

Abstract
Selected aldohexoses (d‐glucose, d‐mannose, and d‐galactose) and aldopentoses (d‐xylose, l‐arabinose, and d‐ribose) are readily available components of biopolymers. Isomerization reactions of these substances are very attractive as carbon‐efficient processes to broaden the portfolio of abundant monosaccharides. This review focuses on the chemocatalytic isomerization of aldoses into the corresponding ketoses as well as epimerization of aldoses at C2. Recent advances in the fields of catalysis by bases and Lewis acids are considered. The emphasis is laid on newly uncovered catalytic systems and mechanisms of carbohydrate transformations.
Keywords: biomass, chemocatalysis, isomerization, Lewis acids, saccharides
1. Introduction
The chemical industry is nowadays dependent on fossil resources such as oil, coal, and gas.1 However, a long‐term outlook predicts biomass to become a main carbon source for chemical manufacture. Therefore, numerous investigations are now focused on the efficient valorization of biomass. Given that carbohydrates represent most of the biomass‐derived organic compounds,2 great attention is currently being paid to transformations of mono‐, oligo‐, and polysaccharides. Noteworthy, the main source of saccharides are polysaccharides, such as cellulose, starch, hemicelluloses, inulin, and so on. The strategy of “platform chemicals”3 considers first the depolymerization of such biopolymers to release the monomers4 and, second, upgrading of these monomers.1b, 5 Remarkably, numerous recent investigations have elaborated protocols for the synthesis of fuels along with commodity and fine chemicals based on monosaccharides.1b, 5, 6
Biopolymers are sources of several aldoses and 2‐ketoses, but seven monosaccharides significantly predominate, namely, d‐glucose, d‐fructose, d‐xylose, l‐arabinose, d‐ribose, d‐mannose, and d‐galactose (Figure 1). d‐Glucose clearly prevails as the major building block of plant biomass.2a From a synthetic point of view, it would be interesting to extend the list of readily available monosaccharides. Isomerization is a well‐known carbon‐efficient way to produce rare monosaccharides based on abundant ones (Figure 1). Moreover, valuable compounds can be produced on the basis of the products of isomerization (Figure 2). For instance, d‐xylose can be converted into d‐xylulose and further into furfural under much milder conditions than those typically used for furfural production.7 Together with 5‐hydroxymethylfurfural (HMF), furfural is a biomass‐derived platform chemical of great interest. d‐Tagatose is a very promising ketose that can be applied as a low‐calorie sweetener and in cosmetic and pharmaceutical formulations. Currently, d‐tagatose is produced from d‐galactose catalyzed by l‐arabinose isomerase.8
Figure 1.
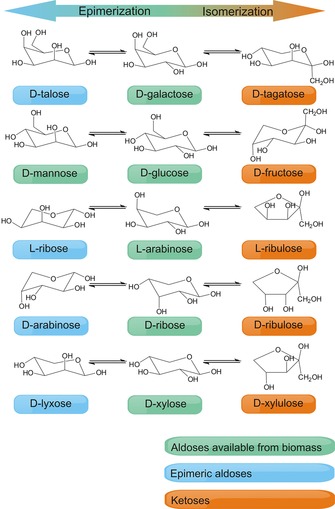
Formulae of aldoses available from biomass, epimeric aldoses, and ketoses.
Figure 2.
Application areas of monosaccharides. A photo of a tree is depicted from pihtahvoya.ru.
Furthermore, epimerization of biomass‐derived monosaccharides at the C2 position gives rise to rare monosaccharides with interesting properties. For example, d‐lyxose, d‐arabinose, and d‐talose can be used as starting materials for the synthesis of antitumor agents.9 The d epimers of saccharides dominate in nature with the exception of l‐arabinose, which is available from hemicelluloses. Consequently, l‐arabinose can be used as a substrate for the synthesis of rare l‐monosaccharides. For instance, epimerization of l‐arabinose enables synthesis of l‐ribose, which is in high demand in medical chemistry for the synthesis of potent agents against the hepatitis B virus as well as the Epstein–Barr virus.10 Considering that epimerases are only active on sugars substituted with phosphate or nucleotide groups, efficient chemocatalytic systems for the direct epimerization of monosaccharides are of upmost importance.11
In addition to isomerization into 2‐ketoses and C2 epimerization processes, the synthesis of other isomers is potentially of industrial relevance. For example, catalytic activity of Ti‐β zeolite was reported for the isomerization of d‐glucose into l‐sorbose with 73 % enantiomeric purity.12 l‐Sorbose is an important intermediate for the production of vitamin C. Currently, l‐sorbose is manufactured from d‐glucose by means of a complex multistep biotechnological process.2a
Isomerization of glucose into fructose is an important example of an isomerization process implemented on an industrial scale.2a, 13 Though fructose is present in nature as a monomer of inulin or levan, the biotechnological production of fructose by glucose isomerization appears to be more economically attractive. Currently, fructose is produced mostly as a part of high fructose syrups (HFSs) employed as sweeteners. The manufacture of HFSs is a multistep process that includes: (1) enzymatic hydrolysis of starch to release glucose; (2) isomerization of glucose in the presence of immobilized d‐xylose ketoisomerase; (3) chromatographic enrichment to produce HFS‐90 containing 90 % fructose and 10 % glucose.2a, 13, 14 The immobilization of d‐xylose isomerase together with the elaboration of the respective separation technology has enabled the continuous commercial production of fructose since the launch of this process in 1967.15 Today, d‐xylose ketoisomerase remains one of the largest biocatalytic processes13 owing to the high demand for sweet HFSs. In 2006, the annual worldwide production of fructose was estimated by Lichtenthaler to be approximately 60 000 metric tons.2a Recently, research interest in fructose has increased, because it is regarded as a key intermediate for the valorization of cellulosic biomass. It was demonstrated that fructose can be readily converted into HMF16 and further into valuable products such as fuels and monomers for biomass‐based polymers.1b, 5, 6, 16d, 17 In this context, the enzymatic production of fructose appears to be expensive owing to the high cost and the low stability of the enzymes, the need for highly pure glucose, and the use of buffer solutions. This has propelled extensive research with the aim to develop suitable chemocatalysts for the isomerization of glucose into fructose.
In this review, we address recent advances in the field of the chemocatalytic isomerization of monosaccharides. The first and the second sections of this review are focused on the isomerization of aldoses into C2 ketoses in the presence of basic and Lewis acidic catalysts, respectively. The third part considers progress made in the epimerization of aldoses.
Noteworthy, herein we mostly focus on aqueous‐phase processes. In recent years, various studies have addressed isomerization with the use of ionic liquids as solvents. This direction of research was previously reviewed by Zakrzewska et al.18 In what follows, monosaccharides mentioned without specifying epimeric configuration refer to the d enantiomers.
2. Isomerization over Basic Catalysts
Bases were the first chemocatalysts that were uncovered for the isomerization of carbohydrates as long ago as 1885. This base‐catalyzed isomerization is also named after the discoverers of this reaction, that is, the Lobry de Bruyn–Alberda van Ekenstein transformation. The isomerization of an aldose results in the formation of a ketose and an epimeric aldose, but the isomeric ketose is usually formed in higher amount (Figure 3).
Figure 3.
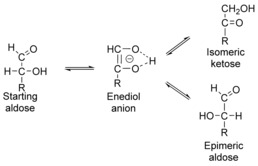
Isomerization of aldoses catalyzed by bases via the enediol anion.19
Early investigations mainly involved the use of soluble alkalis, such as sodium hydroxide or calcium hydroxide, operating at high pH values and room temperature.19, 20 Under these conditions, the reaction suffers from a low rate of isomerization and the formation of numerous acidic byproducts. The formation of acidic compounds is also catalyzed by bases, which promote the isomerization of saccharides into oligomeric acidic products, saccharinic acids, or lactic acid.21 This leads to low carbon efficiency as well as neutralization of the basic catalyst by acidic byproducts. Additionally, dehydration and condensation of the byproducts result in a strong darkening of the reaction solution. This strong coloration is indicated as one of the reasons why alkali catalysts are regarded as inappropriate for fructose production in the food industry.13 Later on, the utilization of organic bases, for example, triethylamine, was shown to improve the selectivity.20a Amines have been confirmed to be efficient catalysts for the isomerization; moreover, the degradation of saccharides in the presence of amines is much slower than that over inorganic bases. In 2001, Angyal summarized the main results on the isomerization of saccharides over soluble bases.22
Table 1 presents an overview of literature data on isomerization over base catalysts. Though isomerization in the presence of soluble bases does not result in a high yield of a product, the yield can be significantly improved by adding borates, boronates,26, 27 or aluminates.28, 35 For instance, Mendicino reported an 85 % yield of fructose by glucose isomerization in the presence of borates and NaOH.26 The yield increases owing to in situ complexation of fructose under basic conditions. Numerous patents have appeared on the isomerization of glucose promoted by complexation of fructose, and this highlights the great commercial interest in such an approach. For instance, good yields of ketoses have been reported in the presence of aluminate resins to facilitate a 72 % yield of fructose36 and in the presence of poly(arylboric acid) resins and NaOH to give fructose in 57 % yield.27 A combination of amines with boric acid gives rise to d‐fructose and d‐tagatose with yields up to 63 and 52 %, respectively.37 More recently, Despax et al. revisited sodium aluminate as a catalyst for glucose isomerization into fructose.29 Fructose yields of 40 and 49 % are reported for aqueous and organic solvents, respectively.
Table 1.
Literature data on catalytic activity of basic catalysts for glucose isomerization into fructose.
| Entry | Reaction conditions | Results[a] | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| catalyst | solvent | T [°C] | glucose concentration [wt %] | glucose/catalyst[b] | X [%] | S [%] | Y [%] | ||
| Soluble alkalis | |||||||||
| 1 | KOH | water | 78 | 0.45 | 8 | 18 | 61 | 11 | 23 |
| 38 | 47 | 18 | |||||||
| 2 | NaOH | water | 90 | 10 | – | 20 | 70 | 14 | 24 |
| 36 | 50 | 18 | |||||||
| 3 | Et3N | water | 100 | 10 | 17.8 | 57 | 54 | 31 | 25 |
| Soluble alkalis with complexing agents | |||||||||
| 4 | NaOH+Na2B4O7 | water | 100 | 0.018 | 0.01 | n.d. | n.d. | 85 | 26 |
| 5 | NaOH+p‐tolyl boric acid | water | 50 | 23.4 | n.d. | n.d. | n.d. | 56 | 27 |
| 6 | NaAlO2 | water | 43 | 12 | 1.1 | n.d. | n.d. | 60–70 | 28 |
| 7 | NaAlO2 | water | 55 | 40 | 3.1 | 59 | 70 | 41 | 29 |
| 8 | NaAlO2 | DMSO/propylene glycol/water | 55 | 40 | 3.1 | 68 | 72 | 49 | 29 |
| Solid catalysts | |||||||||
| 9 | CsA zeolite | water | 95 | 10 | 5 | 34 | 66 | 22 | 30 |
| 10 | NaA zeolite | water | 95 | 10 | 5 | 26 | 72 | 19 | 30 |
| 11 | MgO⋅Al2O3 [c] | water | 95 | 10 | 5 | 30 | 66 | 20 | 30 |
| 12 | Mg0.75Al0.25(OH)2(OH)0.25 | DMF | 80 | 3 | 3 | 50 | 69 | 35 | 31 |
| 13 | Mg0.75Al0.25(OH)2(OH)0.25 [d] | DMF | 100 | 3 | 3 | 42 | 88 | 37 | 32 |
| 14 | Mg0.75Al0.25(OH)2(CO3)0.125 | water | 110 | 10 | 5 | 34 | 89 | 30 | 33 |
| 15 | Fe3O4@SiO2‐TMG[e] | water | 120 | 20 | 4 | 46 | 54 | 25 | 34 |
[a] Conversion (X), selectivity (S), and yield (Y) are given for fructose formation; n.d.=not determined. [b] Mass ratio: initial mass of glucose divided by mass of catalyst. [c] Calcined hydrotalcite with Mg/Al=2.5. [d] Hydrotalcite rehydrated under sonication. [e] Magnet base catalyst: tetramethylguanidine (TMG) immobilized onto silicon dioxide coated magnetic iron oxide.
The reason for the improved yield of fructose in the presence of complexing anions has been discussed in literature. Enhanced yields are explained by complexation of fructose with an anion, for example, borate or aluminate.36 It is well known that borates form more stable complexes with ketoses than with aldoses.38 In situ complexation of fructose enables higher yields owing to a decrease in the ketose concentration, which results in a shift in the glucose–fructose equilibrium towards fructose, and owing to the prevention of fructose degradation as a result of the increased stability of the obtained complexes relative to that of pure fructose.
Current investigations mainly focus on elaborating chemocatalytic processes to substitute the biotechnological process for the isomerization of glucose into fructose. In this respect, solid catalysts are beneficial owing to facile catalyst separation and recycling. Materials such as hydrotalcites,30, 31, 32, 33, 39 immobilized amines,34 zeolites in alkaline‐exchange form,30, 40 mesoporous ordered molecular sieves of the M41S family,41 zirconium carbonate,42 and anion‐exchanged resins43 have been reported as efficient solid base catalysts. In the interest of productivity, the experiments are conducted at elevated temperatures, though saccharides, especially ketoses, are not stable under harsh conditions.34, 44 Therefore, isomerization is usually performed at temperatures not exceeding 110–120 °C. Owing to good solubility, water is a solvent of choice for saccharides, though polar organic solvents, such as DMF,31, 32, 39b DMSO,29 DMSO/ethylene glycol,29 and DMSO/propylene glycol,29 have also been utilized. Interestingly, a solvent can potentially influence the kinetics of isomerization over a solid base, analogously to what was uncovered for glucose isomerization in the presence of NaOH. Thus, the isomerization rate is 2.4 times higher for a water/ethanol (30:70) mixture than for pure water. This can be explained by the greater ionization constant of glucose in the water/ethanol mixture (ionization of glucose as a step of the isomerization mechanism is discussed below).45
The kinetics of isomerization in the presence of solid bases resembles that over soluble bases. Thermodynamics predict a fructose yield of approximately 50 % based on glucose (not taking into account mannose).46 In the presence of epimerases under physiological conditions, the equilibrium of glucose/fructose/mannose corresponds to 41:41:18.47 El Khadem et al. have investigated the isomerization of hexoses in the presence of KOH as a catalyst, and they report different compositions of the final mixture when starting from an aldose, an epimeric aldose, or an isomeric ketose. For instance, the distributions of obtained monosaccharides upon starting from glucose, mannose, and fructose are shown in Table 2. Very similar results are reported for glucose isomerization in the presence of other soluble and solid catalysts, that is, the yield of fructose does not usually exceed 35 %.24, 30, 31, 32, 33, 34, 39a, 39c, 41, 42, 44, 48
Table 2.
Composition of glucose/fructose/mannose mixtures obtained starting from different isomers in the presence of KOH.49
| Starting monosaccharide | Glucose/fructose/mannose | Total amount of monosaccharides [%] |
|---|---|---|
| glucose | 53:39:8 | 87 |
| fructose | 63:31:6 | 80 |
| mannose | 35:25:40 | 99 |
Cation‐exchanged zeolites demonstrate high activity and selectivity for the isomerization of glucose30, 39a as well as disaccharides such as lactose, cellobiose, and maltose.40 Shukla et al. reported that the isomerization of disaccharides is accompanied by hydrolysis and release of monosaccharides. At the same time, degradation of the saccharides over zeolites is much slower than that over soluble NaOH. Thus, the disaccharides degrade as much as 55–62 % in the presence of NaOH, but the substrate decomposes by only 10–13 % over zeolites under the same reaction conditions.40 The catalytic activity decreases in a row: NaA>NaX>NaY, that is, lower Si/Al ratios provide higher concentrations of basic sites and greater activity.30, 40 Considering the exchanged cation, the following series of activity is observed for the isomerization of glucose: Ca2+<Ba2+<Li+<Na+<K+<Cs+.30 Interestingly, A zeolite outperforms X zeolite and Y zeolite in terms of catalytic activity, and a small pore aperture of 4.1 Å does not seem to cause diffusional issues.30 An obstacle for the application of sodium‐exchanged zeolites is rather leaching of sodium into the aqueous medium.30, 39a Nevertheless, NaA zeolite has successfully been tested for the isomerization of glucose under a continuous operation mode. The initial glucose conversion of 20 % drops to 10 % after 25 h on stream. Thereafter, the catalyst demonstrates a constant glucose conversion of 10 % for another 25 h on stream.30
Magnesium–aluminum hydrotalcites have been proven to be efficient catalysts for the isomerization of glucose into fructose. Hydrotalcites are layered double hydroxides with the general formula [Mg1−xAlx(OH)2]x+(Ax/n n−)⋅ m H2O, for which Mg2+ ions are partially substituted by Al3+ in the brucite‐type layers. An− is an interlayer anion, x (0.17<x<0.33) is the fraction of aluminum, and m denotes water molecules of crystallization.50 Hydrotalcites containing HO− or CO3 2− as interlayer anions have been tested for the isomerization reaction. Hydrotalcites in the HO− form exhibit activity superior to materials in the carbonate form because the basicity of the hydroxide ion is higher than that of the carbonate ion. Nevertheless, handling of hydrotalcites in the HO− form requires precaution, as contact of the catalyst with air leads to consumption of CO2 and the formation of carbonates in the interlayer space.39a, 51 Calcination of Mg–Al hydrotalcites results in a MgO–Al2O3 mixture possessing medium‐strong Lewis basic O2−−Mn+ pairs and isolated O2− ions as strong basic sites.52 The presence of strong basic centers appears to be detrimental, as saccharides undergo rapid decomposition in the presence of MgO–Al2O3 oxides, which leads to lower selectivity for fructose.31, 39a Though we have detected some leaching of Mg upon using [Mg1−xAlx(OH)2]x+(CO3)x/2 ⋅ m H2O hydrotalcites for the isomerization of glucose in the aqueous phase, the catalysts can be successfully recycled after calcination and rehydration.24, 33 We have tested commercial Mg6Al2(CO3)(OH)16 ⋅4 H2O for glucose isomerization under continuous conditions and have observed a gradual decrease in catalytic activity. Nevertheless, the catalytic activity can be restored by calcination and rehydration of hydrotalcite.24
Importantly, similar to zeolites, hydrotalcites outperform soluble hydroxides in terms of selectivity. In the presence of hydrotalcites, the number of byproducts is much lower than that formed with the use of soluble bases. Dihydroxyacetone, glyceraldehyde, glycolaldehyde, and lactic acid have been identified as side products that are formed in the presence of [Mg1−xAlx(OH)2]x+(CO3)x/2 ⋅ m H2O hydrotalcites.24, 33
An interlayer anion of hydrotalcites is regarded as an active center for isomerization. Figure 4 demonstrates the structure of [Mg1−xAlx(OH)2]x+(CO3)x/2 ⋅ m H2O hydrotalcites. Hydrotalcites consist of layered nanocrystallites that are a few nanometers in size. Noteworthy, owing to significant electrostatic forces and a network of hydrogen bonds, the interlayer space is unavailable for a neutral substrate such as glucose. Owing to electrostatic attraction, nanocrystallites are organized into polycrystalline primary particles that have diameters of tens to hundreds of nanometers. The primary particles interact with each other to form materials with different morphologies, for example, hydrotalcites with a “sand‐rose” structure, or the materials are formed by stacking of basal planes.33 At the end, the majority of the carbonate anions are no longer accessible to the substrates (Figure 4). Previous studies have suggested that two locations of carbonates provide good access to the active sites. First, the interlayer anions located at the edges of the primary particles are accessible to the substrates.53 Moreover, the anions located at crystal defects of the lamellar structure are also catalytically active.32, 53b, 54 Lee et al. have demonstrated that calcination and sonication‐assisted rehydration of hydrotalcites results in improved catalytic activity of the materials for the isomerization of glucose into fructose. Sonication during rehydration leads to vertical breaking and exfoliation of the hydrotalcite layers. This results in an increased concentration of basic centers and improves catalytic activity for isomerization.32 Later on, we have also found a dependency of glucose conversion on basicity of the hydrotalcites and have proposed protocols for the synthesis of hydrotalcites with a high concentration of basic sites.33 Precipitation in aqueous medium at the isoelectric point of the hydrotalcite (pH 10) gives rise to materials with a “sand‐rose” structure. These materials exhibit a higher concentration of basic sites than materials prepared at pH values below 10 and demonstrate stacking of the basal planes. Alternatively, hydrotalcites with a high concentration of accessible basic sites can be produced by precipitation in aqueous ethanol media.33
Figure 4.
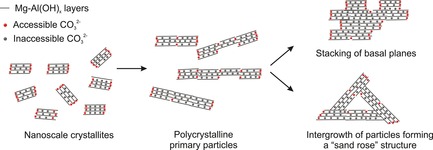
Schematic representation of [Mg1−xAlx(OH)2]x+(CO3)x/2 ⋅ m H2O hydrotalcite structure. Water in the interlayer space is not shown.
Amines have also been studied as catalysts for the isomerization of glucose into fructose.25, 34, 44 The so‐called Maillard reaction is an undesired side process that occurs during isomerization and causes the formation of colored products by the reaction of reducible sugars with primary or secondary amines. Nevertheless, the Maillard reaction proceeds much more slowly for secondary amines than for primary amines, and tertiary amines are not expected to react at all. Liu et al. have observed some darkening of the reaction mixture during glucose isomerization in the presence of secondary and tertiary amines, but the authors explain this by caramelization.25 The reaction mixture can be decolorized by using activated carbon as a sorbent.25 The data published so far on the relative activity of different amines is somewhat controversial. On the one hand, Carraher et al. conclude that HO− ions are active species for catalysis and that an amine is required only for the generation of hydroxide anions upon reaction with water.44 On the other hand, Yang et al. have titrated the reaction mixture with different amines to reach the same pH0 value. Therein, a clear dependence of the reaction rate on the nature of the catalyst is observed.34 Thus, isomerization in the presence of a strong base such as tetramethylguanidine (pK a=21) is quicker than that in the presence of weaker bases, for example, 1‐(3‐aminopropyl)imidazole (pK a=9.63, 6.5) and 1,5,7‐triazabicyclo[4.4.0]dec‐5‐ene (pK a=13.6). Elucidating the role of the amines during isomerization is hampered by the fact that the pH changes during the reaction. As a result of the formation of acidic byproducts, the pH drops by 2–4 units by the end of the isomerization.44 Immobilized amines appear to be stable for glucose isomerization.34 Yang et al. have very recently reported the fabrication of a magnetic base catalyst based on magnetic iron oxide coated with silicon oxide. The surface of the latter is functionalized with (3‐chloropropyl)trimethoxylsilane prior to immobilization of the amines. As a result, magnetic spheres with diameters of approximately 300 nm can be prepared and successfully recycled for isomerization. Interestingly, amines immobilized onto chloromethylated polystyrene resin undergo quick deactivation owing to sorption of byproducts.34 The mechanism of the isomerization of glucose into fructose has been studied by Carraher et al. by using molecular amines as catalysts and water as the solvent. The proposed mechanism consists of the following steps: (1) ionization of glucose in cyclic form to give a glucose anion existing in the open‐chain (acyclic) form; (2) abstraction of a hydrogen atom from C2 to yield an enediol intermediate; (3) formation of a fructose anion in open‐chain form; (4) closing of the cycle and abstraction of a proton from water (Figure 5).
Figure 5.
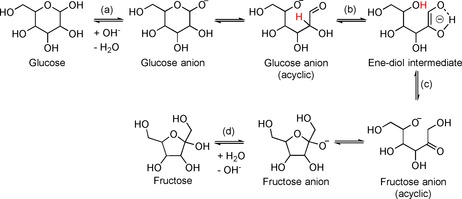
Mechanism of glucose isomerization into fructose catalyzed by amines proposed by Carraher et al.44
Formation of the enediol intermediate has been proven by the presence of an absorption band at λ=286 nm in the UV spectrum of the reaction mixture.25 By using glucose isotopically labelled at the C2 position ([C2‐D]glucose), a kinetic isotopic effect of k H/k D=3.8 is observed. This indicates that abstraction of a hydrogen atom from the C2 atom of glucose (Figure 5, step b) plays a significant role in the overall reaction rate. Carraher et al. have considered two possibilities for implementation of stage b, and they are an intramolecular shift of a hydrogen atom from C2 to O5 (as shown in Figure 5) or bimolecular deprotonation with HO− followed by reprotonation from H2O (not shown). None of the possible mechanisms can be excluded and parallel deprotonation/reprotonation by either intramolecular reaction or by HO− is proposed.44
3. Isomerization over Lewis Acids
The catalytic activity of Lewis acids for the isomerization of monosaccharides was uncovered much later than that of basic catalysts for the same reaction. It is not surprising, taking into account that classic Lewis acids such as AlCl3 and FeCl3 are usually deactivated in the presence of water. In aqueous media, water molecules coordinate to a metal cation and the obtained hydrated cation undergoes partial hydrolysis. As a result, reactions catalyzed by Lewis acids are usually performed under anhydrous conditions to avoid deactivation of the catalyst.63 At the same time, water is a solvent of choice for monosaccharides, as they are highly polar and poorly soluble in organic solvents. The revealed catalytic activity of Lewis acids for the isomerization of carbohydrates in bulk water has prompted enormous research interest. As a result, catalysis by Lewis acids has been studied very intensively for both soluble and solid catalysts. Though a few reviews have already considered isomerization catalyzed by Lewis acids,6a, 63, 64 this research area is developing very quickly. Herein, we summarize the latest findings of this research topic.
Moliner et al. demonstrated for the first time the catalytic efficiency of Sn silicalite with β zeolite topology for the isomerization of glucose into fructose in water.46 This catalyst is also referred to as Sn‐β zeolite. Embedded into the hydrophobic matrix of β zeolite, the tin ions maintain the Lewis acidity to catalyze the isomerization in aqueous media. Even very concentrated solutions of glucose, up to 45 wt %, can be successfully converted.46 Sn‐β has also been shown to be efficient in the isomerization of other carbohydrates such as xylose,7, 55, 56, 65 lyxose,65a ribose,55, 65a arabinose,65a mannose,55, 65a galactose,55, 65a and lactose.56, 66 The structure of the active centers and the mechanism of isomerization have recently been intensively studied by means of spectroscopic and computational tools, with a focus mostly on glucose as a substrate. Glucose and fructose are present in water solution nearly exclusively in the six‐membered (pyranose) and five‐membered (furanose) ring forms.67 Nevertheless, it has been shown by solid‐state NMR and IR spectroscopy that glucose and fructose are adsorbed on Sn‐β in their open‐chain forms. Román‐Leshkov et al. have confirmed that the reaction takes place through an intramolecular hydride shift from the C2 carbon atom of glucose to the C1 position.62 Investigations of the kinetic isotopic effect have revealed that the hydride shift is kinetically relevant.12, 62, 68 The mechanism of the isomerization over Sn‐β can be described stepwise as follows: (1) coordination of glucose to an active site; (2) hydride transfer; (3) desorption of fructose (Figure 6). The ring‐opening and ring‐closing steps do not exhibit significant apparent reaction barriers.69
Figure 6.
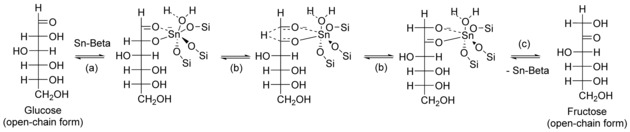
Mechanism of glucose isomerization into fructose by hydride shift proposed by Román‐Leshkov et al.62
A number of studies aimed at elucidating the structure of the active sites of Sn‐β. Boronat et al. suggest the presence of two types of sites for Sn‐β (Figure 7).70 The first type is a dehydrated closed site containing tetracoordinated Sn connected to four O−Si groups. Partial hydrolysis of a closed site generates an open site consisting of a (SiO)3Sn(OH) group adjacent to a silanol group Si(OH). Both open and closed sites can coordinate two molecules of water, which changes the tetrahedral state into an octahedral state (Figure 7). The presence of different sites can be determined spectroscopically, for example, by using solid‐state 119Sn NMR spectroscopy.68a A detailed description of the methods used for the characterization of solid Lewis acids can be found in recently published reviews.71 The role of the open and closed sites for catalysis has been widely discussed. Noteworthy, there is strong evidence that the open sites of TS‐1 are more active than the closed sites for the oxidation of alkanes with hydrogen peroxide.72 Moreover, it has been demonstrated that the open sites of Sn‐β are responsible for activity in the Meerwein–Ponndorf–Verley and Baeyer–Villiger reactions.70, 73 In line with this, computational studies of glucose isomerization suggest that the open sites of Sn‐β are more active than the closed ones.68a, 74 The cooperative action of a Sn metal site and a proton donor is proposed to stabilize the transition state during hydride transfer. Different proton donors are suggested, that is, an adjacent silanol group74, 75 and co‐adsorbed water molecules.75b Additionally, the role of the hydroxy groups attached to the Sn metal center in the (SiO)3Sn(OH) open site as a Brønsted base has been considered. It is suggested that the Brønsted base lowers the energy barrier for the initial deprotonation step.68b In general, stabilization of the hydroxy group on the C2 atom of glucose by the oxygen atom in the first coordination sphere of Sn is proposed.69
Figure 7.
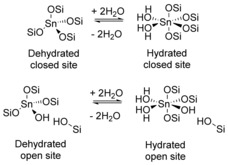
Schematic representation of tin sites present in Sn‐β, as suggested by Boronat et al.70
Rai et al. have performed density functional calculations to reveal the distinction between the open and closed sites.74 Their results suggest that isomerization takes place on the open sites and that the adjacent silanol group participates directly in the hydride‐transfer step. They considered proton transfer from the OH group attached to C2 of glucose to Sn−OH as the first step. Thereafter, glucose in the open‐chain form coordinates in a monodentate fashion toward Sn, whereas the OH group connects to the silanol group through hydrogen bonding, as illustrated in Figure 8. Rai et al. call this transition state a “3 ‘H’ shuttle transition state” to highlight the importance of three hydrogen atoms: (1) a proton atom that is transferred from glucose to the Sn−OH group; (2) a hydrogen atom of a silanol group; (3) a hydride that undergoes hydride transfer from the C1 atom to the C2 atom of glucose. Such coordination facilitates hydride transfer and subsequent proton transfer in a single step with a lower energy of activation. A concerted mechanism, typical of enzymatic catalysis, has been demonstrated to be energetically favorable for isomerization catalyzed by Sn‐β. Interestingly, the same group has also performed calculations for the monodentate coordination of a substrate to a Sn metal center, that is, considering an adjacent silanol group as a spectator (Figure 8).74 In this case, epimerization of glucose into mannose is energetically more favorable than isomerization into fructose. Epimerization through monodentate coordination is predicted to take place through a carbon shift, also known as the Bílik mechanism (see Section 4 for details). Later on, Bermejo‐Deval et al.76 reported experimental evidence supporting the conclusions of Rai et al.74 Bermejo‐Deval et al. have performed cationic exchange of H+ of the silanol groups for Na+ to exclude the possibility that the silanol groups participate in catalysis. Sn‐β‐containing silanol groups in the OH form (Sn‐β) isomerize glucose into fructose (Figure 8, left), whereas the catalyst in the Na+ form (Na‐Sn‐β) catalyzes the epimerization of glucose into mannose (Figure 8, right). The experimental studies have also confirmed the predicted74 change in the reaction mechanism: isomerization over Sn‐β takes place through intramolecular hydride shift, whereas glucose–mannose epimerization in the presence of Na‐Sn‐β occurs through intramolecular carbon shift. Additionally, Sn‐β can be treated with ammonia prior to the catalytic tests, which results in adsorption of NH3 on the open sites (Figure 7). Such a blockage of the open sites causes a dramatic decrease in the catalytic activity, and this confirms the significance of the open sites for catalysis.76 These tendencies are observed for both water and methanol solvents.76 Very recently, Brand et al. have reported further evidence to support the fact that an adjacent silanol group participates in the hydride transfer mechanism. In this case, molecular complexes of Sn (tin silsesquioxanes) have been used to model the open and closed sites of Sn‐β. Results in line with the model depicted in Figure 8 have been acquired.77 To sum up, recent reports suggest that isomerization of glucose into fructose takes place on the open sites of Sn‐β, and a Sn metal site with an adjacent silanol group presents an active site for isomerization (Figure 8).
Figure 8.
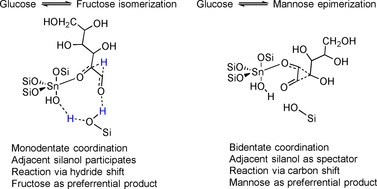
Transition states proposed by Rai et al.74 for glucose isomerization (left) or epimerization (right) in the presence of Sn‐β.
Understanding of structure–activity relationships is a key point for optimization of a solid catalyst. As described above, Sn‐β is the first solid Lewis acid uncovered for isomerization.46 Sn‐β surpasses Ti‐β in terms of catalytic activity.46, 68a Li et al. reported the results of a computational study on metal (M)‐containing BEA (β polymorph A) zeolite considering different metals as active sites in the zeolite matrix, namely, Sn, Ti, Zr, V, Nb, Si, and Ge. The lowest energy barriers for glucose isomerization were found forSn and Zr.69
The role of the hydrophobic matrix in catalysis over Sn‐β has been under discussion. The first reported Sn‐β catalyst46 was prepared according to a method reported by Corma et al.61 to obtain a material containing isolated Sn ions surrounded by a matrix of silicalite with a Sn‐to‐Si molar ratio of approximately 1:100.7, 46 The high hydrophobicity of zeolites results from its high crystallinity and very low defect density. The obtainment of such an ideal structure requires the use of HF as a reagent, because the traditional synthesis of zeolites under alkaline conditions gives rise to hydrophilic materials.66a, 78 Experimental evidence suggests that the hydrophobic matrix of Sn‐β protects the active sites from deactivation through contact with bulk water.46, 79 Osmundsen et al. have comparatively studied the catalytic activity of Sn‐BEA, Sn‐MCM‐41, and Sn‐SBA‐15. Sn‐MCM‐41 and Sn‐SBA‐15 exhibit a hydrophilic nature of the pore walls owing to their amorphous structure, as opposed to hydrophobic Sn‐BEA.79 Interestingly, Sn‐BEA demonstrates good catalytic activity in both water and methanol solvents. Conversely, Sn‐MCM‐41 and Sn‐SBA‐15 exhibit significantly higher catalytic activity in methanol than in water.79 These results indicate that the hydrophobic matrix of β silicalite prevents interaction of the Sn metal centers with bulk water, whereas the active centers of Sn‐MCM‐41 and Sn‐SBA‐15 undergo deactivation in aqueous media.79 Gounder and Davis have studied glucose isomerization in the presence of Ti‐containing solid Lewis acids: Ti‐β‐OH (a material with a high concentration of defects aged in NaOH medium) and Ti‐β‐F (a material with a low concentration of defects aged in fluorine medium). Ti‐β‐F has higher catalytic activity than Ti‐β‐OH, and this has been explained in terms of the hydrophobic and hydrophilic natures of the respective titanosilicates.78 At the same time, solid‐state 119Sn NMR spectroscopy and calorimetry data suggest that the metal centers of Sn‐β and Ti‐β are hydrated under ambient conditions.66a, 68a, 76 Thus, octahedral coordination is typical for the Sn active sites of Sn‐β, even though the active centers are surrounded by a hydrophobic matrix (Figure 7). Consequently, coordination of glucose to Sn proceeds by dehydration of the metal center, transfer of glucose from the aqueous solution into the hydrophobic silicalite pocket, and sorption of glucose. Bai et al. have found that the entropy of glucose transfer from aqueous solution into silicalite is rather large and positive.80 It has also been found that glucose isomerization is accelerated if methanol is used as the solvent instead of water. This effect is explained by the fact that methanol shows better wettability of the hydrophobic zeolite walls than water.66a
Potential diffusion hindrances for penetration of a carbohydrate substrate into the zeolite grain have been considered in a series of investigations. The high catalytic activity of Sn‐β has been explained by its appropriate pore diameter (≈0.7 nm), which does not hinder diffusion of the glucose molecules (0.85 nm in size).78 Gounder and Davis have used the kinetic isotopic effect to prove that glucose isomerization occurs over Ti‐β in the kinetic regime.66a Contrary to zeolites with β topology, tin‐containing zeolites with MFI topology exhibit low catalytic activity for glucose isomerization. This can be explained by the pore diameter of MFI, which is too small at approximately 0.55 nm.46, 65b, 75b, 78 Dapsens et al. have comparatively investigated the catalytic activity of a conventional Sn‐MFI [external surface area (S ext): 49 m2 g−1] versus that of hierarchical mesoporous Sn‐MFI (S ext=133 m2 g−1). Opposite to conventional Sn‐MFI, mesoporous Sn‐MFI is catalytically active for the isomerization of glucose and even disaccharide lactose. Conversion of xylose into xylulose over mesoporous Sn‐MFI is faster than over conventional Sn‐MFI.56 Cho et al. report a similar increase in the reaction rate for xylose isomerization upon using templating synthesis of Sn‐MFI.65c
The majority of recent investigations focus on C2 isomeric ketose as a target product of isomerization. In addition, for nearly every investigation the formation of an epimeric aldose (Figure 1) occurs, though in somewhat lower amounts than the ketose (Table 3, for more details see Section 4). Additionally, Gounder and Davis have uncovered the parallel conversion of d‐glucose over Ti‐β into d‐fructose and l‐sorbose.12 The ratio of k fructose/k sorbose depends on the solvent, and the formation of sorbose predominates in methanol.12, 66a A mechanism occurring through C1–C5 hydride shift has been proposed on the basis of NMR spectroscopy investigation and a study of the reaction kinetics (Figure 9).12
Table 3.
Literature data on the catalytic activity of Lewis acids for the isomerization of aldoses.
| Entry | Reaction conditions | Results[a] | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| catalyst | substrate | substrate concentration [wt %] | T [°C] | substrate/catalyst[b] | substrate/metal[c] | X [%] | S ketose [%] | Y ketose [%] | S epimeric aldose [%] | Y epimeric aldose [%] | ||
| Isomerization using water as solvent | ||||||||||||
| 1 | Sn‐β[d] | glucose | 10[e] | 110 | 1.5 | 50 | 55 | 58 | 32 | 16 | 9 | 46 |
| 2 | Sn‐β[f] | glucose | 10 | 110 | 5 | 926 | 45 | 67 | 30 | 17 | 8 | 55 |
| 3 | meso‐Sn‐MFI | glucose | 3 | 80 | 1.5 | 381 | 35 | 72 | 25 | 3 | 1 | 56 |
| 4 | CrCl3 | glucose | 4.5 | 120 | – | 10 | 52 | 49 | 25 | n.d. | n.d. | 57 |
| 5 | AlCl3 | glucose | 4.5 | 120 | – | 10 | 32 | 83 | 26 | n.d. | n.d. | 57 |
| 6 | SnCl4 | glucose | 4.5 | 120 | – | 10 | 18 | 26 | 5 | n.d. | n.d. | 57 |
| 7 | Sn‐β[f] | mannose | 10 | 110 | 5 | 926 | 75 | 63 | 47 | 18 | 13 | 55 |
| 8 | Sn‐β[f] | galactose | 10 | 110 | 5 | 926 | 38 | 67 | 25 | 5 | 2 | 55 |
| 9 | Sn‐β[d] | xylose | 10 | 100 | 1.2 | 50 | 60 | 45 | 27 | 18 | 11 | 7 |
| 10 | meso‐Sn‐MFI | xylose | 3 | 80 | 1.5 | 458 | 18 | 44 | 8 | trace | trace | 56 |
| 11 | meso‐Sn‐MFI | lactose | 3 | 80 | 1.5 | 201 | 30 | 80 | 24 | n.d. | n.d. | 56 |
| 12 | Sn‐β[f] | ribose | 10 | 110 | 5 | 926 | 69 | 29 | 20 | 37 | 25 | 55 |
| 13 | H‐USY | erythrose | 1.1 | 120 | 1.7 | – | 52 | 75 | 39 | 4 | 2 | 58 |
| Two‐step isomerization using methanol and water as solvents (Figure 10) | ||||||||||||
| 14 | H‐USY | glucose | 3[g] | 120 | 1.7–12.5 | – | 73 | 75 | 55 | n.d. | n.d. | 59 |
| 15 | H‐USY | xylose | 3 | 100 | 1.7–12.5 | – | 65 | 72 | 47 | trace | trace | 60 |
[a] Conversion (X), selectivity (S), and yield (Y). [b] Mass ratio: initial mass of substrate divided by mass of catalyst; n.d.=not determined. [c] Molar ratio of initial substrate amount to amount of Lewis acid. [d] Sn‐β was prepared according to the procedure of Corma et al.61 [e] Up to 45 wt % is possible with nearly the same efficiency. [f] Sn‐β was prepared by grafting of Sn to dealuminated zeolite. [g] Similar results were obtained for solutions with a glucose concentration up to 16.7 wt %.
Figure 9.
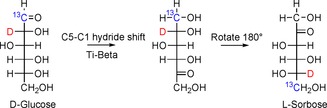
Isomerization of d‐glucose into l‐sorbose catalyzed by Ti‐β through C5–C1 hydride shift.12, 66a
In addition to the products of isomerization, a number of byproducts also form and the mass balances are often not closed. Retro‐aldolization of carbohydrates68b and the formation of lactic acid65a, 68b have been reported as side reactions over solid Lewis acids. In addition, the formation of HMF,65a, 68b furfural,65a and levulinic acid68b is observed. Exactly similar to basic catalysts, the yield of glucose–fructose isomerization does not exceed approximately 35 % (Table 3). In some studies, thermodynamic equilibrium is reached, but at the expense of an overall decrease in mass balance.46, 65a
The stability of a catalyst is a crucial point determining its potential applicability on large scale. Deactivation of Sn‐β during the course of the reaction has been shown.82 Nevertheless, Sn‐β can be regenerated by calcination in air to restore its catalytic activity.7, 46, 55 Filtration tests have demonstrated that the isomerization of glucose is truly heterogeneously catalyzed in the presence of Sn‐β.55 Leaching of Sn from hierarchical Sn‐MFI during isomerization has been reported, though it can be suppressed by using ethanol or methanol as the solvent instead of water.56 Lari et al. have comparatively studied the stabilities of Sn‐containing zeolites with different topologies (e.g., MFI, MOR, BEA, and FAU) for the isomerization of xylose under continuous operation. Two methods of catalyst preparation have been investigated, namely, hydrothermal synthesis and alkaline‐assisted metalation. The hydrophobicity of the zeolites appears to play a key role in the stability of the materials. Thus, hydrothermally prepared Sn‐β demonstrates good stability during 24 h on stream. Deactivation of other materials is observed owing to Sn loss, partial amorphization of the zeolites, restructuring of the Sn active sites, and fouling. The authors draw attention to the chelation capability of xylose, a representative polyol. As a result, significant leaching of Sn is observed for many materials.83
Notably, optimization of the synthetic procedure used to prepare Sn‐β has also attracted significant attention.55, 84 As stated above, the synthesis of hydrophobic Sn‐β requires ageing of the material in the presence of HF for a long period of time. Therefore, the up‐scaling of catalyst synthesis is expected to be challenging owing to the coproduction of dangerous wastes. In recent years, more environmentally benign methods for the synthesis of Sn‐β have been elaborated. Advances in this field have been comprehensively reviewed by Dapsens et al.71b
The use of zeolites in the H+ form as catalysts for isomerization has been suggested by Saravanamurugan et al. They propose a two‐step procedure for the preparation of fructose, as presented in Figure 10. The first step produces methyl fructoside in methanol. The second step involves hydrolysis of methyl fructoside in aqueous media to release fructose. Both steps are catalyzed by the same catalyst, which is a zeolite in hydrogen form. Fructose can be obtained in yields as high as 55 %. Screening of materials suggests that the catalytic activity of H‐USY exceeds that of H‐Y and H‐β. This is explained by an optimal ratio of strong and medium acid sites for H‐USY.59 The same protocol has successfully been applied to the isomerization of xylose into xylulose.60 Remarkably, the isomerization of tetroses, that is, erythrose and threose to erythrulose, efficiently proceeds over an H+ zeolite in aqueous media. Surprisingly, the reaction rate decreases upon using alcohols as solvents instead of water.58
Figure 10.
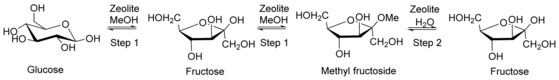
Isomerization of glucose into fructose in a two‐step process catalyzed by a zeolite in the H+ form. Step 1 (in methanol): isomerization of glucose into fructose and formation of methyl fructoside; step 2 (in water): hydrolysis of methyl fructoside releasing fructose.59
In addition to solid Lewis acids, molecular salts are catalytically active for the isomerization of glucose into fructose. According to Tang et al., the catalytic activity of the soluble catalysts decreases in a row: CrCl3>AlCl3>SnCl4.57 Insight into the structure of the active species for CrCl3 has been provided by Choudhary et al. through the use of computational methods and extended X‐ray absorption fine structure (EXAFS). They note that a partially hydrolyzed [Cr(H2O)5(OH)]2+ cation is responsible for the isomerization of glucose into fructose.81 Interestingly, the Cr‐containing metal–organic framework MIL‐101 demonstrates comparatively low catalytic activity for isomerization.85 Tang et al. have demonstrated that [Al(OH)2aq]+ are the active centers for isomerization in the presence of aluminum salts in water.57 Analogously to catalysis by Sn‐β, isomerization of glucose over soluble Lewis acids proceeds by C2–C1 hydride shift as a rate‐limiting step.57, 81 Choudhary et al. have performed a comparative study and have identified a similar reaction mechanism for the isomerization of glucose over solid (Sn‐β) and soluble (AlCl3 and CrCl3) Lewis acids.81 It is suggested that an OH group on the metal center assists in the deprotonation step57, 68b, 81, 86 (Figure 11).
Figure 11.
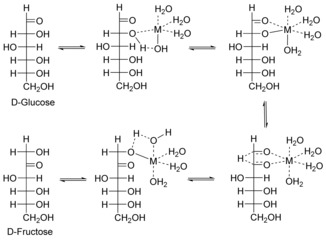
Mechanism of glucose isomerization into fructose in the presence of soluble Lewis acids.57, 68b, 81
An advantage of isomerization in the presence of Lewis acids is compatibility of these catalysts with Brønsted acids. Therefore, one‐pot processes utilizing a combination of these catalysts can be implemented. For example, glucose can be isomerized over a Lewis acid to obtain fructose, and subsequent dehydration of fructose catalyzed by H+ yields HMF.6j, 16d Analogously, a one‐pot process starting from xylose to yield furfural has been reported.7 Interestingly, both solid and soluble Lewis acids have been proposed for such one‐pot reactions. For example, CrCl3,81 AlCl3,87 and lanthanide salts88 have been intensively studied for the one‐pot conversion of glucose into HMF. A number of reports consider combinations of Brønsted acids with solid Lewis acids including Sn‐β,7 Nb2O5 ⋅ n H2O,89 Cr‐based heteropoly acid ionic crystal,90 Ti phosphates,91 and dealuminated β zeolite.92 Nevertheless, there are some concerns regarding the long‐term stability of zeolites in hot water in the presence of Brønsted acids and salts.93
4. Epimerization of Aldoses
Epimerization of aldoses at the C2 atom (Figure 1) has received much less attention than their isomerization into ketoses. In fact, to date only a few chemocatalytic systems have been uncovered for selective epimerization. As mentioned in Sections 2 and 3, epimerization takes place in the presence of basic catalysts, as well as Lewis acids (Table 4). However, in both cases, formation of ketoses predominates (Tables 1, 2–3). If catalyzed by a base, isomerization takes place via an enediol intermediate, as shown in Figure 3. Prevailing formation of ketoses over epimeric aldoses has been explained by de Wit in terms of entropy of activation. Thus, glucose isomerization into fructose requires little reorganization of the intermediate, whereas formation of mannose takes place through rotation around the C2−C3 bond (Figure 3).19 The rotation is connected with substantial reorganization of the water shell, and thus, mannose is formed more slowly than fructose.19
Table 4.
Literature data on the catalytic epimerization of aldoses.
| Entry | Reaction conditions[a] | Results[b] | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| substrate | catalyst | substrate concentration [wt %] | T [°C] | substrate/catalyst[c] | substrate/metal[d] | X [%] | S [%] | Y [%] | ||
| Epimerization by molecular complexes | ||||||||||
| 1 | glucose | Ca(OH)2 | 1.4 | 65 | – | 0.25 | 40 | 53 | 21 | 95 |
| 2 | glucose | Ni/amine | 1.2 | 80 | – | 1 | 29 | 100 | 29 | 96 |
| Catalytic epimerization | ||||||||||
| 3 | glucose | molybdic acid | 17 | 95 | 100 | 100 | 25 | 100 | 25 | 97 |
| 4 | glucose | Sn‐β+sodium borate | 5 | 85 | 2.5 | 100 | 16 | 94 | 15 | 98 |
| 5 | glucose | H3PMo12O40 | 10 | 100 | – | 50 | 31 | 90 | 28 | 99 |
| 6 | glucose | HNbMoO6 | 10 | 120 | 30 | 48 | 33 | 88 | 29 | 100 |
| 7 | mannose | molybdic acid | 17 | 95 | 100 | 100 | 75 | 100 | 75 | 97 |
| 8 | mannose | Sn‐β+sodium borate | 5 | 85 | 2.5 | 100 | 17 | 82 | 14 | 98 |
| 9 | mannose | HNbMoO6 | 10 | 120 | 30 | 48 | 67 | 96 | 64 | 100 |
| 10 | xylose | Sn‐β+sodium borate | 5 | 85 | 2.5 | 100 | 30 | 67 | 20 | 98 |
| 11 | xylose | HNbMoO6 | 10 | 120 | 30 | 57 | 31 | 99 | 31 | 100 |
| 12 | arabinose | HNbMoO6 | 10 | 120 | 30 | 57 | 31 | 76 | 24 | 100 |
| 13 | arabinose | Sn‐β+sodium borate | 5 | 85 | 2.5 | 100 | 34 | 88 | 30 | 98 |
[a] All results are for reactions performed in aqueous media. [b] Conversion (X), selectivity (S), and yield (Y) are given for C2 epimeric aldose. [c] Mass ratio: initial mass of substrate divided by mass of catalyst. [d] Molar ratio of initial substrate amount to amount of metal ions.
Selective epimerization of aldoses can proceed through the formation of molecular complexes. The calcium‐catalyzed epimerization was discovered by Kusin in 1936.94 Much later, this process was revisited and investigated by Yanagihara et al.95, 101 and Angyal.102 It has been suggested that, under alkaline conditions, glucose forms complexes with selected cations, such as Ca2+, La3+, and Nd3+.95, 101, 102 Angyal supposes a rare tetradentate coordination of glucose to the metal center.102 The epimerization catalyzed by metal ions proceeds through rearrangement of the carbon chain, as shown in Figure 12 a. The bond of C3 migrates from C2 to C1 to give rise to an inverted configuration at C2. The Ca‐catalyzed epimerization requires a threo configuration at C3 and C4, which limits the substrate scope of this reaction. More details on this epimerization can be found in a literature survey reported by Angyal.22 In addition, aldoses can be epimerized by reaction systems containing a nickel complex with diamine ligands.103 This epimerization proceeds through formation of a ternary nickel/amine/saccharide complex intermediate103b through the carbon shift illustrated in Figure 12 a.104 The reaction system proves to be feasible for epimerization of a variety of aldose substrates, which was recently comprehensively reviewed by Osanai.104 Herein, we would like to point out an interesting peculiarity of Ni‐catalyzed epimerization. The reaction proceeds smoothly in methanol for different diamine ligands. However, only nickel complexes with hydrophobic ligands (i.e., with alkyl chain lengths >C10) exhibit catalytic activity in aqueous media, whereas complexes with hydrophilic ligands are completely inactive. It has been shown that Ni complexes bearing hydrophobic ligands agglomerate in water to form metallomicelles.96 The hydrophobic environment of these metallomicelles plays a crucial role in catalysis, in line with the recently uncovered high importance of hydrophobic pores of Sn‐β (Section 3).
Figure 12.
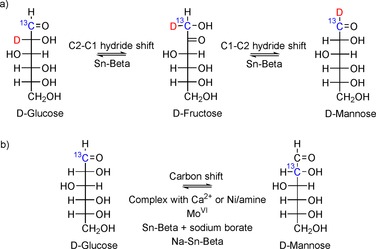
Mechanism of glucose epimerization into mannose by a) carbon shift and b) two successive hydride shifts.
Epimerization through the formation of molecular complexes with Ca2+ or Ni/amines exhibits a number of advantages, including very mild reaction conditions. Indeed, the reaction can be completed at 65 °C in 10–15 min.22, 104 Additionally, epimerization proceeds by complexation, and the yield of the product is limited by the thermodynamics of the complexes, not the saccharides. Consequently, the use of an excess amount of the complexing agent enables thermodynamically predicted yields to be surpassed. For instance, the thermodynamic equilibrium of mannose/glucose is approximately 28:72, but mannose yields above 50 % have been reported in the presence of Ca(OH)2 102 and Ni/amine.103a At the same time, it should be noted that epimerization by molecular complexes utilizes an equimolar or higher amount of the complexing metal with respect to the substrate concentration. If lower amounts of Ca(OH)2 are used, isomerization yielding a ketose predominates in the obtained alkaline medium.22 Brunner and Opitz have reported a significant decrease in the catalytic activity of Ni/amine upon using substoichiometric quantities of catalysts. Thus, mannose yields drop from 47 % for a glucose/nickel molar ratio of 1:1 to 10 % for a glucose/nickel molar ratio of 10:1.105
In this respect, discovery of an efficient molybdenum catalyst by Bílik in the 1970s has attracted much attention. The epimerization takes place under mild reaction conditions, that is, the reaction equilibrium in 10–20 wt % aqueous solution of a substrate is usually reached after 2–6 h at 70–90 °C by utilizing 0.1–0.2 % molybdic acid.106 The reaction is very sensitive to the pH of the solution, and the highest epimerization rate is obtained in the pH range of 1.5 to 3.5. The epimerization follows the carbon‐shift mechanism (Figure 12 a) through coordination of a substrate toward a MoVI dimer. Interestingly, the epimerization of aldoses by carbon shift is sometimes referred to as the “Bílik mechanism” or the “Bílik reaction”. A wide scope of substrates can be epimerized in the presence of molybdenum catalysts, though some limitations for substrates have been reported. The substrate should have a carbon chain length of at least four carbon atoms with hydroxy groups at C2, C3, and C4. Importantly, the Bílik reaction was very quickly scaled up to a pilot plant running in Bratislava. A large number of publications and patents focusing on Bílik epimerization highlight the great commercial importance of this process. Given that the early literature was thoroughly reviewed by Petruš et al.,106 herein, we concentrate on more recent publications. Chethana et al. have performed a computational study explaining the dependence of the rate of epimerization on the pH. They have found that under optimized conditions (pH 1.5–3.5), the concentration of the dimeric MoVI species is maximal.107 Ju et al. have recently reported the high catalytic activity of molybdenum‐based polyoxometalates for the epimerization of glucose. The catalytic activity of H3PMo12O40, Ag3PMo12O40, and Sn0.75PMo12O40 has been demonstrated.99 A one‐pot process combining hydrolysis of starch and glucose/mannose epimerization has been performed by Hricovíniová. An equilibrium mixture of glucose and mannose was obtained.108 Numerous efforts have been made to produce solid catalysts containing molybdenum(VI) species to perform epimerization continuously. For instance, immobilization of molybdate species on ion‐exchange resins has been performed.109 Köckritz et al. have studied the long‐term stability of immobilized molybdates with 800 h on stream. A slow decrease in catalytic activity has been observed mostly because of leaching of the active species into the liquid phase.110 Additionally, heptamolybdate exchanged on quaternary ammonia modified SBA‐15‐type mesoporous silica exhibits good activity and stability.111 Takagaki et al. have recently demonstrated the catalytic activity of the layered niobium molybdates LiNbMoO6 and HNbMoO6. The latter is also efficient for the one‐pot hydrolysis–epimerization of cellobiose, which leads to an equilibrium mixture of glucose and mannose.100 Noteworthy, some authors have reported the reduction of MoVI species during epimerization.99, 110 Nevertheless, the catalyst can be reoxidized upon treatment with dilute H2O2 solution.110, 112
Lewis acids catalyze epimerization, but the selectivity for epimeric aldose is low compared to that of the ketose (Table 3). Bermejo‐Deval et al. suggest that glucose epimerization into mannose over Sn‐β takes place by two consecutive hydrogen shifts (Figure 12 b).76 Notably, this mechanism occurs over the open sites of Sn‐β, that is: (1) if no salt is added to the reaction mixture to exchange the protons of the silanol groups with cations; (2) water or methanol is used as the solvent; (3) fructose is produced as the major product. Computational studies confirm that the energetically favored formation of mannose proceeds over the open sites of Sn‐β through two hydride shifts.113 Choudhary et al. suggest the formation of lyxose during isomerization of xylose to proceed through a similar pathway with the same intermediate for xylulose and lyxose formation.68c
In fact, Sn‐β can be used as a selective catalyst for preferential epimerization if used in combination with salts. Gunther et al. report the selective aqueous‐phase epimerization of glucose, mannose, xylose, and arabinose catalyzed by Sn‐β+sodium borate {with molecular formula Na2[B4O5(OH)4]⋅ 8 H2O}.98, 114 The epimerization takes place through carbon shift (Figure 12 a). Under the same reaction conditions without sodium borate, formation of ketoses dominates over epimerization. It is suggested that complexation of the borate anion with saccharides leads to a change in the reaction mechanism. Formation of the borate–saccharide complexes confined in the pores of Sn‐β has been confirmed by means of solid‐state NMR spectroscopy.98 Calculations performed by using density functional theory suggest that a glucose complex with tetrahedral borate inhibits competitive isomerization.115 More recently, Bermejo‐Deval et al. have demonstrated that epimerization of glucose into mannose through carbon shift also takes place over Na+‐exchanged Sn‐β.76 The postsynthetic exchange of the protons of the silanol groups with Na+ leads to the prevailing formation of mannose in both water and methanol as solvents. This effect has also been suggested by Rai et al., who used a computational approach. If the silanol group adjacent to a Sn metal center does not participate in the transition state, carbon shift is predicted to become a more energetically favorable mechanism (Figure 8 right).74 Nevertheless, sodium‐exchanged Sn‐β is unstable and leaching of Na+ into solution is observed during the course of the reaction.76
5. Conclusions and Outlook
Summarizing the recently published literature on the isomerization of monosaccharides, a few breakthroughs can be highlighted. First, the catalytic performance of solid bases appears to be outstanding compared to that of soluble bases, especially in terms of selectivity for ketoses. Though the catalytic activity of basic catalysts for isomerization has been known for more than a century, the application of basic catalysts on a commercial level has been hindered mostly because of low selectivity. The good catalytic performance of solid bases is indicative of the high potential of these materials. Second, the discovery of Lewis acids with high catalytic activity in the aqueous phase has significantly broadened the range of catalysts for the transformation of monosaccharides. The superior catalytic performance of Sn‐β zeolite is a fascinating example of a chemocatalyzed reaction occurring through a concerted mechanism, analogously to enzymatic catalysis. So far, catalytic systems for the isomerization of d‐glucose into d‐fructose (Sn‐β), l‐sorbose (Ti‐β), and d‐mannose (Sn‐β+sodium borate) have been uncovered. We are convinced that ongoing intensive work in this area will result in new interesting catalytic systems for the isomerization of monosaccharides in the near future.
As an outlook, we would like to put forward the following points. The long‐term stability of catalysts for isomerization is of great interest, as porous catalysts are expected to be influenced by hot water. The chelating effect of saccharides can result in redistribution of metal species. Moreover, strong adsorption of (by)products potentially causes deactivation of the catalysts. Therefore, investigation of the catalytic isomerization processes under continuous conditions will be informative in terms of catalytic activity and selectivity as a function of time on stream. Additionally, the majority of investigations currently report on isomerization leading to equilibrium mixtures of isomers. However, efficient recovery of individual substances from these mixtures is crucial, as recovery and purification processes can be limiting factors for process economy, even if catalysis is very efficient. Finally, insight into the structure of the active species as well as the isomerization mechanisms and reaction networks will facilitate the establishment of structure–activity relationships. We believe that addressing these challenges will accelerate the implementation of newly discovered catalytic processes on a commercial level.
Biographical Information
Irina Delidovich received her diploma in chemistry in 2008 from the Novosibirsk State University. In 2011, she obtained her Ph.D. degree from the Boreskov Institute of Catalysis (BIC) under the supervision of Professor Oxana Taran. During her stay at BIC (2006–2012), she worked on the aldol addition and selective oxidation of carbohydrates. Since 2012, she has been conducting studies on the conversion of cellulosic biomass in the research group of Prof. Palkovits. Her scientific interests include the synthesis and characterization of solid catalysts as well as catalytic transformations of saccharides.

Biographical Information
Regina Palkovits is a Full Professor for Heterogeneous Catalysis & Chemical Technology at RWTH Aachen University. She graduated in Chemical Engineering from the Technical University Dortmund and carried out her Ph.D. studies under the supervision of Prof. Ferdi Schüth at the Max‐Planck‐Institut für Kohlenforschung from 2003–2006. Afterwards, she joined the group of Prof. Bert Weckhuysen at Utrecht University as a postdoctoral fellow. In 2008, she returned as a group leader to the Max‐Planck‐Institut für Kohlenforschung, and since 2010, she has been a professor at RWTH Aachen University.

Acknowledgements
We thank the Alexander von Humboldt Foundation and the Bayer Foundation for financial support. This work was performed as part of the Cluster of Excellence “Tailor‐Made Fuels from Biomass” funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities.
I. Delidovich, R. Palkovits, ChemSusChem 2016, 9, 547.
References
- 1.
- 1a. Dapsens P. Y., Mondelli C., Pérez-Ramírez J., ACS Catal. 2012, 2, 1487–1499; [Google Scholar]
- 1b. Serrano-Ruiz J. C., Luque R., Sepulveda-Escribano A., Chem. Soc. Rev. 2011, 40, 5266–5281; [DOI] [PubMed] [Google Scholar]
- 1c. Rinaldi R., Schüth F., Energy Environ. Sci. 2009, 2, 610–626. [Google Scholar]
- 2.
- 2a. Lichtenthaler F. W. in Biorefineries—Industrial Processes and Products: Status Quo and Future Directions, Vol. 2 (Eds.: B. Kamm, P. R. Gruber, M. Kamm), Wiley-VCH, Weinheim, 2006; [Google Scholar]
- 2b. van Bekkum H., Besemer A. C., Chem. Sustainable Dev. 2003, 11, 11–21. [Google Scholar]
- 3.
- 3a. Bozell J. J., Petersen G. R., Green Chem. 2010, 12, 539–554; [Google Scholar]
- 3b.T. Werpy, G. R. Petersen, Top Value Added Chemicals from Biomass, Vol. I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas, US Department of Energy, Washington, DC, 2004, www.nrel.gov/docs/fy04osti/35523.pdf.
- 4.
- 4a. Mäki-Arvela P., Salmi T., Holmbom B., Willför S., Murzin D. Y., Chem. Rev. 2011, 111, 5638–5666; [DOI] [PubMed] [Google Scholar]
- 4b. Huang Y.-B., Fu Y., Green Chem. 2013, 15, 1095–1111. [Google Scholar]
- 5. Gallezot P., Chem. Soc. Rev. 2012, 41, 1538–1558. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Hara M., Nakajima K., Kamata K., Sci. Technol. Adv. Mater. 2015, 16, 034903; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Kobayashi H., Fukuoka A., Green Chem. 2013, 15, 1740–1763; [Google Scholar]
- 6c. Zhou C.-H., Xia X., Lin C.-X., Tong D.-S., Beltramini J., Chem. Soc. Rev. 2011, 40, 5588–5617; [DOI] [PubMed] [Google Scholar]
- 6d. Van de Vyver S., Geboers J., Jacobs P. A., Sels B. F., ChemCatChem 2011, 3, 82–94; [Google Scholar]
- 6e. Climent M. J., Corma A., Iborra S., Green Chem. 2011, 13, 520–540; [Google Scholar]
- 6f. Mäki-Arvela P., Holmbom B., Salmi T., Murzin D. Y., Catal. Rev. 2007, 49, 197–340; [Google Scholar]
- 6g. Huber G. W., Corma A., Angew. Chem. Int. Ed. 2007, 46, 7184–7201; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7320–7338; [Google Scholar]
- 6h. Corma A., Iborra S., Velty A., Chem. Rev. 2007, 107, 2411–2502; [DOI] [PubMed] [Google Scholar]
- 6i. Huber G. W., Iborra S., Corma A., Chem. Rev. 2006, 106, 4044–4098; [DOI] [PubMed] [Google Scholar]
- 6j. Delidovich I., Leonhard K., Palkovits R., Energy Environ. Sci. 2014, 7, 2803–2830. [Google Scholar]
- 7. Choudhary V., Pinar A. B., Sandler S. I., Vlachos D. G., Lobo R. F., ACS Catal. 2011, 1, 1724–1728. [Google Scholar]
- 8. Oh D.-K., Appl. Microbiol. Biotechnol. 2007, 76, 1–8. [DOI] [PubMed] [Google Scholar]
- 9.Z. Ahmed, E.-J. Biotechnol 2001, DOI: 10.2225/vol4-issue2-fulltext-7.
- 10. Ma T., Pai S. B., Zhu Y. L., Lin J. S., Shanmuganathan K., Du J., Wang C., Kim H., Newton M. G., Cheng Y. C., Chu C. K., J. Med. Chem. 1996, 39, 2835–2843. [DOI] [PubMed] [Google Scholar]
- 11. Samuel J., Tanner M. E., Nat. Prod. Rep. 2002, 19, 261–277. [DOI] [PubMed] [Google Scholar]
- 12. Gounder R., Davis M. E., ACS Catal. 2013, 3, 1469–1476. [Google Scholar]
- 13. Hanover L. M., White J. S., Am. J. Clin. Nutr. 1993, 58, 724–732. [Google Scholar]
- 14.
- 14a. Parker K., Salas M., Nwosu V. C., Biotechnol. Mol. Biol. Rev. 2010, 5, 71–78; [Google Scholar]
- 14b. White J. in Fructose, High Fructose Corn Syrup, Sucrose and Health (Ed.: J. M. Rippe), Springer, New York, 2014, pp. 13–33. [Google Scholar]
- 15. Häusler H., Stütz A. E. in Glycoscience, Vol. 215, Springer, Berlin, 2001, pp. 77–114. [Google Scholar]
- 16.
- 16a. Dashtban M., Gilbert A., Fatehi P., RSC Adv. 2014, 4, 2037–2050; [Google Scholar]
- 16b. Tong X., Ma Y., Li Y., Appl. Catal. A 2010, 385, 1–13; [Google Scholar]
- 16c. Román-Leshkov Y., Chheda J. N., Dumesic J. A., Science 2006, 312, 1933–1937; [DOI] [PubMed] [Google Scholar]
- 16d. Rosatella A. A., Simeonov S. P., Frade R. F. M., Afonso C. A. M., Green Chem. 2011, 13, 754–793. [Google Scholar]
- 17.
- 17a. Delidovich I., Hausoul P. J. C., Deng L., Pfützenreuter R., Rose M., Palkovits R., Chem. Rev. 2016, 116, 1540–1599; [DOI] [PubMed] [Google Scholar]
- 17b. Gandini A., Polym. Chem. 2010, 1, 245–251. [Google Scholar]
- 18. Zakrzewska M. E., Bogel-Łukasik E., Bogel-Łukasik R., Chem. Rev. 2011, 111, 397–417. [DOI] [PubMed] [Google Scholar]
- 19. de Wit G., Kieboom A. P. G., van Bekkum H., Carbohydr. Res. 1979, 74, 157–175. [Google Scholar]
- 20.
- 20a. Parrish F. W., US Patent US 3514327A, 1970;
- 20b. Kooyman C., Vellenga K., De Wilt H. G. J., Carbohydr. Res. 1977, 54, 33–44; [Google Scholar]
- 20c. El Khadem H. S., Ennifar S., Isbell H. S., Carbohydr. Res. 1987, 169, 13–21. [Google Scholar]
- 21. De Bruijn J. M., Kieboom A. P. G., van Bekkum H., Starch/Staerke 1987, 39, 23–28. [Google Scholar]
- 22. Angyal S. J. in Glycoscience, Vol. 215, Springer, Berlin, 2001, pp. 1–14. [Google Scholar]
- 23. de Bruijn J. M., Kieboom A. P. G., van Bekkum H., Recl. Trav. Chim. Pays-Bas 1987, 106, 35–43. [Google Scholar]
- 24. Delidovich I., Palkovits R., Catal. Sci. Technol. 2014, 4, 4322–4329. [Google Scholar]
- 25. Liu C., Carraher J. M., Swedberg J. L., Herndon C. R., Fleitman C. N., Tessonnier J.-P., ACS Catal. 2014, 4, 4295–4298. [Google Scholar]
- 26. Mendicino J. F., J. Am. Chem. Soc. 1960, 82, 4975–4979. [Google Scholar]
- 27. Barker S. A., Somers P. J., Hatt B. W., (Böhringer Mannheim) US 3875140, 1973.
- 28. Shaw A. J., Tsao G. T., Carbohydr. Res. 1978, 60, 376–382. [Google Scholar]
- 29. Despax S., Estrine B., Hoffmann N., Le Bras J., Marinkovic S., Muzart J., Catal. Commun. 2013, 39, 35–38. [Google Scholar]
- 30. Moreau C., Durand R., Roux A., Tichit D., Appl. Catal. A 2000, 193, 257–264. [Google Scholar]
- 31. Yu S., Kim E., Park S., Song I. K., Jung J. C., Catal. Commun. 2012, 29, 63–67. [Google Scholar]
- 32. Lee G., Jeong Y., Takagaki A., Jung J. C., J. Mol. Catal. A 2014, 393, 289–295. [Google Scholar]
- 33. Delidovich I., Palkovits R., J. Catal. 2015, 327, 1–9. [Google Scholar]
- 34. Yang Q., Zhou S., Runge T., J. Catal. 2015, 330, 474–484. [Google Scholar]
- 35. Shaw III A. J., Tsao G. T., Carbohydr. Res. 1978, 60, 327–325. [Google Scholar]
- 36. Rendleman J. A., Hodge J. E., Carbohydr. Res. 1979, 75, 83–99. [Google Scholar]
- 37. Hicks K. B., (US Agriculture) US 4273922A, 1981.
- 38. van den Berg R., Peters J. A., van Bekkum H., Carbohydr. Res. 1994, 253, 1–12. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Lecomte J., Finiels A., Moreau C., Starch/Staerke 2002, 54, 75–79; [Google Scholar]
- 39b. Takagaki A., Ohara M., Nishimura S., Ebitani K., Chem. Commun. 2009, 6276–6278; [DOI] [PubMed] [Google Scholar]
- 39c. Weisgerber L., Palkovits S., Palkovits R., Chem. Ing. Tech. 2013, 85, 512–515. [Google Scholar]
- 40. Shukla R., Verykios X. E., Mutharasan R., Carbohydr. Res. 1985, 143, 97–106. [Google Scholar]
- 41. Souza R. O. L., Fabiano D. P., Feche C., Rataboul F., Cardoso D., Essayem N., Catal. Today 2012, 195, 114–119. [Google Scholar]
- 42. Son P., Nishimura S., Ebitani K., React. Kinet. Mech. Catal. 2014, 111, 183–197. [Google Scholar]
- 43. Lv L., Guo X., Bai P., Shuo Z., Asian J. Chem. 2015, 27, 2774–2778. [Google Scholar]
- 44. Carraher J. M., Fleitman C. N., Tessonnier J.-P., ACS Catal. 2015, 5, 3162–3173. [Google Scholar]
- 45. Vuorinen T., Sjöström E., Carbohydr. Res. 1982, 108, 23–29. [Google Scholar]
- 46. Moliner M., Román-Leshkov Y., Davis M. E., Proc. Natl. Acad. Sci. USA 2010, 107, 6164–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. King-Morris M. J., Serianni A. S., Carbohydr. Res. 1986, 154, 29–36. [DOI] [PubMed] [Google Scholar]
- 48. Simonov A. N., Pestunova O. P., Matvienko L. G., Parmon V. N., Russ. Chem. Bull. Int. Ed. 2005, 54, 1967–1972. [Google Scholar]
- 49. El Khadem H. S., Ennifar S., Isbell H. S., Carbohydr. Res. 1989, 185, 51–59. [Google Scholar]
- 50. Cavani F., Trifirò F., Vaccari A., Catal. Today 1991, 11, 173–301. [Google Scholar]
- 51.
- 51a. Nishimura S., Takagaki A., Ebitani K., Green Chem. 2013, 15, 2026–2042; [Google Scholar]
- 51b. Miyata S., Clays Clay Miner. 1983, 31, 305–311. [Google Scholar]
- 52. Debecker D. P., Gaigneaux E. M., Busca G., Chem. Eur. J. 2009, 15, 3920–3935. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Roelofs J. C. A. A., van Dillen A. J., de Jong K. P., Catal. Today 2000, 60, 297–303; [Google Scholar]
- 53b. Abelló S., Medina F., Tichit D., Pérez-Ramírez J., Groen J. C., Sueiras J. E., Salagre P., Cesteros Y., Chem. Eur. J. 2005, 11, 728–739; [DOI] [PubMed] [Google Scholar]
- 53c. Winter F., Xia X., Hereijgers B. P. C., Bitter J. H., van Dillen A. J., Muhler M., de Jong K. P., J. Phys. Chem. B 2006, 110, 9211–9218. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Climent M. J., Corma A., Iborra S., Epping K., Velty A., J. Catal. 2004, 225, 316–326; [Google Scholar]
- 54b. Chimentão R. J., Abelló S., Medina F., Llorca J., Sueiras J. E., Cesteros Y., Salagre P., J. Catal. 2007, 252, 249–257. [Google Scholar]
- 55. Dijkmans J., Gabriels D., Dusselier M., de Clippel F., Vanelderen P., Houthoofd K., Malfliet A., Pontikes Y., Sels B. F., Green Chem. 2013, 15, 2777–2785. [Google Scholar]
- 56. Dapsens P. Y., Mondelli C., Jagielski J., Hauert R., Perez-Ramirez J., Catal. Sci. Technol. 2014, 4, 2302–2311. [Google Scholar]
- 57. Tang J., Guo X., Zhu L., Hu C., ACS Catal. 2015, 5, 5097–5103. [Google Scholar]
- 58. Saravanamurugan S., Riisager A., Catal. Sci. Technol. 2014, 4, 3186–3190. [Google Scholar]
- 59. Saravanamurugan S., Paniagua M., Melero J. A., Riisager A., J. Am. Chem. Soc. 2013, 135, 5246–5249. [DOI] [PubMed] [Google Scholar]
- 60. Paniagua M., Saravanamurugan S., Melian-Rodriguez M., Melero J. A., Riisager A., ChemSusChem 2015, 8, 1088–1094. [DOI] [PubMed] [Google Scholar]
- 61.
- 61a. Corma A., Nemeth L. T., Renz M., Valencia S., Nature 2001, 412, 423–425; [DOI] [PubMed] [Google Scholar]
- 61b. Camblor M. A., Corma A., Valencia S., J. Mater. Chem. 1998, 8, 2137–2145. [Google Scholar]
- 62. Román-Leshkov Y., Moliner M., Labinger J. A., Davis M. E., Angew. Chem. Int. Ed. 2010, 49, 8954–8957; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9138–9141. [Google Scholar]
- 63. Román-Leshkov Y., Davis M. E., ACS Catal. 2011, 1, 1566–1580. [Google Scholar]
- 64.
- 64a. Caratzoulas S., Davis M. E., Gorte R. J., Gounder R., Lobo R. F., Nikolakis V., Sandler S. I., Snyder M. A., Tsapatsis M., Vlachos D. G., J. Phys. Chem. C 2014, 118, 22815–22833; [Google Scholar]
- 64b. Moliner M., Dalton Trans. 2014, 43, 4197–4208. [DOI] [PubMed] [Google Scholar]
- 65.
- 65a. Holm M. S., Pagan-Torres Y. J., Saravanamurugan S., Riisager A., Dumesic J. A., Taarning E., Green Chem. 2012, 14, 702–706; [Google Scholar]
- 65b. Lew C. M., Rajabbeigi N., Tsapatsis M., Microporous Mesoporous Mater. 2012, 153, 55–58; [Google Scholar]
- 65c. Cho H. J., Dornath P., Fan W., ACS Catal. 2014, 4, 2029–2037. [Google Scholar]
- 66.
- 66a. Gounder R., Davis M. E., J. Catal. 2013, 308, 176–188; [Google Scholar]
- 66b. Ren L., Guo Q., Kumar P., Orazov M., Xu D., Alhassan S. M., Mkhoyan K. A., Davis M. E., Tsapatsis M., Angew. Chem. Int. Ed. 2015, 54, 10848–10851; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10998–11001. [Google Scholar]
- 67. Lambert J. B., Lu G., Singer S. R., Kolb V. M., J. Am. Chem. Soc. 2004, 126, 9611–9625. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a. Bermejo-Deval R., Assary R. S., Nikolla E., Moliner M., Román-Leshkov Y., Hwang S.-J., Palsdottir A., Silverman D., Lobo R. F., Curtiss L. A., Davis M. E., Proc. Natl. Acad. Sci. USA 2012, 109, 9727–9732; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68b. Choudhary V., Pinar A. B., Lobo R. F., Vlachos D. G., Sandler S. I., ChemSusChem 2013, 6, 2369–2376; [DOI] [PubMed] [Google Scholar]
- 68c. Choudhary V., Caratzoulas S., Vlachos D. G., Carbohydr. Res. 2013, 368, 89–95. [DOI] [PubMed] [Google Scholar]
- 69. Li Y.-P., Head-Gordon M., Bell A. T., ACS Catal. 2014, 4, 1537–1545. [Google Scholar]
- 70. Boronat M., Concepción P., Corma A., Renz M., Valencia S., J. Catal. 2005, 234, 111–118. [Google Scholar]
- 71.
- 71a. Corma A., García H., Chem. Rev. 2003, 103, 4307–4366; [DOI] [PubMed] [Google Scholar]
- 71b. Dapsens P. Y., Mondelli C., Perez-Ramirez J., Chem. Soc. Rev. 2015, 44, 7025–7043. [DOI] [PubMed] [Google Scholar]
- 72. Khouw C. B., Davis M. E., J. Catal. 1995, 151, 77–86. [Google Scholar]
- 73. Corma A., Renz M., Collect. Czech. Chem. Commun. 2005, 70, 1727–1736. [Google Scholar]
- 74. Rai N., Caratzoulas S., Vlachos D. G., ACS Catal. 2013, 3, 2294–2298. [Google Scholar]
- 75.
- 75a. Yang G., Pidko E. A., Hensen E. J. M., ChemSusChem 2013, 6, 1688–1696; [DOI] [PubMed] [Google Scholar]
- 75b. Li G., Pidko E. A., Hensen E. J. M., Catal. Sci. Technol. 2014, 4, 2241–2250. [Google Scholar]
- 76. Bermejo-Deval R., Orazov M., Gounder R., Hwang S.-J., Davis M. E., ACS Catal. 2014, 4, 2288–2297. [Google Scholar]
- 77. Brand S. K., Labinger J. A., Davis M. E., ChemCatChem 2016, 8, 121–124. [Google Scholar]
- 78. Gounder R., Davis M. E., AIChE J. 2013, 59, 3349–3358. [Google Scholar]
- 79. Osmundsen C. M., Holm M. S., Dahl S., Taarning E., Proc. R. Soc. London Ser. A 2012, 468, 2000–2016. [Google Scholar]
- 80. Bai P., Tsapatsis M., Siepmann J. I., Langmuir 2012, 28, 15566–15576. [DOI] [PubMed] [Google Scholar]
- 81. Choudhary V., Mushrif S. H., Ho C., Anderko A., Nikolakis V., Marinkovic N. S., Frenkel A. I., Sandler S. I., Vlachos D. G., J. Am. Chem. Soc. 2013, 135, 3997–4006. [DOI] [PubMed] [Google Scholar]
- 82. Rajabbeigi N., Torres A. I., Lew C. M., Elyassi B., Ren L., Wang Z., Je Cho H., Fan W., Daoutidis P., Tsapatsis M., Chem. Eng. Sci. 2014, 116, 235–242. [Google Scholar]
- 83. Lari G. M., Dapsens P.-Y., Scholz D., Mitchell S., Mondelli C., Pérez-Ramírez J., Green Chem. 2016, 18, 1249–1260. [Google Scholar]
- 84.
- 84a. Chang C.-C., Wang Z., Dornath P., Je Cho H., Fan W., RSC Adv. 2012, 2, 10475–10477; [Google Scholar]
- 84b. Kang Z., Zhang X., Liu H., Qiu J., Yeung K. L., Chem. Eng. J. 2013, 218, 425–432. [Google Scholar]
- 85. Akiyama G., Matsuda R., Sato H., Kitagawa S., Chem. Asian J. 2014, 9, 2772–2777. [DOI] [PubMed] [Google Scholar]
- 86. Mushrif S. H., Varghese J. J., Vlachos D. G., Phys. Chem. Chem. Phys. 2014, 16, 19564–19572. [DOI] [PubMed] [Google Scholar]
- 87. Pagán-Torres Y. J., Wang T., Gallo J. M. R., Shanks B. H., Dumesic J. A., ACS Catal. 2012, 2, 930–934. [Google Scholar]
- 88. Zhao S., Guo X., Bai P., Lv L., Asian J. Chem. 2014, 26, 4537–4542. [Google Scholar]
- 89. Nakajima K., Baba Y., Noma R., Kitano M., Kondo J. N., Hayashi S., Hara M., J. Am. Chem. Soc. 2011, 133, 4224–4227. [DOI] [PubMed] [Google Scholar]
- 90. Yi X., Delidovich I., Sun Z., Wang S., Wang X., Palkovits R., Catal. Sci. Technol. 2015, 5, 2496–2502. [Google Scholar]
- 91. Nakajima K., Noma R., Kitano M., Hara M., J. Mol. Catal. A 2014, 388–389, 100–105. [Google Scholar]
- 92. Otomo R., Yokoi T., Kondo J. N., Tatsumi T., Appl. Catal. A 2014, 470, 318–326. [Google Scholar]
- 93. Gardner D. W., Huo J., Hoff T. C., Johnson R. L., Shanks B. H., Tessonnier J.-P., ACS Catal. 2015, 5, 4418–4422. [Google Scholar]
- 94.A. Kusin, Ber 1936, 69, 1041–1049, DOI: 10.1002/cber.19360690524.
- 95. Yanagihara R., Osanai S., Yoshikawa S., Chem. Lett. 1990, 19, 2273–2276. [Google Scholar]
- 96. Osanai S., Yanagihara R., Uematsu K., Okumura A., Yoshikawa S., J. Chem. Soc. Perkin Trans. 2 1993, 1937–1940. [Google Scholar]
- 97. Bilik V., Chem. Zvesti 1972, 26, 183–186. [Google Scholar]
- 98. Gunther W. R., Wang Y., Ji Y., Michaelis V. K., Hunt S. T., Griffin R. G., Román-Leshkov Y., Nat. Commun. 2012, 3, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ju F., VanderVelde D., Nikolla E., ACS Catal. 2014, 4, 1358–1364. [Google Scholar]
- 100. Takagaki A., Furusato S., Kikuchi R., Oyama S. T., ChemSusChem 2015, 8, 3769–3772. [DOI] [PubMed] [Google Scholar]
- 101. Yanagihara R., Soeda K., Shiina S., Osanai S., Yoshikawa S., Bull. Chem. Soc. Jpn. 1993, 66, 2268–2272. [Google Scholar]
- 102. Angyal S. J., Carbohydr. Res. 1997, 300, 279–281. [Google Scholar]
- 103.
- 103a. Tanase T., Shimizu F., Yano S., Yoshikawa S., J. Chem. Soc. Chem. Commun. 1986, 1001–1003; [Google Scholar]
- 103b. Tanase T., Shimizu F., Kuse M., Yano S., Hidai M., Yoshikawa S., Inorg. Chem. 1988, 27, 4085–4094. [Google Scholar]
- 104. Osanai S. in Glycoscience, Vol. 215 (Ed.: A. E. Stütz), Springer, Berlin, 2001, pp. 43–76. [Google Scholar]
- 105. Brunner H., Opitz D., J. Mol. Catal. A 1997, 118, 273–282. [Google Scholar]
- 106. Petruš L., Petrušová M., Hricovíniová Z. in Glycoscience, Vol. 215 (Ed.: A. E. Stütz), Springer, Berlin, 2001, pp. 15–41. [Google Scholar]
- 107. Chethana B. K., Lee D., Mushrif S. H., J. Mol. Catal. A 2015, 410, 66–73. [Google Scholar]
- 108. Hricovíniová Z., Tetrahedron: Asymmetry 2011, 22, 1184–1188. [Google Scholar]
- 109.
- 109a. Dobler W., Ernst H., Paust J., (BASF) US 4778531A, 1988;
- 109b. Dobler W., Paust J., US 5015296A, 1991.
- 110. Köckritz A., Kant M., Walter M., Martin A., Appl. Catal. A 2008, 334, 112–118. [Google Scholar]
- 111. Stockman R., Dekoninck J., Sels B. F., Jacobs P. A., Stud. Surf. Sci. Catal. 2005, 156, 843–850. [Google Scholar]
- 112. Vercauteren R. L. M., Elseviers M., (Cerestar) US 5773606A, 1998.
- 113. Christianson J. R., Caratzoulas S., Vlachos D. G., ACS Catal. 2015, 5, 5256–5263. [Google Scholar]
- 114. Gunther W. R., Duong Q., Román-Leshkov Y., J. Mol. Catal. A 2013, 379, 294–302. [Google Scholar]
- 115. Chethana B. K., Mushrif S. H., J. Catal. 2015, 323, 158–164. [Google Scholar]