

Abstract
Ixazomib is the first oral proteasome inhibitor to be investigated in the clinic. This clinical study assessed whether the pharmacokinetics of ixazomib would be altered if administered after a high‐calorie, high‐fat meal. In a 2‐period, 2‐sequence, crossover study design, adult patients with advanced solid tumors or lymphoma received a 4‐mg oral dose of ixazomib as immediate‐release capsules on day 1 without food (fasted, administered following an overnight fast) or with food (fed, following consumption of a high‐calorie, high‐fat meal), followed by another dose on day 15 in the alternate food intake condition (fasted to fed or fed to fasted). Twenty‐four patients were enrolled; of these, 15 were included in the pharmacokinetic‐evaluable population. Administration of ixazomib after a high‐fat meal reduced both the rate and extent of absorption of ixazomib. Under fed conditions, the median time to peak plasma concentration (Tmax) of ixazomib was delayed by approximately 3 hours compared with administration in the fasted state (1.02 hours vs 4.0 hours), and there was a 28% reduction in total systemic exposure (area under the curve, AUC) and a 69% reduction in peak plasma concentration (Cmax). Together, the results support the administration of ixazomib on an empty stomach, at least 1 hour before or at least 2 hours after food. These recommendations are reflected in the United States Prescribing Information for ixazomib (clinicaltrials.gov identifier NCT01454076).
Keywords: multiple myeloma, ixazomib, pharmacokinetics, food effects
The proteasome inhibitor ixazomib is the first oral, small‐molecule inhibitor of the 20S proteasome.1 The drug substance is administered as a stable citrate ester, designated as ixazomib citrate, formulated as immediate‐release capsules. Under physiological conditions, ixazomib citrate undergoes rapid hydrolysis to the biologically active boronic acid, ixazomib.1, 2, 3 Phase 1 studies have shown single‐agent ixazomib to be generally well tolerated in patients with multiple myeloma (MM),4, 5 AL amyloidosis,6 lymphoma,7 and solid tumors,8 with encouraging evidence of preliminary activity, particularly in patients with MM and AL amyloidosis.4, 5, 6 These early‐phase studies determined the recommended phase 2/3 weekly dose (dosing on days 1, 8, and 15 of each 28‐day cycle) of ixazomib to be 4 mg,4, 5, 6 and a population pharmacokinetic (PK) analysis has demonstrated the feasibility of administering a fixed dose rather than using body‐surface‐area‐based dosing.9 On the basis of these early‐phase data,4, 5, 6 a 4‐mg weekly oral dose of ixazomib was selected for use in phase 3 clinical development in patients with relapsed and/or refractory MM (TOURMALINE‐MM1), in patients with newly diagnosed MM (TOURMALINE‐MM2), as maintenance therapy in patients with MM (TOURMALINE‐MM3, TOURMALINE‐MM4), and in patients with relapsed and/or refractory primary systemic AL amyloidosis (TOURMALINE‐AL1). In these ongoing clinical trials, as well as all ixazomib efficacy trials to date, ixazomib is administered on an empty stomach (at least 1 hour before or at least 2 hours after food). On November 20, 2015, the United States (US) Food and Drug Administration (FDA) granted approval for the use of ixazomib in combination with lenalidomide and dexamethasone for the treatment of patients with MM who have received at least 1 prior therapy,10 based on data from the TOURMALINE‐MM1 trial.
Ixazomib is highly soluble and has moderate permeability (Takeda, data on file); hence, it is considered to be a Biopharmaceutics Classification System (BCS) class 3 compound.11 Ixazomib is rapidly absorbed, with a median time to peak plasma concentration (Tmax) of approximately 1 hour postdose.4 After weekly multiple dosing, ixazomib had a long terminal half‐life of 3.6 to 11.3 days, and a day 15 accumulation ratio (day 15 area under the curve from time 0 to 168 hours postdose [AUC]0‐168/day 1 AUC0‐168) of 2.0.4 Ixazomib also exhibits dose‐ and time‐independent PK.9 The absolute oral bioavailability of ixazomib among 226 adult cancer patients, following administration as immediate‐release capsules, was estimated to be 60%.9 Ixazomib also showed rapid absorption in a phase 1 study of patients with MM, with a Tmax of 1 hour (range 0.5‐8.0 hours).4
Ingestion of food causes several physiological gastrointestinal effects, including delayed gastric emptying, changes in gastrointestinal pH, and increased splanchnic blood flow,12 that can alter the rate and/or extent of absorption of orally administered antineoplastic agents. For example, large increases in the bioavailability of lapatinib (a tyrosine kinase inhibitor) have been observed in healthy volunteers and patients with advanced solid tumors when doses were administered with a high‐fat breakfast, which could have an impact on safety.13
Because food may change the bioavailability of a drug, a food‐effect study is typically conducted in clinical development to assess the PK of the drug administered following ingestion of a standardized high‐calorie, high‐fat meal, representing the setting of maximum possible perturbation of oral bioavailability in the postprandial state. Based on US FDA guidance, the recommended design for a food‐effect bioavailability study is a randomized, balanced, 2‐treatment (fed vs fasted), 2‐period, 2‐sequence crossover study, with each dose separated by an adequate washout period.14
The effects of food on the PK of ixazomib have not been reported previously, and, prior to the present study, patients in all ixazomib clinical studies have been instructed to take ixazomib on an empty stomach (at least 1 hour before or at least 2 hours after food). This phase 1, open‐label, multicenter, PK study assessed whether the PK properties of a single dose of ixazomib are altered when the drug is administered after a high‐calorie, high‐fat meal.
Methods
Patients
The study protocol and protocol amendments were approved by the institutional review boards at all participating centers. All patients provided written informed consent, and the trial was conducted according to the stipulations set out in the Declaration of Helsinki and International Conference on Harmonization Guideline for Good Clinical Practice. The study was registered at www.clinicaltrials.gov as NCT01454076.
Adult patients with histologically or cytologically confirmed metastatic and/or advanced solid tumor malignancies or lymphoma (Eastern Cooperative Oncology Group [ECOG] performance status 0 or 1), for which no effective standard treatment was available, were included. Patients with advanced solid tumors had radiographically measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1.15 Patients with lymphoma had radiographically or clinically measurable disease, defined as at least 1 measurable tumor mass >1.5 cm in the long axis and >1.0 cm in the short axis not previously irradiated or grown since previous irradiation as defined by International Working Group (IWG) criteria.16
All patients had an absolute neutrophil count (ANC) ≥1.25 × 109/L and a platelet count >100 × 109/L (ANC ≥1.0 × 109/L and platelet count >75 × 109/L for patients with lymphoma or Waldenström's macroglobulinemia with an underlying malignant bone marrow involvement), total bilirubin <1.5 times the upper limit of the normal range (ULN), alanine transaminase or aspartate transaminase ≤2.5 times the ULN, and calculated creatinine clearance >60 mL/min. Patients with grade >2 peripheral neuropathy, or any comorbid systemic illness or other severe concurrent disease that, in the judgment of the investigator, would have made the patient inappropriate for entry into the study or interfered significantly with the proper assessment of safety and toxicity of the prescribed regimens were not eligible. Patients who received systemic treatment, within 14 days before study enrollment, with strong inhibitors of CYP1A2 or CYP3A or strong CYP3A inducers or who used Ginkgo biloba or St. John's wort were also not eligible. During cycle 1 of the study (the PK cycle), moderate CYP1A2 and CYP3A inhibitors were also prohibited.
Study Design
The primary objective of this study was to characterize the effect of a high‐fat meal on the single‐dose PK of ixazomib. Secondary objectives included characterizing the safety and tolerability of oral ixazomib in patients with advanced solid tumors or lymphoma. The primary endpoint was the ratio of geometric mean peak plasma concentration (Cmax) and AUC0‐216 of ixazomib administered with food versus without food and 90% confidence intervals (CIs).
A randomized, 2‐period, 2‐sequence, crossover study design was used (Figure 1). In cycle 1, patients received a single 4‐mg dose of oral ixazomib (as a single immediate‐release capsule) on day 1 without food (fasted; sequence 1) or with food (fed; sequence 2), followed by a second dose on day 15 in the alternate food intake condition (fasted to fed, or fed to fasted). After completion of the PK cycle (ie, cycle 2 and beyond), ixazomib was administered as a 4‐mg dose on an empty stomach on days 1, 8, and 15 of each 28‐day cycle.
Figure 1.

Ixazomib food‐effect study design. Arrows indicate dosing or sampling days. In cycle 2 and subsequent cycles (all 28‐day cycles), all patients received only ixazomib, 4 mg daily, on days 1, 8, and 15. Ixazomib was administered for a maximum of 12 cycles unless disease progression or unacceptable toxicity occurred. The starting dose for cycle 2 was 4 mg, with the option of dose escalation to 5.3 mg at cycle 4 and beyond. PK, pharmacokinetics.
In the fasted condition, ixazomib was administered following an overnight fast of approximately 10 hours. In the fed condition, following an overnight fast (∼10 hours), patients started the recommended high‐fat meal at least 30 minutes before ixazomib administration on day 1 or day 15. It was expected that a patient should ideally require no more than 30 minutes to finish at least 75% of the meal. However, if a patient required an additional 15 to 30 minutes to finish the meal (total consumption time <1 hour), this was acceptable provided that the start of the meal was not more than 1 hour before the ixazomib dose. The ixazomib dose was to be taken with approximately 8 oz (240 mL) of water, no earlier than 30 minutes after the start of the meal. In both the fasted and fed conditions, no food was allowed for 4 hours after dosing. Water was permitted as desired, except for 1 hour before and 1 hour after ixazomib administration.
The test meal was high calorie (∼850 calories) and high fat (∼60% of total calories from fat) with the following approximate composition: 2 fried or scrambled eggs, 2 strips of bacon, 2 slices of toasted bread with ∼1 teaspoon of butter, 60 to 120 g hash browns (fried potato), and 240 to 280 mL whole milk. The approximate nutritional content of the test high‐fat meal was 55.6 g fat, 55 g carbohydrate, and 31 g protein. Substitutions to the content of the test meal could be made based on a patient's dietary restrictions provided a nutritionist ensured that the test meal contained a similar number of calories from protein, carbohydrate, and fat and had comparable meal volume and viscosity as the protocol‐specified meal.
Assessments
Blood samples for PK analysis were collected at the following prespecified time points during cycle 1 after ixazomib administration on day 1 and day 15: predose and 0.5, 1, 1.5, 2, 3, 4, 8, 24, 48, 72, 96, 168, 192, and 216 hours postdose. Plasma ixazomib concentrations were measured using a validated liquid chromatography/tandem mass spectrometry assay. A reversed‐phase gradient method, running at a flow rate of 0.3 mL/min on a Fortis Phenyl, 2.1 × 50 mm, 5 μm column, provided sample stacking and separation for the analyte, with a retention time of 1.85 (±0.3) minutes. Ixazomib and the internal standard (13C9‐ixazomib) were ionized in the positive ion spray mode and detected through multiple reaction monitoring of mass transition pairs at 343.1→109.0 m/z and 352.1→115.0 m/z, respectively. Assay linearity was achieved over a concentration range of 0.5 to 500 ng/mL for ixazomib. Intrarun precision for ixazomib in human plasma samples ranged from 4.4% to 6.0% coefficient of variation (CV), with a bias of –2.0% to 0.5%.
Adverse events (AEs) were evaluated throughout and up to 30 days after the last dose of study medication or the start of subsequent antineoplastic therapy and were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.
Statistical Analyses
All patients who received at least 1 dose of ixazomib were included in the safety population. The PK‐evaluable population included all patients who received the protocol‐specified dosing regimen during cycle 1, had consumed at least 75% of the standard high‐fat meal, did not receive any excluded concomitant medication through the completion of PK sampling, and had sufficient concentration‐time data to permit reliable estimation of PK parameters by noncompartmental analysis methods. Patients who were not PK‐evaluable were replaced.
Ixazomib plasma PK parameters were calculated using noncompartmental methods with Phoenix WinNonlin version 6.2 (Pharsight, St. Louis, MO). PK parameters were summarized using descriptive statistics. For the food‐effect estimation, the ratios of geometric mean AUC0‐216 and Cmax for the fed versus fasted state and associated 2‐sided 90%CIs were calculated by an analysis of variance (ANOVA) on log‐transformed values fitting terms for treatment condition (fasted or fed), sequence, and period as fixed effects. Patient within sequence was treated as a random effect in the model. After log transformation, AUC0‐216 and Cmax were analyzed separately. Point estimates (least‐squares [LS] means) and adjusted 90%CIs for the difference of LS means between treatment (fasted or fed) were calculated and then exponentially back‐transformed to provide point and CI estimates for the ratios of interest.
Results
Patients and Treatment Exposure
A total of 24 patients were enrolled. Key baseline patient and disease characteristics are shown in Table 1. At data cutoff (August 4, 2014), 23 patients (96%) had discontinued the study, and 1 patient (4%) was ongoing. The most common reason for discontinuation was progressive disease (11 patients, 46%); additional reasons were placement in hospice care (n = 2; 8%), AEs (n = 7; 29%), withdrawal by the patient (n = 1; 4%), and withdrawal by the principal investigator (n = 1; 4%). The reason for discontinuation was not provided for 1 patient.
Table 1.
Patient Baseline Demographics and Disease Characteristics
| Overall (N = 24) | |
|---|---|
| Median age, years (range) | 63 (46‐85) |
| Male, n (%) | 13 (54) |
| Race, n (%)a | |
| Caucasian | 18 (75) |
| African American | 4 (17) |
| Asian | 0 |
| Other/not reported | 2 (8) |
| Mean body weight, kg (range) | 74.1 (45.1‐112) |
| Disease type, n (%) | |
| Colorectalb | 7 (29) |
| Endometrial | 5 (21) |
| Non‐small‐cell lung cancer | 2 (8) |
| Esophageal | 1 (4) |
| Peripheral T‐cell lymphoma NOS | 1 (4) |
| Small‐cell lung cancer | 1 (4) |
| Waldenström macroglobulinemia | 1 (4) |
| Other | 6 (25) |
| Disease stage, n (%) | |
| II | 1 (4) |
| IV | 19 (79) |
| IVA | 1 (4) |
| Not available | 1 (4) |
| Missing | 2 (8) |
| ECOG performance status, n (%) | |
| 0 | 4 (17) |
| 1 | 20 (83) |
| Median time since initial diagnosis, months (range)c | 27 (5‐81) |
| Prior antineoplastic therapy, n (%) | 24 (100) |
| Prior radiation therapy, n (%) | 15 (63) |
| Prior surgical procedure, n (%) | 19 (79) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; NOS, not otherwise specified.
Not reported for 1 patient.
Includes 4 patients with colon cancer, 2 patients with rectal cancer, and 1 patient with colorectal cancer.
n = 23.
At data cutoff, patients had received a median of 2 cycles of ixazomib (range 1‐16). Four patients (17%) received ≥4 cycles of therapy, including 2 who received ≥13 cycles.
The most frequently reported concomitant medications were opioids (71%), laxatives (71%), antiemetics and antinauseants (63%), other analgesics and antipyretics (63%), vitamins A and D, including combinations of the two (54%), nonsteroidal anti‐inflammatory and antirheumatic products (54%), and intravenous solution additives (50%). Six patients (25%) received antihistamines for systemic use, and 6 patients (25%) received direct‐acting antivirals; 4 patients received acyclovir (2 for AEs, 1 for shingles prophylaxis, and 1 for medical history of cold sores), 1 patient received famciclovir for AEs, and 1 patient received valacyclovir for a medical history of herpes outbreaks.
Pharmacokinetics
Fifteen patients were included in the PK‐evaluable population. Seven patients were enrolled in sequence 1 (fasted state on day 1, fed state on day 15), and 8 patients in sequence 2 (fed state on day 1, fasted state on day 15). The mean ixazomib plasma concentration‐time profiles after administration with or without a high‐fat meal are shown in Figure 2. The median Tmax was 1.02 hours under fasted conditions and 4.0 hours under fed conditions (Table 2). Geometric mean values for Cmax and AUC0‐216 were both lower when ixazomib was administered under fed conditions versus the fasted state. The least‐squares geometric mean ratios (90%CIs) for Cmax and AUC0‐216 were 0.31 (0.21‐0.45) and 0.72 (0.58‐0.89), respectively. A period effect was observed in the ANOVA analysis for Cmax and AUC0‐216, indicating higher exposures in period 2 versus period 1 (ratio of period 2 AUC to period 1 AUC estimated as 2.21). However, in the statistical analysis, this was accounted for during estimation of the least‐squares geometric mean ratios and the 90%CIs. Day 1 geometric mean (%CV) terminal half‐life values for ixazomib under fasted (N = 4) and fed (N = 4) conditions were 189 (23) hours and 198 (25) hours, respectively.
Figure 2.
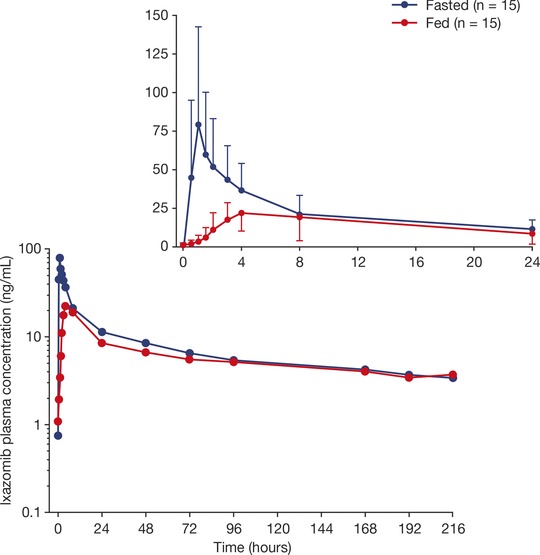
Mean plasma concentration‐time profiles of ixazomib under fasted and fed conditions (n = 15). The inset shows the mean plasma ixazomib concentrations over the first 24 hours after dosing. Error bars in the inset figure indicate standard deviation.
Table 2.
Plasma PK Parameters of Ixazomib Under Fed and Fasted Conditions
| Parameter | Fasted (Reference) (n = 15) | Fed (Test) (n = 15) | Least‐Squares Geometric Mean Ratio (90%CI) (Test/Reference) |
|---|---|---|---|
| Median Tmax, hours (range) | 1.02 (0.48‐4.0) | 4.0 (1.93‐8.03) | |
| Geometric mean Cmax, ng/mL (%CV) | 77.0 (57) | 22.8 (54) | 0.31 (0.21‐0.45) |
| Geometric mean AUC0‐216, h · ng/mL (%CV) | 1470 (50) | 999 (79) | 0.72 (0.58‐0.89) |
Abbreviations: AUC0‐216, area under the plasma concentration‐time curve from 0 to 216 hours postdose; CI, confidence interval; Cmax, maximum observed plasma concentration; PK, pharmacokinetic; Tmax, first time of Cmax.
Safety
All 24 patients received at least 1 dose of ixazomib and were included in the safety population. All patients had at least 1 treatment‐emergent AE (TEAE), and 13 (54%) had at least 1 grade ≥3 TEAE (Table 3). Any‐grade drug‐related TEAEs were seen in 20 patients (83%); the most common were vomiting (58%), nausea (50%), and fatigue (25%) (Table 4). Five patients (21%) had at least 1 grade ≥3 drug‐related TEAE; these were anemia (2 patients; 8%), dehydration, fatigue, peripheral edema, and vomiting (1 patient each; 4%) (Table 4). There were no grade 4 drug‐related TEAEs.
Table 3.
Safety Profile
| AE, n (%) | Safety Population (N = 24) |
|---|---|
| Any AE | 24 (100) |
| Any drug‐related AE | 20 (83) |
| Any grade ≥3 AE | 13 (54) |
| Any drug‐related grade ≥3 AE | 5 (21) |
| Any serious AE | 12 (50) |
| Any drug‐related serious AE | 2 (8) |
| AE leading to discontinuation, n (%) | 7 (29) |
| On‐study deaths, n (%) | 3 (13) |
Abbreviation: AE, adverse event.
Table 4.
Most Common Any‐Grade (≥5% of Patients) and All Grade ≥3 (≥1 Patient) Drug‐Related AEs
| Any‐Grade Drug‐Related AE, n (%) | Safety Population (N = 24) |
|---|---|
| Vomiting | 14 (58) |
| Nausea | 12 (50) |
| Fatigue | 6 (25) |
| Rasha | 5 (21) |
| Anemia | 4 (17) |
| Weight loss | 4 (17) |
| Decreased appetite | 3 (13) |
| Diarrhea | 3 (13) |
| Asthenia | 2 (8) |
| Chills | 2 (8) |
| Constipation | 2 (8) |
| Thrombocytopenia | 2 (8) |
| Grade ≥3 Drug‐Related AE, n (%) | |
| Anemia | 2 (8) |
| Dehydration | 1 (4) |
| Fatigue | 1 (4) |
| Peripheral edema | 1 (4) |
| Vomiting | 1 (4) |
Abbreviation: AE, adverse event.
Includes follicular rash, intermittent dark skin lesion, macular rash, maculopapular rash, pruritus.
Six patients (25%) had a TEAE characterized as a rash. These were characterized as macular rash, follicular rash, maculopapular rash, pruritus, or intermittent dark skin lesions. All rash TEAEs were grade 1 except for 1 case of grade 2 maculopapular rash. Most rash TEAEs resolved with no intervention, although of the 5 cases that were considered related to ixazomib, treatment was held due to rash in 1 patient. One patient had 2 cases of peripheral neuropathy; 1 case of grade 1 peripheral neuropathy at cycle 1 day 5 (C1D5), which resolved the following day, and 1 case of unrelated grade 1 peripheral neuropathy at cycle 1 day 14, which resolved 1 week later. There were no other cases of peripheral neuropathy. Two patients had 3 cardiovascular AEs (atrial fibrillation, hypotension, superior vena cava syndrome), all of which were considered unrelated to treatment.
One patient (4%) experienced TEAEs resulting in dose reduction. Seven patients (29%) had a TEAE resulting in study drug discontinuation. The most commonly reported TEAEs leading to dose modification were vomiting (8% of patients) and disease progression of endometrial cancer (8%).
Of 12 patients who had a serious AE, it was determined to be drug‐related in 2 patients (8%). Drug‐related serious AEs included dehydration in 1 patient, and dermatitis and facial edema in another patient. Three patients died on study, 2 due to endometrial cancer and 1 due to rectal cancer. None of the deaths was considered related to ixazomib.
Discussion
This study was conducted to determine the effect of a high‐calorie, high‐fat meal on the rate and extent of absorption of the oral proteasome inhibitor ixazomib in patients with cancer. The study was designed in accordance with the guidance provided by the US FDA, which recommends the use of a randomized, balanced, 2‐treatment (fed vs fasted), 2‐period, 2‐sequence, crossover study in which each treatment is separated by an adequate washout period. Due to the long terminal half‐life of ixazomib,4 in the present study, ixazomib was not administered to patients on day 8 during cycle 1 to ensure adequate washout between the first and second periods of PK characterization on day 1 and day 15, respectively. This study was conducted after a single dose because ixazomib has linear PK. At the start of the study, patients were allowed only 30 minutes to finish their meal based on the FDA guidance. However, most of the terminal cancer patients were finding it difficult to finish the high‐fat, high‐calorie meal within 30 minutes. Hence, the study was amended to allow patients an additional 15 to 30 minutes to finish the meal (total consumption time <1 hour).
The results of this study constitute the first report of the effects of food on the PK of ixazomib. Collectively, the data indicate that a high‐fat meal reduced both the rate and extent of absorption of ixazomib. Under fed conditions, the median Tmax for ixazomib was delayed by approximately 3 hours compared with administration in the fasted state (1.02 hours vs 4.0 hours), and there was a 28% reduction in the AUC and a 69% reduction in Cmax. These observations are consistent with ixazomib being a BCS class 3 molecule.11, 17 Although the magnitude of the observed negative food effect of a high‐fat meal on AUC was modest, the potential clinical relevance of this 28% reduction in total systemic exposure cannot be disregarded, as it would effectively decrease ixazomib exposures at the 4‐mg starting dose to those observed following administration of the first reduced dose level of 3 mg. Based on preliminary reports of the exposure‐response relationships for single‐agent ixazomib in relapsed/refractory MM,18 this represents a reduction in exposure that falls on the dynamic portion of the exposure‐safety and exposure‐clinical benefit relationships. Accordingly, to be consistent with dosing conditions in the completed and ongoing phase 1 to phase 3 clinical trials and maximize oral bioavailability, it is recommended that ixazomib be administered on an empty stomach, at least 1 hour before or at least 2 hours after food. These findings are reflected in the US Prescribing Information for ixazomib.10
The safety profile of oral ixazomib was generally consistent with previous studies of ixazomib in patients with MM, AL amyloidosis, lymphoma, and solid tumors.5, 6, 7, 8 , 19, 20 Also in line with previous ixazomib studies,5, 6, 7, 8 , 19, 20 but in contrast with studies with the proteasome inhibitor bortezomib,21, 22 the incidence of peripheral neuropathy was low, with only 1 case of drug‐related, reversible grade 1 peripheral neuropathy reported. Importantly, in this study, AEs appeared generally manageable with dose modifications and/or supportive care.
Conclusions
Overall, the results of this study support the administration of oral ixazomib on an empty stomach (at least 1 hour before or at least 2 hours after food) in all ixazomib clinical studies, including the ongoing phase 3 studies in MM (TOURMALINE‐MM1, TOURMALINE‐MM2, TOURMALINE‐MM3, and TOURMALINE‐MM4) and AL amyloidosis (TOURMALINE‐AL1). The findings of this analysis are reflected in the Dosing and Administration section of the US Prescribing Information for ixazomib.10
Acknowledgments
This work was funded by Millennium Pharmaceuticals, Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. Writing support during the development of the manuscript was provided by Jane Saunders of FireKite, an Ashfield company, part of UDG Healthcare plc, which was funded by Millennium Pharmaceuticals and complied with Good Publication Practice 3 ethical guidelines (Battisti WP, et al. Ann Intern Med. 2015;163:461–464).
Declaration of Competing Interests
Neeraj Gupta, Michael J. Hanley, Karthik Venkatakrishnan, and Bingxia Wang are employees of Millennium Pharmaceuticals, Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. Ai‐Min Hui is a former employee of Millennium Pharmaceuticals, Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. Sunil Sharma, Alberto Bessudo, and John Nemunaitis have no conflicts of interest to declare.
This work has been presented at the following meeting: Poster presented at the 19th Congress of the European Hematology Association, Milan, Italy, June 12‐15, 2014.
References
- 1. Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70(5):1970–1980. [DOI] [PubMed] [Google Scholar]
- 2. Chauhan D, Tian Z, Zhou B, et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011;17(16):5311–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee EC, Fitzgerald M, Bannerman B, et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B‐cell and plasma cell malignancies. Clin Cancer Res. 2011;17(23):7313–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar SK, Bensinger WI, Zimmerman TM, et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124(7):1047–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richardson PG, Baz R, Wang M, et al. Phase 1 study of twice‐weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014;124(7):1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Merlini G, Sanchorawala V, Jeffrey ZA, et al. Long‐term outcome of a phase 1 study of the investigational oral proteasome inhibitor (PI) ixazomib at the recommended phase 3 dose (RP3D) in patients (Pts) with relapsed or refractory systemic light‐chain (AL) amyloidosis (RRAL). Blood. 2014;124(21):3450.25293779 [Google Scholar]
- 7. Assouline SE, Chang J, Cheson BD, et al. Phase 1 dose‐escalation study of IV ixazomib, an investigational proteasome inhibitor, in patients with relapsed/refractory lymphoma. Blood Cancer J. 2014;4:e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith DC, Kalebic T, Infante JR, et al. Phase 1 study of ixazomib, an investigational proteasome inhibitor, in advanced non‐hematologic malignancies. Invest New Drugs. 2015;33(3):652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gupta N, Zhao Y, Hui AM, Esseltine DL, Venkatakrishnan K. Switching from body surface area‐based to fixed dosing for the investigational proteasome inhibitor ixazomib: a population pharmacokinetic analysis. Br J Clin Pharmacol. 2014;79(5):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. United States Food & Drug Administration . NINLARO (ixazomib) capsules, for oral use. United States Prescribing Information, November 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208462lbl.pdf.
- 11. Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–420. [DOI] [PubMed] [Google Scholar]
- 12. Singh BN, Malhotra BK. Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin Pharmacokinet. 2004;43(15):1127–1156. [DOI] [PubMed] [Google Scholar]
- 13. Koch KM, Reddy NJ, Cohen RB, et al. Effects of food on the relative bioavailability of lapatinib in cancer patients. J Clin Oncol. 2009;27(8):1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. US Food and Drug Administration . Guidance for Industry. Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Considerations. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM154838.pdf. Accessed July 16, 2015. [Google Scholar]
- 15. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. [DOI] [PubMed] [Google Scholar]
- 16. Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. [DOI] [PubMed] [Google Scholar]
- 17. Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60(6):717–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta N, Labotka R, Liu G, Hui AM, Venkatakrishnan K. Exposure‐safety‐efficacy analysis of oral ixazomib citrate (MLN9708) in relapsed/refractory multiple myeloma (MM): phase 3 dose selection for maintenance post autologous stem cell transplant [abstract]. Haematologica. 2014;99:364. [Google Scholar]
- 19. Gupta N, Goh YT, Min CK, et al. Pharmacokinetics and safety of ixazomib plus lenalidomide‐dexamethasone in Asian patients with relapsed/refractory myeloma: a phase 1 study. J Hematol Oncol. 2015;8:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open‐label phase 1/2 study. Lancet Oncol. 2014;15(13):1503–1512. [DOI] [PubMed] [Google Scholar]
- 21. Jagannath S, Barlogie B, Berenson J, et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004;127(2):165–172. [DOI] [PubMed] [Google Scholar]
- 22. Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. [DOI] [PubMed] [Google Scholar]