

Abstract
Poor postnatal growth after preterm birth does not match the normal rapid growth in utero and is associated with preterm morbidities. Insulin‐like growth factor 1 (IGF‐1) axis is the major hormonal mediator of growth in utero, and levels of IGF‐1 are often very low after preterm birth. We reviewed the role of IGF‐1 in foetal development and the corresponding preterm perinatal period to highlight the potential clinical importance of IGF‐1 deficiency in preterm morbidities.
Conclusion
There is a rationale for clinical trials to evaluate the potential benefits of IGF‐1 replacement in very preterm infants.
Keywords: Development, Foetus, Insulin‐like growth factor 1, Metabolism, Preterm infant
Abbreviations
- BPD
Bronchopulmonary dysplasia
- IGF‐1
Insulin‐like growth factor 1
- IGFBP
Insulin‐like growth factor binding proteins
- MRI
Magnetic resonance imaging
- ROP
Retinopathy of prematurity
- VEGF
Vascular endothelial growth factor
Key notes.
Poor postnatal growth after preterm birth does not match the normal rapid growth in utero and is associated with preterm morbidities.
We reviewed the role of insulin‐like growth factor (IGF‐1) in foetal development and the corresponding preterm perinatal period to highlight its potential clinical importance in preterm morbidities.
There is a rationale for clinical trials to evaluate the potential benefits of IGF‐1 replacement in very preterm infants.
Introduction
Insulin‐like growth factor 1 (IGF‐1) is a 70‐amino acid single peptide mitogenic hormone, similar in structure to proinsulin, which stimulates systemic body growth in many different species. IGF‐1 is produced in most organs, but the liver is the main source of circulating IGF‐1 1, 2, 3. The IGF‐1/insulin axis is central to the control of metabolism, promoting growth and cell survival for almost every cell and organ in the body. Both insulin and IGF‐1 promote cellular uptake of glucose. Insulin, which has mainly metabolic functions, primarily targets organs that are involved in storing energy and are mediated by activation of the insulin receptor, such as adipose tissue, muscle and the liver. IGF‐1, which predominantly mediates growth and differentiation, initiates intracellular signalling through multiple pathways by binding to its specific tyrosine kinase receptor, the IGF‐1 receptor 4. The IGF‐1 receptor is present in many cell types in many tissues 5, including nerve and brain cells, vascular endothelial cells, skeletal muscle, lungs, cartilage, bones, liver, kidneys, skin and hematopoietic cells 6, 7, 8. Approximately 98% of IGF‐1 is bound to one of the seven binding proteins Insulin‐like growth factor binding proteins (IGFBP). IGFBP‐3, which is the most common, accounts for 80% of the IGFBP bound IGF‐1. In the circulation, IGF‐1/IGFBP‐3 may be bound into a ternary complex with an acid labile subunit 9.
In the perinatal period, the levels of IGF‐I and IGFBP‐3 are low and the levels of IGFBP‐1 are high, compared to older children and adults 10. At the same time, the acid labile subunit is often undetectable in foetal serum at 27 weeks of gestation 11. One of the acid labile subunit's main roles is the extension of IGF‐1 half‐life by protecting it from degradation and preventing the passage of IGF‐1 to the extravascular compartment. Hence, binding to proteins in the perinatal period is different than in older children and adults, as is IGF‐1 availability.
IGF‐1 function
Insulin‐like growth factor 1 plays a key role in maintaining homoeostasis, increasing progenitor cell potential and improving physiologic performance under both rest and stress conditions and is a potent inhibitor of programmed cell death 12, 13. In addition, IGF‐1 has insulin‐like effects and is involved in glucose and lipid metabolism 14. In the foetus, IGF‐1 and insulin, which promotes IGF‐1 production 14, rather than growth hormone is the main driver of growth 15. IGF‐1‐dependent growth is mediated by the glucose–insulin axis, which allows a rapid response to nutritional fluctuations 16, and concentrations of IGF‐1 normally rise throughout mid–late gestation to support the accelerated growth that normally occurs in the third trimester in utero 8, 16, 17.
IGF‐1 mechanism of action
Insulin‐like growth factor 1 regulates cell differentiation and proliferation through modulation of cellular DNA synthesis 12 and is one of the most potent natural activators of the protein kinase B (PKB or Akt) signalling pathway, which stimulates cell proliferation. A key IGF‐1 pathway is regulated by phosphatidylinositol‐3 kinase (PI3K) and its downstream partner, the mammalian target of rapamycin (mTOR), which upregulates Akt driving growth. IGF‐1 also binds to the insulin receptor at lower affinity than to the IGF‐1 receptor, to activate the insulin receptor at approximately 0.01 times the potency of insulin. Part of this signalling may be via IGF‐1 receptor/insulin receptor heterodimers 14.
IGF‐1 is a major growth hormone in the foetus
There is abundant genetic and experimental evidence suggesting that the IGF‐1 system is a very important endocrine determinant of foetal growth 18. Although IGF‐2 has some effect on foetal and placental development 19, 20, 21, direct measurements of IGF‐1 and IGF‐2 support the predominant role of IGF‐1 in foetal growth. Human foetal serum IGF‐1 concentrations in utero from 15 to 37 weeks of gestational age correlate with foetal weight and bone length 20, 21, 22, 23.
Umbilical cord IGF‐1 concentrations reflect foetal IGF‐1 levels at birth and correlate with birth weight. Cord serum IGF‐1 concentrations are lower at preterm birth and following intrauterine growth restriction. In contrast, serum IGF‐2 concentrations do not correlate with foetal length 22, 24. IGF‐1 is also detectable in many foetal tissues from the first trimester 25, 26, suggesting that IGF‐1 even plays a role in early foetal development. However, the increase in circulating IGF‐1 during the period of foetal life that corresponds to the third trimester, suggests that IGF‐1 is more important in the later months of foetal growth (Fig. 1).
Figure 1.
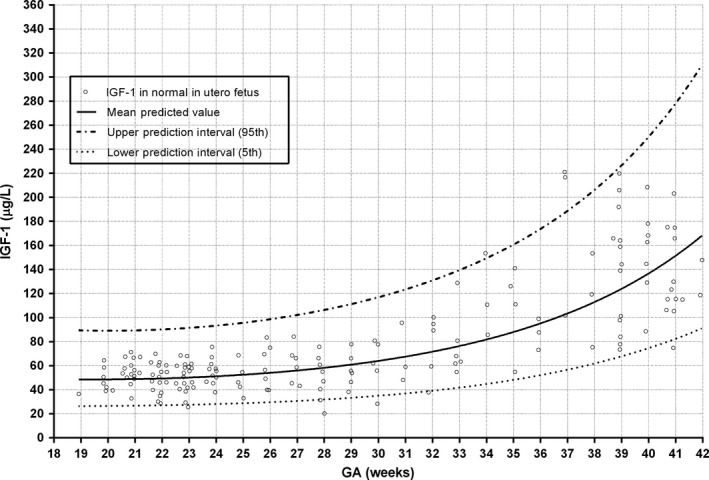
Mean foetal blood IGF‐1 concentrations (samples obtained by cordocentesis from normal pregnancies measured by RIA) double from 18 to 42 weeks of gestational age (GA) (n = 174) (213, 257). This figure combines data from Lasarre et al. and Bang et al. on IGF1 levels in utero.
Organ‐specific roles of IGF‐1 in the foetus
Animal studies of IGF‐1 in growth and metabolism
Extensive evidence for the growth promoting effects of IGF‐1 in utero comes from mice, with selected deletions of IGF‐1 and the IGF‐1 receptor. IGF‐1 and IGF‐1 receptor null mice are severely growth restricted (40–65%) 27, 28 and die shortly after birth due to respiratory failure from lack of lung and diaphragmatic muscle development. They also have generalised organ hypoplasia, including the skin, bones and brain, and exhibit neurologic deficits 28.
Manipulation of IGF‐1 production in various tissues indicates endocrine as well as autocrine/paracrine IGF‐1 effects on growth 29, 30. Laboratory studies show that IGF‐1 promotes glucose uptake in peripheral tissues 31, 32. This was demonstrated in a liver‐specific IGF‐1 deficient mouse model, which showed a 75% reduction in circulating IGF‐1 with insulin insensitivity in muscle. Treatment of these animals with IGF‐1 reduced hyperinsulinemia and improved insulin sensitivity, providing evidence for IGF‐1 as an important component of overall insulin action in peripheral tissues 33.
Human genetic studies of IGF‐1 and growth in utero
In humans, genetic defects of IGF‐1 and IGF‐1 receptor are rarely seen in viable infants, but four case reports of polymorphisms of the IGF‐1 gene in patients have described intrauterine growth restriction or being born small for gestational age as well as microcephaly, sensorineural deficits, developmental delay and metabolic abnormalities 34, 35. The severity of the foetal growth restriction is illustrated with birth weight standard deviation scores (SDS) ranging from −2.5 to −3.5 (Table 1) 36, 37, 38, 39, 40.
Table 1.
SDS scores of birth weight, length, head circumference and presence of microcephaly, developmental delay and adiposity in patients with various homozygous IGF‐1 defects 36, 37, 38, 39
| Woods et al. 37 | Bonapace et al. 38 | Walenkamp et al. 39 | Netchine et al. 36 | |
|---|---|---|---|---|
| Birth weight SDS | −3.9 | −4.0 | −2.5 | −2.5 |
| Birth length SDS | −5.4 | −6.5 | −3.0 | −3.7 |
| Birth head circ. SDS | −4.9 | −7.5 | −2.5 | |
| Microcephaly | Yes | Yes | Yes | |
| Developmental delay | Yesa | Yesb | Yesc | Yesd |
| Adiposity | No | Increased abdominal | ||
| Fat mass | No |
Moderately delayed motor development and behavioural difficulties, with hyperactivity and a short attention span.
Delayed psychomotor development and poor responses to sound were noted.
Severe mental retardation (IQ <40) and motor unrest.
Wechsler Intelligence Scale for Children III testing showed a 70–75 development quotient.
Evaluating growth hormone genetic defects also helps our understanding of the critical role of IGF‐1 versus growth hormone in foetal growth. In the full‐term infant, postnatal production of IGF‐1 is predominantly growth hormone dependent, but this is not the case in utero. Children with growth hormone deficiency or growth hormone insensitivity are normal size at birth. However, after birth, growth hormone axis defects lead to IGF‐1 deficiency and growth restriction. Children who are born preterm do not respond with increased growth‐to‐growth hormone in the perinatal period 8, 15.
IGF‐1/insulin regulation in the foetus and newborn
During foetal life, serum IGF‐1 concentration is regulated by the nutrient supply from the mother and the foetus is continuously supplied with a diet rich in carbohydrates and amino acids and low in fat 41, 42.
Energy is mainly produced by glycolysis, and the capacity to generate energy through more efficient oxidative phosphorylation of fat is poor. In the foetus, fatty acids are stored mainly in liver and adipose tissue instead 43. Immediately after birth, a brief period of starvation occurs before milk, which is high in fat and low in carbohydrates, is ingested.
The regulation of the growth hormone/IGF‐1/insulin axis in the foetus and the newborn infant is very different to that observed during childhood and adult life (Fig. 2A, B).
Figure 2.

(A) Describes the relationship between insulin and the GH/IGF‐1 axis in childhood and in (B) preterm newborn. GH, growth hormone; GHBP, growth hormone‐binding protein; GH receptor, growth hormone receptor; BP1, IGF binding protein 1; IGF‐1, insulin growth factor 1, BPs, IGF binding proteins, ALS, acid labile subunit, IGFR, IGF‐1 receptor.
In childhood, although hepatic IGF‐1 generation is driven by the growth hormone, insulin has a permissive role at the hepatic growth hormone receptor. Insulin also regulates hepatic production of IGFBP‐1, which is an inhibitor of IGF bioactivity. When nutrition is poor, reduced levels of portal insulin will lead to impaired IGF‐1 production and increased levels of IGFBP‐1. However, in the preterm newborn infant, hepatic IGF‐1 generation is not controlled by the growth hormone and nutrition and insulin play more important roles. The acid labile subunit levels are reduced, and this means that much of the circulating IGF‐1 is in binary complexes. Insulin through IGFBP‐1 plays an important role in regulating IGF‐1 bioactivity. Finally, there is evidence that in the newborn infant, IGF‐1, signalling through the IGF1 receptor, may have a role in maintaining pancreatic beta cell function 44.
The adult pattern of growth hormone regulation of hepatic IGF‐1 production is not present in the foetus and only emerges gradually in the term newborn infant, reflecting the appearance of growth hormone‐binding protein in the circulation. Full growth hormone regulation of IGF‐1 production is not established until the second year of postnatal life. The role of growth hormone in the foetus and newborn infant is not completely characterised and levels may be high in the preterm infant. Growth hormone may regulate metabolism through its effects on lipolysis and ketogenesis. It has been suggested that when growth hormone acts through nonhepatic growth hormone receptors, with or without tissue specific IGF‐1 generation, it may play a role in the early development of the brain, pancreas and other organs.
Insulin is the primary regulator of hepatic IGF‐1 generation in the foetus and the newborn infant, and through insulin, it is augmented by the direct and indirect effects of nutrients, such as glucose and protein.
As well as regulating hepatic IGF‐1 production, insulin inversely regulates the hepatic production of IGFBP‐1, which in turn inhibits IGF‐1 bioactivity. Low levels of insulin are associated with reduced hepatic IGF‐1 generation and increased levels of IGFBP‐1, leading to reduced IGF‐1 bioactivity. Free IGF‐1 levels and IGF‐1 bioavailability are thought to be critical to the development of pancreatic beta cell mass and subsequent insulin secretion in newborn infants. Islet cell‐specific IGF receptor knockouts develop insulin deficiency within the first few days of postnatal life. In humans, birth is followed by a period of beta cell apoptosis, which is thought to be induced by reductions in placental IGF‐2. This is followed by re‐establishment of beta cell insulin secretion linked to nutritional and gut peptide (GLP‐1) secretion. The complex interactions between insulin and IGF‐1 are critical to understanding subsequent IGF‐1‐linked preterm complications 45, 46, 47, 48.
IGF‐1 and adipose tissue in the foetus
Adipose tissue, which is a complex and highly active metabolic endocrine organ, is a target for both IGF‐1 and insulin. It responds to signals from hormones and the central nervous system and secretes growth factors and adipokines, such as leptin and adiponectin, with important endocrine functions 49. Dysfunctional adipose tissue that has too much or too little fat mass has been associated with insulin insensitivity and type 2 diabetes 50, 51. At 24 weeks of gestational age, a foetus contains very little fat 52 and the third trimester is a period of intense adipogenesis, involving proliferation and mitotic expansion of committed pre‐adipocytes. This is followed by terminal differentiation, which leads to activation of lipid and carbohydrate metabolism genes.
Mature adipocytes contain a single large lipid droplet. In a study of preterm infants, the lipid content of white adipose tissue increased from 184 to 547 mg/g adipose tissue, which was consistent with a larger proportion of pre‐adipocytes in less mature infants 53. In vitro human pre‐adipocytes contain half as much insulin receptor as IGF‐1 receptor protein, while in mature adipocytes, 10 times more insulin receptor than IGF‐1 receptor protein is found. Thus, adipocyte differentiation is characterised by an increased insulin receptor/IGF‐1 receptor ratio, which indicates that insulin sensitivity increases with increasing gestational age 54. In keeping with this, foetal tissues are thought to be less responsive to insulin until 28–30 weeks of gestation 55. Thus, it appears that IGF‐1‐dependent proliferation and the differentiation of pre‐adipocytes must take place before insulin‐dependent storage of fat can proceed.
IGF‐1 and foetal brain development
The developing brain requires very large amounts of energy and animal studies provide evidence that endogenous brain IGF‐1 serves to augment brain glucose utilisation during development 56. In contrast, insulin does not seem to be involved in the regulation of brain metabolism 57. As well as increasing brain glucose uptake and energy metabolism, in vivo and in vitro studies show that IGF‐1 is important for normal growth and development of the central nervous system 58. IGF‐1 influences brain cell proliferation, apoptosis, myelination, neurogenesis, maturation and differentiation (58, 59,S61). IGF‐1 losses in genetic experiments involving mice affected all aspects of brain development, resulting in a 35% reduction in brain volume (S61). In contrast, mice overexpressing IGF‐1 had a 9–55% increase in brain volume, reflecting an increase in cell size and cell number in most brain areas (S62,S63).
All the reported IGF‐1 gene defects in viable humans have been associated with microcephaly and developmental delay (36, 40,S64–66) (Table 1). There is improved general growth with IGF‐1 treatment, including brain growth, which is reflected in increased head circumferences in children with growth hormone insensitivity syndrome (S67,S68). The anti‐apoptotic and neuronal and white matter rescue effects of exogenous IGF‐1 in the immature brain have been evaluated after defined insults such as hypoxia–ischaemia and induced inflammation in animal models (S69). The effects of supplementary treatment with IGF‐1 on long‐term brain development in an animal model mimicking very preterm birth have not yet been determined.
IGF‐1 and foetal lung development
In mice, IGF‐1 has been shown to be critical during prenatal lung growth and organogenesis (S70–72). The cells responsible for the local synthesis of IGF‐1 in the lung are type II pneumocytes, alveolar macrophages and mesenchymal cells (S73,S74). Recent studies have shown that IGF‐1 mRNA expression in the lung occurs predominantly during foetal life and decreases before birth, becoming barely detectable in the neonatal lung. Prenatal IGF‐1 mutant mice have thickened mesenchyme, alterations in cellular matrix deposition, thinner smooth muscles and dilated blood vessels. The addition of IGF‐1 to embryonic IGF‐1 deficient lungs cultured ex vivo increases airway septa remodelling and distal epithelium maturation (S70). Mice with a homozygous null mutation of the IGF‐1 gene have atelectatic lungs and high postnatal mortality due to lung collapse and respiratory failure (28,S75,S76).
IGF‐1 and foetal cardiovascular system development
The evidence for IGF‐1 control of neonatal vascular growth mostly comes from studies of vascular development in the eye. IGF‐1 is a critical nonoxygen‐regulated factor in retinal neurovascular development (S77–79). A lack of IGF‐1 in knockout mice prevented normal retinal vascular growth, despite the presence of vascular endothelial growth factor (VEGF), which is important to vessel development (S80) (Fig. 3). Low in vitro levels of IGF‐1/IGF receptor binding prevent VEGF‐induced activation of protein kinase B (Akt) and mitogen‐activated protein kinase, which are critical for endothelial cell proliferation and survival (S80).
Figure 3.
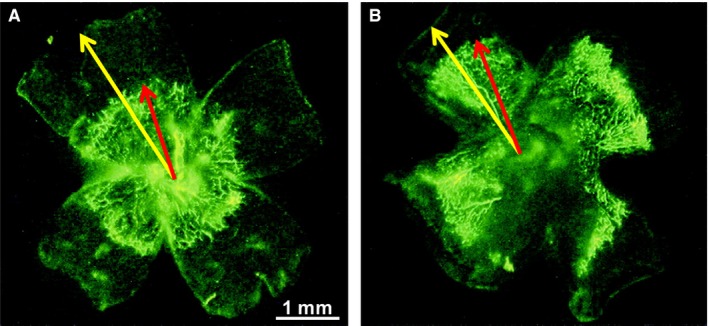
Effect of IGF‐I inhibition on vascular growth. Mice were perfused with fluorescein dextran at postnatal day 5, eyes were enucleated, and retinas were examined in flat mount. There was significantly retarded vascular growth (perfused vessels seen as bright green) in the retinas of the IGF‐I−/− mice (A) compared with littermate IGF‐I+/+ controls with normal IGF‐I levels (B). The distance from the optic nerve to the vessel front (red arrow) vs optic nerve to periphery (yellow arrow) was 58 ± 4.8% for IGF‐I−/− retinas vs 70.3 ± 5.8% for IGF‐I+/+ controls (p < 0.001), indicating that IGF‐I is critical for normal vascular development and that low IGF‐I in the neonatal period could cause retardation of vascular growth. Figure by Hellström et al., with permission from PNAS (S80).
Cell culture and explant studies have shown that VEGF‐A and IGF‐1 differ in their ability to stabilise newly formed blood vessels and endothelial cell tubes. On its own, VEGF‐A fails to support an enduring vascular response. In contrast to VEGF, IGF‐1 stabilises newly developing blood vessels, a function that is dependent on Erk activity and associated with prolonged Erk activation (S81). There are very few studies that provide genetic evidence for the role of growth hormone and IGF‐1 in foetal retinal vascularisation in humans. However, one clinical report notes that when there were defects in the growth hormone/IGF‐1 axis, there was significantly less retinal vascularisation, demonstrated by a lower number of vascular branching points than in the reference group of 100 normal controls (S82).
In vitro studies of foetal rat cardiomyocytes have shown that IGF‐1 inhibits apoptotic signalling of cardiomyocytes (S83). It has also been shown that IGF‐I treatment of foetal lambs in late gestation was associated with increased myocyte volume in male but not female lambs (S84).
Loss of IGF‐1 after preterm birth
After very preterm birth, there is a precipitous decline in serum IGF‐1 concentrations (Fig. 4), which are on average 10 ng/mL compared to the average of >50 ng/mL measured in utero at a postmenstrual age of 23–30 weeks (23, 60,S85,S86).
Figure 4.
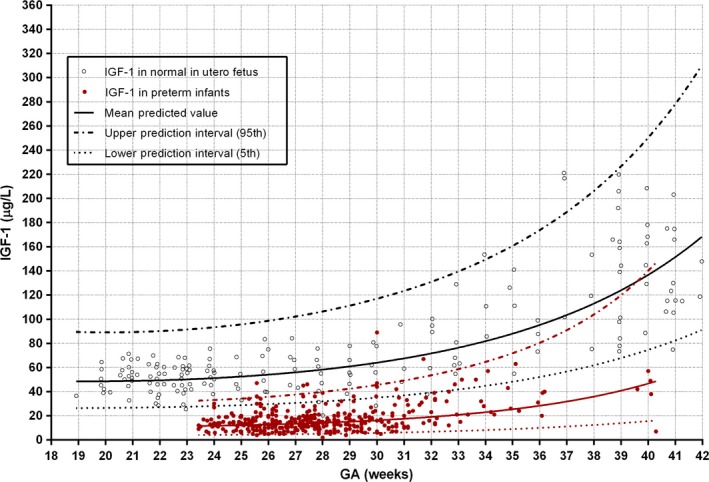
Normal intrauterine IGF‐1 concentrations obtained from the umbilical cord with cordocentesis over 18–42 weeks of gestational age (GA) (black circles) (n = 174) 23, 60 compared to preterm infants of matched postmenstrual ages (red dots) (S86,S87).
At IGF‐1 levels of 10 ng/mL, VEGF signalling is impaired (S86–88). The concentration of IGF‐1 remains very low after preterm birth, resulting in an increasing disparity over time between IGF‐1 serum concentrations in preterm infants and the corresponding in utero levels. This fall in IGF‐1 levels is likely to be multifactorial, including the impact of altered nutrient availability, insulin action, hypoxia and inflammatory cytokines (S86,S89).
Factors influencing IGF‐1 concentrations in preterm infants
In adults and children, the secretion of IGF‐1 is generally dependent on nutrient availability, thereby ensuring that growth is appropriate for the nutrient supply (S90). In this setting, IGF‐1 appears to be a clinically useful indicator of nutritional status (S91), with concentrations increasing with protein intake independent of total caloric consumption (S92,S93). However, in very preterm infants increases in protein and nutrient intake are not associated with early postnatal growth or with increases in IGF‐1 concentration before 30 weeks of postmenstrual age, indicating deficient nutrient utilisation (S94). In addition to under‐nutrition and malnutrition, hypoxia (S89), inflammation (S86) and genetic factors and other hormones, such as T4, cortisol and sex steroids, influence plasma IGF‐1 concentrations (S95).
Deficient energy metabolism
Normally, the newborn infant rapidly develops the pathways for fatty acid oxidation in mitochondria in liver, muscle, kidney cortex and brown adipose tissue (S96). Preterm birth is associated with delayed postnatal activation of mitochondrial oxidative phosphorylation and impaired switch from glycolytic to oxidative metabolism (S97–99). Thus, the energy status of a preterm neonate is not just dependent on energy intake, but to what extent, nutrients can be assimilated and converted to energy.
Loss of IGF‐1 supply
During the third trimester, the foetus swallows significant amounts of amniotic fluid, which contains higher concentrations of IGF‐1 than cord blood during gestation or at delivery (S100). Human milk, especially colostrum, also contains IGF‐1, although IGF‐1 levels drop by 80% by 3 days of age (S101). Although preterm infants may be given trophic feeds of maternal milk/colostrum, the volumes are usually very small and maternal expression of colostrum may be difficult during the immediate postnatal period. Therefore, the loss of IGF‐1 sources in amniotic fluid and colostrum/milk may contribute to lower IGF‐1 levels.
Critical illness
In foetuses as well as in older children, hypoxia reduces circulating IGF‐1 (S89,S102). Variations in oxygenation with episodes of both hypoxia and hyperoxia are common during the first weeks of life in preterm neonates (S103). Preterm delivery may be precipitated by infection and inflammatory markers in cord blood are associated with reduced IGF‐1 concentrations (S86). Acute and chronic hypoxia and inflammation also increase IGFBP‐1 concentration, which inhibits IGF‐1 actions. In addition, very preterm infants are susceptible to infection, which in adults decreases circulating IGF‐1 concentrations (S104).
Clinical impact of low IGF‐1 levels on the preterm infant
Low IGF‐1 levels and growth retardation
In the preterm infant, the early neonatal period is a critical time for growth and early growth is associated with better short‐term and long‐term outcomes (S105). Studies have shown that despite improvements in nutrient delivery, postnatal growth remains poor, particularly in the first month of life of extremely preterm infants (S106). This period is associated with low IGF‐1 levels and this impaired growth may not be entirely reversible, affecting adult body composition and metabolic processes (S107–108). In preterm infants, IGF‐1 concentrations are positively correlated with birth weight, length, head circumference and ponderal index, a measure of leanness (17,S109). In extremely premature infants, there is also a relationship between persistently low IGF‐1 concentrations and slow physical growth (16,S110–114).
As shown in Figure 5, preterm infants have extrauterine growth retardation from birth until around 30–32 weeks of postmenstrual age, when growth accelerates, according to data from four studies of longitudinal postnatal weight SDS of extremely preterm infants (S115–118).
Figure 5.
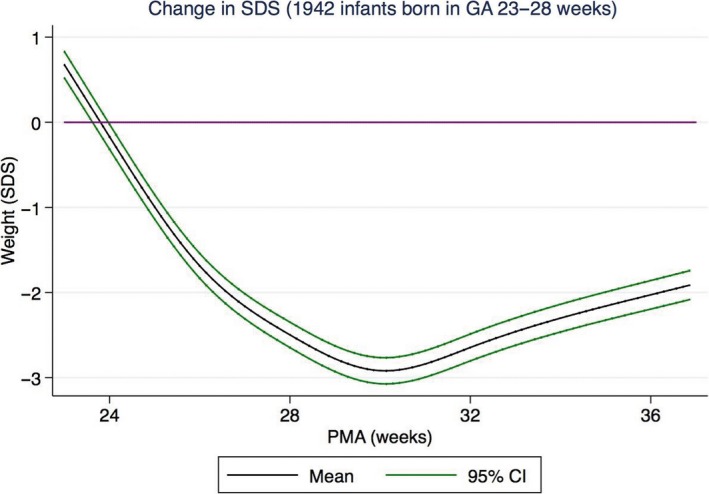
Change in weight SDS with increasing postmenstrual age (PMA) illustrating phases of growth retardation and catch‐up in preterm infants compared to undisturbed intrauterine growth (purple line) (S112). Data from 1942 infants born at GA 23–28 weeks from North America and Sweden. Figure courtesy of Susanna Klevebro.
Insulin‐like growth factor 1 concentrations in preterm infants correlate inversely with standard deviation scores for postnatal weight loss during the growth retardation phase of up to about 30 weeks of postmenstrual age and positively with weight gain during the later catch‐up phase after 30 weeks of postmenstrual age (S94). IGF‐1 concentration is only positively associated with calorie intake when IGF‐1 concentrations are higher, with later catch‐up growth after 30 weeks of postmenstrual age (S94).
Early low IGF‐1 concentrations are associated with later development of obesity. When preterm children reach 5 years of age, they have normal bone mass but less lean mass and increased fat mass compared to full‐term controls (S119). High early postnatal IGF‐1 concentration positively predicts lumbar spine bone mass. Even in moderate to late preterm infants, with gestational ages of 32–36 weeks at birth, postnatal IGF‐1 concentrations correlate with early accelerated postnatal growth (S114).
Low IGF‐1 levels and complications of preterm birth
A prolonged duration of low serum IGF‐1 in extremely premature infants is strongly associated with increased risk of multiple major neonatal morbidities, which have a significant impact on long‐term health (S79,S86–88). These include bronchopulmonary dysplasia (BPD), retinopathy of prematurity (ROP), poor brain development and generalised growth retardation.
Low IGF‐1 levels and impaired brain development in preterm infants
Preterm infants are born in a critical period of brain development (S120) with increases in brain volume and complexity being most marked from 25 to 40 weeks of gestation in utero (S121) (Fig. 6).
Figure 6.

Developmental processes and growth in normal brain development during gestation and after birth. Figure by Lagercrantz with permission from Cambridge University Press (S121).
There is evidence linking low postnatal IGF‐1 concentrations in preterm infants with poor brain development. With the development of magnetic resonance imaging (MRI) neuroimaging, the impact of preterm birth on the brain can be seen.
Postnatal head circumference has been correlated to serum IGF‐1 concentrations during postnatal life in preterm born infants (S122) (Fig. 7).
Figure 7.
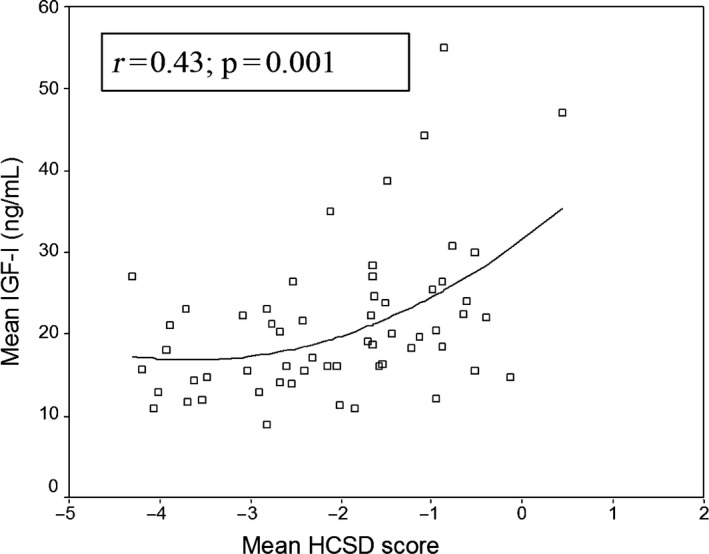
HCSDS at PMA 31 weeks correlate significantly with mean serum IGF‐1 levels from birth to PMA 31 weeks among 58 preterm children, Figure by Löfqvist et al. with permission from Pediatrics (S122).
In addition, mean IGF‐1 concentrations between birth and a postmenstrual age of 35 weeks positively and significantly correlate with total brain volume, un‐myelinated white matter volume, grey matter volume and cerebellar volume, as estimated by volumetric MRI in a cohort of 52 extremely preterm infants (S87). In this same cohort, a slower rate of increase in IGF‐1 concentrations during early postnatal life correlated with an increased risk of subnormal mental development 5 at the corrected age of 24 months, evidenced by Mental Developmental Index scores of <85 (S123).
IGF‐1 and pulmonary development in preterm infants
Preterm infants have incomplete lung development and in extremely preterm infants inadequate postnatal lung maturation combined with postnatal lung injury results in BPD. BPD is a serious complication, affected infants may require oxygen supplements for months to years and some have persistent lung dysfunction throughout adulthood. Despite advances in neonatal care, BPD continues to affect a significant proportion (25–42%) of extremely premature infants and is a major contributor to long‐term respiratory impairment and extended hospitalisation (S124–127). BPD has been characterised by impaired alveolar or microvascular development (S128–130). In addition, in extremely premature neonates, lower postnatal IGF‐1 concentrations during the first weeks after birth have been correlated with later development of BPD independent of gestational age and birth weight standard deviation score (S131,S132). Preclinical interventional studies in rodent model suggest that restoring IGF‐1 concentration may prevent or treat BPD, as IGF‐1 was shown to have beneficial effects on inflammatory lung injury and developmental repair (S133).
The relationship between IGF‐1 and nutritional intake has been shown to differ in premature infants with and without BPD (S134), consistent with increased calorie needs in children with BPD (S135). In preterm infants without BPD, IGF‐1 correlated positively with protein intake and caloric intake over the 3 days before sample collection and with weight change over the previous week (r = 0.46). In contrast, infants with BPD only showed a significant correlation between IGF‐1 and weight change (S134) and none with IGF‐1 and nutrient intake. Animal studies have added additional insights. IGF‐1‐treatment in chronically hypoxic rats resulted in more weight gain than vehicle‐treated rats. These results suggest that IGF‐1 treatment promotes anabolism under chronic hypoxic conditions (S136).
IGF‐1 and vascular development in the preterm infant: risks of ROP and cardiovascular disease
At preterm birth, the vascular system is not fully developed. The contribution of low IGF‐1 levels to poor postnatal vascular development in preterm babies is best documented in the setting of early retinopathy and long‐term risks of cardiovascular disease.
Preterm birth has been associated with overall cardiovascular mortality and morbidity in young adulthood: a nationwide Swedish study showed that individuals born before 32 weeks of postmenstrual age had a nearly twofold increased risk of cerebrovascular disease (S137,S138).
Other data are also consistent with low IGF‐1 linked with preterm birth affecting many vascular beds. For example, low neonatal IGF‐1/IGFBP‐1 ratios and severe ROP have been associated with higher blood pressure in 4‐year‐old children born very preterm, suggesting that low early IGF‐1 concentrations could predict future cardiovascular morbidity (S139).
As previously noted, most evidence about vascular development in association with IGF‐1 comes from studies of the eye because of the problems of ROP, a common morbidity of prematurity that has been associated with lifelong poor visual acuity and visual disorders, including blindness (S140,S141). A review estimated that in 2010, approximately 20 000 infants worldwide had become blind or severely visually impaired from ROP, with a further 12 300 being visually impaired (S142). The retina of an infant is incompletely vascularised at preterm birth. Postnatal retinal vascular growth is then compromised by low IGF‐1 concentrations and low VEGF concentrations compared to those in utero. As a result, vascularisation is delayed and the retina becomes hypoxic. Neovascularisation and blindness can ensue (S79,S80,S88,S143). Serum IGF‐1 concentrations and duration of low IGF‐1 correlate strongly with severity of ROP (S79,S80,S144,S145). An interventional preclinical study of oxygen‐induced retinopathy found that mice who received IGF‐1 treatment developed less retinopathy (p = 0.00001), which supports the possible role of supplemental IGF‐1 in preventing ROP (S146).
Altered glucose metabolism in the preterm infant
Although insulin is the primary regulator of glucose metabolism, IGF‐1 also influences glucose homoeostasis. During foetal life, glucose is the major fuel substrate. In foetal lambs, glucose availability largely determined foetal IGF‐1 secretion, supporting the hypothesis that IGF‐1 is involved in foetal glucose homoeostasis (S147,S148).
Following preterm birth, energy requirements increase and nutrient intake is very often inadequate so energy requirements are not met. In addition, glucose metabolism is frequently deranged by low IGF‐1 levels. Hyperglycaemia during the first weeks of life is strongly associated with morbidities such as ROP (S149), which is similar to diabetic retinopathy in many respects. It has been hypothesised that early insulin replacement in preterm infants could reduce hyperglycaemia and promote anabolism by increasing IGF‐1 concentrations and that this could have beneficial impact on morbidity, mortality and growth. Pilot data have supported this hypothesis, with early insulin treatment in the first week of life increasing IGF‐1 concentrations to improve longitudinal growth (S150). A substudy demonstrated that early intervention with insulin was related to increased IGF‐1 concentrations at 28 days and that low IGF‐1 concentrations were associated with hyperglycaemia, increased morbidity and reduced growth (S132). Children born preterm, as opposed to full term, have aspects of metabolic syndrome, including decreased insulin sensitivity and altered adiposity (S151,S152). This is consistent with animal studies that have shown the relationship between low circulating IGF‐1 and insulin insensitivity 33.
In the 1990s, it was first shown (S153,S154) that IGF‐1 concentrations in preterm infants correlated with nutritional status and adequacy of nutrient intake. The relationship between nutrient intake and IGF‐1 concentrations in preterm infants was confirmed (S93,S94,S111), although the association between dietary protein intake IGF‐1 concentrations and long‐term outcomes of preterm infants need to be further explored (S108).
Rationale for IGF‐1 replacement studies
IGF‐1 deficiency is well documented in the preterm population, especially in those with intrauterine growth retardation or in those born small for gestational age (S94). Animal genetic studies and human gene defects in the IGF‐1 system have illustrated the consequences of low levels of IGF‐1 on foetal development 27, 36. There are long‐term pathological consequences of early IGF‐1 deficits in the preterm population that are difficult to reverse, such as cardiovascular disease (S139) or poor brain development 20. Thus, early replacement of IGF‐1 has both short‐term and long‐term potential benefits to many organ systems.
IGF‐1 treatment studies to date
An important question in a population with inadequate nutrition is whether IGF‐1 treatment can improve nutrient utilisation. Intravenous infusions of IGF‐1 given to rats fed a diet with 50% of the calories they needed were associated with an increase in body weight, without a change in food intake, indicating improved food utilisation. A lower rate of urea excretion was also seen, and this was presumably due to decreased protein breakdown. No hypoglycaemia was observed (S155). IGF‐1 treatment in burn patients in a catabolic state with high metabolic needs caused increased body weight and decreased protein breakdown (S156). In addition, head injury patients in a catabolic state benefitted from IGF‐1 infusion and aggressive nutrition compared to aggressive nutrition alone, with more than a doubling of the number of patients with improvement of outcome from poor to good (S157). In a dose‐dependent manner, IGF‐1 treatment after hypoxic ischaemic brain injury in adult rats reduced neuronal loss by providing trophic support within most cerebral structures (S158).
Some adverse effects have been associated with intermittent subcutaneous IGF‐1 treatment. Laron syndrome patients and patients with short stature are given daily or twice daily subcutaneous doses of IGF‐1. In some patients, hypoglycaemia has occurred, which has been associated with injection site lipohypertrophy and tonsillar hypertrophy and headache (S159). None of these adverse effects have been observed to date when providing short‐term replacements of IGF‐1 in preterm infants to normal in utero concentrations with continuous infusion (S160). An ongoing phase II trial will determine the safety, feasibility and potential benefit and risks of IGF‐1 replacement with IGF‐1/IGFBP‐3 in extremely premature infants.
Conclusion
Following preterm birth, serum IGF‐1 concentrations fall and remain low for weeks to months. This may result in the suppression of development during this period, corresponding to the third trimester of pregnancy, with a loss of IGF‐1 action during the period of rapid rise in utero, which occurs at a postmenstrual age of 22–40 weeks. There have been many reports of correlations between low IGF‐1 and complications of prematurity. These complications resembled many developmental abnormalities of growth seen with IGF‐1 gene defects in experimental animals and in humans. These included abnormalities of general growth and metabolic derangement, such as insulin insensitivity, lung immaturity and retinal immaturity – leading to ROP – and brain developmental abnormalities leading to deficits in cognitive function. Results of animal and human studies indicate that IGF‐1 treatment has the potential to have an important effect on the nutrient assimilation, growth and development of infants born preterm. Clinical trials are required to determine the risks and benefits of IGF‐1 replacement in very preterm infants.
Conflict of Interest
Preventing retinopathy of prematurity by administering insulin‐like growth factor 1 is covered by a patent owned by Premacure AB, Uppsala, Sweden. AH and LEHS are medical consultants for Shire Pharmaceuticals who own Premacure AB. AH, CL, DL, IP‐H and A‐LH own shares in Premalux AB, a company with a financial interest in Premacure AB.
Funding
This work was supported by a European Commission FP7 project 305485 PREVENT‐ROP grant to all of the authors.
Supporting information
Data S1 References S61–160 can be found in the supporting information online.
References
- 1. D'Ercole AJ, Stiles AD, Underwood LE. Tissue concentrations of somatomedin C: further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc Natl Acad Sci U S A 1984; 81: 935–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han VK, D'Ercole AJ, Lund PK. Cellular localization of somatomedin (insulin‐like growth factor) messenger RNA in the human fetus. Science 1987; 236: 193–7. [DOI] [PubMed] [Google Scholar]
- 3. Schwander JC, Hauri C, Zapf J, Froesch ER. Synthesis and secretion of insulin‐like growth factor and its binding protein by the perfused rat liver: dependence on growth hormone status. Endocrinology 1983; 113: 297–305. [DOI] [PubMed] [Google Scholar]
- 4. Blakesley VA, Scrimgeour A, Esposito D, Le Roith D. Signaling via the insulin‐like growth factor‐I receptor: does it differ from insulin receptor signaling? Cytokine Growth Factor Rev 1996; 7: 153–9. [DOI] [PubMed] [Google Scholar]
- 5. Bondy CA, Werner H, Roberts CT Jr, LeRoith D. Cellular pattern of insulin‐like growth factor‐I (IGF‐I) and type I IGF receptor gene expression in early organogenesis: comparison with IGF‐II gene expression. Mol Endocrinol 1990; 4: 1386–98. [DOI] [PubMed] [Google Scholar]
- 6. Rinderknecht E, Humbel RE. The amino acid sequence of human insulin‐like growth factor I and its structural homology with proinsulin. J Biol Chem 1978; 253: 2769–76. [PubMed] [Google Scholar]
- 7. Pollak MN, Schernhammer ES, Hankinson SE. Insulin‐like growth factors and neoplasia. Nat Rev Cancer 2004; 4: 505–18. [DOI] [PubMed] [Google Scholar]
- 8. Randhawa R, Cohen P. The role of the insulin‐like growth factor system in prenatal growth. Mol Genet Metab 2005; 86: 84–90. [DOI] [PubMed] [Google Scholar]
- 9. Rajaram S, Baylink DJ, Mohan S. Insulin‐like growth factor‐binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev 1997; 18: 801–31. [DOI] [PubMed] [Google Scholar]
- 10. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. Insulin‐ like growth factors. N Engl J Med 1997; 336: 633–40. [DOI] [PubMed] [Google Scholar]
- 11. Barrios V, Pozo J, Munoz MT, Buno M, Argente J. Normative data for total and free acid‐ labile subunit of the human insulin‐like growth factor‐binding protein complex in pre‐ and full‐term newborns and healthy boys and girls throughout postnatal development. Horm Res 2000; 53: 148–53. [DOI] [PubMed] [Google Scholar]
- 12. LeRoith D, Werner H, Beitner‐Johnson D, Roberts CT Jr. Molecular and cellular aspects of the insulin‐like growth factor I receptor. Endocr Rev 1995; 16: 143–63. [DOI] [PubMed] [Google Scholar]
- 13. Fernandez M, Sanchez‐Franco F, Palacios N, Sanchez I, Fernandez C, Cacicedo L. IGF‐I inhibits apoptosis through the activation of the phosphatidylinositol 3‐kinase/Akt pathway in pituitary cells. J Mol Endocrinol 2004; 33: 155–63. [DOI] [PubMed] [Google Scholar]
- 14. LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin‐like growth factor 1. Nat Clin Pract Endocrinol Metab 2007; 3: 302–10. [DOI] [PubMed] [Google Scholar]
- 15. Murray PG, Clayton PE. Endocrine control of growth. Am J Med Genet C Semin Med Genet 2013; 163C: 76–85. [DOI] [PubMed] [Google Scholar]
- 16. Kajantie E, Dunkel L, Rutanen EM, Seppala M, Koistinen R, Sarnesto A, et al. IGF‐I, IGF binding protein (IGFBP)‐3, phosphoisoforms of IGFBP‐1, and postnatal growth in very low birth weight infants. J Clin Endocrinol Metab 2002; 87: 2171–9. [DOI] [PubMed] [Google Scholar]
- 17. Chiesa C, Osborn JF, Haass C, Natale F, Spinelli M, Scapillati E, et al. Ghrelin, leptin, IGF‐1, IGFBP‐3, and insulin concentrations at birth: is there a relationship with fetal growth and neonatal anthropometry? Clin Chem 2008; 54: 550–8. [DOI] [PubMed] [Google Scholar]
- 18. Gluckman PD, Harding JE. The physiology and pathophysiology of intrauterine growth retardation. Horm Res 1997; 48(Suppl. 1): 11–6. [DOI] [PubMed] [Google Scholar]
- 19. Fowden AL. The insulin‐like growth factors and feto‐placental growth. Placenta 2003; 24: 803–12. [DOI] [PubMed] [Google Scholar]
- 20. Zhang S, Zhai G, Wang J, Shi W, Zhang R, Chen C. IGF‐II expression and methylation in small for gestational age infants. J Pediatr Endocrinol Metab: JPEM 2015; 28: 613–8. [DOI] [PubMed] [Google Scholar]
- 21. Napoli F, Di Iorgi N, Bagnasco F, Cangemi G, D'Amico B, Boschetti M, et al. Growth factors and metabolic markers in cord blood: relationship to birth weight and length. J Biol Regul Homeost Agents 2014; 28: 237–49. [PubMed] [Google Scholar]
- 22. Ashton IK, Zapf J, Einschenk I, MacKenzie IZ. Insulin‐like growth factors (IGF) 1 and 2 in human foetal plasma and relationship to gestational age and foetal size during midpregnancy. Acta Endocrinol 1985; 110: 558–63. [DOI] [PubMed] [Google Scholar]
- 23. Lassarre C, Hardouin S, Daffos F, Forestier F, Frankenne F, Binoux M. Serum insulin‐like growth factors and insulin‐like growth factor binding proteins in the human fetus. Relationships with growth in normal subjects and in subjects with intrauterine growth retardation. Pediatr Res 1991; 29: 219–25. [DOI] [PubMed] [Google Scholar]
- 24. Gluckman PD, Johnson‐Barrett JJ, Butler JH, Edgar BW, Gunn TR. Studies of insulin‐like growth factor ‐I and ‐II by specific radioligand assays in umbilical cord blood. Clin Endocrinol 1983; 19: 405–13. [DOI] [PubMed] [Google Scholar]
- 25. Han VK, Lund PK, Lee DC, D'Ercole AJ. Expression of somatomedin/insulin‐like growth factor messenger ribonucleic acids in the human fetus: identification, characterization, and tissue distribution. J Clin Endocrinol Metab 1988; 66: 422–9. [DOI] [PubMed] [Google Scholar]
- 26. Roberts CT Jr, Lasky SR, Lowe WL Jr, Seaman WT, LeRoith D. Molecular cloning of rat insulin‐like growth factor I complementary deoxyribonucleic acids: differential messenger ribonucleic acid processing and regulation by growth hormone in extrahepatic tissues. Mol Endocrinol 1987; 1: 243–8. [DOI] [PubMed] [Google Scholar]
- 27. Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin‐like growth factors in embryonic and postnatal growth. Cell 1993; 75: 73–82. [PubMed] [Google Scholar]
- 28. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin‐like growth factor I (Igf‐1) and type 1 IGF receptor (Igf1r). Cell 1993; 75: 59–72. [PubMed] [Google Scholar]
- 29. Yakar S, Rosen CJ, Beamer WG, Ackert‐Bicknell CL, Wu Y, Liu JL, et al. Circulating levels of IGF‐1 directly regulate bone growth and density. J Clin Invest 2002; 110: 771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci U S A 2008; 105: 19378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moses AC, Young SC, Morrow LA, O'Brien M, Clemmons DR. Recombinant human insulin‐like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes 1996; 45: 91–100. [DOI] [PubMed] [Google Scholar]
- 32. Scavo LM, Karas M, Murray M, Leroith D. Insulin‐like growth factor‐I stimulates both cell growth and lipogenesis during differentiation of human mesenchymal stem cells into adipocytes. J Clin Endocrinol Metab 2004; 89: 3543–53. [DOI] [PubMed] [Google Scholar]
- 33. Yakar S, Liu JL, Fernandez AM, Wu Y, Schally AV, Frystyk J, et al. Liver‐specific igf‐1 gene deletion leads to muscle insulin insensitivity. Diabetes 2001; 50: 1110–8. [DOI] [PubMed] [Google Scholar]
- 34. Arends N, Johnston L, Hokken‐Koelega A, van Duijn C, de Ridder M, Savage M, et al. Polymorphism in the IGF‐I gene: clinical relevance for short children born small for gestational age (SGA). J Clin Endocrinol Metab 2002; 87: 2720. [DOI] [PubMed] [Google Scholar]
- 35. Vaessen N, Janssen JA, Heutink P, Hofman A, Lamberts SW, Oostra BA, et al. Association between genetic variation in the gene for insulin‐like growth factor‐I and low birthweight. Lancet 2002; 359: 1036–7. [DOI] [PubMed] [Google Scholar]
- 36. Netchine I, Azzi S, Le Bouc Y, Savage MO. IGF1 molecular anomalies demonstrate its critical role in fetal, postnatal growth and brain development. Best Pract Res Clin Endocrinol Metab 2011; 25: 181–9. [DOI] [PubMed] [Google Scholar]
- 37. Woods KA, Camacho‐Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin‐like growth factor I gene. N Engl J Med 1996; 335: 1363–7. [DOI] [PubMed] [Google Scholar]
- 38. Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin‐like growth factor 1 (IGF1) deficiency. J Med Genet 2003; 40: 913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walenkamp MJ, van der Kamp HJ, Pereira AM, Kant SG, van Duyvenvoorde HA, Kruithof MF, et al. A variable degree of intrauterine and postnatal growth retardation in a family with a missense mutation in the insulin‐like growth factor I receptor. J Clin Endocrinol Metab 2006; 91: 3062–70. [DOI] [PubMed] [Google Scholar]
- 40. Netchine I, Azzi S, Houang M, Seurin D, Perin L, Ricort JM, et al. Partial primary deficiency of insulin‐like growth factor (IGF)‐I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab 2009; 94: 3913–21. [DOI] [PubMed] [Google Scholar]
- 41. Battaglia FC. Principal substrates of fetal metabolism: fuel and growth requirements of the ovine fetus. Ciba Found Symp 1978; 1: 57–74. [DOI] [PubMed] [Google Scholar]
- 42. Rao PN, Shashidhar A, Ashok C. In utero fuel homeostasis: lessons for a clinician. Indian J Endocrinol Metab 2013; 17: 60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Girard J, Duee PH, Ferre P, Pegorier JP, Escriva F, Decaux JF. Fatty acid oxidation and ketogenesis during development. Reprod Nutr Dev 1985; 25: 303–19. [DOI] [PubMed] [Google Scholar]
- 44. Kulkarni RN, Holzenberger M, Shih DQ, Ozcan U, Stoffel M, Magnuson MA, et al. Beta‐cell‐specific deletion of the IGF1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta‐cell mass. Nat Genet 2002; 31: 111–5. [DOI] [PubMed] [Google Scholar]
- 45. Frystyk J, Ritzel RA, Maubach J, Busing M, Luck R, Klempnauer J, et al. Comparison of pancreas‐transplanted type 1 diabetic patients with portal‐venous versus systemic‐venous graft drainage: impact on glucose regulatory hormones and the growth hormone/insulin‐like growth factor‐I axis. J Clin Endocrinol Metab 2008; 93: 1758–66. [DOI] [PubMed] [Google Scholar]
- 46. van Haeften TW, Twickler TB. Insulin‐like growth factors and pancreas beta cells. Eur J Clin Invest 2004; 34: 249–55. [DOI] [PubMed] [Google Scholar]
- 47. Ogilvy‐Stuart AL, Hands SJ, Adcock CJ, Holly JM, Matthews DR, Mohamed‐Ali V, et al. Insulin, insulin‐like growth factor I (IGF‐I), IGF‐binding protein‐1, growth hormone, and feeding in the newborn. J Clin Endocrinol Metab 1998; 83: 3550–7. [DOI] [PubMed] [Google Scholar]
- 48. Iniguez G, Ong K, Bazaes R, Avila A, Salazar T, Dunger D, et al. Longitudinal changes in insulin‐like growth factor‐I, insulin sensitivity, and secretion from birth to age three years in small‐ for‐gestational‐age children. J Clin Endocrinol Metab 2006; 91: 4645–9. [DOI] [PubMed] [Google Scholar]
- 49. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004; 89: 2548–56. [DOI] [PubMed] [Google Scholar]
- 50. Tsatsoulis A, Mantzaris MD, Bellou S, Andrikoula M. Insulin resistance: an adaptive mechanism becomes maladaptive in the current environment – an evolutionary perspective. Metabolism 2013; 62: 622–33. [DOI] [PubMed] [Google Scholar]
- 51. Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol 2006; 17: 162–9. [DOI] [PubMed] [Google Scholar]
- 52. Ziegler EE, O'Donnell AM, Nelson SE, Fomon SJ. Body composition of the reference fetus. Growth 1976; 40: 329–41. [PubMed] [Google Scholar]
- 53. Kuipers RS, Luxwolda MF, Offringa PJ, Martini IA, Boersma ER, Dijck‐Brouwer DAJ, et al. Gestational age dependent content, composition and intrauterine accretion rates of fatty acids in fetal white adipose tissue. Prostaglandins Leukot Essent Fatty Acids 2012; 86: 13–20. [DOI] [PubMed] [Google Scholar]
- 54. Bäck K, Arnqvist HJ. Changes in insulin and IGF‐I receptor expression during differentiation of human preadipocytes. Growth Horm IGF Res 2009; 19: 101–11. [DOI] [PubMed] [Google Scholar]
- 55. Hill DE. Fetal effects of insulin. Obstet Gynecol Annu 1982; 11: 133–49. [PubMed] [Google Scholar]
- 56. Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA. Insulin‐like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci U S A 2000; 97: 10236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baskin DG, Figlewicz DP, Woods SC, Porte D Jr, Dorsa DM. Insulin in the brain. Ann Rev Physiol 1987; 49: 335–47. [DOI] [PubMed] [Google Scholar]
- 58. O'Kusky J, Ye P. Neurodevelopmental effects of insulin‐like growth factor signaling. Front Neuroendocrinol 2012; 33: 230–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Joseph D'Ercole A, Ye P. Expanding the mind: insulin‐like growth factor I and brain development. Endocrinology 2008; 149: 5958–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bang P, Westgren M, Schwander J, Blum WF, Rosenfeld RG, Stangenberg M. Ontogeny of insulin‐like growth factor‐binding protein‐1, ‐2, and ‐3: quantitative measurements by radioimmunoassay in human fetal serum. Pediatr Res 1994; 36: 528–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 References S61–160 can be found in the supporting information online.