B). These data indicate that Snap29 localization to KTs is concomitant with that of Spc105R and is preceded by enrichment of Snap29 and possibly Spc105R in a perinuclear region.
To analyze Snap29 localization at the ultrastructural level, we performed correlative light and cryoelectron microscopy (CLEM) in S2 cells. In cells in interphase, Snap29 is excluded from the nucleus, consistent with our immunofluorescence analysis (Appendix Fig S2A). In contrast, in cells in pro‐metaphase, we detected signal associated with the fenestrated nuclear envelope (NE), the perinuclear endoplasmic reticulum (ER), and other cytoplasmic membrane organelles (Fig 1H). In addition, we found a prominent portion of the labeling associated with KTs (Fig 1H–J). At higher magnification, we observe that the portion of signal at the KT labels the electron‐dense outer KT area, but not the MTs connected to it (Fig 1J). Finally, we detect signal that is associated with electron‐dense material scattered in the nucleoplasm and in the cytoplasm (Fig 1H and I, arrowheads). This evidence confirms that Snap29 is present at KTs and indicates that it also associates membrane and non‐membrane nuclear compartments in mitotic cells.
Snap29 localizes to KTs of dividing cells also in Drosophila epithelial tissue. Immunofluorescence analysis shows the presence of Snap29 at KTs of dividing cells in the wing imaginal disk, a proliferating larval organ (Fig 2A). To identify genes required for Snap29 localization at the KT, we expressed RNAi hairpins for KT factors in vivo in wing imaginal disks using the GAL4 driver Bxms1096. We then analyzed Snap29 localization at KT in dividing cells. We found that Snap29 localization to KT is lost in tissues depleted of Mis12, Spc105R, or Ndc80 and Nuf2, two components of the Ndc80 complex. Localization is normal in tissue similarly depleted of the Drosophila RZZ component Zw10 (Fig 2A, quantification in B). A similar effect was observed by depletion in S2 cells with double‐stranded RNAs (dsRNA; Appendix Fig S3A–D). Snap29 localization at KTs is also not affected by treatment with the MT depolymerizing drug colcemid (Appendix Fig S3E and F). KT localization independent of MTs is in agreement with our localization data that did not highlight association with MTs. Overall, these results indicate that Snap29 recruitment to the KT depends on KMN network components, but not on the RZZ complex or MTs.
Figure 2. Snap29 localization at KTs in vivo depends on the N‐terminal SNARE domain and on outer KT components.
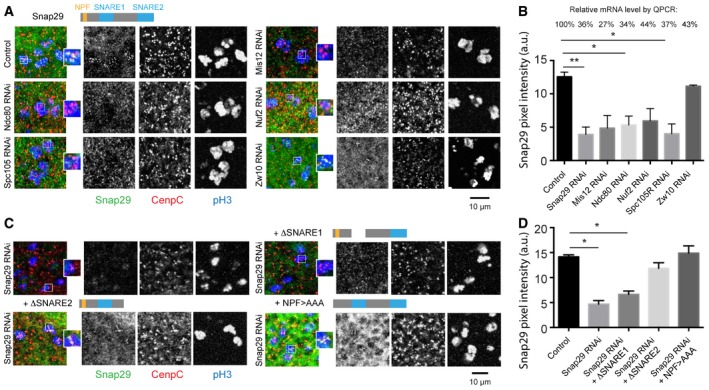
- Comparable maximum projections of confocal sections of the dorsal wing pouch portion of third‐instar larval imaginal disks. A cartoon of full‐length Snap29 is shown above the control panel. Anti‐pH3 labels mitotic cells and CenpC marks KTs.
- Quantification of the Snap29 signal as in (A), considering > 30 KTs per sample. Relative mRNA expression of the downregulated genes versus control, measured by qPCR, is shown above the graph. Note that efficient depletion of Zw10 does not affect Snap29 localization to KTs.
- Comparable maximum projections of confocal sections of dorsal wing pouches in which Snap29 has been downregulated and the indicated Snap29 form overexpressed. Cartoons of the expressed Snap29 mutant forms are shown above the panels. Anti‐pH3 labels mitotic cells and CenpC marks KTs.
- Quantification of Snap29 as in (C), considering > 28 KTs per sample. Note that expression of a form of Snap29 lacking the SNARE1 domain does not rescue Snap29 localization to KTs in Snap29 RNAi cells.
To identify which part of Snap29 is required for recruitment to KTs, we expressed Snap29 forms lacking the NPF motif, or either of the two SNARE domains, in wing disk cells depleted of endogenous Snap29. We have previously shown that expression of full‐length Snap29 can rescue the lethality associated with a mutation in the Drosophila Snap29 locus and that neither of the mutant forms can provide full rescue (Morelli et al, 2014). Despite this, expression of Snap29 with a mutated NPF (NPF > AAA) or lacking the SNARE domain 2 (ΔSNARE2) in Snap29‐depleted cells leads to restoration of Snap29 localization to KT, while expression to similar levels of a form of Snap29 lacking SNARE domain 1 (ΔSNARE1) does not (Fig 2C, quantified in D). These data indicate that the first SNARE domain of Snap29 mediates recruitment to the outer KT.
Snap29 controls mitotic progression
To test whether Snap29 acts during cell division, we analyzed the phenotype of S2 cells expressing histone 2B‐GFP and mCherry‐α‐tubulin (Erhardt et al, 2008), upon knockdown of Snap29 (dsSnap29 cells) by time‐lapse microscopy. A large proportion of dsSnap29 cells did not form a metaphase plate and, less frequently, showed chromosome alignment and segregation defects, and formation of tripolar spindles and micronuclei, compared to control cells (Fig 3A and B arrowheads, quantification in C; Movies EV1 and EV2). These data indicate that Snap29 is required for correct mitotic progression of S2 cells.
Figure 3. Snap29 depletion affects mitotic progression.
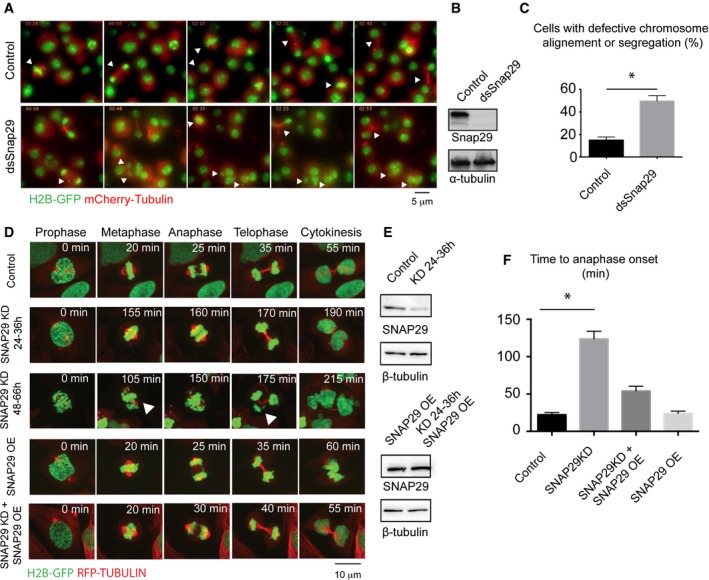
- Selected frames of time‐lapse imaging of control S2 cells or cells treated for 96 h with dsRNA against the central portion of Snap29. Note that depleted cells do not display a recognizable metaphase plate, or fail to segregate all chromosomes correctly, or form tripolar spindles or micronuclei (arrowheads; arrowheads in control point to cells dividing normally).
- Western blotting of extract from control cells and cells depleted of Snap29 (dsSnap29) relative to the experiment in (A).
- Quantification of the mitotic phenotype of control and dsSnap29‐depleted cells, based on time‐lapse imaging of an average of > 28 individual cells per sample. The mean with standard error of the mean (SEM) is shown, and P‐value is determined by two‐tailed t‐test considering all defects together. *P ≤ 0.05.
- Selected frames of time‐lapse imaging of control U2OS cells and U2OS cells treated for the indicated time. Note that the depleted cell at 48–66 h display altered metaphase plate and chromosome segregation (arrowheads).
- Western blot analysis of extracts relative to the experiments in (D).
- Quantification of the time from prophase to entry in anaphase based on analysis of > 20 individual cells/sample by time‐lapse microscopy. The mean with standard error of the mean (SEM) is shown, and P‐values are determined by Kruskal–Wallis test with Dunn's multiple comparison analysis. *P ≤ 0.05. OE: overexpression.
To assess whether Snap29 is required for mitosis in also human cells, we depleted SNAP29 with siRNAs in U2OS cells expressing histone 2B‐GFP and RFP‐α‐tubulin and analyzed mitotic events by time‐lapse microscopy. Similarly to S2 cells, compared to control, SNAP29 knocked‐down (SNAP29 KD) cells divide with chromosomes incorrectly aligned to the metaphase plate or segregate aberrantly, eventually generating daughter cells with one or more micronuclei (Fig 3D and E; Movies EV3, EV4 and EV5). 59 ± 20% of SNAP29 KD cells form micronuclei (average n = 28 cells/experiment), while only 6 ± 3% of control cells do so (average n = 32 cells/experiment), indicating a significant presence of mitotic defects (P‐value: 0.037). In addition, cells exhibit an extensive prophase to metaphase delay (Fig 3D and E, quantification in F). These phenotypes are rescued by expression in KD cells of a siRNA‐resistant SNAP29, which per se does not alter mitosis (Fig 3D and E, quantified in F; Movie EV6), indicating that they are specific to SNAP29. Overall, the activity of Snap29 during cell division appears conserved in human cells and required for normal mitotic progression.
SNAP29 regulates MT attachment formation or stabilization
As observed in U2OS cells, SNAP29 KD HeLa cells display mitotic defects and nuclear fragmentation (Appendix Fig S4A–C). Thus, we used HeLa cells to investigate further SNAP29 activity in mitosis. Because cell division delays can be due to triggering of SAC activity, we first assessed SAC response by treating control and depleted HeLa cells with the MT depolymerizing drug nocodazole. We found cell arrest in both samples, indicating that the SAC is functional in depleted cells. In continuous presence of nocodazole, SNAP29 KD cells exit from mitosis earlier than control cells (Appendix Fig S4D), suggesting either that the SAC is inefficient, or that other factors that control mitotic timing are affected by SNAP29 depletion.
Because mitotic delay and segregation defects can originate from defective chromosome attachment to MTs, we determined stability of attachment upon cold‐induced MT depolymerization (cold shock). Compared to control cells in metaphase, SNAP29 KD cells present a high number of KTs that are not attached to MTs. Most of these are positive for MAD1, a SAC component that is removed upon attachment, and are not aligned to metaphase plate (Fig 4A, quantified in B). We also measured inter‐KT distances as a readout of KT tension generated by MT attachment. Compared to control cells, we found significantly reduced inter‐KT distances in not aligned chromosomes of SNAP29 KD cells (Fig 4D, quantified in D), indicating that SNAP29 KD are unable to stabilize MT binding to KTs.
Figure 4. SNAP29 controls MT binding to KTs in HeLa cells.
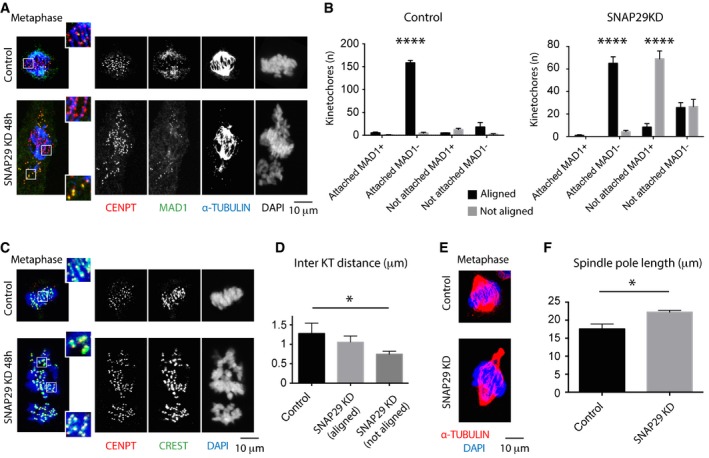
- Single confocal sections of HeLa cells at metaphase treated for 8 min at 4°C and immunostained to reveal the presence of unattached kinetochores. CENPT labels all KTs, while MAD1 identifies unattached ones. Enlargements of boxed areas displaying both chromosomes aligned and not aligned at the metaphase plate.
- Quantification of the number of KTs based on attachment, position, and MAD1 positivity. Each point per category represents one experiment in which 10 random KTs/cell of 20 cells/sample were counted. Note that in control cells, most of the KTs are attached, MAD1‐negative, and aligned to the metaphase plate, while in SNAP29KD cells half of the KTs are unattached, MAD1‐positive, and not aligned to the metaphase plate.
- Single confocal sections of HeLa cells. CREST and CENPT were used to label the centromere region and associated KTs, respectively. Enlargements of boxed areas displaying both chromosomes aligned and not aligned at the metaphase plate are shown.
- Average inter‐KT distance based on 10 chromosomes/cell and 10 cells/sample. Note that both aligned and not aligned KTs have a shorter inter‐KT distance compared to aligned control KTs.
- Single confocal sections of HeLa cells at metaphase stained to visualize the mitotic spindle and DNA.
- Averaged spindle pole length based on > 20 cells with bipolar spindles/sample.
Consistent with failure to form or stabilize MT binding to KTs, SNAP29 KD cells show evident MT spindle defects. While the vast majority of control HeLa cells forms a bipolar spindle (90.2%, with 9.8% tripolar spindles; n = 102 cells), SNAP29KD cells frequently form monopolar spindles (24%, with 75% bipolar spindles and 1% tripolar spindles; n = 100 cells). In addition, bipolar spindles of SNAP29KD cells are often abnormally elongated, compared to bipolar spindles of control cells (Fig 4A and E, quantified in F), a phenotype also observed upon depletion of other KT components (DeLuca et al, 2002). These data reveal that MTs do not bind stably to KTs in HeLa cells with low SNAP29 content.
SNAP29 regulates outer KT formation
Because the KMN network is crucial for MT binding (Cheeseman et al, 2006; Ciferri et al, 2007), we next tested whether in SNAP29 KD cells KMN components are present at KTs. Compared to control cells, mitotic SNAP29 KD cells arrested by nocodazole incubation harbor equal amount of HEC1 (Human Ndc80) and reduced levels of DSN1 or NSL1 (two Mis12 complex components) at KTs (Fig 5A and B, quantified in F). In sheer contrast, KNL1 and ZWINT are not found at KTs in SNAP29 KD cells (Fig 5C, quantification in F), despite the fact that overall amounts of KNL1 and ZWINT are not significantly changed (Fig 5D). These data indicate that SNAP29 is required for KNL1 and ZWINT localization to outer KTs, which are crucial for formation of the MT‐interacting surface.
Figure 5. SNAP29 is required for KT integrity in HeLa cells.
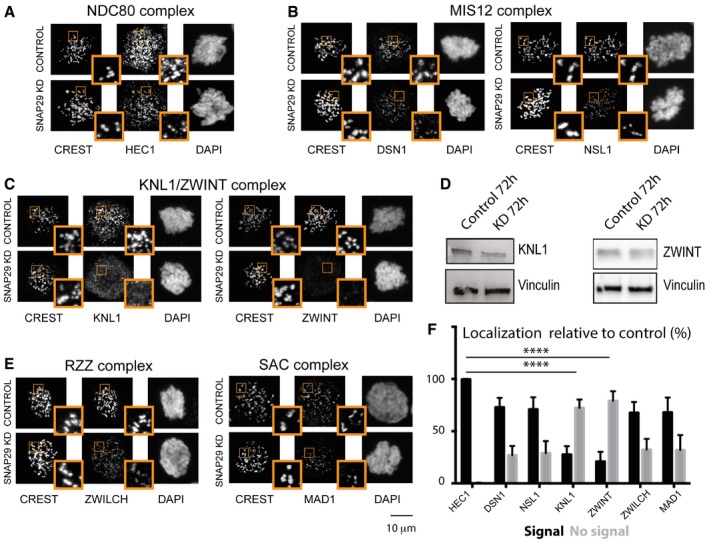
-
A–CMax projection of single control and SNAP29 KD HeLa cells treated with 2 mM thymidine for 24 h, released in normal medium for 8 h, and arrested in prometaphase with nocodazole for 2 h.
-
DImmunoblotting of protein extracts from HeLa cells treated as indicated.
-
EMax projection of single control and SNAP29 KD HeLa cells treated with 2 mM thymidine for 24 h, released in normal medium for 8 h, and arrested in prometaphase with nocodazole for 2 h.
-
FQuantification of three independent experiments evaluating the localization of the KT factors shown in (A–C and E), relative to CREST. 100 KTs from 10 control cells and 200 KTs from 10 SNAP29 KD cells per sample were identified by CREST staining. The mean with standard error of the mean (SEM) is shown, and P‐values are obtained by two‐way ANOVA with Sidak's multiple comparison analysis relative to localization of HEC1, which is not lost on SNAP29KD cells. ****P ≤ 0.0001.
Interestingly, we find that localization to the KTs of the RZZ component ZWILCH and the SAC component MAD1 is still present in SNAP29 KD cells, despite very low levels of KNL1 and ZWINT at KTs (Fig 5E, quantification in F). Consistent with this, in SNAP29 KD cells lacking ZWINT or the KNL1 interactor BUB1, a considerable amount of MAD1 is found at KTs (Appendix Fig S5A and B). In addition, SNAP29 KD cells with almost normal KT MAD1 levels display reduced amounts of the RZZ component ZW10 (Appendix Fig S5C). In agreement with recent reports (Caldas et al, 2015; Silió et al, 2015), these data indicate that MAD1 and the RZZ complex can be recruited to KTs in a SNAP29 and KNL1‐independent manner, possibly supporting SAC activity in SNAP29KD cells.
KNL1 localization depends on SNAP29
To determine how SNAP29 supports KT formation, we measured KNL1 localization to KTs in SNAP29 KD cells overexpressing siRNA‐resistant SNAP29 mutant forms. We observed that a full‐length human SNAP29, a functional form of Drosophila Snap29 (Morelli et al, 2014), or a human SNAP29 mutant form lacking the first SNARE domain (SNARE1), all rescue KNL1 localization to KTs in SNAP29 KD cells, while a mutant lacking the second SNARE domain (SNARE2) is less efficient (Fig 6A and B; quantification in D; Appendix Fig S6A and B). Thus, KNL1 Snap29 ability to recruit KNL1 to KTs is conserved and requires the C‐terminal SNARE domain.
Figure 6. Snap29 mutants prevent recruitment of KNL1 to KTs and cause its ectopic accumulation.
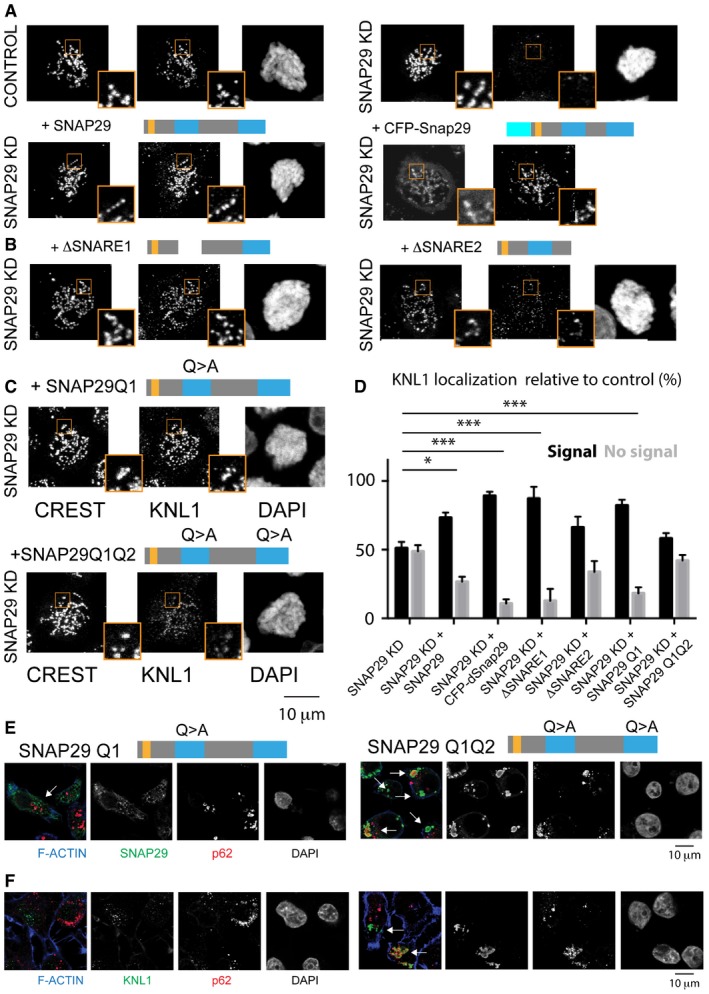
-
A–CKNL1 localization analysis in control, SNAP29 KD, and SNAP29 KD expressing full‐length human SNAP29, CFP‐tagged, full‐length Drosophila Snap29 (CFP‐Snap29), or the mutant forms schematized above the panels. Max projections of single cells in prophase are shown.
-
DQuantification of three independent experiments evaluating the localization of KNL1 relative to CREST. 200 KTs from 10 control cells and 200 KTs from 10 SNAP29 KD cells per sample were identified by CREST staining. The mean with standard error of the mean (SEM) is shown, and P‐values are obtained by two‐way ANOVA with Dunnett's multiple comparison analysis relative to localization of KNL1 in SNAP29KD cells. *P ≤ 0.05; ***P ≤ 0.001
-
E, FSingle confocal sections of HeLa cells in interphase overexpressing the indicated constructs. In cells expressing SNAP29 Q1 Q2, SNAP29 (E) and KNL1 (F) accumulate in large compartments that are often positive for p62 (arrows).
To assess whether KNL1 recruitment to KTs is linked to the membrane fusion function of SNAP29, we overexpressed mutant forms of SNAP29 with Q to A mutations in the center of the first or of both SNARE domains (SNAP29 Q1 and SNAP29 Q1 Q2, respectively; Appendix Fig S6B). Similar mutations have been reported to prevent disassembly of SNARE complexes by N‐ethylmaleimide sensitive factor (NSF) and α‐snap, trapping them at fusions sites (Scales et al, 2001). Consistent with this, upon overexpression of SNAP29 Q1 Q2 (and to a much lesser extent, of SNAP29 Q1), we observed large cytoplasmic accumulations of SNAP29, which are positive for ubiquitin and for the autophagy adapter p62 (Fig 6E; Appendix Fig S6C), as well as formation of large cytoplasmic membrane aggregates (Appendix Fig S6D). Such alterations are consistent with impairment of membrane fusion and with sequestering of mutant SNAP29 to membrane compartments that carry autophagy cargoes. Importantly, expression of SNAP29 Q1 Q2 in depleted cells rescues KNL1 localization to KTs poorly, while expression of SNAP29 Q1 does so comparably to wild‐type SNAP29 (Fig 6C, quantified in D). These data indicate that trapping SNAP29 at membranes prevents its function at KTs. Strikingly, upon overexpression of SNAP29 Q1 Q2, we observe accumulation of KNL1 in p62‐positive compartments (Fig 6F) and cytoplasmic accumulation of ZW10 and of the Ska complex component SKA1 (Appendix Fig S6E and F), despite the fact that overall amounts of these KT proteins are not significantly changed (Appendix Fig S6B). In contrast, other components of the outer KT, such as NSL1, MAD1, HEC1, or CREST, are unaffected by overexpression of SNAP29 Q1 Q2 (Appendix Fig S6E; EM TV unpublished observations). Cytoplasmic KNL1 accumulation is never observed in SNAP29 KD cells (Appendix Fig S6G). However, we find that KNL1 and SNAP29 colocalize in the cytoplasm of cells overexpressing GFP‐SNAP29 in interphase and they can be coprecipitated (Appendix Fig S6H and I). Overall, recruitment to KTs of KNL1, but not its association to SNAP29, depends on release of SNAP29 from SNARE complexes.
Snap29 controls tissue architecture in vivo
In agreement with the established association between chromosomal instability and tumorigenesis (Janssen et al, 2011), loss of Snap29 might result in alteration of the architecture of tissues and organs. To assess whether this is the case, we studied Drosophila epithelial organs predominantly composed of cells homozygous for the mutant Snap29 B6‐21 (Snap29 mutant disk), which encodes a truncated form of Snap29 lacking SNARE2 (Morelli et al, 2014). Consistent with our data in tissue culture cells, Snap29 mutant disks are devoid of Spc105R (Appendix Fig S7A and B), enriched in pH3‐positive cells in pro‐metaphase (Appendix Fig S7C and D), and present multiple small pH3‐positive foci (Appendix Fig S7D, arrowheads), possibly representing fragmented nuclei. Overall, these data confirm that KT formation requires the C‐terminal part of Snap29 in vivo.
Compared to wild‐type disks, Snap29 mutant disks also present epithelial architecture defects characterized by appearance of mesenchymal‐like F‐actin‐rich cells (Appendix Fig S7A and B, arrowheads) (Morelli et al, 2014). Disks that are trans‐heterozygous for null mutations in Syx17 or Vamp7, which act with Snap29 in autophagy, do not display epithelial alteration (Morelli et al, 2014) and localize Snap29 at KTs (Appendix Fig S7E and F), ruling out their involvement in Snap29 localization at KTs and a function of autophagy in tissue formation. In contrast, altered epithelial architecture is a reported consequence of aberrant cell division in Drosophila imaginal disks. In fact, such phenotype has been shown to correlate with appearance of aneuploid, unpolarized, and apoptotic cells, and with activation of Drosophila tumor‐associated signaling pathways, such as Jak/Stat and JNK (Dekanty et al, 2012). Consistent with this, Snap29 mutant disks show mislocalization of the apical and basolateral polarity determinants aPKC and Dlg and increased expression of JAK/STAT (Morelli et al, 2014), and JNK signaling reporters Puc‐LacZ (Appendix Fig S7C and D). In addition, compared to control, Snap29 mutant eye disks also contain numerous apoptotic cells (Fig 7E and F). Block of apoptosis by expression of the bacterial inhibitor p35, which per se does not alter development (Fig 7G), reduces the presence of apoptotic cells and strongly increases appearance of unpolarized F‐actin‐rich cells (Fig 7H, arrowheads). This indicates that loss of Snap29 in a developing epithelium initiates a process of aberrant tissue formation that is counteracted by apoptosis. Interestingly, both Snap29 mutant eye disks and Snap29 mutant eye disks expressing p35 are able to survive and grow for weeks, when allografted in the abdomen of host flies (Appendix Fig S7F and I). Despite the fact that Snap29 mutant tissue does not invade the host, as it is the case of mutant of tumor suppressor genes (Caussinus & Gonzalez, 2005), loss of Snap29 might, at least partially, echo aspects of tumorigenesis.
Figure 7. Drosophila Snap29 activity supports tissue formation in imaginal disks.
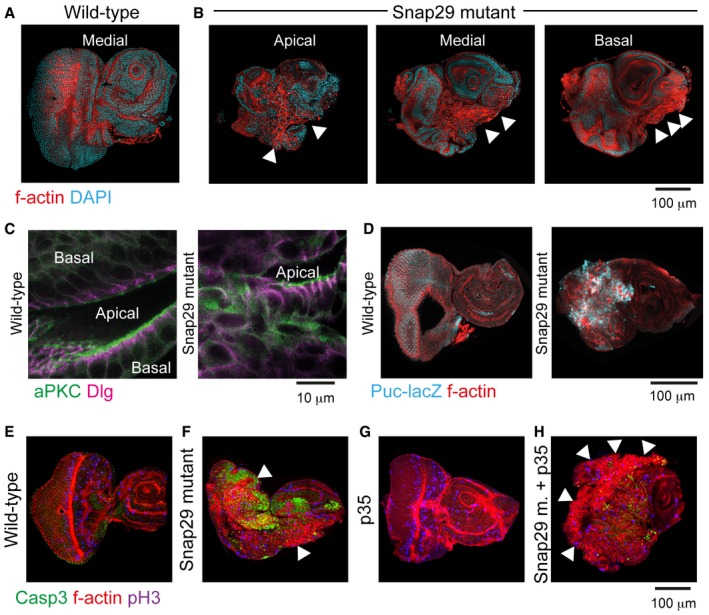
-
A, BConfocal sections of wild‐type and Snap29 mutant eye‐antennal imaginal disks (eye disk portion to the left) of L3 larvae. Disks were labeled with phalloidin and DAPI to visualize the subcortical F‐actin cytoskeleton and nuclei, respectively. Arrowheads in (B) point to actin‐rich unpolarized cells in apical, medial, and basal sections of the same disk.
-
CHigh magnification of a single confocal cross‐section of wild‐type and Snap29 mutant eye disk tissues. Two facing folds of the tissue are separated by the apical lumen. aPKC and Dlg mark the apical and lateral plasma membrane domains, respectively. In Snap29 mutant, tissue the apical–basal polarity is disrupted.
-
DSingle confocal cross‐sections of wild‐type and Snap29 eye‐antennal disks, expressing puckered‐LacZ (Puc‐lacZ), a reporter of JNK signaling activation.
-
E–HSingle confocal cross‐sections of wild‐type and Snap29 eye‐antennal disks (E, F), and of otherwise wild‐type and Snap29 eye‐antennal disks expressing the bacterial apoptosis inhibitor p35 (G, H). Anti‐activated caspase‐3 (Casp3) marks apoptotic cells.
Similar to imaginal disks, Snap29 localizes to KTs of dividing cells in the follicular epithelium (FE), the monolayer encasing the germ line of the adult ovary (Appendix Fig S8A). Upon expression in vivo of Snap29 dsRNAs, which leads to strong Snap29 downregulation in the Drosophila FE, the epithelium appears disorganized and multilayered (Appendix Fig S8B and C). Together, these in vivo data indicate that Snap29 supports tissue formation in multiple Drosophila epithelia.
Discussion
Our data uncover an essential and conserved step of KT formation that occurs in prophase and requires Snap29. Such step controls localization of Knl1 (and ZWINT in human cells) to KTs (see model in Appendix Fig S9). Snap29 and the RZZ components Rod and Zw10 are known act in membrane transport between the Golgi apparatus and the ER (Willett et al, 2013), and the RZZ complex shares similarity with ER tethering complexes (Çivril et al, 2010; Wainman et al, 2012). Strikingly, the autophagosome, which depends on Snap29 for fusion to lysosomes (Itakura et al, 2012; Takats et al, 2013; Morelli et al, 2014), is formed de novo using Golgi and ER components and engages MTs for dynein‐directed transport to lysosomes (Maday et al, 2012; Ge et al, 2014), evoking tantalizing similarities between aspects of membrane trafficking and KT formation. Overall, our data support the possibility that Snap29, Knl1, and the RZZ complex might act at the KT similarly to tethering and fusion complexes existing on membranes, which need to stabilize MTs during trafficking events. Such scenario might imply common ancestry, and underscored also the existence of protozoa that divide with KTs associated with the nuclear membrane that is itself attached to MT fibers (Gómez‐Conde et al, 2000).
Ectopic recruitment of Knl1 and possibly RZZ and SKA complex components at sites of SNAP29 Q1 Q2 trapping predicts that the trafficking and KT functions of Snap29 are interconnected, perhaps because of the existence of a common cellular pool of Snap29. A possibility that awaits further investigation is that SNARE domains of Snap29 interact with KT proteins directly. Interestingly, the C‐terminal part of Knl1, Zwint, and Mis12 and Nnf1 all contain multiple coiled‐coil regions. These are placed at the interaction surface between the Mis12, Ndc80, and Knl1 complexes (Schittenhelm et al, 2009; Kiyomitsu et al, 2011; Krenn et al, 2014; Petrovic et al, 2014), exactly where we see Snap29 located by super‐resolution microscopy. In mammalian cells, the Snap29 paralog Snap25 binds Zwint, and Snap29 has been found in Zwint immunoprecipitations (Lee et al, 2002; van Vlijmen et al, 2008; Hutchins et al, 2010). Thus, Snap29 could dock to the Mis12 complex with a SNARE domain and could stabilize interactions with Knl1 and Zwint with the second C‐terminal SNARE domain. Interestingly, no Zwint homolog has been found in Drosophila, suggesting that in flies Snap29 could substitute for Zwint. The ability of KNL1 to interact with a SNAP29 that cannot be released from SNARE complex, both suggest that the interaction of KNL1 with SNAP29 might occur on the side of the SNARE domain that is not occupied by a syntaxin or a Vamp. These data also suggest that SNAP29 could act on Knl1 also prior to nuclear entry and KT localization.
Our evidence in vivo indicates that SNAP29 function could support tissue development by ensuring faithful chromosome segregation and that such activity is crucial in cells that become resistant to apoptosis. Based on this, we predict that loss of SNAP29 could be selected in cancers with highly unstable genomes. In addition, rare congenital syndromes such as CEDNIK, Roberts syndrome, and primary microcephaly (MCPH) are caused by mutations in Snap29 and other genes encoding proteins that regulate mitosis, respectively (Musio et al, 2004; Sprecher et al, 2005; Genin et al, 2012), overall suggesting that ability to cope with defective mitotic cells is major process for tissue development and homeostasis.
Materials and Methods
Cell cultures and treatments
Mycoplasma‐free S2 cells (gift from B. Mellone) and Mycoplasma‐free S2‐HB2‐GFP‐mCherry‐α‐tubulin cells (gift from Sylvia Erhardt) were cultured in Schneider's medium (Gibco) supplemented with 1% glutamine and 20% fetal bovine serum (FBS) at 28°C. To depolymerize MTs, S2 cells were incubated with 0.5 μg/ml colcemid (Sigma‐Aldrich) for 2 h.
The Mycoplasma‐free U2OS‐H2B‐GFP‐α‐tubulin‐mCherry cell line (gift from Letizia Lanzetti), stably expressing a GFP‐tagged histone H2B (H2B) and a mCherry‐tagged α‐tubulin, was cultured in DMEM (Gibco) supplemented with 2 mM l‐glutamine and 10% FBS at 37°C with 5% CO2 and with the addition of 0.5 mg/ml G418 and 1 μg/ml puromycin. Mycoplasma‐free HeLa cells were provided by the IFOM cell culture facility. They were cultured in DMEM (Gibco) supplemented with 2 mM l‐glutamine and 10% FBS at 37°C with 5% CO2. To depolymerize MTs, HeLa cells were incubated with 100 or 200 ng/ml of nocodazole. Thymidine treatment to synchronize cells has been performed according to Zhang et al (2014). Cells were then collected for immunofluorescence analysis or immunoprecipitation. The cold‐shock assay was performed as in Serio et al (2011). Subsequently, cells were fixed and processed for immunofluorescence analysis. To detect membrane organelles, we have treated live cells with 5 μg/ml FM1‐43 (Life Technology) following manufacturer instructions.
Experiments with flies
Flies were maintained on standard media. All experiments were performed at 25°C. Genotypes for the experiments listed in Figures are in Appendix Table S1. Mutant eye disks were generated as in Morelli et al (2014) and tumor allograft as in Rossi and Gonzalez (2015). Flies strains used from Bloomington Drosophila Stock Center are as follows: w, BxMS1096‐GAL4 (8860), UAS‐Snap29RNAi (25862), UAS‐Ndc80RNAi (38260), UAS‐Nuf2RNAi (36725), UAS‐Mis12RNAi (38535 and 35471), UAS‐Spc105R RNAi (35466). Flies strains used from Vienna Drosophila RNAi Center (VDRC) are as follows: UAS SNAP29RNAi (107947) and UAS‐RodRNAi (19152). Other strains used are: w; FRT42, Snap29B6‐21; UAS‐ΔSNARE1; UAS‐ΔSNARE2; UAS‐NPF > AAA, Syx17LL06330, Df(2R)BSC132, Vamp7G7738, Df(3L)Exel8098 (Morelli et al, 2014), UAS‐p35 (gift from S. Cohen); and Puc‐LacZ (gift from H. Jasper) and traffic jam‐GAL4 (gift from V. Riechmann).
Immunostainings
Imaginal disks, ovaries, S2 cells, and HeLa cells were fixed and stained as in Kobia et al (2014); Morelli et al (2014). Cells, disks, and ovaries were mounted on slides using Mowiol Mounting Medium (Calbiochem) or glycerol 70%.
For Drosophila tissues and S2 cell experiments, primary antibodies against the following antigens were used: rabbit anti‐Snap29 1:1,000 (Morelli et al, 2014), mouse anti‐phospho‐histone H3 1:1,000 (Abcam), rabbit anti‐cleaved caspase‐3 1:200 (Cell Signaling), rat anti‐α‐tubulin 1:100 (AbD Serotec), chicken anti‐Cid 1:1,000 (gift from B. Mellone), mouse anti‐γ‐tubulin 1:100 (Sigma‐Aldrich), rat anti‐Incenp 1:400 (gift from K. McKim), chicken anti‐Ndc80 1:200 (gift from T. Maresca), guinea pig anti‐CenpC 1:5,000 (gift from S. Erhardt), rabbit anti‐aPKC 1:1,000 (Sigma), sheep anti‐Spc105R 1:1,000 (gift from D. Glover), mouse anti‐lamin A (Developmental Studies Hybridoma Bank—DSHB), mouse anti‐Dlg 1:100, and mouse anti‐βGal 1:25 (DSHB). For human cells, the following primary antibodies were used: human anti‐CREST 1:1,000, mouse anti‐DSN1 1:1,000, mouse anti‐NSL1 1:1,000, rabbit anti‐CENPT 1:1,000, rabbit anti‐KNL1 1:1,000, rabbit anti‐ZWILCH 1:1,000, mouse anti‐MAD1 1:1,000, sheep anti‐BUB1 1:100 (all gifts from A. De Antoni); rabbit anti‐ZWINT 1:1,000, rabbit anti‐ZW10 1:500, rabbit anti‐SKA1 1:100 (all gifts from A. Musacchio); and goat anti‐lamin A/C 1:50 (Santa Cruz), mouse anti‐HEC1 1:1,000 (Abcam), mouse anti‐p62 1:1,000 (Abnova), mouse anti‐ubiquitin (FK2) 1:100 (ENZO), DAPI 1:1,000 (Sigma). Alexa‐conjugated secondary antibodies (Invitrogen), rabbit Atto594 (Sigma), Alexa 555‐WGA (Invitrogen), and Phalloidin‐TRITC (Sigma) were used. For all confocal imaging, we used a Leica microscope with ×40/NA 1.25 or ×63/NA 1.4 oil lenses. For super‐resolution images, we used a Leica TCS SP8 STED microscope with ×100/NA 1.4. Images were edited with Adobe Photoshop and ImageJ and assembled with Adobe Illustrator.
CLEM
3 × 106 growing S2 cells were plated on Matek previously coated with polyornithine (Sigma‐Aldrich) and let adhere for 2 h. Cells were fixed with 4% PFA + 0.05% glutaraldehyde in Hepes 0.15 M adjusted to pH 7.2–7.4 for 5 min and then fixed again with 4% PFA in Hepes 0.15 M adjusted to pH 7.2–7.4 three times for 10 min. Cells were quickly washed three times with Hepes 0.2 M and incubated with blocking solution (0.005 g/ml BSA, 0.001 g/ml saponin, 0.0027 g/ml NH4Cl in Hepes 0.2 M) for 30 min. Cells were then incubated for 2 h with primary antibodies (anti‐Snap29 1:1,000, anti‐pH3 1:1,000) diluted in blocking solution, washed three times with Hepes 0.2 M, and incubated with secondary antibodies diluted in blocking solution and diluted in PBS 1:10. Cells were finally washed three times with Hepes 0.2 M. Imaging was performed using a DeltaVision Elite imaging system (Applied Precision) driven by softWoRx software and equipped with a phase‐contrast 60× oil immersion objective (Olympus, NA 1.25). Images were edited with ImageJ. Sample were processed for CLEM according to Beznoussenko and Mironov (2015) using nanogold‐anti‐rabbit Fab', Kit Gold Enhance EM (GEEM) for signal enhancement.
Protein extraction, Western blots, and immunoprecipitations
Cells were collected, homogenized, and incubated for 20 min on ice in 1 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X‐100, 1% deoxycholate, 0.1% SDS, and protease inhibitors 1:200 (Calbiochem). Lysates were cleared by centrifugation. Supernatants were recovered and quantified, separated by SDS–PAGE, and transferred to nitrocellulose by standard methods. Primary antibodies used were rabbit anti‐SNAP29 (1:500), mouse anti‐KNL1 1:30, rabbit anti‐KNL1 1:1,000, rabbit anti‐ZWINT 1:1,000, rabbit anti‐ZW10 1:500, mouse anti‐DSN1 1:1,000, rabbit anti‐SKA1 1:1,000 (all gifts from A. Musacchio), mouse anti‐β‐tubulin 1:8,000, and mouse anti‐vinculin 1: 8,000 (Amersham). Secondary antibodies used were anti‐rabbit and anti‐mouse 1:8,000 (Amersham) and anti‐mouse Trueblot 1:1,000 (Roche). Immunoblots were visualized with SuperSignal West Pico/Femto Chemiluminescent Substrate (Thermo Scientific) using Chemidoc (Bio‐Rad). S2 cell immunoprecipitations were performed as in Morelli et al (2014). The antibodies used were the rabbit anti‐Snap29, rabbit anti‐Ndc80 (gift from P. Somma) as positive control, and rabbit anti‐yeast Mad2 (gift from A. Ciliberto) as negative control. HeLa cell immunoprecipitations were performed in high salt JS buffer (Tris–HCl pH 7.6, NaCl 150 mM, glycerol 20%, 0.5% NP‐40, MgCl2 2 mM, Na pyrophosphate 0.1 M pH 7.5, PMSF 0.1 M in ethanol, Na vanadate 0.5 M pH 7.5 in Hepes, NaF 0.5 M) with addition of protease inhibitors 1:200 (Calbiochem) and phosphatase inhibitor 1:100 (Sigma). Five micrograms of anti‐SNAP29 and anti‐DSN1 was used every 1 mg of extract.
Double‐stranded RNA interference in S2 cells
A cDNA library from S2 cells was used as template to amplify regions of the genes of interest using the following T7‐ and T3‐tagged primers:
T3‐Snap29 5′‐taatacgactcactatagggagaAACCCAGGAGGTGGGTAAG‐3′
T7‐Snap29 5′‐aattaaccctcactaaagggagaATGTTATCCAGCAATTCATTTTG‐3′
T3‐Zw10 5′‐taatacgactcactatagggagaCCGGACATATTTCTGGAGGA‐3′
T7‐Zw10 5′‐attaaccctcactaaagggagaTGATGGTCTCGTAGCACTCG‐3′
T3‐Rod 5′‐taatacgactcactatagggagaTGGTGGAGATCATGGCTAAC‐3′
T7‐Rod 5′‐attaaccctcactaaagggagaCCTTGGCGCTTTCAATTTG‐3′
T3‐Zwilch 5′‐taatacgactcactatagggagaAACTCTCATTGAAAATAGCTACC‐3′
T7‐Zwilch 5′‐attaaccctcactaaagggagaCACATTGGAAGAGCATACTAAA‐3′
PCR products were transcribed in vitro with T3 and T7 polymerases (Promega) according to the manufacturer's instructions. To generate double‐stranded RNAs (dsRNA), T3 and T7 transcripts were annealed at 68°C for 15 min and at 37°C for 30 min. For dsRNA treatments, we starved S2 cells for 30 min in serum‐free medium. We then added medium with 20% serum and dsRNA at the concentration of 15 μg/106 cells and incubated for 96 h.
RT–PCR was performed as described in Morelli et al (2014). The following primers were used:
Ndc80 F 5′‐TGGAGAAGAGGGAGAAGCAG‐3′ Ndc80 R 5′‐GTAGATCCTCGTTCCGTTGC‐3′ Rod F 5′‐CCAAGGGAAAGAATCAGGAAA‐3′ Rod R 5′‐AGCTAAGCGGATCGTTTTCA‐3′ Zw10 F 5′‐CGAAGTGGCAAACGATCC‐3′ Zw10 R 5′‐TGCGGTCCATTAGTTTGACA‐3′ Zwilch F 5′‐ACATTTGTCCGCCGAACTT‐3′ Zwilch R 5′‐GCTCCTTGACCATCTCCTTG‐3′ Spc105R F 5′‐GCCATCGAACTCCTTTGAGA‐3′ Spc105R R 5′‐ATTCCTCGTGGCACTATGCT‐3′ Nuf2 F 5′‐ATGGCGTTATCAGTCGAAATT‐3′ Nuf2 R 5′‐TCGCAGCTCTGTCACTTGACT‐3′.
siRNA silencing
0.25–0.35 × 106 cells were plated in six‐well plate and left in the incubator for 24 h. The mix composed of 30 pmoles per 0.25 × 106 cells of siRNA specific for Snap29 (5′‐GAC AAG AUG GAC CAA GAU‐3′) targeting between the nucleotides 285 and 304 (D‐011935‐04‐0005, Dharmacon) and lipofectamine (RNAiMax, Invitrogen) was prepared and added to cells, following the manufacturer's instruction. Cells were collected at different time points (mostly 48 and 72 h) after transfection and processed for further analysis.
Production of human SNAP29 forms
A human Snap29 cDNA encoding a siRNA‐resistant RNA (pCMV.SPORT6‐HumanSNAP29‐R) was generated mutagenizing the siRNA target sequence in the human Snap29 cDNA in order to maintain the same amino acid sequence of the wild‐type protein. The resulting mutated sequence is as follows: 5′‐GAT AAA ATG GAT CAG GAC‐3′. pCMV.SPORT6‐Human‐Snap29 containing Snap29 cDNA cloned between EcoRI and XhoI was used as template for mutagenesis by nested PCR. We performed a first PCR using FOR‐HSNAP29 primer 5′‐GATCGAATTCTTCAGCTTACCCTAAGAGCTAC‐3′, containing the restriction site sequence for EcoRI and the first 18 nucleotides of the Snap29 cDNA sequence, and the REV‐HSNAP29R 5′‐ATTTCAAGTCCTGATCCATTTTATCCACCAT‐3′, which contains the mutagenized siRNA target sequence. The second PCR was performed using FOR‐HSNAP29R 5′‐ATGGTGGATAAAATGGATCAGGACTTGAAGAT‐3′, containing the mutagenized sequence, and REV‐HSNAP29 5′‐CCGCTCGAGTCAGAGTTGTCGAACTTTTCTTTCTG‐3′, containing the last 18 nucleotides of Human‐Snap29 cDNA and the XhoI restriction site sequence. The two PCR products were used as template for a third PCR performed with the FOR‐HSNAP29 and REV‐HSNAP29 to obtain the entire Human‐Snap29 cDNA sequence mutagenized in the indicated region. The PCR product was cloned into pCMV.SPORT6 to obtain the pCMV.SPORT6‐HumanSNAP29‐R.
SNAP29 ΔSNARE1, ΔSNARE2, Q1, and Q1Q2 mutants were generated using HumanSNAP29‐R as template. To generate the ΔSNARE1 insert, two regions of Snap29 cDNA were amplified with the following primers: ForSNAP29 5′‐GATCGAATTCGTTTCCCAGACTGAGAGCCGCGC‐3′ and RevΔSN1 5′‐GCTCTTAATGCTATTGATGGCGACCCCAACCTT‐3′, ForΔSN1 5′‐AAGGTTGGGGTCGCCATCAATAGCATTAAGAGC‐3′ and RevSNAP29 5′‐GATCTCTAGAATCACAATAGAGTGGAAATCCG‐3′, respectively. The two PCR products were used as templates for a second PCR using ForSNAP29 and RevSNAP29 as primers. To generate the ΔSNARE2 insert, Snap29 cDNA was amplified with the following primers: ForSNAP29 and RevΔSN1 5′‐GATCTCTAGAATCACAATAGAGTGGAAATCCGTCTGTCTTCAAGTACTCATGGCAGA‐3′. To generate the Q1 insert, two regions of Snap29 cDNA were amplified with the following primers: ForSNAP29 5′‐GATCGAATTCGTTTCCCAGACTGAGAGCCGCGC‐3′ and RevQ1 5′‐ACTCCTCGCGCACGGGCGAGCTCCT‐3′, ForQ1 5′‐AGGAGCTCGCCCGTGCGCGAGGAGT‐3′ and RevSNAP29 5′‐GATCTCTAGAATCACAATAGAGTGGAAATCCG‐3′, respectively. The two PCR products were used as templates for a second PCR using For SNAP29 and RevSNAP29 as primers. To generate the Q1Q2 insert, Q1 Snap29 cDNA was amplified with the following primers: ForSNAP29 and RevQ2 5′‐AAGAATGTCATCTGCCTCCTCAATTTCTGT‐3′, ForQ2 5′‐ACAGAAATTGAGGAGGCAGATGACATTCTT‐3′ and RevSNAP29. The two PCR products were used as templates for a second PCR using ForSNAP29 and RevSNAP29 as primers. Finally, to generate GFP‐SNAP29, HumanSNAP29‐R was cloned EcoRI/BamHI into pEGFP1‐C1.
For rescue or overexpression experiments, a mix composed of the relevant vector alone or mixed with the siRNA specific for Snap29 and Lipofectamine 2000 was prepared following the manufacturer's instruction (Invitrogen). Five hours after transfection, fresh medium was added and cells were incubated at 37°C in 5% CO2. Cells were collected 24–48 h after transfection.
Production of anti‐humanSNAP29
To produce a GST‐humanSNAP29 for pAb production, we generated an insert by PCR using pCMVSPORT6.Human‐snap29 as template and the following primers:
GST‐hSNAP29 BamHI 5′‐CGCCGGATCCACCATGTCAGCTTACCCTAAAAGCTAC‐3′
GST‐hSNAP29 XhoI 5′‐CCGCTCGAGTCAGAGTTGTCGAACTTTTCTTTCTG‐3′
The PCR product was inserted using BamHI and XhoI into pGEX‐GST. IPTG induced expression of GST‐hSNAP29, and purification was performed by the IFOM antibody service facility according to standard protocols.
In both cases, purified proteins were used for rabbit immunizations (Eurogentec). Sera affinity purification was performed by the IFOM antibody service facility, using AminoLink® Kit (Biotechnology).
Time‐lapse analyses
Control and dsSnap29 S2‐HB2‐GFP‐mCherry‐α‐tubulin cells were recorded using a DeltaVision Elite imaging system (Applied Precision) equipped with a phase‐contrast 60×/NA 1.25 oil immersion objective (Olympus). Cells were plated into glass‐bottomed dishes (Matek) and placed onto a sample stage within an incubator chamber set at 28°C. Images were acquired using 8‐ and 80‐ms exposure for the GFP and mCherry RFP, respectively, every 2 min for 3 h and keeping the laser intensity at 2% for GFP and at 5% for mCherry. Z‐stacks of images collected every 0.5 μm for a total of 10 μm were collected. We acquired automatically 20 fields and 20 stacks using a high‐precision motorized stage.
Control and SNAP29 KD U2OS‐HB2‐GFP‐RFP‐α‐tubulin cells were recorded using a Spinning Disk confocal microscope (PerkinElmer UltraVIEW VoX). Cells were plated into glass‐bottomed dishes (Matek) and placed onto a sample stage within an incubator chamber (Okolab) set at 37°C in 5% CO2. Images were captured with a 40× N.A. = 1.3 oil immersion objective using 120‐ and 150‐ms exposure for the GFP and RFP, respectively, every 5 min for 8 h and keeping the laser intensity at minimum to avoid phototoxicity. Z‐stacks of three images each were collected every 4.5 μm for a total of 9 μm. We acquired automatically seven fields and 17 stacks using a high‐precision motorized stage.
For mitotic timing experiments, U2OS‐HB2‐GFP‐RFP‐α‐tubulin or HeLa cells were recorded using a widefield microscope (Olympus IX‐81). Samples were placed on different wells of the same plate and imaged simultaneously. The plate was placed onto a sample stage within an incubator chamber (Okolab) set at 37°C in an atmosphere of 5% CO2. Images of U2OS‐HB2‐GFP‐RFP‐α‐tubulin cells were captured with a 40 ×/N.A. = 60 objective using 250‐ms exposure for the GFP every 5 min for 48 h. Z‐stacks of images collected every 1.5 μm for a total thickness around 4.5 μm. Six different fields per sample were acquired. For HeLa cells, 16 different fields per sample were captured with a LUCPLANFL 20×/N.A. 0.45 objective using 50‐ms exposure for the bright field every 5 min for 48 h.
Measurements
All experiments were repeated at least three times for quantification and the mean with standard error of the mean (SEM) is shown. P‐values are as follows: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. Quantifications were performed with ImageJ and Prism was used for statistical analyses. Sample sizes and statistical methods are detailed in the figure legends.
Quantification of Snap29 localization at KTs in Drosophila tissue and in S2 cells in knockdown experiments was performed using a plugin developed in house. Briefly, a mask was drawn automatically around the CenpC signal of pH3‐positive cells and the Snap29 signal colocalizing with CenpC was quantified. In colcemid S2 experiments, a mask was drawn manually on pH3‐positive nuclei and the masked Snap29 signal was quantified.
The time to anaphase onset in HeLa cells was computed from chromosome condensation to beginning of chromosome segregation. Adaptation time in nocodazole‐treated HeLa cells was determined by the time passing from the moment in which a cell rounds up due mitotic entry to the moment in which the same cell reverts to substrate adhesion without dividing, a trait that signals exit from mitosis. In this experiment, the recording begins immediately after the addition of nocodazole.
Quantification of KT status in control and depleted HeLa cells in metaphase subjected to cold shock was performed by sorting individual KTs identified by CENPT signal by position (aligned to the metaphase plate or not), MAD1 status (positive or negative), and proximity of tubulin signal (attached or not).
Measurement of inter‐KT distances in HeLa cells was performed using an ImageJ plugin developed in house. Briefly, the line selection tool was used to manually draw a straight line between the brightest spots of CENPC signal lined on CREST signal representing KT pairs. The resulting fluorescence intensity profile of the CENPC was analyzed using the “Find Peak” plugin (http://fiji.sc/Find_Peaks) to automatically find the peaks. The peak‐to‐peak distance represents the inter‐KT distance.
To measure spindle lengths, the threshold tool was used to manually set a level to segment the image and extract the position of the spindle poles. An ellipse was fitted to each segmented spindle pole pairs. The major axis of the fitted ellipse represents the spindle length.
Quantifications of KT localizations in HeLa cells were performed by masking a 1.5 μm by 0.5 μm volume across z‐stacks of three images encompassing the bulk KT signal identified by CREST (HeLa). The corresponding signal in the KT marker channel was catalogued on the basis of pixel intensity.
The measurement of the average area Snap29 mutant tissue before and after allograft was performed using ImageJ.
Author contributions
TV and EM conceived the study. GVB and AAM performed the CLEM analysis. ET performed the allograft experiment. EM and VM performed all the other experiments. TV analyzed the data and wrote the manuscript with help from all other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Acknowledgements
We thank A. Oldani and S. Barozzi for help with imaging, G. Ossolengo for help with biochemistry, M. Gualtieri for help with the allograft experiment, and E. Martini and A. Corno for help with quantifications and statistics. We thank the Bloomington Stock center, the DHSB, and the VDRC for reagents. We are indebted to A. Musacchio, B. Mellone, A. De Antoni, A. Ciliberto, M. Mapelli, P. De Wulf, M. Foiani, M. Gonzalez‐Gaitan, L. Lanzetti, and D. Bilder for discussion. Work in T.V. laboratory is supported by Telethon Italia Investigator grant #GGP13225 and by AIRC (Associazione Italiana Ricerca sul Cancro) Investigator grant #15954. EM is supported by an AIRC fellowship and VM is supported by a fellowship from Fondazione Umberto Veronesi.
The EMBO Journal (2016) 35: 2223–2237
References
- Beznoussenko GV, Mironov AA (2015) Correlative video‐light‐electron microscopy of mobile organelles. Methods Mol Biol 1270: 321–346 [DOI] [PubMed] [Google Scholar]
- Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE (2005) Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr Biol 15: 856–861 [DOI] [PubMed] [Google Scholar]
- Caldas GV, Lynch TR, Anderson R, Afreen S, Varma D, DeLuca JG (2015) The RZZ complex requires the N‐terminus of KNL1 to mediate optimal Mad1 kinetochore localization in human cells. Open Biol 5: 150160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E, Gonzalez C (2005) Induction of tumor growth by altered stem‐cell asymmetric division in Drosophila melanogaster. Nat Genet 37: 1125–1129 [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson‐Kubalek EM, Desai A (2006) The conserved KMN network constitutes the core microtubule‐binding site of the kinetochore. Cell 127: 983–997 [DOI] [PubMed] [Google Scholar]
- Ciferri C, Musacchio A, Petrovic A (2007) The Ndc80 complex: hub of kinetochore activity. FEBS Lett 581: 2862–2869 [DOI] [PubMed] [Google Scholar]
- Çivril F, Wehenkel A, Giorgi FM, Santaguida S, Fonzo AD, Grigorean G, Ciccarelli FD, Musacchio A (2010) Structural analysis of the RZZ complex reveals common ancestry with multisubunit vesicle tethering machinery. Structure 18: 616–626 [DOI] [PubMed] [Google Scholar]
- Dekanty A, Barrio L, Muzzopappa M, Auer H, Milan M (2012) Aneuploidy‐induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc Natl Acad Sci USA 109: 20549–20554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Moree B, Hickey JM, Kilmartin JV, Salmon ED (2002) hNuf2 inhibition blocks stable kinetochore‐microtubule attachment and induces mitotic cell death in HeLa cells. J Cell Biol 159: 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Musacchio A (2012) Structural organization of the kinetochore–microtubule interface. Curr Opin Cell Biol 24: 48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, Mellone BG, Betts CM, Zhang W, Karpen GH, Straight AF (2008) Genome‐wide analysis reveals a cell cycle‐dependent mechanism controlling centromere propagation. J Cell Biol 183: 805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Baskaran S, Schekman R, Hurley JH (2014) The protein‐vesicle network of autophagy. Curr Opin Cell Biol 29: 18–24 [DOI] [PubMed] [Google Scholar]
- Genin A, Desir J, Lambert N, Biervliet M, Van Der Aa N, Pierquin G, Killian A, Tosi M, Urbina M, Lefort A, Libert F, Pirson I, Abramowicz M (2012) Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum Mol Genet 21: 5306–5317 [DOI] [PubMed] [Google Scholar]
- Gómez‐Conde E, Mena‐López R, Hernndez‐Jaúregui P, González‐Camacho M, Arroyo R (2000) Trichomonas vaginalis: chromatin and mitotic spindle during mitosis. Exp Parasitol 96: 130–138 [DOI] [PubMed] [Google Scholar]
- Hohenstein AC, Roche PA (2001) SNAP‐29 is a promiscuous syntaxin‐binding SNARE. Biochem Biophys Res Commun 285: 167–171 [DOI] [PubMed] [Google Scholar]
- Holt GD, Snow CM, Senior A, Haltiwanger RS, Gerace L, Hart GW (1987) Nuclear pore complex glycoproteins contain cytoplasmically disposed O‐linked N‐acetylglucosamine. J Cell Biol 104: 1157–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Varoqueaux F, Wiederhold K, Takamori S, Urlaub H, Fasshauer D, Jahn R (2006) Identification of SNAP‐47, a novel Qbc‐SNARE with ubiquitous expression. J Biol Chem 281: 17076–17083 [DOI] [PubMed] [Google Scholar]
- Hutchins JRA, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A et al (2010) Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328: 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Mizushima N (2012) The hairpin‐type tail‐anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH (2011) Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333: 1895–1898 [DOI] [PubMed] [Google Scholar]
- Kang J, Bai Z, Zegarek MH, Grant BD, Lee J (2011) Essential roles of snap‐29 in C. elegans . Dev Biol 355: 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsani KR, Karess RE, Dostatni N, Doye V (2008) In vivo dynamics of Drosophila nuclear envelope components. Mol Biol Cell 19: 3652–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiseleva E, Rutherford S, Cotter LM, Allen TD, Goldberg MW (2001) Steps of nuclear pore complex disassembly and reassembly during mitosis in early Drosophila embryos. J Cell Sci 114: 3607–3618 [DOI] [PubMed] [Google Scholar]
- Kiyomitsu T, Murakami H, Yanagida M (2011) Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol Cell Biol 31: 998–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblich JA, Lehner CF (1993) Synergistic action of Drosophila cyclins A and B during the G2‐M transition. EMBO J 12: 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobia F, Duchi S, Deflorian G, Vaccari T (2014) Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic notch signaling. Mol Oncol 8: 207–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Overlack K, Primorac I, van Gerwen S, Musacchio A (2014) KI motifs of human Knl1 enhance assembly of comprehensive spindle checkpoint complexes around MELT repeats. Curr Biol 24: 29–39 [DOI] [PubMed] [Google Scholar]
- Lee H‐K, Safieddine S, Petralia RS, Wenthold RJ (2002) Identification of a novel SNAP25 interacting protein (SIP30). J Neurochem 81: 1338–1347 [DOI] [PubMed] [Google Scholar]
- Maday S, Wallace KE, Holzbaur ELF (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196: 407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli E, Ginefra P, Mastrodonato V, Beznoussenko GV, Rusten TE, Bilder D, Stenmark H, Mironov AA, Vaccari T (2014) Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila . Autophagy 10: 2251–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musio A, Mariani T, Montagna C, Zambroni D, Ascoli C, Ried T, Vezzoni P (2004) Recapitulation of the Roberts syndrome cellular phenotype by inhibition of INCENP, ZWINT‐1 and ZW10 genes. Gene 331: 33–40 [DOI] [PubMed] [Google Scholar]
- Petrovic A, Mosalaganti S, Keller J, Mattiuzzo M, Overlack K, Krenn V, De Antoni A, Wohlgemuth S, Cecatiello V, Pasqualato S, Raunser S, Musacchio A (2014) Modular assembly of RWD domains on the Mis12 complex underlies outer kinetochore organization. Mol Cell 53: 591–605 [DOI] [PubMed] [Google Scholar]
- Rapaport D, Lugassy Y, Sprecher E, Horowitz M (2010) Loss of SNAP29 impairs endocytic recycling and cell motility. PLoS One 5: e9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki TM, Rizki RM (1983) Blood cell surface changes in Drosophila mutants with melanotic tumors. Science 220: 73–75 [DOI] [PubMed] [Google Scholar]
- Rossi F, Gonzalez C (2015) Studying tumor growth in Drosophila using the tissue allograft method. Nat Protoc 10: 1525–1534 [DOI] [PubMed] [Google Scholar]
- Rotem‐Yehudar R, Galperin E, Horowitz M (2001) Association of insulin‐like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem 276: 33054–33060 [DOI] [PubMed] [Google Scholar]
- Santaguida S, Musacchio A (2009) The life and miracles of kinetochores. EMBO J 28: 2511–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Saegusa K, Sato K, Hara T, Harada A, Sato K (2011) Caenorhabditis elegans SNAP‐29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells. Mol Biol Cell 22: 2579–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Yoo BY, Scheller RH (2001) The ionic layer is required for efficient dissociation of the SNARE complex by alpha‐SNAP and NSF. Proc Natl Acad Sci USA 98: 14262–14267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schardt A, Brinkmann BG, Mitkovski M, Sereda MW, Werner HB, Nave K‐A (2009) The SNARE protein SNAP‐29 interacts with the GTPase Rab 3A: implications for membrane trafficking in myelinating glia. J Neurosci Res 87: 3465–3479 [DOI] [PubMed] [Google Scholar]
- Schittenhelm RB, Chaleckis R, Lehner CF (2009) Intrakinetochore localization and essential functional domains of Drosophila Spc105. EMBO J 28: 2374–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio G, Margaria V, Jensen S, Oldani A, Bartek J, Bussolino F, Lanzetti L (2011) Small GTPase Rab5 participates in chromosome congression and regulates localization of the centromere‐associated protein CENP‐F to kinetochores. Proc Natl Acad Sci USA 108: 17337–17342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silió V, McAinsh Andrew D, Millar Jonathan B (2015) KNL1‐Bubs and RZZ provide two separable pathways for checkpoint activation at human kinetochores. Dev Cell 35: 600–613 [DOI] [PubMed] [Google Scholar]
- Sprecher E, Ishida‐Yamamoto A, Mizrahi‐Koren M, Rapaport D, Goldsher D, Indelman M, Topaz O, Chefetz I, Keren H, O'brien TJ, Bercovich D, Shalev S, Geiger D, Bergman R, Horowitz M, Mandel H (2005) A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet 77: 242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M, Yang B, Yoo JS, Huang B, Shen M, Yu S, Luo Y, Scheller RH (1998) Three novel proteins of the syntaxin/SNAP‐25 family. J Biol Chem 273: 34171–34179 [DOI] [PubMed] [Google Scholar]
- Su Q, Mochida S, Tian JH, Mehta R, Sheng ZH (2001) SNAP‐29: a general SNARE protein that inhibits SNARE disassembly and is implicated in synaptic transmission. Proc Natl Acad Sci USA 98: 14038–14043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovacs AL, Hegedus K, Juhasz G (2013) Autophagosomal Syntaxin17‐dependent lysosomal degradation maintains neuronal function in Drosophila . J Cell Biol 201: 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkei Z, Przewloka MR, Ladak Y, Albadri S, Sossick A, Juhasz G, Novak B, Glover DM (2012) Spatiotemporal dynamics of Spc105 regulates the assembly of the Drosophila kinetochore. Open Biol 2: 110032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vlijmen T, Vleugel M, Evers M, Mohammed S, Wulf PS, Heck AJ, Hoogenraad CC, van der Sluijs P (2008) A unique residue in rab3c determines the interaction with novel binding protein Zwint‐1. FEBS Lett 582: 2838–2842 [DOI] [PubMed] [Google Scholar]
- Wainman A, Giansanti MG, Goldberg ML, Gatti M (2012) The Drosophila RZZ complex – roles in membrane trafficking and cytokinesis. J Cell Sci 125: 4014–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski J, Caldwell V, Paumet F (2012) A novel function for SNAP29 (Synaptosomal‐Associated Protein of 29 kDa) in mast cell phagocytosis. PLoS One 7: e49886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Kudlyk T, Pokrovskaya I, Schonherr R, Ungar D, Duden R, Lupashin V (2013) COG complexes form spatial landmarks for distinct SNARE complexes. Nat Commun 4: 1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SH, Xu Y, Zhang T, Griffiths G, Lowe SL, Subramaniam VN, Seow KT, Hong W (1999) GS32, a novel Golgi SNARE of 32 kDa, interacts preferentially with syntaxin 6. Mol Biol Cell 10: 119–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Lischetti T, Nilsson J (2014) A minimal number of MELT repeats supports all the functions of KNL1 in chromosome segregation. J Cell Sci 127: 871–884 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File