

Abstract
SOX9 is a master transcription factor that regulates development and stem cell programs. However, its potential oncogenic activity and regulatory mechanisms that control SOX9 protein stability are poorly understood. Here, we show that SOX9 is a substrate of FBW7, a tumor suppressor, and a SCF (SKP1/CUL1/F‐box)‐type ubiquitin ligase. FBW7 recognizes a conserved degron surrounding threonine 236 (T236) in SOX9 that is phosphorylated by GSK3 kinase and consequently degraded by SCFFBW 7α. Failure to degrade SOX9 promotes migration, metastasis, and treatment resistance in medulloblastoma, one of the most common childhood brain tumors. FBW7 is either mutated or downregulated in medulloblastoma, and in cases where FBW7 mRNA levels are low, SOX9 protein is significantly elevated and this phenotype is associated with metastasis at diagnosis and poor patient outcome. Transcriptional profiling of medulloblastoma cells expressing a degradation‐resistant SOX9 mutant reveals activation of pro‐metastatic genes and genes linked to cisplatin resistance. Finally, we show that pharmacological inhibition of PI3K/AKT/mTOR pathway activity destabilizes SOX9 in a GSK3/FBW7‐dependent manner, rendering medulloblastoma cells sensitive to cytostatic treatment.
Keywords: cisplatin, FBW7, medulloblastoma, metastasis, SOX9
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Medulloblastoma is one of the most common malignant childhood brain tumors with a survival rate of 70% following aggressive surgery, radiation, and chemotherapy (Gottardo & Gajjar, 2006). Survivors of the disease often demonstrate severe side effects from radiotherapy with neurological sequelae or psychological problems. New treatment options are needed not only to cure high‐risk patients but also to alleviate therapy‐induced side effects. Among the promising strategies employed to address these issues are the continuous refinement of molecular subtyping criteria and the development of genetically driven medulloblastoma models, which together are used as platforms to dissect the molecular biology and identify mechanisms of response and resistance based on pathway activity status. To date, medulloblastoma can be classified into four distinct molecular subgroups: WNT, SHH, Group 3 and Group 4 (Taylor et al, 2012). This categorization arguably provides a reliable foundation for predicting patient prognosis and outcome, as well as development of individualized targeted treatments. Nonetheless, it is also increasingly evident that molecular heterogeneity within a defined molecular subgroup of medulloblastoma accounts for variability in the efficacy of targeted treatments (as exemplified in Kool et al, 2014; Morrissy et al, 2016).
Our prior expression profiling analysis of human medulloblastoma specimens revealed that SOX9 (sex‐determining region Y (SRY)‐box 9) is commonly elevated in WNT and SHH medulloblastoma (Swartling et al, 2012). SOX9 is a member of the high‐mobility group (HMG)‐box class of transcription factors and controls establishment of broad transcriptional programs regulating stem cell properties, differentiation, proliferation, and survival (Adam et al, 2015; Larsimont et al, 2015). In embryonal development, SOX9 transcriptional activity plays a crucial role in regulating the development of all germ layers (reviewed in Jo et al, 2014), including the maintenance and differentiation of neural stem cells in the developing brain (Stolt et al, 2003; Scott et al, 2010). Furthermore, both phosphorylation (by protein kinases A and G at S64 and S181) and SUMOylation of SOX9 protein have been reported to positively influence SOX9's transactivation in chondrocytes (Huang et al, 2000) and in neural crest development (Taylor & Labonne, 2005). Deregulation of SOX9, including its overexpression and/or cooperation with other oncoproteins, contributes to evolvement of central cancer hallmarks in various tumor types (Matheu et al, 2012). For instance, SOX9 has been reported to collaborate with the transcription factor SNAI2 (SLUG) to promote breast cancer metastasis through epithelial‐to‐mesenchymal (EMT) reprogramming (Guo et al, 2012). In another case, SOX9 enhances KRAS‐driven pancreatic acinar malignant transformation (Kopp et al, 2012), possibly through SOX9‐mediated protection of GLI1 from β‐TrCP‐mediated protein degradation (Deng et al, 2015). In sonic hedgehog (SHH)‐driven basal cell carcinogenesis, SOX9 was further shown to be a critical downstream effector of WNT/β‐catenin signaling for maintenance of the tumor‐initiating cell population (Larsimont et al, 2015).
In medulloblastoma, our previous work demonstrated that SOX9 cooperates with the MYCN oncogene to promote tumor formation and penetrance (Swartling et al, 2012). While there is an abundant interest for SOX9 in initiating and driving oncogenicity of various malignancies, comparatively little is known regarding how SOX9 activity is regulated at the post‐translational level. Work from de Crombrugghe's laboratory provided evidence for proteasomal‐dependent degradation of SOX9 (Akiyama et al, 2005) by the E6‐AP/UBE3A HECT ubiquitin ligase in chondrocytes (Hattori et al, 2013). In cancer cells, we previously showed that activation of protein kinase G II (PKGII) phosphorylates and downregulates SOX9 protein leading to cell cycle arrest and increased neuronal differentiation of glioma cells (Swartling et al, 2009).
FBW7, a member of the WD40‐repeat‐containing F‐box protein family, functions as a substrate recognition component of a multi‐subunit ubiquitin ligase known as SCF (SKP1/CUL1/F‐box) (Welcker & Clurman, 2008). Located on chromosome 4q32, the FBW7 gene is alternatively spliced into three isoforms, α (nucleus), β (cytoplasmic), and γ (enriched in the nucleolus) (Welcker & Clurman, 2008). FBW7 is best known for its function in regulating the stability of numerous oncoproteins including cyclin E, MYC, JUN, and NOTCH1, among others (Davis et al, 2014). FBW7 binds substrates through interaction of key residues located on the surface of its β‐propeller, formed by its WD40 repeat domains, and a short motif in the substrate known as the Cdc4 phosphodegron (CPD), commonly phosphorylated by glycogen synthase kinase 3 (GSK3) or other kinases on serine (S) and/or threonine residues (T) (CPD consensus: S/TPPXS/T/E/D) (Orlicky et al, 2003). In line with its role as a master oncoprotein suppressor, FBW7 inactivation in cancers by genetic deletion, loss of expression, or somatic mutations is thought to directly contribute to tumor development and progression (Tan et al, 2008; Wang et al, 2012b). Despite the indisputable function of FBW7 in carcinogenesis, the full set of target substrates remains to be discovered and knowledge of how FBW7 status influences and contributes to the progression of specific malignancies is limited. This has also hampered efforts to target the downstream oncogenic effectors in FBW7‐inactivated tumors.
Here, we investigate the post‐translational regulation of SOX9 in medulloblastoma and identify SOX9 as a novel FBW7 substrate for ubiquitylation and proteasomal degradation in response to phosphorylation by GSK3 kinase. Degradation of SOX9 through the GSK3/FBW7 pathway likely represents an important mechanism for regulation of SOX9 activity in medulloblastoma as increased SOX9 protein level promotes migration, metastasis, and treatment resistance. FBW7 is frequently mutated in SHH medulloblastoma tumors and transcriptionally downregulated across all other patient subgroups. Accordingly, we observe a strong relationship between FBW7 loss‐of‐function and increased SOX9 protein levels in all medulloblastoma subgroups. Transcriptional profiling of medulloblastoma cells with stabilized SOX9 revealed differential expression of genes promoting metastasis through epithelial‐to‐mesenchymal (EMT) molecular reprogramming and genes directly associated with cisplatin resistance. Strikingly, our results provide evidence that targeting the PI3K/AKT/mTOR pathway, which correlates with poor prognosis in medulloblastoma (Kool et al, 2014), destabilizes SOX9 protein in a GSK3/FBW7‐dependent manner thus further sensitizing medulloblastoma cells to cisplatin therapy.
Results
SOX9 interacts with FBW7α through a conserved degron motif phosphorylated by GSK3
To systematically identify new FBW7 substrates, we previously performed quantitative mass spectrometry using FBW7 knockout (KO) HCT116 cells combined with directed searches for proteins with FBW7 phosphodegron (CPD) motifs (Arabi et al, 2012). Enrichment of SOX9 protein in the nuclear fraction of the HCT116 FBW7‐KO cells (Fig EV1A) and the presence of a canonical interaction motif (S/TPPXS/T) at position threonine 236 (T236) (Fig 1A) indicated that SOX9 could be a substrate for FBW7, and most likely the alpha isoform that resides in the nucleus. Analysis of the SOX9 protein sequence and its evolutionary conservation across species revealed that the amino acid motif spanning T236 to T240 resembled the high‐affinity consensus CPD found in other established FBW7α substrates (e.g., cyclin E, MYC, and JUN) (Figs 1A and EV1B).
Figure EV1. SOX9 interacts with FBW7α through its conserved degron motif phosphorylated by GSK3.
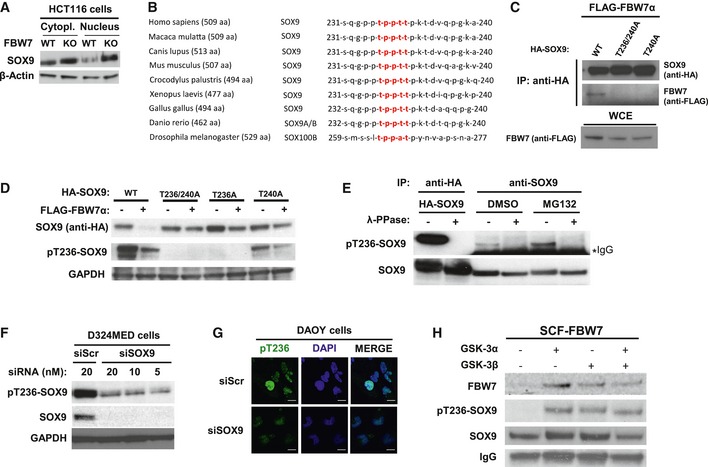
- Western blotting of total SOX9 protein levels in the cytoplasmic and nuclear fractions of HCT116‐FBW7 WT versus KO cells. The β‐actin was used as a loading control.
- General evolutionary conservation for SOX9 amino acid sequence surrounding the human CPD motif (highlighted in red) of threonine 236–240 across species.
- Western blotting of FLAG‐FBW7α eluted from the immunoprecipitated HA‐SOX9 wild‐type (WT) or CPD mutants (‐T236/240A and ‐T240A). The HA‐SOX9‐WT and the CPD mutant constructs were transiently co‐expressed for 24 h with FLAG‐FBW7α in HEK293 prior to immunoprecipitation with anti‐HA antibody. Equal protein expression of FBW7α across the HEK293 cells transfected with different SOX9 constructs was assessed by immunoblotting of the whole‐cell extract.
- Co‐expression of FBW7α with HA‐SOX9 WT or various other CPD mutant constructs (‐T236/240A, ‐T236A, or ‐T240A) in HEK293 cells. Whole‐cell lysates were collected 24 h following transfection for Western blotting of the total exogenous and the phosphorylated SOX9 proteins using anti‐HA and our pT236‐SOX9 antibody, respectively. Immunoblot of GAPDH protein was used to indicate protein loading in each lane.
- Detection of both exogenous and endogenous phosphorylated SOX9 protein from SOX9 immunoprecipitates. HA‐SOX9‐transfected or non‐transfected HEK293 cells were used as sources for exogenous and endogenous SOX9 protein, respectively. Following SOX9 immunoprecipitation with either anti‐HA (for exogenous) or anti‐SOX9 (for endogenous) antibody, the resulting immunoprecipitates were divided and either treated with λ‐phosphatase or left untreated prior to gel electrophoresis and immunoblotting with pT236‐SOX9 antibody. The SOX9 protein blot shows the total protein level present in each sample. Treatment of HEK293 with proteasome inhibitor MG132 (10 μM) increased the level of phosphorylated SOX9.
- Immunoblots of endogenous pT236 and total SOX9 protein 24 h following transfection of D324MED medulloblastoma cell line with either non‐targeting scramble RNA (siScr) or increasing concentrations of siRNA against SOX9. GAPDH protein was used to indicated protein loading for each sample
- Representative immunofluorescence staining depicting high intensity of pT236‐SOX9 (Alexa Fluor 488; green) staining in the nucleus (counterstained with DAPI; blue) in Daoy medulloblastoma cells. Transfection of Daoy cells with 20 nM siSOX9 depleted the nuclear staining of pT236‐SOX9. Images were taken using a 40× objective. Scale bar: 20 μm.
- Bead‐immobilized IVT HA‐SOX9 WT were subjected to in vitro kinase reaction with 1 unit of recombinant active GSK3α, GSK3β, or their combination (i.e., 0.5 unit for each isoform) for 90 min at 37°C prior to elution and gel electrophoresis. The SOX9 blot shows total SOX9 protein eluted from the beads from each in vitro kinase reaction.
Figure 1. SOX9 interacts with FBW7α through a conserved degron motif phosphorylated by GSK3.
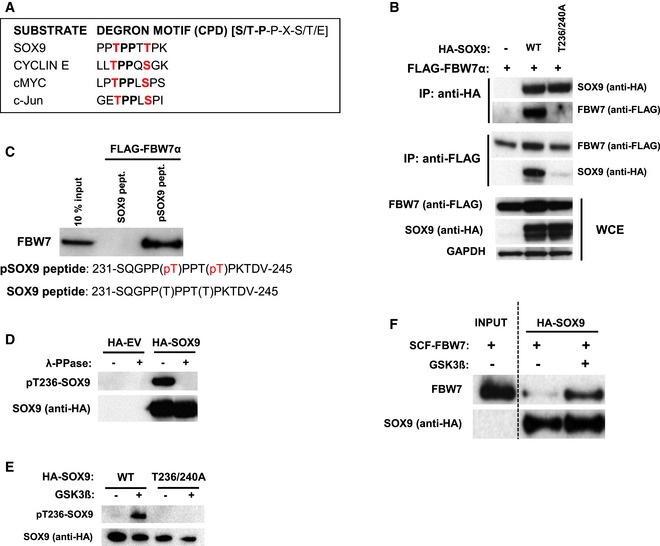
- General sequence alignment of SOX9 Cdc4 phosphodegron (CPD) motif against other high‐affinity motifs present in previously established FBW7α substrates including cyclin E, cMYC, and c‐Jun.
- Immunoblot analysis of the immunoprecipitated HA‐SOX9 or FLAG‐FBW7α and their pull‐down products. Western blots from whole‐cell extract (WCE) of the transfected HEK293 show the levels of exogenous HA‐SOX9 or FLAG‐FBW7α proteins. Cells were treated with 10 μM MG132 for 4 h prior to harvesting and immunoprecipitation. Blots are representative of three independent experiments.
- FBW7α in vitro binding assay. FBW7α was eluted from the agarose bead‐bound SOX9 peptide (encompassing amino acids 231‐245), which had been incubated with the recombinant SCFFBW7α for 1 h at 37°C. The agarose bead‐bound peptide contains either the non‐phosphorylated SOX9 amino acid sequence (SOX9 peptide) or the threonine phosphorylated amino acids (pSOX9 peptide). The input (10%) show the level of the supplemented recombinant SCFFBW7α in the in vitro binding reaction. Blot is representative of two independent experiments.
- Representative Western blots from three independent repeats of pT236‐SOX9 from lysates of HEK293 transfected with either HA‐EV or HA‐SOX9. Each lysate was divided and left untreated or subjected to lambda phosphatase (λ‐PPase) treatment for 1 h at 37°C prior to gel electrophoresis. SOX9 blot show the presence of SOX9 protein in both the untreated and the phosphatase‐treated SOX9‐transfected cell lysates.
- Bead‐immobilized in vitro‐translated (IVT) HA‐SOX9 wild‐type (WT) or T236/240 mutant were either untreated or subjected to in vitro GSK3 kinase reaction for 90 min at 37°C prior to elution and gel electrophoresis. SOX9 immunoblots are representative of three independent experiments.
- GSK3‐mediated phosphorylation of threonine 236 promoted SOX9 interaction with recombinant SCFFBW7α in vitro. IVT HA‐SOX9 immobilized on beads were either untreated or phosphorylated with GSK3 as described in (E) prior to further incubation with recombinant SCFFBW7α for in vitro binding assay. Immunoblot show FBW7α protein in the in vitro binding reaction (input) and FBW7α eluted from the untreated and GSK3‐phosphorylated SOX9. Blots are representative of three independent experiments.
To examine whether FBW7α interacts with SOX9 and to determine which amino acids may participate in the binding, we mutated T236, T240, or both T236/240 in SOX9 to alanine and assessed FBW7α binding. Whereas wild‐type (WT) SOX9 immunoprecipitated with FBW7α, all three SOX9 mutants (T236A, T240A, and T236/240A) failed to bind FBW7α (Figs 1B and EV1C). As FBW7α interacts with phosphorylated substrates, we next analyzed whether phosphorylation of the SOX9 CPD sequence is required for the interaction. Synthetic peptides comprising the SOX9 motif surrounding residues T236 and T240 (amino acids 231–245) were immobilized on beads and tested for their binding to FBW7α. Only the phosphorylated peptide bound recombinant FBW7α in vitro (Fig 1C), suggesting that phosphorylation of the CPD motif triggers the interaction of SOX9 with FBW7α.
To further explore SOX9 CPD phosphorylation, we generated a phospho‐antibody against the SOX9 231SQGPPpTPPTpTPKTDV245 peptide. Importantly, using this tool, we were able to detect both SOX9‐WT and SOX9‐T240A, but not SOX9‐T236A or T236/240A by immunoblot analysis (Fig EV1D), implying that the phospho‐antibody primarily detects pT236‐SOX9. Phosphatase treatment of immunoprecipitated exogenous and endogenous SOX9 demonstrated that the pT236‐SOX9 antibody specifically detects phosphorylated SOX9 (Figs 1D and EV1E). The specificity of the pT236‐SOX9 antibody was further validated by immunoblotting and immunofluorescence staining following RNAi‐mediated SOX9 depletion in the medulloblastoma cell lines D324MED and Daoy (Fig EV1F and G). Given that GSK3 phosphorylates the central threonine position of the CPD in many FBW7α substrates, we next analyzed whether GSK3 also phosphorylates SOX9. Using purified recombinant GSK3α and GSK3β, we found that GSK3 kinase directly phosphorylates in vitro‐translated (IVT) SOX9‐WT but not the SOX9‐T236/240A mutant (Figs 1E and EV1H). Finally, to investigate whether GSK3‐mediated phosphorylation of SOX9 triggers its interaction with FBW7α, we incubated GSK3‐phosphorylated SOX9 with purified recombinant SCFFBW7α complex. As shown in Figs 1F and EV1H, GSK3 indeed promoted the binding of SOX9 to SCFFBW7α in vitro. Altogether, these results suggest that GSK3‐mediated phosphorylation of SOX9 on T236 is required for FBW7α to bind SOX9.
FBW7α promotes SOX9 ubiquitylation and degradation in a GSK3‐dependent manner
In order to determine whether FBW7α regulates SOX9 protein stability, each FBW7 isoform was specifically depleted using siRNAs (as previously described in van Drogen et al, 2006) and endogenous SOX9 protein abundance was monitored. This experiment revealed that depletion of the nuclear isoform, FBW7α, increased the steady state and the half‐life of SOX9 (Fig 2A). This was consistent with the reduced turnover and nuclear enrichment of SOX9 in the HCT116 FBW7‐KO colon cancer cells as compared to HCT116 FBW7‐WT cells (Figs EV1A and EV2A). No significant changes in SOX9 mRNA levels were observed upon FBW7α depletion, consistent with a post‐translational mode of SOX9 regulation by FBW7 in these cells (Fig EV2B). Furthermore, FBW7α and SOX9 were observed to co‐localize in the nucleus and ectopic expression of FBW7α reduced SOX9 protein levels in a dose‐dependent manner (Fig 2B and C). Knockdown of FBW7α also increased both the steady state level and stability of SOX9 protein in different cancer cell lines derived from breast, brain, and colon (Fig EV2C and D). Consistent with the inability of the SOX9‐T236/240A mutants to interact with FBW7α (Figs 1B and EV1C), we found that forced expression of FBW7α did not significantly change the level or stability of either SOX9‐T236A or T236/240A as compared to SOX9‐WT which was rapidly eliminated when co‐expressed with FBW7α (Figs 2D and EV1D). In addition, the proteasome inhibitor MG132 prevented FBW7α‐mediated depletion of SOX9‐WT (Figs 2D and EV2E), strongly suggesting that FBW7α targets SOX9 for proteasomal degradation.
Figure 2. FBW7α promotes SOX9 protein turnover in a GSK3‐dependent manner through ubiquitin–proteasome system.
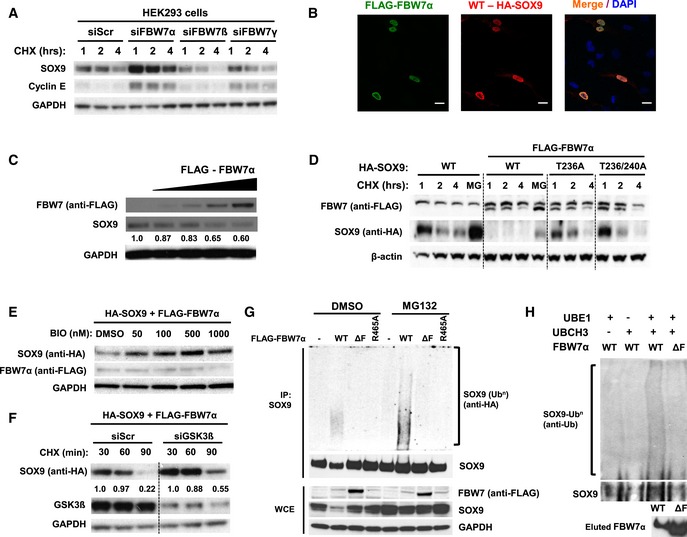
- Endogenous SOX9 protein turnover in the presence of cycloheximide following RNAi depletion of FBW7α, FBW7β, or FBW7γ. HEK293 cells were transfected with 20 nM of scrambled siRNA or siRNAs specifically targeting FBW7α, FBW7β, or FBW7γ for 72 h prior to chase with the addition of 100 ng/ml cycloheximide. Immunoblot of cyclin E, an established SCFFBW7 substrate, was used to assess the efficacy of RNAi‐mediated depletion of FBW7 protein. GAPDH served as a protein loading control.
- Immunofluorescence staining of D324MED medulloblastoma cells showing nuclear co‐localization of HA‐SOX9 WT (Alexa Fluor 568; red) and FLAG‐FBW7α (Alexa Fluor 488; green). The cell nuclei were counterstained with DAPI (blue). Images are representative of multiple fields taken at 40× objective magnification. Scale bar indicates 20 μm.
- Endogenous SOX9 protein levels following transfection (24 h) of 100, 250, 500, and 1,000 ng of plasmid expressing FLAG‐FBW7α. Changes in SOX9 protein levels were analyzed relative to GAPDH levels using ImageJ. The blots shown are representative of three independent experiments.
- Expression of FLAG‐FBW7α enhance HA‐SOX9‐WT protein turnover but not HA‐SOX9‐T236A or HA‐SOX9‐T236A/T240A protein turnover over a 4‐h cycloheximide (100 ng/ml) chase in HEK293 cells. β‐actin protein served as a loading control. The blots shown are representative of four independent experiments.
- Treatment of HEK293 cells with increasing concentration of the GSK3α/β inhibitor BIO, increase HA‐SOX9‐WT protein level in a dose‐dependent manner. HA‐SOX9‐WT and FLAG‐FBW7α were co‐expressed in HEK293 cells prior to treatment with different concentrations of BIO for 4 h. Whole‐cell lysates were collected for Western blotting with anti‐HA (SOX9) and anti‐FLAG (FBW7) antibodies. GAPDH served as a protein loading control. The blots shown are representative of two independent experiments.
- RNAi depletion of GSK3β attenuate FBW7α‐induced HA‐SOX9‐WT turnover in HEK293 cells. HA‐SOX9‐WT protein turnover was examined following 48 h depletion of siGSK3b (20 nM) in the presence of cycloheximide (100 ng/ml). Immunoblots with GSK3β antibodies demonstrated depletion of GSK3β protein with the siRNA. Changes in SOX9 protein levels were analyzed relative to GAPDH levels using ImageJ. The blots shown are representative of three independent experiments.
- FLAG‐FBW7α‐WT expression promote poly‐ubiquitylation of endogenous SOX9 in HEK293 cells. Expression of FBW7α lacking the F‐box domain (ΔF), or containing R465A mutation did not induce SOX9 poly‐ubiquitylation in vivo. Ubiquitylation assay was performed under denaturing condition to disrupt non‐covalently linked ubiquitin as described in the Materials and Methods. Expression of different FLAG‐FBW7α constructs and endogenous SOX9 protein were examined in the whole‐cell lysates. GAPDH protein was used as a loading control. The blots shown are representative of four independent experiments.
- Reconstitution of SOX9 poly‐ubiquitylation by FLAG‐FBW7α in vitro. Immobilized IVT HA‐SOX9 WT was incubated with FLAG‐FBW7α‐WT or the ΔF mutant for 60 min at 37°C. Reaction mixture lacking the UbE1, an E1 ubiquitin‐activating enzyme, or UbcH3, an E2 ubiquitin‐conjugating enzyme, served as a negative control for the in vitro reaction. SOX9 poly‐ubiquitylation was assessed following elution of the protein from the immobilized beads under denaturing condition as described in Materials and Methods. The blots shown are representative of three independent experiments.
Figure EV2. FBW7α promotes SOX9 protein turnover in a GSK3‐dependent manner through the ubiquitin–proteasome system.
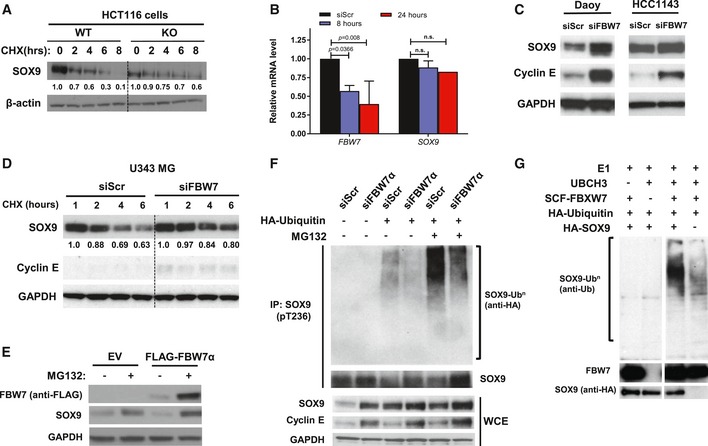
- Endogenous SOX9 protein turnover in HCT116‐FBW7 WT and KO cells over the course of 8 h following the addition of 100 ng/ml cycloheximide. β‐Actin indicates total protein loading for each sample.
- Quantitative PCR analysis of FBW7 and SOX9 mRNA transcripts in Daoy at 8 and 24 h following transfection with 20 nM siFBW7. The FBW7 and SOX9 mRNAs were adjusted to the B2M mRNA prior to being expressed relative to the siScr control. Data are expressed as mean + standard deviation from two independent experiments, with statistical significance determined by multiple‐comparison two‐way ANOVA with Bonferroni's post‐test.
- Immunoblots of endogenous SOX9 protein in medulloblastoma cell line Daoy and breast cancer cell line HCC1143 following depletion of FBW7 by RNAi (20 nM) for 48 h. Accumulation of cyclin E is used to assess the efficiency of FBW7 knockdown. GAPDH immunoblot is shown as a loading control.
- Cycloheximide chase of endogenous SOX9 protein over the course of 6 h in glioma cell line U343MG. The cells were transfected with either non‐targeting (siScr) or FBW7‐specific siRNA for 72 h prior to experiments. Immunoblots of cyclin E, established SCFFBW7 substrate, indicated the efficacy of siFBW, while GAPDH protein was used as total protein loading control for each sample.
- Western blotting of endogenous SOX9 protein level upon treatment with 10 μM MG132. HEK293 cells were transfected with 1 μg FLAG‐FBW7α for 24 h prior to treatment with the proteasome inhibitor. Whole‐cell lysates were collected 4 h following MG132 treatment for gel electrophoresis and immunoblotting. GAPDH protein was immunoblotted to indicate total protein loading for each sample.
- RNAi depletion of FBW7α decreased endogenous SOX9 ubiquitylation in HEK293. The cells were transfected with either scramble (siScr) or siFBW7α for 72 h prior to assessment of endogenous SOX9 ubiquitylation in the absence or presence of MG132. Endogenous SOX9 protein was immunoprecipitated under denaturing condition (1% SDS) from the whole‐cell lysate using the pT236‐SOX9 antibody and eluted as described in Materials and Methods. Total SOX9, cyclin E, and GAPDH proteins in the whole‐cell lysate were immunoblotted.
- Reconstitution of ubiquitylation reaction in vitro using bead‐immobilized IVT HA‐SOX9‐WT and recombinant, active human SCFFBW7α. Reaction mixture lacking the UbcH3, SCFFBW7α, and IVT HA‐SOX9‐WT served as control for the experiments. Ubiquitylation was assessed following elution of IVT HA‐SOX9‐WT from the bead. Immunoblots of SOX9 and FBW7 proteins present in the eluted fraction are shown.
Given that GSK3 promotes FBW7–SOX9 interaction (Fig 1F), we next investigated whether inhibition of GSK3 activity interfered with FBW7α‐mediated SOX9 degradation. As expected, treatment of cells with the GSK3 inhibitor (BIO) or depletion of GSK3 using siRNA significantly attenuated degradation of SOX9 by FBW7α (Fig 2E and F).
Finally, we assessed whether SCFFBW7α mediates the ubiquitylation of SOX9. Expression of FBW7α‐WT promoted the formation of high molecular weight SOX9‐ubiquitin conjugates, whereas expression of F‐box‐deleted (ΔF) FBW7 (which can bind protein substrates but not the SCF core ligase) or FBW7 with a WD40 domain mutant (R465A) (which binds the SCF core but lacks ability to interact with protein substrates) was unable to support SOX9 poly‐ubiquitylation in vivo (Fig 2G). Supporting these results, depletion of FBW7α by siRNA significantly reduced SOX9 ubiquitylation in cells (Fig EV2F). Finally, we reconstituted SOX9 ubiquitylation in vitro using purified recombinant proteins. As shown in Figs 2H and EV2G, only when the SCFFBW7α ubiquitin ligase was present in the reaction, SOX9 was efficiently ubiquitylated. Taken together, these results show that SCFFBW7α ubiquitylates and targets pT236‐SOX9 for proteasomal degradation in a GSK3‐dependent manner.
SOX9 protein stabilization correlates with low levels of FBW7α in medulloblastoma patients
Using whole‐exome sequencing and expression analysis, we observed that FBW7 missense and nonsense mutations occur in approximately 11% of adult SHH subtype cases (Kool et al, 2014). The majority of the missense mutations target distinct arginine residues that have been shown to be directly involved in substrate binding and degradation (Fig 3A) (Akhoondi et al, 2007; Thompson et al, 2007; Forbes et al, 2015). We confirmed that SOX9 is degraded in medulloblastoma cells by ectopic expression of FBW7α‐WT in medulloblastoma‐derived Daoy cells, whereas arginine mutant FBW7α (R465A or R479A) was deficient in SOX9 degradation (Fig 3B). We also demonstrated that only FBW7α‐WT significantly interacted with SOX9, whereas the FBW7α arginine mutations identified in primary medulloblastoma specimens did not (Fig 3A and C). As mentioned above, depletion of FBW7 using siRNAs increased SOX9 protein levels in Daoy medulloblastoma cells (Fig EV2C).
Figure 3. SOX9 protein stabilization correlates with low levels of FBW7α in medulloblastoma patients.
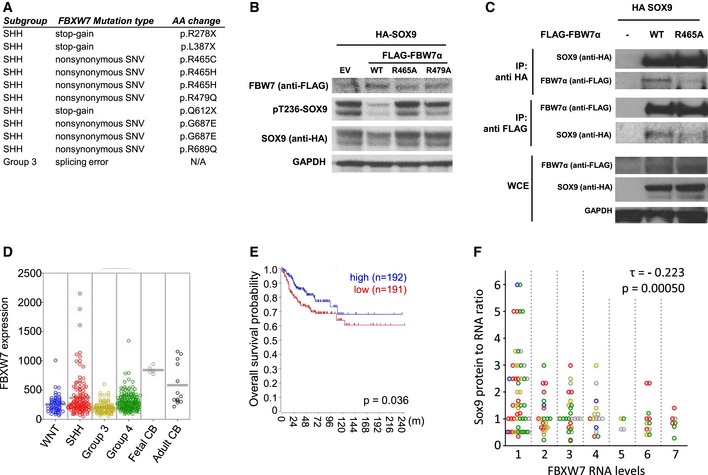
- FBW7 mutations across medulloblastoma molecular subgroup as identified by exome sequencing. In the SHH medulloblastoma subgroup, FBW7 mutations occurred approximately 11% in a cohort containing 133 SHH medulloblastoma cases (Kool et al, 2014) and can be categorized as stop‐gain (n = 3) and non‐synonymous (n = 7). One case of splicing error mutation is detected in the Group 3 medulloblastoma.
- Immunoblotting of HA‐SOX9‐WT protein following expression of FLAG‐FBW7α constructs including wild‐type (WT), R465A or R479A mutation in Daoy cells. The cells were transfected with equal amounts of HA‐SOX9‐WT and FLAG‐FBW7α, and the whole‐cell lysates were collected 24 h following the transfection. GAPDH expression was used as a loading control. The blots shown are representative of two independent experiments.
- Co‐immunoprecipitation of HA‐SOX9 with either wild‐type (WT) or R465A‐FLAG‐FBW7α. Unlike the wild‐type protein, FLAG‐FBW7α containing arginine 465 mutation did not significantly co‐immunoprecipitate with HA‐SOX9 (IP with anti‐HA antibody) and vice versa (IP with anti‐FLAG antibody). HA‐SOX9 and FLAG‐FBW7α‐WT or R465A mutant were assessed in the whole‐cell extract (WCE) of the transfected HEK293 cells with GAPDH protein used to indicate total protein loading.
- Comparison of FBW7 mRNA expression across the medulloblastoma subgroups (WNT, SHH, Group 3, and Group 4) in a cohort containing 423 medulloblastoma cases. Expression of FBW7 in both normal fetal (n = 5) and adult cerebellum (n = 13) was used for comparison against the medulloblastoma.
- Overall survival analysis of medulloblastoma patients based upon FBW7 expression level. Analysis was performed on 383 out of the 423 cases from which the survival data were available. Cohort was divided into high (blue line)‐ and low‐FBW7 (red line)‐expressing subgroup using FBW7 median expression as group classifier. A log‐rank test was used to show differences between groups.
- Correlation analysis between SOX9 protein/mRNA and FBW7 mRNA expression level from medulloblastoma tissue microarray consisting of 142 tissue samples. SOX9 protein level was scored following immunohistochemistry staining, while SOX9 and FBW7 mRNA expression was analyzed by the RNAscope as described in Materials and Methods. A negative correlation was determined by Kendall's correlation test (τ = −0.223, P < 0.001) between SOX9 protein/mRNA and FBW7 mRNA expression. Affiliation of samples with medulloblastoma subgroups is indicated by different colored circle (blue = WNT, red = SHH, yellow = Group 3, green = Group 4, gray = undetermined).
Source data are available online for this figure.
To explore whether FBW7 may be transcriptionally downregulated in primary medulloblastoma specimens, we used a large combined set of gene expression profiles from primary medulloblastomas and analyzed overall FBW7 expression levels (using the R2 database: http://r2.amc.nl). As compared to both normal fetal (n = 5) and adult cerebellum (n = 13), FBW7 transcript levels were significantly lower in all medulloblastoma molecular subgroups (Fig 3D). In light of these findings, we further examined whether FBW7 expression could be linked to clinical outcome in medulloblastoma. Indeed, FBW7 significantly separated medulloblastoma tumors according to overall survival when the expression data were dichotomized into high vs. low FBW7 groups, as evaluated by log‐rank analysis of Kaplan–Meier curves (Fig 3E). Using a best cutoff scanning approach, we also found a statistically significant correlation between low FBW7 groups and worse outcome in the SHH molecular subtype (Fig EV3A). Similar trends were found in the Group 3 and Group 4 medulloblastoma molecular subtypes (Fig EV3A), albeit not statistically significant upon correcting for multiple testing.
Figure EV3. SOX9 protein stabilization correlates with low levels of FBW7α in medulloblastoma patients.
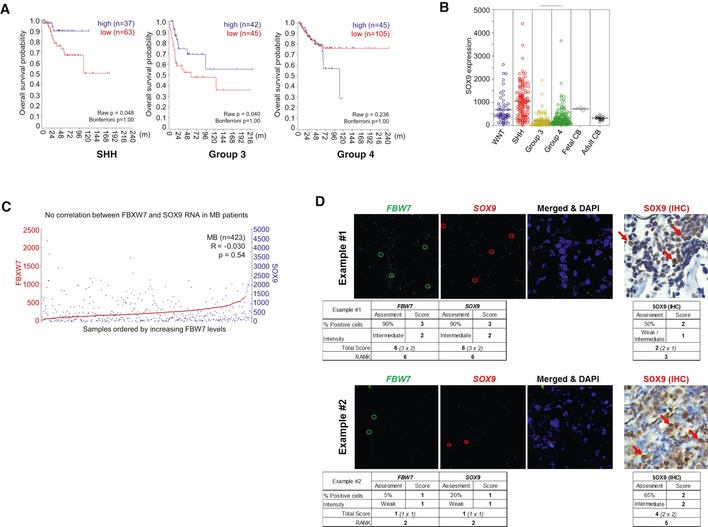
- Overall survival in the different MB molecular subgroups of patients with low vs. high FBW7 expression. After Bonferroni correction, the results were not significant in any of the MB subgroups (Bonferroni P = 1.000).
- SOX9 expression in the four molecular subgroups of MB (423 tumors). Adult (n = 13) and fetal (n = 5) cerebellums were used as controls. ANOVA: P = 2.7e‐05.
- FBW7 and SOX9 mRNA levels do not correlate with each other in a cohort of 423 SHH patients (R = −0.030; P = 0.54).
- Examples of FBW7 (green circles) and SOX9 RNAscope (red circles) images and corresponding SOX9 IHC protein staining (red arrows). Overall tumor expression analyses were performed based upon the “percentage of positive cells” and the staining “intensity” from at least three different image fields by two independent, blinded subjects for each tumor as described in detail in Materials and Methods. The overall score was subsequently converted to “Rank” for correlation analysis of SOX9 protein:RNA ratios against FBW7 expression.
We next used the combined set of gene expression profiles to analyze SOX9 mRNA levels in 423 medulloblastoma samples. In accordance with our previous report (Swartling et al, 2012), SOX9 mRNA levels were found elevated in the SHH cases as compared to both fetal and normal adult cerebellum (Fig EV3B). Both the Group 3 and the Group 4 tumors have lower SOX9 mRNA levels relative to normal fetal and adult cerebellum tissues (Fig EV3B). Additionally, FBW7 mRNA expression did not correlate with SOX9 mRNA expression across the medulloblastoma subgroups (Fig EV3C). In order to test whether a correlation exists between FBW7 expression and SOX9 protein levels, we performed tissue microarray (TMA) analysis using an independent cohort of 142 medulloblastoma tumor specimens, encompassing all four molecular subgroups. Using a novel multiplex RNA in situ hybridization technique, the RNAscope technology (Wang et al, 2012a), we evaluated FBW7 and SOX9 mRNA levels and compared them to SOX9 protein levels as measured using immunohistochemistry (Fig EV3D). Notably, when SOX9 expression was scored using the ratio of SOX9 protein/mRNA, a significant negative correlation with FBW7 expression was observed regardless of medulloblastoma subgroup (Fig 3F). Collectively, these data show that SOX9 is a substrate of FBW7 in medulloblastoma cells and its accumulation is associated with downregulation of FBW7 in primary medulloblastoma tumors, potentially contributing to medulloblastoma malignancy.
FBW7α suppresses the migration and metastasis of SOX9‐driven medulloblastoma cells
To further examine the biological functions of FBW7α‐mediated repression of SOX9 in medulloblastoma, we transduced Daoy cells with SOX9 and doxycycline‐inducible FBW7α (i.e., Daoy‐SOX9/T‐FBW7α). Doxycycline treatment resulted in a dose‐dependent suppression of phospho‐T236 and total SOX9 protein levels in these cells (Fig EV4A). To assess the consequence of FBW7α‐mediated control of exogenous SOX9 on proliferation, we measured cell‐doubling time in parental versus SOX9‐transduced Daoy with or without doxycycline‐induced FBW7α expression. Interestingly, constitutive expression of SOX9 in cultured Daoy cells prolonged cell‐doubling time compared to parental cells, and FBW7α induction reversed this effect (Fig EV4B). Induction of FBW7α was not associated with changes in cell death as analyzed by PARP and caspase‐3 cleavage (Fig EV4C and data not shown). Orthotopic engraftment of these cells into nude mice resulted in complete mortality within 45 days post‐injection in the absence of doxycycline treatment. By comparison, feeding the mice with doxycycline‐containing chow (and hence, inducing FBW7α) significantly increased overall survival (P = 0.0285; Fig 4A). Postmortem examination of the mouse brain from the Daoy‐SOX9 group revealed not only aggressive tumor at the sites of injection in the cerebellum but also metastatic spread in the ventricles, brain stem, and parts of the forebrain. Mice treated with doxycycline (Daoy‐SOX9/T‐FBW7α), on the other hand, presented a constrained tumor at the site of injection in the cerebellum (Fig 4B). Assessment of cell proliferation by immunohistochemistry showed a slight increase in Ki67‐positive cells in the doxycycline‐treated mice compared to untreated mice (Fig EV4D). No significant difference in the levels of SOX9 staining was evident in the Daoy tumors with constitutive expression of SOX9 from mice treated with or without doxycycline. Interestingly, SOX9 protein staining was found to be suppressed in the center part of FBW7α‐induced (+ doxycycline) tumors (Fig EV4E–H, black arrow). Migrating droplets of cells were only found in tumors without FBW7α‐induction (Fig EV4E1 and G2). We also performed immunohistological staining of tumors with or without FBW7α expression to check for differences in cell death (cleaved caspase‐3) or in differentiation markers of gliogenesis (GFAP) and neurogenesis (Syp and TuJ1), but no clear differences between the different groups of mice were found (not shown).
Figure EV4. FBW7α circumvents SOX9‐induced cell migration and metastasis in medulloblastoma.
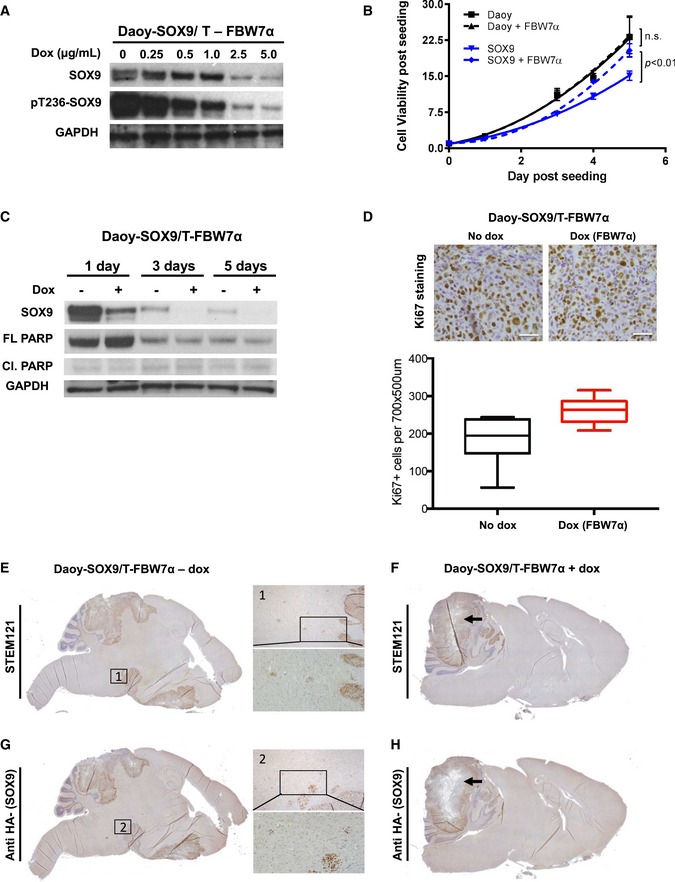
- Immunoblots of WCE from SOX9‐expressing Daoy cells with dox‐inducible FBW7α expression. FBW7α expression was induced with different levels of dox for 24 h prior to cell harvesting for Western blotting. GAPDH immunoblot is shown as a loading control.
- Assessment of changes in Daoy cell viability upon constitutive expression of SOX9 and doxycycline induction of FBW7α. Changes in cell viability are expressed relative to day 0, at the time of seeding. Cell‐doubling time compared to parental cells (Daoy T d = 31.47 h vs. SOX9 T d = 35.67 h), and FBW7α induction reversed this effect (SOX9 + FBW7α T d = 30.20 h).
- Time‐course Western blotting profiling of Daoy cells constitutively expressing SOX9 upon doxycycline induction of FBW7α.
- Quantification of Ki67‐positive cells in SOX9‐expressing Daoy cells with dox‐inducible FBW7α tumors (+ and − dox). Representative IHC stainings used for quantification are shown. Scale bars: 50 μm. Box‐and‐whisker plots displaying the quantification of Ki67‐positive cells. The horizontal line inside each box represents the median value, lower and upper box borders display the 25th and 75th percentiles, respectively, and the interval between the two whiskers include non‐outlier values (min to max).
- IHC staining targeting human‐specific epitope (STEM121) identifying transplanted human medulloblastoma cells in SOX9‐expressing Daoy cells without FBW7α expression. Between the primary tumor and the metastatic compartment human tumor cell “droplets” are evident, as seen in the marked region 1.
- IHC targeting human‐specific epitope (STEM121), identifying transplanted human medulloblastoma cells in SOX9‐expressing Daoy cells with dox‐induced FBW7α expression. Black arrow indicates the center part of the tumor (similar to the region indicated by the black arrow in H).
- Antibody targeting HA epitope identifying HA‐SOX9 levels in SOX9‐expressing Daoy cells without FBW7α expression. Between the primary tumor and the metastatic compartment human SOX9‐positive tumor cell “droplets” are evident, as seen in the marked region 2.
- Antibody targeting HA epitope identifying HA‐SOX9 levels in SOX9‐expressing Daoy cells with dox‐induced FBW7α expression. Black arrow indicates the center part of the tumor (similar to F) with reduced SOX9 expression to be compared to center regions of the tumor in (G).
Figure 4. FBW7α counteract SOX9‐induced cell migration and metastasis in medulloblastoma.
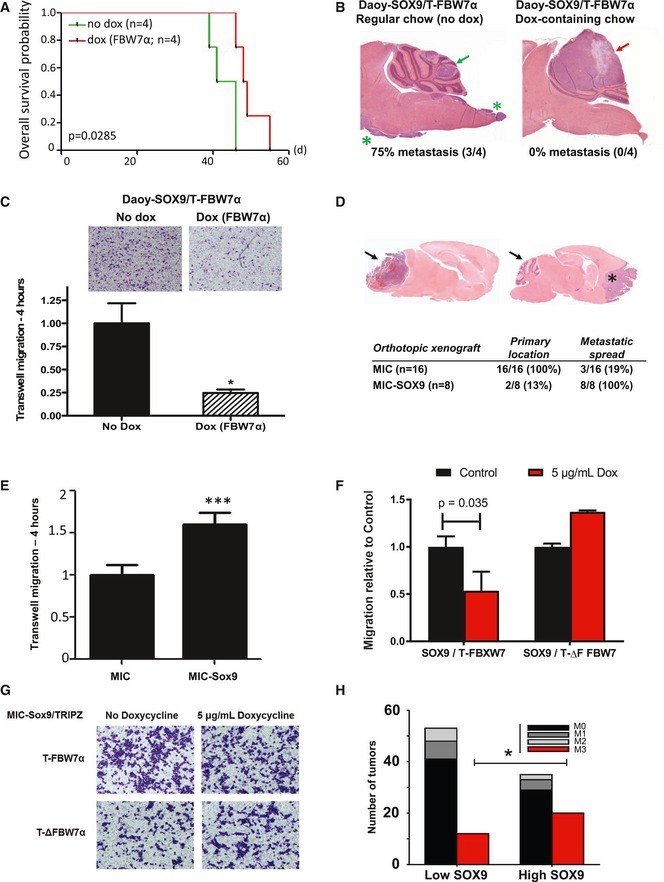
- Overall survival analysis of mice bearing Daoy cells expressing SOX9 with doxycycline (dox)‐inducible FBW7α (Daoy‐SOX9/T‐FBW7α). The dox‐preconditioned or the control cells (105 cells) were orthotopically xenografted to the cerebellum of 6‐week‐old athymic nude Foxn1 nu mice, which were continuously fed with either dox‐containing chow (red line) or regular chow food (green line), respectively. Mice xenografted with Daoy‐SOX9/T‐FBW7α cells and fed continuously with dox‐containing chow (red line) survived longer (P = 0.0285) than the control group (green line).
- Representative hematoxylin and eosin (H&E) staining of medulloblastoma tumors arising from the orthotopic xenograft experiment described in (A). Both the control (non‐dox‐treated) and the dox‐treated mice developed primary intracerebellar tumors (green and red arrows, respectively). SOX9‐expressing Daoy tumors disseminated to the spinal cord (3/4; green asterisks), while the group continuously receiving dox‐containing chow showed no metastatic spread (0/4).
- Representative bright‐field images of migrating Daoy‐SOX9/T‐FBW7α cells in the absence or presence of dox. Daoy‐SOX9/T‐FBW7α cells were either maintained in regular culture or preconditioned with dox prior to Transwell experiments. The cells (2 × 105 cells/insert) were seeded and allowed to migrate across 5‐μm filter pore toward the laminin‐coated Transwell for 4 h prior to fixation and staining with crystal violet. Histogram quantification of cell migration was performed by measuring the crystal violet absorbance of the stained migrating cells and presented as mean + standard deviation from two independent experiments, each containing two technical replicates.
- Representative H&E staining of primary (arrow) and disseminated tumors (asterisk) arising from orthotopic xenograft of parental and SOX9‐expressing medulloblastoma‐initiating cells (MIC and MIC‐Sox9, respectively). Quantification of primary and metastatic tumors arising from both of MIC (n = 16) and MIC‐SOX9 (n = 8) is shown in the table (P = 0.0000136).
- Quantitative comparison of MIC and MIC‐SOX9 cells migrating across 5‐μm filter pore toward the laminin‐coated Transwell for 4 h. Quantification of cell migration was performed as described in (C) and presented as mean + standard error of the mean from six replicates.
- Transwell migration of SOX9‐expressing MICs with dox‐inducible FBW7α or dominant negative ΔF FBW7α. The cells were either maintained in regular cell culture media or preconditioned dox‐media prior to assessment of in vitro Transwell migration as described in (C). Data are mean + standard deviation from two independent experiments, each containing two technical replicates. Statistical analysis comparing the effect of dox‐induction of WT or ΔF FBW7 was carried out using unpaired, two‐tailed Student's t‐test.
- Representative bright‐field images of (F).
- Correlation between clinical metastatic staging and SOX9 protein across the TMA of 142 cases of human medulloblastoma. The SOX9 protein levels were determined by IHC in Fig 3F. Tumors with high SOX9 protein level frequently presented with the M3 metastatic staging (*P = 0.038, Fisher's exact test) and spread to the spinal cord (red bars). Analysis was carried out by comparing patients with high SOX9 protein rank (rank 5–7) versus patients with low SOX9 protein rank (rank 1–3).
We reasoned that the observed tumor containment in response to induction of FBW7α in vivo was due to suppression of SOX9‐induced cell motility. To test this, we performed Transwell migration assays comparing SOX9‐transduced cells with Daoy cells co‐expressing SOX9 and FBW7α. As shown in Fig 4C, induction of FBW7α (+ dox) potently suppressed cell motility (> 70%, P < 0.05) in SOX9‐expressing Daoy cells (Fig 4C). In accordance, we recapitulated these findings using our previously developed medulloblastoma‐initiating cells (MICs) derived from MYCNT58A‐transduced postnatal day 0 cerebellum mouse neural stem cells (Swartling et al, 2012). Orthotopic transplantation of the MICs led to intracerebral tumors (16 out of 16), while only three (18.75%) of these disseminated and formed tumors in the frontal base of the forebrain. Strikingly, forced expression of SOX9 in these MICs (MIC‐SOX9) increased the rate of metastasis to the forebrain to 100% (8 out of 8) (Fig 4D), and only two (25%) of the transplanted MIC‐SOX9 cells formed intracerebellar tumors at the site of injection (Fig 4D). In vitro Transwell assays again confirmed that SOX9 overexpression increased the motility of MICs (Fig 4E) and that expression of FBW7α, but not its dominant negative form ∆F‐FBW7α, reduced the SOX9‐induced cell motility (Fig 4F and G). Finally, to determine whether SOX9 protein correlates with metastasis, we went back to our TMA cohort of 142 medulloblastoma patients and correlated SOX9 protein levels with metastasis at diagnosis as determined by pathological grading of metastasis (M1–M4). No M4 cases were included in this cohort. We found that SOX9 protein levels correlated with a significant increase in the M3 metastasis stage (P < 0.038), indicating spread of tumor cells to the brain stem and spinal cord (Zeltzer et al, 1999; Fig 4H).
SOX9 stabilization promotes a metastasis‐supportive program in medulloblastoma
To examine the biological function of SOX9 degradation for medulloblastoma progression in more detail, we generated doxycycline‐inducible SOX9‐WT or stable SOX9‐T236/240A human primary MB002 cells (which have low levels of endogenous SOX9). Immediate transcriptional changes following an 8‐h induction of SOX9‐WT or SOX9‐T236/240A were assessed relative to the MB002 cells transduced with an empty vector (EV) using RNA sequencing (RNA‐Seq). Induction of SOX9‐WT and SOX9‐T236/T240A generated similar transcriptional profiles (R = 0.61), with SOX9 itself being one of the genes most differentially expressed and significantly upregulated (Fig 5A). Hierarchical clustering of the most variable genes across these samples alone or in the context of human medulloblastoma signature genes (Northcott et al, 2011) showed that the different cell lines formed individual clusters, with SOX9‐WT and SOX9‐T236/240A being more similar to each other compared to the EV‐transduced cells (Fig EV5A). Comparison of the RNA‐Seq‐derived transcription profiles to human medulloblastoma molecular subgroups (Northcott et al, 2011) revealed that all three MB002 cell lines were classified as Group 3 tumors.
Figure 5. RNA‐Seq profiling of MB002 cells expressing SOX9‐WT and FBW7‐insensitive SOX9 mutant.
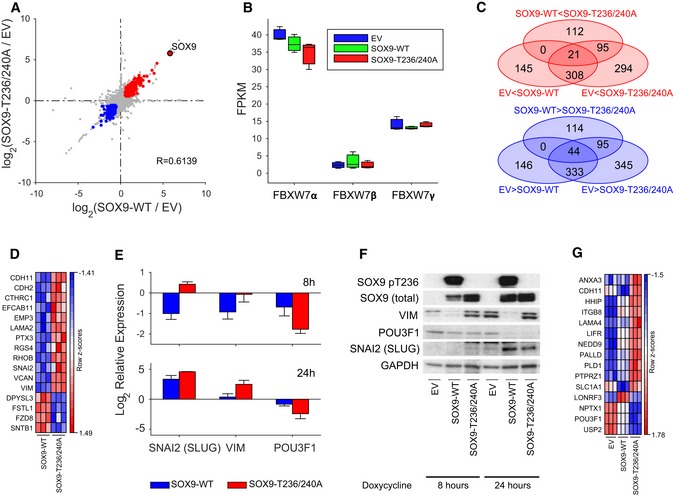
- Scatter plot depicting the relationship between transcriptional changes measured as log2 FC between EV to SOX9‐WT (x‐axis) and EV to SOX9‐T236/T240A (y‐axis). Each dot represents one transcript, with colored dots indicating transcripts upregulated (log2 FC > 0.585; P < 0.05; red dots) or downregulated (log2 FC < −0.585; P < 0.05; blue dots) in both comparisons. The transcript corresponding to the SOX9 gene is highlighted as a red filled dot. Indicated R‐value indicates Spearman's rank correlation coefficient between the two FC profiles.
- Box‐and‐whisker plot displaying the sample specific expression of three FBW7 isoforms: FBW7α (NM_033632), FBW7β (NM_018315), and FBW7γ (NM_001013415'). Each box represents the expression distribution over three replicates. The horizontal line inside each box represents the median value, lower and upper box borders display the 25th and 75th percentiles, respectively, and the interval between the two whiskers includes all non‐outlier values.
- Venn diagram illustrating the overlap of differentially expressed (FC > 1.5, P < 0.05) transcripts between any of the three comparisons: EV to SOX9‐WT (upregulated: 145 + 308 + 21; downregulated 146 + 333 + 44), EV to SOX9‐T236/T240A (upregulated: 294 + 308 + 95 + 21; downregulated 345 + 333 + 95 + 44), or SOX9‐WT to SOX9‐T236/T240A (upregulated: 112 + 95 + 21; downregulated 114 + 95 + 44). Each oval represents the number of transcripts either upregulated or downregulated in the respective comparison, where the direction of differential expression is indicated by the inequality sign in the label. Numbers in overlapping regions signify the number of transcripts commonly differentially expressed in all contributing comparisons.
- Heat map of the row‐wise z‐scores of 16 EMT hallmark genes in SOX9‐WT and SOX9‐T236/T240A samples. Heat map was generated using the GenePattern software (Reich et al, 2006).
- Quantitative PCR (qPCR) of SNAI2 (SLUG), VIM, and POU3F1 (OCT6) (normalized to GAPDH levels) in MB002 cells stably transduced with EV (set to 1), SOX9‐WT, and SOX9‐T236/240A. Cells were induced for 8 or 24 h with doxycycline. The data shown represents mean expression values and error bars indicate the standard error of the mean.
- Western blot of SOX9, HA‐SOX9, pSOX9‐T236, SNAI2 (SLUG), VIM, and POU3F1 (OCT6) in MB002 cells stably transduced with EV, SOX9‐WT, and SOX9‐T236/240A. Cells were induced for 24 and 48 h with doxycycline, and GAPDH was used as a loading control.
- Heat map of the row‐wise z‐scores of 14 pro‐metastasis genes differentially expressed between SOX9‐WT and SOX9‐T236/T240A samples and previously identified as SOX9 target genes (Kadaja et al, 2014; Oh et al, 2014; Larsimont et al, 2015). Heat map was generated using the GenePattern software (Reich et al, 2006).
Figure EV5. Hierarchical clustering, MB subgroup classification, and GSEA of transcriptional profiles in MB002 cells overexpressing SOX9‐WT or SOX9‐T236/240A.
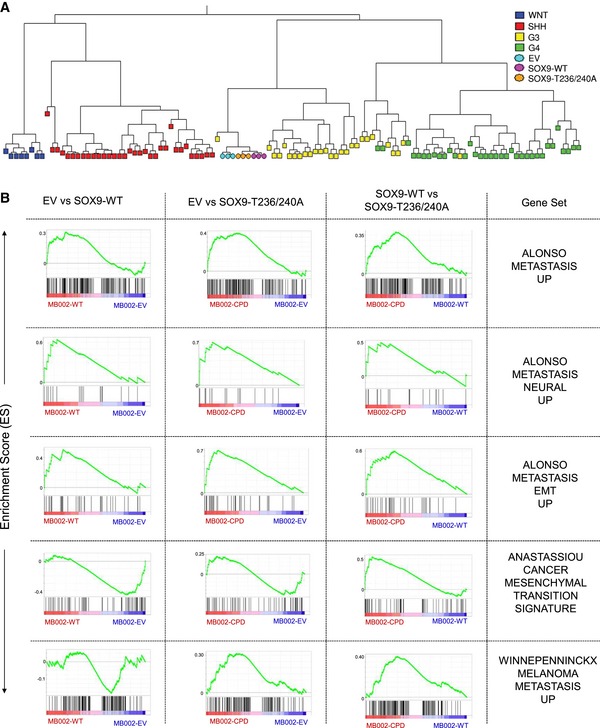
- Hierarchical clustering of 103 human medulloblastoma samples with known molecular subgroups (Northcott et al, 2011) together with triplicates each of MB002 cells transduced with EV (blue circles), SOX9‐WT (orange circles), or SOX9‐T236/240A (purple circles). The clustering was performed on the 3,167 signature genes most variably expressed between the four MB subgroups. Extraction of signature genes, hierarchical clustering, and plotting of the dendrogram were performed using the metagene code for cross‐platform, cross‐species projection of transcription profiles (Tamayo et al, 2007).
- Gene sets with significant enrichment in SOX9‐expressing MB002 cells. A GSEA on a list of 19 gene sets related to metastasis, migration, or EMT (Table EV5) was performed for each pairwise comparison of MB002 EV, SOX9‐WT, and SOX9‐T236/240A. The figure shows the five gene sets (rows) with significant positive enrichment in either SOX9‐WT against EV (left column), SOX9‐T236/240A against EV (middle column), or SOX9‐T236/240A against SOX9‐WT (right column).
Analysis of the FBW7 transcripts in the MB002 cells revealed that the alpha isoform is the most abundant transcript, while both the beta and the gamma isoforms are expressed at significantly lower levels (Fig 5B). Notably, neither expression of SOX9‐WT nor that of SOX9‐T236/240A significantly affected the endogenous level of the different FBW7 transcripts (Fig 5B).
Of the transcripts significantly upregulated with SOX9 expression when compared to EV, 38% of these transcripts (i.e., 329 out of 863) were shared between the WT and the FBW7‐resistant SOX9 mutant (Fig 5C, red Venn diagram). Likewise, there were 377 out of 963 significantly downregulated transcripts (constituting 39%) that were shared between SOX9‐WT and SOX9‐T236/240A (Fig 5C, blue SOX9‐T236/240A as compared to SOX9‐WT Venn diagram).
Despite the similar transcriptional changes induced by SOX9‐WT and T236/240A, detailed comparison of RNA‐Seq profiling revealed subsets of differentially expressed transcripts among these cells (Fig 5C). Specifically, there were 207 among 228 transcripts that were exclusively upregulated and 209 out of 253 transcripts that were exclusively downregulated with the expression of SOX9‐T236/240A in MB002 (Fig 5C, red Venn and blue Venn, respectively; Table EV1). Twenty‐one transcripts (Fig 5D, upper panel; Table EV2) were significantly upregulated in SOX9‐WT but also showed a further significant increase in SOX9‐T236/240A as compared to EV (EV < SOX9‐WT < SOX9‐T236/240A). Conversely, 44 transcripts (Fig 5D, upper panel; Table EV2) were significantly downregulated more potently in SOX9‐T236/240A as compared to SOX9‐WT (SOX9‐T236/240A < SOX9‐WT < EV). To explore the functional significance of SOX9 accumulation in medulloblastoma, we next assessed the biological processes influenced by SOX9 in MB002 cells. First, we performed gene set overlap analysis of differentially expressed genes in response to induction of SOX9‐WT or SOX9‐T236/240A as well as between SOX9‐WT and SOX9‐T236/240A. Interestingly, among the HALLMARK gene sets, EMT genes were among the top 10 signatures upregulated in SOX9‐WT or SOX9‐T236/240A as compared to EV. The EMT functional category was also found to be the most significantly upregulated gene set in SOX9‐T236/240A as compared to SOX9‐WT (Table EV4). At the individual gene level, we identified several EMT hallmark factors (12 upregulated; four downregulated) including SNAI2 and VIM from our transcriptome profiles that were differentially upregulated or downregulated, respectively, in SOX9‐T236/240A compared to SOX9‐WT (Fig 5D). To confirm these results at the molecular level, SOX9 was induced for 8 and 24 h with doxycycline and analyzed for expression of SNAI2 and VIM. Quantitative real‐time PCR (qRT–PCR) and immunoblot analysis demonstrated elevated mRNA and protein levels in the SOX9‐T236/240A mutant cells as compared to EV and the SOX9‐WT cells (Fig 5E and F). Thus, SOX9 and more potently stabilized SOX9 appear to promote expression of genes linked to EMT reprogramming in medulloblastoma. As EMT promotes migration/metastasis, these profiling data are well in line with the findings that SOX9 degradation controls migration and metastatic spread in medulloblastoma (Fig 4). To further characterize these aforementioned processes in more detail, we performed targeted GSEA on 19 preselected gene sets related to metastasis, migration, and EMT (Table EV5). Strikingly, this analysis revealed that in addition to EMT, metastasis signatures were found with significant enrichment among upregulated genes in SOX9‐T236/240A and/or SOX9‐WT (Fig EV5B).
To assess the change in expression of high‐confidence SOX9 targets following induction of SOX9‐WT or SOX9‐T236/240A, we next compared differentially expressed genes to a list of genes putatively bound and regulated by SOX9 according to previous SOX9 ChIP studies (Kadaja et al, 2014; Oh et al, 2014; Larsimont et al, 2015). Importantly, we identified 24 genes that were documented as direct SOX9 target genes and overlapped with significantly upregulated or downregulated genes, respectively, in SOX9‐T236/240A as compared to SOX9‐WT (P = 0.0068) (Table EV3). This also included the recently described SOX9 target gene POU3F1 (OCT6) (Larsimont et al, 2015), which was found to be repressed at both the mRNA and protein level in SOX9‐WT and more potently in SOX9‐T236/240A mutant cells after 8 and 24 h of doxycycline treatment (Fig 5E and F). Interestingly, of these 24 genes, at least 14 have previously been recognized as pro‐metastasis factors (e.g., PLD1, LAMA4, and NEDD9) again arguing for a significant role of SOX9 in promoting medulloblastoma tumor migration and metastasis (Nagato et al, 2005; Park & Min do, 2011; Speranza et al, 2012) (Fig 5G). Altogether, these results support a role for SOX9 stabilization as driver of medulloblastoma progression through orchestrating a metastasis‐promoting transcriptional program.
SOX9 confers cisplatin resistance in medulloblastoma
Given that EMT reprogramming, stemness, and metastasis are linked to drug resistance in cancer cells, we next asked whether SOX9 abundance influences the response of medulloblastoma to cisplatin, a commonly used chemotherapeutic drug in standard medulloblastoma treatment. Using the doxycycline‐inducible SOX9 model system, we found that SOX9 expression reduced the efficacy of cisplatin treatment not only in medulloblastoma MB002 and Daoy cells, but also in medulloblastoma‐initiating cells (Fig 6A). Quantitative comparison of the cisplatin IC50 values from these experiments showed between 2‐ and 15‐fold increase in cisplatin resistance upon SOX9 overexpression (Fig 6A). Importantly, dox‐induced expression of FBW7α in SOX9 expressing Daoy and MICs improved sensitivity to cisplatin by approximately 2‐fold (Fig 6B). In order to test whether medulloblastoma cells with elevated levels of SOX9 are more resistant to cisplatin treatment in vivo, we transplanted mice with parental, SOX9‐WT‐transduced, or SOX9‐T236/240A‐transduced Daoy cells. Importantly, cisplatin treatment for 14 days significantly prolonged survival in Daoy parental cells, whereas Daoy cells transduced with SOX9‐WT or stable mutant SOX9 did not significantly respond to the cisplatin therapy (Figs 6C and EV6).
Figure 6. SOX9 confers cisplatin resistance in medulloblastoma.
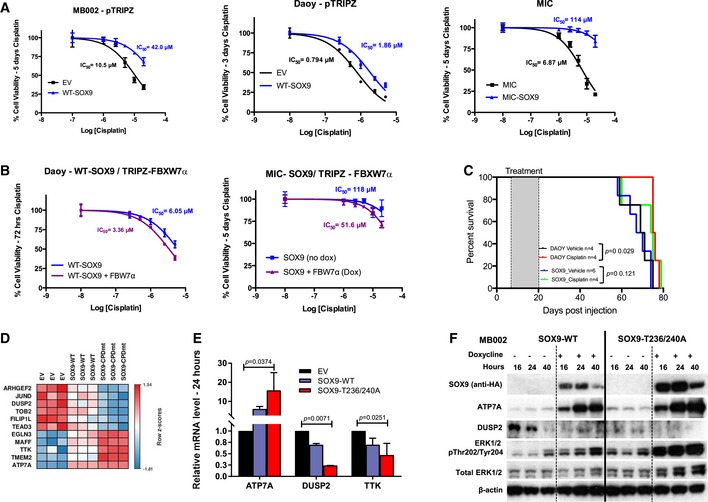
- Cisplatin dose–response curves of MB002 cells (left), Daoy cells (middle), and MICs (right) in the absence (EV) or presence of SOX9 as analyzed by alamarBlue assay. Cells were preconditioned with doxycycline to induce expression of SOX9 (or EV) prior to treatment with increasing concentrations of cisplatin. The IC50 were calculated following 5 (MB002 and MIC) or 3 days (Daoy) of treatment. Data are mean + standard deviation from three independent repeats, each containing five technical replicates.
- Cisplatin dose–response curves of SOX9‐expressing Daoy cells (left) and MICs (right) in the absence or presence of FBW7α. Experiments and data analysis were performed as described in (A).
- Overall survival analysis of mice bearing Daoy cells or Daoy cells expressing dox‐inducible SOX9 treated with cisplatin. The dox‐preconditioned cells (105 cells) were orthotopically xenografted to nude Foxn1 nu mice and left for 1 week prior to being treated with vehicle control or cisplatin (2 mg/kg) intraperitoneally every other day for a total of six doses.
- Heat map of the row‐wise z‐scores of 11 genes associated with cisplatin resistance in MB002 cells expressing Sox9‐WT or Sox9‐T236/T240A. Heat map was generated using the GenePattern software.
- Quantitative analysis of ATP7A, DUSP2, and TTK mRNAs in MB002 cells following expression of SOX9‐WT or SOX9‐T236/240A. Total RNA was collected 24 h following doxycycline treatment, from which cDNA was generated for qPCR. Data are mean mRNA level (normalized to B2M transcript) + standard deviation from three independent experiments with statistical significance determined by multiple‐comparison two‐way ANOVA with Bonferroni's post‐test.
- Time‐course Western blotting of HA‐SOX9, ATP7A, DUSP2, ERK1/2 pThr202/Tyr204, and total ERK1/2 in MB002 cells following doxycycline induction of either EV, SOX9‐WT, or SOX9‐T236/240A. GAPDH was used as a loading control.
Figure EV6. SOX9 confers cisplatin resistance in medulloblastoma.
Overall survival of nude Foxn1 nu mice orthotopically engrafted with Daoy cells expressing SOX9‐T236/240A treated with cisplatin (red curve) or a vehicle control (blue curve). The experiment was carried out as described in Fig 6C.
In an attempt to characterize the molecular mechanisms underlying SOX9‐mediated cisplatin resistance, we re‐assessed our MB002 RNA‐Seq profiling data (Fig 5) for genes previously linked to cisplatin resistance (Galluzzi et al, 2012). Among the significantly differentially expressed genes between EV and SOX9‐WT or SOX9‐WT and SOX9‐T236/240A (21 and 44 genes upregulated or downregulated, respectively) (Fig 5D), we identified 11 genes associated with cisplatin resistance (Fisher's exact test P < 0.00001) (Fig 6D). Increased expression of the copper transporter ATP7A and decreased expression of DUSP2 and TTK were verified by quantitative RT–PCR and immunoblotting (Fig 6E and F). ATP7A has been reported to confer resistance via cytoplasmic sequestration of cisplatin (Galluzzi et al, 2012), whereas repression of the phosphatase DUSP2, a negative regulator of ERK1/2 activity, has been shown to abrogate drug‐induced apoptosis and increase cisplatin resistance via upregulation of drug‐resistance genes (e.g., MDR1 and GRP78) (Lin et al, 2011). Together, these results show that SOX9 promotes cisplatin resistance in medulloblastoma, possibly by upregulating ATP7A and downregulating DUSP2 expression.
PI3K/AKT/mTOR inhibitors sensitize medulloblastoma cells to cisplatin by targeting SOX9 through FBW7
Given the fact that PI3K/AKT signaling antagonizes GSK3 activity (Cross et al, 1995; Cheng et al, 2009), we reasoned that hyperactivation of the PI3K pathway could stabilize SOX9 protein. Strikingly, recurrent mutations affecting PI3K/AKT/mTOR signaling and phosphorylation of AKT and S6K are strongly associated with poor outcome in adult SHH medulloblastoma (Kool et al, 2014), and this pathway is commonly activated in clinically refractory and/or recurrent medulloblastoma (Buonamici et al, 2010). To explore the possibility that pharmacological inhibition of PI3K/AKT/mTOR signaling could potentially sensitize medulloblastoma to cisplatin treatment by targeting SOX9 for degradation through GSK3‐FBW7α, we utilized three pharmacological inhibitors (AZD8186, AZD5363, and AZD2014) to inhibit PI3K/AKT/mTOR activities in Daoy cells (Fig 7A). Specific inhibition of PI3K/AKT/mTOR signaling was confirmed by immunoblotting with a panel of pathway activity markers in medulloblastoma cells treated with each respective drug (Fig EV7A). Immunoblotting with GSK3α pSer21 and GSK3β pSer9 antibodies, established markers for the PI3K/AKT phospho‐inactivated GSK3, revealed decreased levels upon inhibitor treatment (Fig EV7A), suggesting the presence of active GSK3. Notably, expression of FBW7α in the presence of these inhibitors resulted in more potent reduction of total and pT236 phosphorylated SOX9 protein (Fig 7A). Importantly, cycloheximide chase experiments demonstrated enhanced SOX9 degradation in response to PI3K/mTOR inhibition (AZD2014 and AZD8186), and depletion of FBW7α completely rescued inhibitor‐induced turnover of SOX9 (Figs 7B and EV7B). Importantly, co‐treatment of medulloblastoma cells (Daoy and MB002) with cisplatin and AZD2014 translated to a potent synergistic repression of cell viability (Fig 7C; CI (combination index) < 1.0). The drug synergism was also reproduced by combining cisplatin with other PI3K/mTOR inhibitors currently used in clinical trials including BEZ‐235 and KU‐0063794 (Fig EV7C).
Figure 7. Increased SOX9 turnover via PI3K/AKT/mTOR inhibition sensitizes medulloblastoma cells to cisplatin treatment.
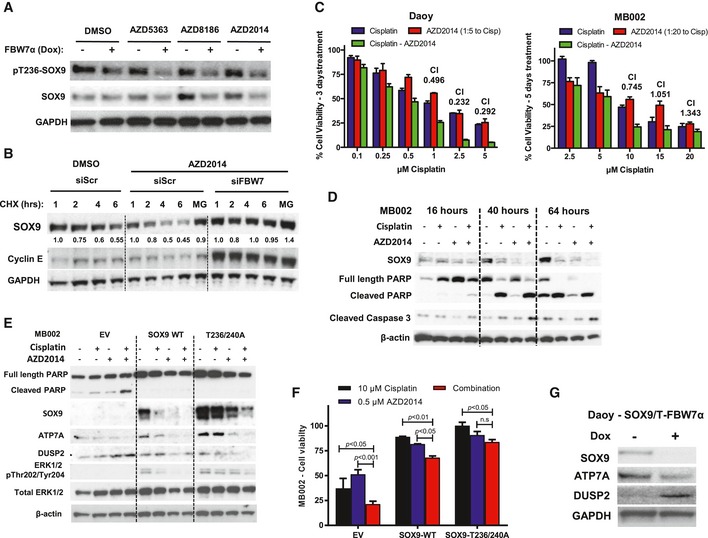
- Western blotting of pT236‐SOX9 and total SOX9 following treatment of Daoy cells with a panel of inhibitors targeting PI3K/AKT/mTOR pathway. The whole‐cell lysates were collected for gel electrophoresis following 6‐h treatment with 1 μM of AZD5363, AZD8186, or AZD2014. The blots shown are representative of three independent repeats.
- Western blotting of endogenous SOX9 protein turnover in the presence of PI3K/mTOR dual inhibitor, AZD2014. Daoy cells were transfected with either siScr or siFBW7 for 48 h prior to further treatment with 1 μM AZD2014. SOX9 protein turnover was examined 2 h following the inhibitor treatment by cycloheximide chase assay. Immunoblotting of cyclin E protein was used to indicate the efficacy of FBW7 knockdown. Changes in SOX9 protein level were quantified relative to GAPDH protein using ImageJ. The blots shown are representative of three independent experiments.
- Quantification of resazurin‐based Daoy and MB002 cell viability following treatment with cisplatin (blue bars), AZD2014 (red bars), or their combination (green bars). Daoy cells were treated for 72 h, while MB002 cells were subjected to 5‐day course of treatment. The cell viability was calculated relative to the vehicle‐treated cells. The synthetic lethality combination index (CI) for each treatment was calculated from the mean of three independent experiments using the Compusyn software. Error bars indicate standard deviation.
- Time‐course Western blotting of endogenous SOX9 and apoptotic markers including cleaved PARP and caspase‐3 following MB002 treatment with cisplatin, AZD2014, or their combination. β‐Actin was used as a loading control. The blots shown are representative of three independent experiments.
- Western blotting profiling of MB002 cells expressing EV, SOX9‐WT, or SOX9‐T236/240A following 48‐h treatment with cisplatin, AZD2014, and their combination. The set of blots shown is representative of three independent experiments.
- Quantification of cell viability of MB002 cells expressing EV, SOX9‐WT, or SOX9‐T236/240A following 5‐day treatment with either 10 μM cisplatin (black bars), 0.5 μM AZD2014 (blue bars), or their combination (red bars). Data are mean + standard deviation from three independent experiments, from which statistical significance was analyzed by two‐way ANOVA multiple comparisons with Bonferroni's post‐test.
- Immunoblots of ATP7A and DUSP2 protein following doxycycline treatment of Daoy‐SOX9/T‐FBW7α cells. The cells were either untreated or incubated with 5 μg/ml doxycycline for 72 h to induce FBW7a prior to being harvested for gel electrophoresis. The blots shown are representative of two independent experiments.
Figure EV7. SOX9 confers cisplatin resistance and targeting of SOX9 through PI3K/AKT/mTOR inhibition increases sensitivity to cisplatin treatment.
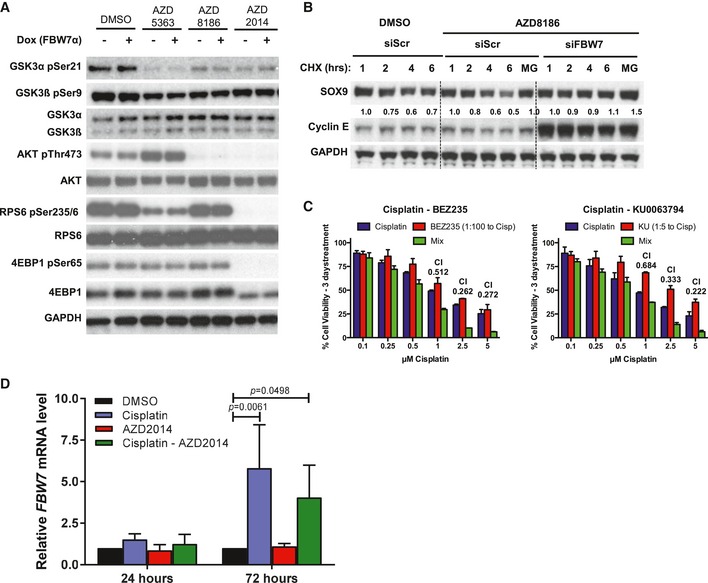
- Immunoblots of PI3K/AKT/mTOR pathway activity to assess inhibition upon treatment of Daoy cells with 1 μM AZD5363, AZD8186, or AZD2014.
- Western blotting of cycloheximide chase from Daoy cells treated with 1 μM PI3K inhibitor AZD8186. Cells were transfected with either non‐targeting or FBW7‐specific siRNA for 48 h prior to cycloheximide chase. siRNA depletion of FBW7 leads to stabilization of SOX9 protein and was calculated using Compusyn software.
- Cytotoxicity assay for Daoy medulloblastoma cells treated with a combination of cisplatin with (i) BEZ235 or (ii) KU0063794. The synthetic lethality combination index (CI) for each treatment was calculated using Compusyn software. Data are expressed as mean + standard deviation from two independent experiments.
- Quantitative PCR analysis of endogenous SOX9 mRNAs in MB002 following 24 and 72 h treatment with cisplatin, AZD2014, or their combination. The mRNAs levels were normalized relative to GAPDH and expressed relative to the DMSO‐treated samples. Data are expressed as mean + standard deviation from two independent experiments, with statistical significance determined by multiple‐comparison two‐way ANOVA with Bonferroni's post‐test.
At the molecular level, degradation of endogenous SOX9 protein using the combination of cisplatin and PI3K/mTOR inhibitor resulted in increased apoptosis as shown by cleaved PARP and caspase‐3 (Fig 7D). Interestingly, we also observed that cisplatin treatment alone reduced SOX9 protein levels (Fig 7D, lanes 6 and 10 vs. lanes 5 and 9, respectively) in MB002 cells, indicating that cisplatin‐induced DNA damage may initially stimulate FBW7α‐mediated degradation of SOX9. In agreement with this possibility, we found that FBW7α mRNA expression increased from 1.5‐fold to fivefold during the course of 24 and 72 h of cisplatin treatment (Fig EV7D). Also, co‐treatment of MB002 cells with cisplatin and PI3K/mTOR inhibitor (AZD2014) further enhanced depletion of SOX9 protein as compared to treatment with either drug alone (Fig 7D, lanes 8 and 12 vs. lanes 6 and 10, respectively). Altogether, these results suggest that SOX9 is targeted for degradation by FBW7α in response to cisplatin treatment and SOX9 degradation is further stimulated with therapeutic drugs inhibiting PI3K/AKT/mTOR signaling.
To characterize in more detail the role of SOX9 abundance and sensitivity to these drugs, we subjected MB002 cells expressing either SOX9‐WT or the SOX9‐T236/240A mutant to treatment with each individual drug or their combination. The FBW7‐insensitive SOX9‐T236/240A mutant remained stable in response to cisplatin treatment compared to wild‐type SOX9 (Fig 7E, lane 10 vs. lane 6). As expected, the AZD2014 treatment stimulated degradation of SOX9 protein and the combination treatment (cisplatin+AZD2014) was more efficient in depleting SOX9‐WT as compared to the SOX9‐T236/240A mutant (Fig 7E). Interestingly, the AZD2014 inhibitor alone, as well as the combination treatment, also reduced SOX9‐T236/240A protein to some extent, indicating that AZD2014 may repress SOX9 independently of FBW7 (Fig 7E). To this end, we examined whether SOX9 abundance affects drug response and cell viability in EV, SOX9‐WT, or SOX9‐T236/240A MB002 cells. As shown in Fig 7E, expression of either SOX9‐WT or SOX9‐T236/240A increased cisplatin resistance as measured by cleaved PARP and alamarBlue assay, respectively (Fig 7E and F). Nonetheless, SOX9‐WT‐expressing cells remained significantly sensitized, albeit to a lesser degree than EV‐expressing MB002, to the cisplatin/AZD2014 combination treatment (Fig 7F), strongly arguing for FBW7‐mediated degradation of SOX9 influences cisplatin sensitivity in these cells.
To further explore the underlying mechanism(s) of how SOX9 stabilization promotes resistance to cisplatin, we next analyzed ATP7A and DUSP2 protein levels in EV, SOX9‐WT, and SOX9‐T236/240A MB002 cells treated with single drugs or their combination. Indeed, we found that the combination treatment significantly attenuated upregulation of ATP7A expression in SOX9‐WT cells compared to SOX9‐T236/240A (Fig 7E, compare lane 8 vs. lane 5 and lane 12 vs. lane 9). Furthermore, the combination treatment also increased DUSP2 expression in MB002 cells (Fig 7E, compare lane 1 vs. lane 4), and in agreement with its role in repressing ERK1/2 activity, we observed reduced ERK1/2 phosphorylation in both SOX9‐WT and SOX9‐T236/240A cells treated with the drug combination as such the effect being less pronounced in the cells expressing FBW7‐insensitive SOX9 (Fig 7E, compare lanes 5–8 vs. lanes 9–12). Supporting these results, we also found that the increased sensitivity to cisplatin upon induction of FBW7α in Daoy‐expressing SOX9 (Fig 6B) was associated with reduced expression of ATP7A and increased DUSP2 protein levels (Fig 7G).
On the basis of these findings, we suggest that inactivation of PI3K/AKT/mTOR signaling sensitizes medulloblastoma cells to cisplatin treatment by stimulating GSK3/FBW7‐mediated SOX9 proteasomal degradation (Fig 8).
Figure 8. Schematic model of FBW7α‐mediated regulation of SOX9 in medulloblastoma cells.
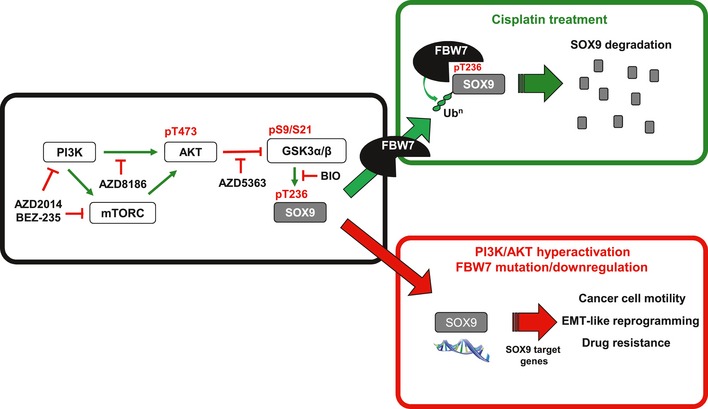
SCFFBW 7α together with GSK3 regulate SOX9 protein levels through the ubiquitin–proteasome system. FBW7α mutations, or transcriptional downregulation, lead to stabilization of SOX9 protein and SOX9‐driven EMT‐like reprogramming, migration and drug resistance in medulloblastoma. Small‐molecule inhibitors of the PI3K/mTOR pathway can be used to stimulate GSK3/FBW7α‐mediated SOX9 turnover and may provide a novel strategy to target SOX9‐driven medulloblastoma.
Discussion
Stabilization of oncoproteins caused by dysregulation of SCF substrate adaptors, like F‐box proteins, is an important driver of malignant brain tumors (Hede et al, 2014). To provide a more complete understanding of how the tumor suppressor FBW7 functions and controls tumorigenesis, we previously employed a proteomic screening approach designed to discover novel SCFFBW7 substrates (Arabi et al, 2012). One of the candidates identified in this screen was the transcription factor SOX9. Here, we show that the SCFFBW7α ubiquitin ligase targets SOX9 for proteasomal degradation and provide experimental evidence of a role for FBW7 in modulating medulloblastoma malignancy and cisplatin resistance by controlling SOX9 proteolysis.
As phosphorylation is the dominant mechanism controlling the interaction and subsequent ubiquitylation of substrates by F‐box proteins (Skaar et al, 2013), we first investigated how FBW7‐mediated degradation of SOX9 is regulated. Using a phospho‐specific SOX9 antibody, we showed that GSK3α/β phosphorylates T236 in SOX9 which resides within a conserved SOX9 CPD motif. The phosphorylation of SOX9 by GSK3 is a prerequisite for the FBW7 to interact, ubiquitylate, and degrade SOX9 in vivo and in vitro. Knockdown of individual FBW7 transcripts showed that SOX9 specifically accumulates upon depletion of the FBW7α splice form which is the primary isoform responsible for SOX9 degradation in cells.
In the brain, SOX9 is required for development and lineage commitment and it is essential for gliogenesis while blocking neural differentiation (Scott et al, 2010; Martini et al, 2013). Consistent with its function in maintaining neural stem cells downstream of SHH, we previously reported that SOX9 expression is high in SHH/WNT medulloblastoma subtypes (Swartling et al, 2012). Recent large‐scale sequencing efforts providing a comprehensive map of mutations in medulloblastoma defined this brain tumor as a heterogenous malignancy (Northcott et al, 2012). Here, we examined the relationship between FBW7 and SOX9 in medulloblastoma in detail. We identify recurrent FBW7 gene mutations in the adult SHH medulloblastoma subgroup and demonstrate that FBW7 expression is frequently downregulated in the other medulloblastoma patient subgroups. Importantly, we find that when FBW7 is functionally inactivated by mutation or reduced expression, SOX9 protein is upregulated demonstrating their inverse association in clinical samples and tumor‐derived cell lines. In line with the notion that inactivation of FBW7 contributes to medulloblastoma malignancy through deregulation of SOX9, we demonstrate the requirement of SOX9 for tumor migration and metastasis and, further, that forced expression of FBW7α attenuates these SOX9‐driven processes. Metastasis at diagnosis correlates with tumors with adverse outcome and characterizes medulloblastoma into a high‐risk category (Zeltzer et al, 1999). The findings that FBW7 is transcriptionally downregulated and mutated in medulloblastoma and that elevated SOX9 levels correlate with poor outcome, tumor cell dissemination, and metastasis in clinical samples and brain tumor xenograft models suggest that SOX9 stabilization through loss of FBW7α could predict metastasis and may be used as a biomarker for medulloblastoma aggressiveness. Interestingly, SOX9 is frequently upregulated in many other malignancies, and recent reports reveal that deregulation of SOX9 promotes critical cancer processes by modulating the malignant transcriptome (Jo et al, 2014). Thus, it is likely that SOX9 stabilization through loss of FBW7 is a common feature also in other cancer types. Indeed, we found that FBW7 depletion stabilizes SOX9 in several different tumor‐derived cell lines of different origins. Noteworthy, Pine and coworkers also identified SOX9 as a target of FBW7 and found that SOX9 is actively degraded by FBW7 in response to DNA damaging agents in various cancer types (Hong et al, 2016).
Although not fully understood, SOX9 acts in concert with other oncogenic transcription factors and co‐factors involved in normal brain or brain cancer development, several of which directly or indirectly promote SOX9 expression, including Gli (Bagheri‐Fam et al, 2006). SOX9 is a downstream target of Gli1 and a positive‐feedback loop between Gli1 and SOX9 was shown in pancreatic ductal carcinoma (Deng et al, 2015), involving SOX9‐mediated degradation of β‐TrCP, another SCF ubiquitin ligase that targets Gli1 for proteasomal degradation (Deng et al, 2015). Thus, it is possible that functional inactivation of FBW7 and consecutive SOX9 stabilization may further promote Gli1 protein stability and SOX9/Gli1‐driven processes including stemness and self‐renewal properties. This is in agreement with our previous findings that SOX9 increases self‐renewal capacity of medulloblastoma‐initiating cells (MICs) (Swartling et al, 2012). Consistently, we have observed that forced expression of FBW7 suppresses the formation of SOX9‐induced MIC spheres in culture (unpublished data). Moreover, SOX9 is a direct target of Notch1 and mediates Notch‐induced mesenchymal features in lung adenocarcinoma (Capaccione et al, 2014) and neural lineage determination (Martini et al, 2013). Although our data suggest that defective SOX9 degradation directly promotes medulloblastoma pathogenesis, at this stage we cannot exclude the possibility that other FBW7 target substrates such as Notch1 and c‐Jun may also contribute to medulloblastoma progression. For instance, deletion of FBW7 in the mouse brain has been shown to impair neural stem cell differentiation and increase progenitor cell death, phenotypes attributed to deficient degradation of the FBW7 target substrates Notch1 and c‐Jun, respectively (Hoeck et al, 2010). Activated Notch1 can induce SHH‐driven medulloblastoma when overexpressed in p53‐deficient mice (Natarajan et al, 2013); however, Notch2 seems to maintain these tumors and is also the Notch member elevated in primary human medulloblastoma (Fan et al, 2004).
To assess the physiological significance of altered SOX9 stability in medulloblastoma, we studied how SOX9‐WT or the FBW7‐insensitive mutant influenced the transcriptional profiles in medulloblastoma MB002 cells. Strikingly, we found that the genes differentially expressed and upregulated in SOX9‐expressing cells and between the SOX9‐WT and the T236/240A mutant overlap significantly with previously known hallmark EMT factors. Furthermore, gene set enrichment analyses demonstrated that the transcriptional changes correlated significantly with gene signatures found to be upregulated in cancer metastasis and EMT processes. One of the genes, palladin, is further known to promote the expression of vimentin (Brentnall et al, 2012) that was also found to be elevated in medulloblastoma cells expressing the stable SOX9 mutant. Interestingly, AKT drives cell migration and invasion in part by phosphorylating both palladin and vimentin (Xue & Hemmings, 2013). Whether SOX9 is actively involved in regulating this process is currently unknown. Importantly, among the genes differentially regulated between the SOX9‐WT and stable SOX9 mutant, a significant number of previously reported SOX9 targets genes were found, including a majority of which have previously been reported as pro‐metastatic genes, such as SNAI2/SLUG. SOX9 has been reported to work in concert with SNAI2/SLUG and trigger an auto‐regulatory gene expression program involved in inducing and sustaining the mammary stem cell state (Guo et al, 2012). A more direct relationship between SNAI2/SLUG and SOX9 was recently found in human lung carcinoma (Luanpitpong et al, 2015), where SLUG and SOX9 were shown to interact in the cytoplasm, and upon SNAI2/SLUG depletion, SOX9 was degraded. However, the ubiquitin ligase responsible for SOX9 degradation was not identified in this study. In our profiling studies, SNAI2/SLUG was found to be rapidly upregulated in response to SOX9 induction.
One of the major findings in this study is that the GSK3‐SOX9‐FBW7 regulatory axis is controlled by the PI3K/AKT/mTOR signaling cascade. The PI3K/AKT/mTOR pathway bridges many signaling pathways downstream of receptor tyrosine kinases (RTKs) and controls various critical processes in cancer progression (Fruman & Rommel, 2014). Inclusion of drugs targeting this pathway was recently suggested to be an option for combination therapies in several malignancies including medulloblastoma (Kool et al, 2014). Importantly, our data show that targeting the PI3K/AKT/mTOR pathway destabilizes SOX9 in medulloblastoma cells with functional FBW7, and thus further sensitizes tumor cells to the chemotherapeutic drug cisplatin. A correlation between loss or reduced FBW7 expression and cisplatin resistance through EMT has previously been reported in non‐small‐cell lung cancer cell lines (Yu et al, 2013). Here, we found that increased or stabilized SOX9 protein reduced cisplatin treatment efficacy in medulloblastoma, both in vitro and in vivo. Related to this process, we identified multiple genes associated with cisplatin resistance that may be directly controlled by SOX9, including ATP7A and DUSP2. Importantly, SOX9‐induced changes in expression of ATP7A and DUSP2 were found to be reversed following destabilization of SOX9 by targeting the PI3K/AKT/mTOR signaling pathway with pharmacological compounds. In the future, it will be important to explore whether SOX9 can be destabilized in vivo by drugs targeting PI3K/AKT/mTOR activity and whether medulloblastoma patients with functional FBW7 are more sensitive to drugs targeting this pathway.
In summary, as illustrated in Fig 8, our study of SOX9 regulation by FBW7 provides insights into how loss of FBW7 might contribute to medulloblastoma malignancy by promoting SOX9‐driven migration, metastasis, and drug resistance. These findings uncover a new molecular mechanism for the post‐translational regulation of SOX9 stability through the GSK3‐SOX9‐FBW7 regulatory axis that opens up new strategies for targeting SOX9 in malignant brain tumors. For example, the use of selective inhibitors targeting PI3K/AKT/mTOR signaling could be a potential point of intervention for patients with recurrent, chemoresistant medulloblastoma with functional FBW7. In addition, development of specific SOX9 inhibitors, or compounds attenuating SOX9 expression, may also potentially be used for selective killing of FBW7‐deficient, metastasis‐prone, and drug‐resistant medulloblastoma cells.
Materials and Methods
Cell culture
Daoy medulloblastoma cell lines were obtained from the American Type Culture Collection. Cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% FBS (Gibco) and 1% L‐glutamine (Gibco).
MB002 cells have been described previously as derivative of primary medulloblastoma tumor with large‐cell histology and gene expression markers consistent with Group 3 medulloblastoma (Bandopadhayay et al, 2014). MB002 cells were maintained in culture media made up of 1:1 (v/v) DMEM/F‐12 (Gibco) and Neurobasal‐A media (Gibco, #10888‐022) supplemented with HEPES, sodium pyruvate, non‐essential amino acid, L‐glutamine, B27 (Gibco, #12587‐10), 20 ng/ml EGF (Peprotech; #AF‐100‐15), 20 ng/ml basic fibroblast growth factor (Peprotech; #AF‐100‐18b), heparin (07980; Stem Cell), and 10 ng/ml leukemia inhibitory factor (LIF; LIF1010; Millipore). Medulloblastoma‐initiating cells (MICs) derived from postnatal, day 0, mouse cerebellar neural stem cells—transduced with MYCNT58A alone or those which were further transduced with SOX9 (MIC‐SOX9) from previously published study (Swartling et al, 2012)—were cultured in Neurobasal‐A media (Gibco, #10888‐022) supplemented with L‐glutamine, B27 (Gibco, #12587‐10), 20 ng/ml EGF (Peprotech; #AF‐100‐15), and 20 ng/ml basic fibroblast growth factor (Peprotech; #AF‐100‐18b).
Lentiviruses and generation of stable cell lines
To generate stable doxycycline‐inducible FBW7α, SOX9wt, and SOX9‐T236/240A cell lines, lentiviruses were first generated in HEK293T cells by packing the specified pTRIPZ lentivector with the packaging plasmids, psPAX2 and pMD2.G. Supernatants containing viral particles (48–96 h post‐transfection) were pooled and used to infect the target cells in the presence of freshly added hexadimethrine bromide (8 mg/ml). Seventy‐two hours post‐infection, the transduced cells were subsequently selected and maintained in their appropriate culture medium in the presence of puromycin (0.125–0.5 μg/ml).
Drug treatment and cell viability assay
cis‐diammineplatinum (II) chloride (cisplatin) was obtained from Sigma‐Aldrich, while small‐molecule inhibitors of PI3K and/or mTOR including AZD5363, AZD8186, AZD2014, KU‐0063794, and BEZ‐235 were purchased from Selleckchem. For assessment of cell viability, the cells were treated with doxycycline when appropriate, counted, and seeded into 96‐well plate for 24 h prior to drug treatment. Following 72–120 h of drug treatment, cell viabilities were measured by adding alamarBlue reagent (Life Technologies) into each well of 96‐well plate.
Immunoprecipitation, SDS–PAGE, and Western blotting
For biochemical analyses of protein interactions and levels, the cells were lysed in NP‐40 lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% NP‐40) supplemented with both protease (Complete Mini; Roche) and phosphatase (PhosSTOP; Roche) inhibitors. Immunoprecipitation of protein complexes from cell lysate was performed using 1 μg of antibodies (rabbit anti‐FLAG for exogenous FLAG‐FBW7α, or rabbit anti‐SOX9 or mouse anti‐HA for endogenous and exogenous SOX9, respectively), which subsequently was immobilized with Sepharose G beads (GE Life Science) and eluted under reducing–denaturing condition in SDS sample buffer at 95°C for 5 min. Proteins were separated by SDS–PAGE, blotted onto PVDF membrane, and detected by immunoblotting using the appropriate antibodies. The list of antibodies and their sources can be found in the Appendix.
Generation of antibodies against SOX9 pT236&240
Rabbit polyclonal antibody specific against SOX9 pT236&240 was produced by Innovagen AB (Lund, Sweden; Project # 92528). Synthetic phospho‐peptide (CQSQGPP(pT)PPT(pT)PKTDV‐NH2) identical to the phosphorylated SOX9 at Thr236&240 was used as an antigen to immunize New Zealand white rabbit. Following four rounds of immunization, sera were collected and subjected to peptide affinity purification against the non‐phosphorylated peptide (CQSQGPPTPPTTPKTDV‐NH2) to acquire the SOX9 pT236&240‐specific antibody.
In vitro kinase and binding assay
Full length SOX9‐WT and SOX9‐T236&240A was synthesized in vitro using TnT T7 Quick Coupled Transcription/Translation kit from Promega (L1170) according to the manufacturer's instructions. Following the synthesis, the proteins were immunoprecipitated and immobilized on Sepharose beads prior to being used for either in vitro kinase or binding assay. For the kinase assay, the immobilized SOX9 proteins were incubated with a complete GSK3 reaction buffer for 90 min at 37°C. Complete GSK3 reaction buffer consists of 50 mM Tris–HCl pH 7.4, 1 mM EGTA, 150 mM NaCl, 0.1% β‐mercaptoethanol, 2 mM ATP, and 1 U of recombinant, active human GSK3β (Abcam, AB60863). For in vitro binding assay, Sepharose bead‐immobilized full‐length SOX9 protein or synthetic peptide was resuspended in 50 mM Tris–HCl pH 7.4 and 150 mM NaCl buffer in the presence of recombinant, active SCFFBW7 complex (Millipore, 23‐030) under constant gentle agitation at 37°C for 1 h. Following the incubation, the supernatant was aspirated and the beads were washed three times with NP‐40 lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% NP‐40) prior to elution under reducing–denaturing condition in SDS sample buffer at 95°C for 5 min for SDS–PAGE.
Ubiquitylation assay
For in vivo ubiquitylation assay, cells were transfected with the indicated plasmids and/or siRNA (siFBW7 for knockdown experiment) and treated with MG132 (20 μM) for 4 h prior to harvest and extraction in 1% SDS and 10 mM N‐ethylmaleimide (Sigma) at 95°C for 10 min. Protein extracts were subsequently sonicated and diluted ten times in NP‐40 lysis buffer followed by immunoprecipitation of total SOX9 protein and gel electrophoresis. For in vitro ubiquitylation assay, Sepharose bead‐immobilized full‐length SOX9 protein was resuspended in ubiquitylation reaction buffer (25 mM Tris–HCl pH 7.6; 5 mM MgCl2; 100 mM NaCl) containing 2 mM ATP, 1 mM DTT, 200 nM UbE1 (Boston Biochem, E‐305), 500 nM His6‐UbcH3 (Boston Biochem, E2‐610), 0.5 μg recombinant, active SCFFBW7 complex (Millipore, 23‐030), and 5 μg ubiquitin (Boston Biochem, E2‐100H). In vitro reactions were performed under gentle agitation at 37°C for 2 h prior to termination of reaction with the addition of 10 mM N‐ethylmaleimide. The beads were subsequently washed three times with 50 mM HEPES pH 7.5/150 mM NaCl containing 10% glycerol, 1% Triton X‐100, 1 mM EDTA, and 1 mM EGTA prior to being eluted in SDS sample buffer for SDS–PAGE.
In vitro cell migration assay
Cell migration was performed using Transwell inserts (5‐μm pores, Millipore, ECM220) according to the manufacturer's instructions. Briefly, 200,000 cells suspended in serum‐free medium were added into Transwell insert and were let to migrate through the pores of the Transwell membrane toward the external laminin or fibronectin coating for 4 h. Cells that migrated were fixed and stained with 0.5% crystal violet, from which images were taken. For quantitative analysis, the stained cells were re‐solubilized in 10% acetic acid solution, from which the crystal violet absorbance was measured at 590 nm. For each independent experiment, two replicates per condition were used.
Immunohistochemistry and RNAscope assay on tissue sections and TMAs
Tissue microarrays (TMAs) were generated as previously described (Korshunov et al, 2010). Brain tumors were isolated, dehydrated, embedded in paraffin, and immunostained together with the TMAs as previously reported (Swartling et al, 2012) using ABC kits from Vector Labs. Antibodies against SOX9 (Millipore), Ki67 (Abcam) and STEM121 (TaKaRa) were used in 1:500, 1:2,000 and 1:1,000 dilution, respectively. RNAscope experiments were performed on TMA sections according to the manufacturer's instructions (Advanced Cell Diagnostics) using probes against human SOX9 (ACD‐300031‐C2) and FBW7 (ACD‐300031‐C3). During the sectioning of the TMAs, some of the tumor samples were lost and some of the samples had less than 10% of the tissue left. These were not scored and are marked as N/A (not available) in the Source Data for Fig 3. The samples in the TMAs were judged one by one and for each channel separately to avoid selection bias.
Under the microscope, specific RNA signals appear as discrete, bright fluorescent spots in or around the DAPI‐stained nuclei (Fig EV3D). To score “RNA intensity” in each tumor specimen, we quantified the number of RNA spots in each positive cell (indicating the relative number of RNA molecules per cell). The total number of RNA fluorescent spots was manually counted from at least three different image fields by two independent, blinded individuals, followed by adjustment to the number of DAPI‐stained and RNA fluorescent signal‐positive nuclei. RNA intensity was scored 0 for negative, 1 for low (< 3 spots/cell) to 3 (more than 10 spots/cell) accordingly. Similarly, the “percentage of tumor cells expressing the RNA” was analyzed by estimating the proportion of DAPI‐stained nuclei with a positive RNA fluorescent signal in a given field (i.e., 0–100%). The average percentage (or RNA positivity) was calculated and likewise converted to scores: 0 for negative, 1 for low (< 20%), two for intermediate (20–80%), and three for high (more than 80%). To grade the overall RNA expression in a given tumor specimen, the “RNA intensity” score was multiplied with the score of the proportion of RNA‐positive cells, generating the possible expression scores of 0, 1, 2, 3, 4, 6, or 9. In order to ensure linear distribution, the resulting multiplication scores were converted to “ranks” from 1 to 7, respectively. This assessment is illustrated in detail in sample images in Fig EV3D.
Similar to RNA, an overall SOX9 protein score was calculated and converted to ranks (Fig EV3D). Finally, to analyze the correlation between SOX9 and FBW7, “SOX9 protein/RNA” ratio was used to account for variability in the levels of SOX9 transcript across different medulloblastoma molecular subgroups. It is known that the WNT and the SHH subgroups have elevated levels of SOX9 mRNA transcript (Swartling et al, 2012), probably as a consequence of activation of the WNT or SHH pathway. Therefore, the calculated SOX9 protein and RNA scores were first translated to rank (1–7; see Table 1), and subsequently used to obtain SOX9 protein/RNA. Details on scoring and ranking for SOX9 and FBW7 are exemplified in Fig EV3D with circles showing RNA spots and arrows pointing to SOX9 protein‐positive cells.
Table 1.
Conversion of the final scores of protein or mRNA expression into RANK
| SCORE | 0 | 1 | 2 | 3 | 4 | 6 | 9 |
| RANK | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
The correlation between ranked FBW7 mRNA expression and SOX9 protein or the SOX9 protein/mRNA ratio, respectively, was investigated using Kendall's rank correlation coefficient τB. The values of τ and p depicted in Fig 3F indicate the correlation coefficient and P‐value of the comparison, respectively.
RNA‐Seq preprocessing
MB002 empty vector, MB002 SOX9‐WT, and MB002 SOX9‐T236/240A cells were treated with 100 ng/ml doxycycline in triplicate for 8 h. Cells were lysed in TRIzol (Life Technologies) and RNA was prepared using Qiagen RNeasy Mini Kit (Qiagen) according to the manufacturer's recommendations. Sequencing of the RNA extracted from MB002 empty vector, MB002 SOX9‐WT, and MB002 SOX9‐T236/240A cells was conducted using the Ion Proton™ System for Next‐Generation Sequencing and performed at NGI, Science for Life Laboratory, Uppsala Biomedicinska Centrum (BMC), Sweden. All RNA sequence reads were mapped against the human genome assembly GRCh37/hg19 (Lander et al, 2001) using STAR v2.4.0 (Dobin et al, 2013). The alignment was performed with a two‐pass approach, where splice junctions discovered during a first alignment guided the forming of a second, final alignment (Veeneman et al, 2016). Unmapped reads were realigned using Bowtie2 v2.2.3 (Langmead & Salzberg, 2012) before duplicate reads were removed with Piccard v1.127 (Guo et al, 2012). The transcriptomes were then assembled and expression profiles quantified with Cufflinks v2.2.1 (Trapnell et al, 2010). Comparisons of transcriptomes and their expression levels were performed using Cuffcompare and Cuffdiff (Trapnell et al, 2013). Both the alignment and the quantification utilized RefSeq data downloaded from UCSC (Karolchik et al, 2004).
Differential expression analysis
The differential expression results produced by Cuffdiff were subsequently processed using the R package CummeRbund (Goff et al, 2013). Fold changes (FCs) were recalculated for each transcript after setting all expression values equal to FPKM+1 to avoid infinity results. Transcripts were considered significantly differentially expressed, if the recalculated FC > 1.5 and the adjusted P‐value p < 0.05.
Gene set overlap and gene set enrichment analysis
Gene set overlap analyses (GSO) were performed using the respective tool on the Gene Set Enrichment Analysis webpage (http://software.broadinstitute.org/gsea/index.jsp) using the predefined gene set databases (Subramanian et al, 2005). Gene set enrichment analyses (GSEAs) were performed on the GenePattern server (Reich et al, 2006) using the respective module and using either precompiled gene set databases or gene sets manually downloaded from the GSEA webpage above. Statistical significance in the GSEAs was performed using permutations of the gene sets rather than samples.
Medulloblastoma subgroup classification
Classification of RNA‐Seq‐derived transcription profiles into the four MB molecular subgroups (WNT, SHH, G3, G4) was performed using the metagene code for cross‐platform, cross‐species projection of transcription profiles (Tamayo et al, 2007). Raw CEL files for a total of 103 MB samples with known subgroup affiliation (eight WNT cases, 33 SHH cases, 27 Group 3 cases, and 35 Group 4 cases) (Northcott et al, 2011) were downloaded from ArrayExpress (E‐GEOD‐21140). CEL files were processed in the Affymetrix Expression Console using the Robust Multichip Average (RMA) method for background correction, quantile normalization, and probe summarization (Bolstad et al, 2003). To facilitate comparability between RNA‐Seq and microarray identifiers, all gene names were translated using the HUGO Gene Nomenclature Committee (Gray et al, 2015). The expression profiles of the 103 MB samples were used to establish metagene classification signatures based on genes, which exhibited a FC > 5 and an absolute difference in expression of at least 600 between the means of any two subgroups. Subgroup affiliation of MB002 empty vector, MB002 SOX9‐WT, and MB002 SOX9‐T236/240R cells was then determined by a hierarchical clustering of the MB002 cell lines together with MB samples using the expression of metagenes. The result of the clustering was additionally verified by a Support Vector Machine (SVM) classification (data not shown).
Comparison to putative SOX9 target genes
Putative SOX9 target genes identified from three murine SOX9 transcription factor ChIP studies were downloaded (Kadaja et al, 2014; Oh et al, 2014; Larsimont et al, 2015) and matched to human orthologs using the BioMart software tool (http://www.ensembl.org/biomart/martview) and human gene names translated using the HUGO Gene Nomenclature Committee (Gray et al, 2015). All three studies investigated expression of putative target genes in the presence as well as conditional ablation/deletion of SOX9, producing a list of target genes proposedly activated or repressed by SOX9. After mapping of orthologs and gene name translation, the study by Larsimont et al resulted in a list of 259 genes putatively activated by SOX9 and 201 genes putatively repressed by SOX9. From the study by Kadaja et al, 60 genes activated by and 12 genes repressed by SOX9 were extracted, while the study by Oh et al proposed 180 genes activated by and six genes repressed by SOX9. Putative target genes activated by SOX9 were mapped to genes significantly upregulated in SOX9‐T236/240A as compared to WT, while target genes putatively repressed by SOX9 were mapped to genes significantly downregulated in SOX9‐T236/240A as compared to SOX9‐WT.
Quantitative real‐time PCR (qRT–PCR)
To perform the qRT–PCR, RNA was first isolated using TRIzol reagent (Invitrogen, Life Technologies), followed by RNeasy Mini Kit (Qiagen). A total of 50 ng of the purified RNA was then used for cDNA synthesis (SuperScript VILO cDNA Synthesis Kit; Invitrogen, Life Technologies). Before preparing the PCR, the cDNA was diluted ten times and 4 μl was used for each PCR. Quantitative PCRs were carried out using SYBR Green or TaqMan probes as described in the Appendix. The sequence for each primer and probe can be found in the Appendix. The data were analyzed with ΔΔCt method and presented as relative expression.
Mice xenograft and cisplatin treatment
Tumor formation was monitored following stereotactic injection of 105 medulloblastoma cells. Tumor cells were injected into the cerebellum of 6‐ to 9‐week‐old athymic nude Foxn1 nu mice (obtained from Harlan Laboratories) as previously described (Swartling et al, 2012). All animal experiments were performed in accordance with local animal ethics committees. One week after transplantation, mice were treated intraperitoneally with cisplatin (2 mg/kg in 10% β‐HP‐cyclodextrin; Sigma‐Aldrich) or vehicle (10% β‐HP‐cyclodextrin), for a total of six doses (administered every other day). Experimental endpoint for the treatment study was 80 days.
Statistical analysis
Data are presented as mean ± SD from two or more independent experiments. Statistical analyses were performed using the statistical package GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA; http://www.graphpad.com) using either log‐rank (Mantel–Cox) analysis, one‐way analysis of variance (ANOVA), or two‐way analysis of variance (ANOVA) with Bonferroni's post hoc test when appropriate. Correlation analyses were performed in MATLAB (MATLAB and Statistics Toolbox Release 2015b, The MathWorks, Inc., Natick, Massachusetts, USA) using either the Spearman rank correlation coefficient or Kendall rank correlation coefficient. Significance of overlap analyses between gene sets was determined in MATLAB using Fisher's exact test. Survival analyses were performed in the R2: Genomics analysis and visualization platform using the implemented LogRank test.
Data availability
Primary data
Kool et al (2014); for gene expression values, GSE49243.
The MB002 RNA‐Seq profiling data are available from NCBI with an access record of GSE84320.
Referenced data
Information about putative SOX9 target genes was obtained directly from the cited articles and/or the respective supplementary information (Kadaja et al, 2014; Oh et al, 2014; Larsimont et al, 2015). Raw CEL files with MB expression data from Northcott et al (2011) were downloaded from ArrayExpress (E‐GEOD‐21140).
Author contributions
The overall study was conceived and designed by ASR, FJS, and OS, with important contributions from VS. ASR and AB performed the majority of the in vitro experiments. SB prepared tumor samples for RNA‐Seq and planned and performed in vivo studies and experimental treatments. SB and GR performed immunohistochemistry and subsequent analysis. GR and VS performed and analyzed RNAscope assays on TMAs. HW analyzed the RNA‐Seq data and performed the majority of bioinformatic analysis including clustering and GSEA of gene with important help from AS. HW, DTWJ, and MK performed and analyzed genes and protein expressions in the clinical cohorts. Y‐JC, SMP, AK and MDT provided human medulloblastoma cohorts and tissue microarrays. AM designed primers and performed qPCR experiments. ASR, AM, MČ, DK, SH and CS generated various plasmid constructs and retroviruses used in this study. SH and AS provided technical support. ASR, VS, and HW analyzed the data and performed statistical analyses. ASR, VS, FJS, and OS wrote the manuscript with significant contributions from CS, SMP, and MK.
Conflict of interest
O Sangfelt and C Spruck report receiving a research grant from Astra Zeneca for a separate project. The other authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
Source Data for Figure 3
Acknowledgements
This work was supported by research grants from the Swedish Childhood Cancer Foundation (FJS, OS, ASR), the Swedish Cancer Society (FJS, OS), the Swedish Research Council (FJS, OS), the European Research Council under Horizon 2020 (Project No. 640275, Medulloblastoma ‐ ERC‐2014‐STG) (FJS), the Ragnar Söderberg Foundation (FJS), the Swedish Society of Medicine (FJS), the Åke Wiberg Foundation (FJS), Science for Life Laboratory (FJS, OS), Åke Olssons Stiftelse (OS), Radiumhemmets Forskningsfonder (OS, ASR), Karolinska Institute Foundations (OS, AB), and Worldwide Cancer Research (FJS). We thank Bert Vogelstein (Johns Hopkins University) for providing HCT116 FBXW7 KO/WT cells and Peter Koopman (Institute for Molecular Bioscience, University of Queensland) for providing pcDNA3.1 HA‐SOX9 plasmid. We also acknowledge technical support from the National Genomics Infrastructure (NGI)/Uppsala Genome Center and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. Work performed at NGI/Uppsala Genome Center has been funded by RFI/VR and the Science for Life Laboratory, Sweden. We further acknowledge support with imaging from the BioVis facility at the Science for Life Laboratory, Uppsala University, Sweden, and Janne Lehtiö for proteomic analysis at the Science for Life Laboratory, Karolinska Institute, Stockholm, Sweden.
The EMBO Journal (2016) 35: 2192–2212
Contributor Information
Fredrik J Swartling, Email: fredrik.swartling@igp.uu.se.
Olle Sangfelt, Email: olle.sangfelt@ki.se.
References
- Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, Fuchs E (2015) Pioneer factors govern super‐enhancer dynamics in stem cell plasticity and lineage choice. Nature 521: 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller‐Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P et al (2007) FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 67: 9006–9012 [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kamitani T, Yang X, Kandyil R, Bridgewater LC, Fellous M, Mori‐Akiyama Y, de Crombrugghe B (2005) The transcription factor Sox9 is degraded by the ubiquitin‐proteasome system and stabilized by a mutation in a ubiquitin‐target site. Matrix Biol 23: 499–505 [DOI] [PubMed] [Google Scholar]
- Arabi A, Ullah K, Branca RM, Johansson J, Bandarra D, Haneklaus M, Fu J, Aries I, Nilsson P, Den Boer ML, Pokrovskaja K, Grander D, Xiao G, Rocha S, Lehtio J, Sangfelt O (2012) Proteomic screen reveals Fbw7 as a modulator of the NF‐kappaB pathway. Nat Commun 3: 976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri‐Fam S, Barrionuevo F, Dohrmann U, Gunther T, Schule R, Kemler R, Mallo M, Kanzler B, Scherer G (2006) Long‐range upstream and downstream enhancers control distinct subsets of the complex spatiotemporal Sox9 expression pattern. Dev Biol 291: 382–397 [DOI] [PubMed] [Google Scholar]
- Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, Yu F, Vue N, Gibson WJ, Paolella BR, Mitra SS, Cheshier SH, Qi J, Liu KW, Wechsler‐Reya R, Weiss WA et al (2014) BET bromodomain inhibition of MYC‐amplified medulloblastoma. Clin Cancer Res 20: 912–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R (2012) Arousal of cancer‐associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One 7: e30219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, Yu Q, Ostrom L, Fordjour P, Anderson DL, Monahan JE, Kelleher JF, Peukert S, Pan S, Wu X, Maira SM et al (2010) Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med 2: 51ra70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaccione KM, Hong X, Morgan KM, Liu W, Bishop JM, Liu L, Markert E, Deen M, Minerowicz C, Bertino JR, Allen T, Pine SR (2014) Sox9 mediates Notch1‐induced mesenchymal features in lung adenocarcinoma. Oncotarget 5: 3636–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CC, Uchiyama Y, Hiyama A, Gajghate S, Shapiro IM, Risbud MV (2009) PI3K/AKT regulates aggrecan gene expression by modulating Sox9 expression and activity in nucleus pulposus cells of the intervertebral disc. J Cell Physiol 221: 668–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of Glycogen‐Synthase Kinase‐3 by Insulin‐Mediated by Protein‐Kinase‐B. Nature 378: 785–789 [DOI] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, Clurman BE (2014) Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26: 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Vanderbilt DB, Lin CC, Martin KH, Brundage KM, Ruppert JM (2015) SOX9 inhibits beta‐TrCP‐mediated protein degradation to promote nuclear GLI1 expression and cancer stem cell properties. J Cell Sci 128: 1123–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Drogen F, Sangfelt O, Malyukova A, Matskova L, Yeh E, Means AR, Reed SI (2006) Ubiquitylation of cyclin E requires the sequential function of SCF complexes containing distinct hCdc4 isoforms. Mol Cell 23: 37–48 [DOI] [PubMed] [Google Scholar]
- Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D, Brat DJ, Perry A, Eberhart CG (2004) Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res 64: 7787–7793 [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ (2015) COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43: D805–D811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13: 140–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G (2012) Molecular mechanisms of cisplatin resistance. Oncogene 31: 1869–1883 [DOI] [PubMed] [Google Scholar]
- Goff L, Trapnell C, Kelley D (2013) CummeRbund: Analysis, exploration, manipulation, and visualization of Cufflinks high‐throughput sequencing data. R package version 2.14.0
- Gottardo NG, Gajjar A (2006) Current therapy for medulloblastoma. Curr Treat Options Neurol 8: 319–334 [DOI] [PubMed] [Google Scholar]
- Gray KA, Yates B, Seal RL, Wright MW, Bruford EA (2015) Genenames.org: the HGNC resources in 2015. Nucleic Acids Res 43: D1079–D1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer‐Hardi U, Bell G, Tam WL, Mani SA, van Oudenaarden A, Weinberg RA (2012) Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148: 1015–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T, Kishino T, Stephen S, Eberspaecher H, Maki S, Takigawa M, de Crombrugghe B, Yasuda H (2013) E6‐AP/UBE3A protein acts as a ubiquitin ligase toward SOX9 protein. J Biol Chem 288: 35138–35148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hede SM, Savov V, Weishaupt H, Sangfelt O, Swartling FJ (2014) Oncoprotein stabilization in brain tumors. Oncogene 33: 4709–4721 [DOI] [PubMed] [Google Scholar]
- Hoeck JD, Jandke A, Blake SM, Nye E, Spencer‐Dene B, Brandner S, Behrens A (2010) Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c‐Jun. Nat Neurosci 13: 1365–1372 [DOI] [PubMed] [Google Scholar]
- Hong X, Liu W, Song R, Shah JJ, Feng X, Tsang CK, Morgan KM, Bunting SF, Inuzuka H, Zheng XFS, Shen Z, Sabaawy HE, Liu LX, Pine SR (2016) SOX9 is targeted for proteasomal degradation by the E3 ligase FBW7 in response to DNA damage. Nucleic Acids Res doi:10.1093/nar/gkw748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhou X, Lefebvre V, de Crombrugghe B (2000) Phosphorylation of SOX9 by cyclic AMP‐dependent protein kinase A enhances SOX9's ability to transactivate a Col2a1 chondrocyte‐specific enhancer. Mol Cell Biol 20: 4149–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo A, Denduluri S, Zhang B, Wang Z, Yin L, Yan Z, Kang R, Shi LL, Mok J, Lee MJ, Haydon RC (2014) The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis 1: 149–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaja M, Keyes BE, Lin M, Pasolli HA, Genander M, Polak L, Stokes N, Zheng D, Fuchs E (2014) SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev 28: 328–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32: D493–D496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, Milde T, Bourdeaut F, Ryzhova M, Sturm D, Pfaff E, Stark S, Hutter S, Seker‐Cin H, Johann P, Bender S et al (2014) Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell 25: 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP 4th, Pan FC, Akiyama H, Wright CV, Jensen K, Hebrok M, Sander M (2012) Identification of Sox9‐dependent acinar‐to‐ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22: 737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H, Sturm D, Wittmann A, Schottler A, Felsberg J, Reifenberger G, Rutkowski S, Scheurlen W, Kulozik AE, von Deimling A, Lichter P, Pfister SM (2010) Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol 28: 3054–3060 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K et al (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsimont JC, Youssef KK, Sanchez‐Danes A, Sukumaran V, Defrance M, Delatte B, Liagre M, Baatsen P, Marine JC, Lippens S, Guerin C, Del Marmol V, Vanderwinden JM, Fuks F, Blanpain C (2015) Sox9 controls self‐renewal of oncogene targeted cells and links tumor initiation and invasion. Cell Stem Cell 17: 60–73 [DOI] [PubMed] [Google Scholar]
- Lin SC, Chien CW, Lee JC, Yeh YC, Hsu KF, Lai YY, Lin SC, Tsai SJ (2011) Suppression of dual‐specificity phosphatase‐2 by hypoxia increases chemoresistance and malignancy in human cancer cells. J Clin Invest 121: 1905–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luanpitpong S, Li J, Manke A, Brundage K, Ellis E, McLaughlin SL, Angsutararux P, Chanthra N, Voronkova M, Chen YC, Wang L, Chanvorachote P, Pei M, Issaragrisil S, Rojanasakul Y (2015) SLUG is required for SOX9 stabilization and functions to promote cancer stem cells and metastasis in human lung carcinoma. Oncogene 35: 2824–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini S, Bernoth K, Main H, Ortega GD, Lendahl U, Just U, Schwanbeck R (2013) A critical role for Sox9 in Notch‐induced astrogliogenesis and stem cell maintenance. Stem Cells 31: 741–751 [DOI] [PubMed] [Google Scholar]
- Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, Canamero M, Bujanda L, Schedl A, Cheah KS, Skotheim RI, Lothe RA, Lopez de Munain A, Briscoe J, Serrano M, Lovell‐Badge R (2012) Oncogenicity of the developmental transcription factor Sox9. Cancer Res 72: 1301–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FM, Ramaswamy V, Lindsay PE, Jelveh S, Donovan LK, Wang X, Luu B, Zayne K, Li Y, Mayoh C, Thiessen N, Mercier E, Mungall KL et al (2016) Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529: 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagato S, Nakagawa K, Harada H, Kohno S, Fujiwara H, Sekiguchi K, Ohue S, Iwata S, Ohnishi T (2005) Downregulation of laminin alpha4 chain expression inhibits glioma invasion in vitro and in vivo . Int J Cancer 117: 41–50 [DOI] [PubMed] [Google Scholar]
- Natarajan S, Li Y, Miller EE, Shih DJ, Taylor MD, Stearns TM, Bronson RT, Ackerman SL, Yoon JK, Yun K (2013) Notch1‐induced brain tumor models the sonic hedgehog subgroup of human medulloblastoma. Cancer Res 73: 5381–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29: 1408–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, Pfister SM (2012) Medulloblastomics: the end of the beginning. Nat Rev Cancer 12: 818–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh CD, Lu Y, Liang S, Mori‐Akiyama Y, Chen D, de Crombrugghe B, Yasuda H (2014) SOX9 regulates multiple genes in chondrocytes, including genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters. PLoS One 9: e107577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F (2003) Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112: 243–256 [DOI] [PubMed] [Google Scholar]
- Park MH, Min do S (2011) Quercetin‐induced downregulation of phospholipase D1 inhibits proliferation and invasion in U87 glioma cells. Biochem Biophys Res Commun 412: 710–715. [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006) GenePattern 2.0. Nat Genet 38: 500–501 [DOI] [PubMed] [Google Scholar]
- Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro MV, Booth S , Gao B, Cheah KS, Lovell‐Badge R, Briscoe J (2010) SOX9 induces and maintains neural stem cells. Nat Neurosci 13: 1181–1189 [DOI] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M (2013) Mechanisms and function of substrate recruitment by F‐box proteins. Nat Rev Mol Cell Biol 14: 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speranza MC, Frattini V, Pisati F, Kapetis D, Porrati P, Eoli M, Pellegatta S, Finocchiaro G (2012) NEDD9, a novel target of miR‐145, increases the invasiveness of glioblastoma. Oncotarget 3: 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M (2003) The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev 17: 1677–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartling FJ, Ferletta M, Kastemar M, Weiss WA, Westermark B (2009) Cyclic GMP‐dependent protein kinase II inhibits cell proliferation, Sox9 expression and Akt phosphorylation in human glioma cell lines. Oncogene 28: 3121–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, Grimmer MR, Lau J, Chesler L, Perry A, Phillips JJ, Taylor MD, Weiss WA (2012) Distinct neural stem cell populations give rise to disparate brain tumors in response to N‐MYC. Cancer Cell 21: 601–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo P, Scanfeld D, Ebert BL, Gillette MA, Roberts CW, Mesirov JP (2007) Metagene projection for cross‐platform, cross‐species characterization of global transcriptional states. Proc Natl Acad Sci USA 104: 5959–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Sangfelt O, Spruck C (2008) The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett 271: 1–12 [DOI] [PubMed] [Google Scholar]
- Taylor KM, Labonne C (2005) SoxE factors function equivalently during neural crest and inner ear development and their activity is regulated by SUMOylation. Dev Cell 9: 593–603 [DOI] [PubMed] [Google Scholar]
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123: 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I (2007) The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 204: 1825–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA‐seq. Nat Biotechnol 31: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeneman BA, Shukla S, Dhanasekaran SM, Chinnaiyan AM, Nesvizhskii AI (2016) Two‐pass alignment improves novel splice junction quantification. Bioinformatics 32: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y (2012a) RNAscope: a novel in situ RNA analysis platform for formalin‐fixed, paraffin‐embedded tissues. J Mol Diagn 14: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, Wei W (2012b) Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett 586: 1409–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8: 83–93 [DOI] [PubMed] [Google Scholar]
- Xue G, Hemmings BA (2013) PKB/Akt‐dependent regulation of cell motility. J Natl Cancer Inst 105: 393–404 [DOI] [PubMed] [Google Scholar]
- Yu HG, Wei W, Xia LH, Han WL, Zhao P, Wu SJ, Li WD, Chen W (2013) FBW7 upregulation enhances cisplatin cytotoxicity in non‐small cell lung cancer cells. Asian Pac J Cancer Prev 14: 6321–6326 [DOI] [PubMed] [Google Scholar]
- Zeltzer PM, Boyett JM, Finlay JL, Albright AL, Rorke LB, Milstein JM, Allen JC, Stevens KR, Stanley P, Li H, Wisoff JH, Geyer JR, McGuire‐Cullen P, Stehbens JA, Shurin SB, Packer RJ (1999) Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol 17: 832–845 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
Source Data for Figure 3
Data Availability Statement
Primary data
Kool et al (2014); for gene expression values, GSE49243.
The MB002 RNA‐Seq profiling data are available from NCBI with an access record of GSE84320.
Referenced data
Information about putative SOX9 target genes was obtained directly from the cited articles and/or the respective supplementary information (Kadaja et al, 2014; Oh et al, 2014; Larsimont et al, 2015). Raw CEL files with MB expression data from Northcott et al (2011) were downloaded from ArrayExpress (E‐GEOD‐21140).