

Abstract
A novel laboratory setup for combined operando X‐ray diffraction and Raman spectroscopy of catalytic solids with online product analysis by gas chromatography is presented. The setup can be used with a laboratory‐based X‐ray source, which results in important advantages in terms of time‐on‐stream that can be measured, compared to synchrotron‐based experiments. The data quality was much improved by the use of a relatively high‐energy MoKα radiation instead of the more conventional CuKα radiation. We have applied the instrument to study the long‐term deactivation of Co/TiO2 Fischer–Tropsch synthesis (FTS) catalysts. No sign of Co sintering or bulk oxidation was found during the experiments. However, part of the metallic Co was converted into cobalt carbide (Co2C), at elevated pressure (10 bar). Furthermore, graphitic‐like coke species are clearly formed during FTS at atmospheric pressure, whereas at elevated pressure fluorescence hampered the interpretation of the measured Raman spectra.
Keywords: carbon, cobalt, Fischer-Tropsch synthesis, Raman spectroscopy, X-ray diffraction
Introduction
Heterogeneous catalysis plays an important role in the production of many chemicals, fuels, and consumer products.1 However, despite their widespread application, the relation between the structure of a catalyst and its activity and/or selectivity is not (yet) known in some important cases. It is for this lack of knowledge that operando techniques have become popular since the introduction of the term operando spectroscopy in the field of catalysis.2, 3, 4, 5, 6 Operando characterization means that the characterization not only happens under reaction conditions, but the catalytic activity is measured simultaneously by using, for example, a gas chromatograph (GC) or a mass spectrometer (MS). Operando experiments are considered to be essential because both the (local) structure and composition of the catalyst can change if it is taken out of the reactor.
Herein we introduce the design and application of an operando instrument that can be used for simultaneous X‐ray diffraction (XRD) and Raman spectroscopy measurements of catalytic materials under working conditions, at elevated temperatures and pressures, and product stream analysis with a GC. Whereas the operando setup is applicable to a wide variety of (gas‐phase) reactions and catalyst systems, we will here showcase its application to the Co/TiO2 Fischer–Tropsch synthesis (FTS) catalyst that was discussed previously.7, 8
Operando XRD is in itself not a new concept in catalysis.9, 10, 11 It has been used extensively to study different FTS catalyst systems, usually combined with X‐ray absorption near‐edge structure (XANES) or extended X‐ray absorption fine structure (EXAFS) spectroscopy.12, 13, 14, 15, 16, 17, 18 In addition, Raman spectroscopy has previously been combined with both XRD and XANES or EXAFS, under reaction conditions.19 However, all of these studies have been performed by using synchrotron radiation as the source of the X‐ray beam. On the one hand, this has certain advantages in terms of beam intensity that enables researchers to obtain high‐quality XRD/EXAFS data with very high angular or spectral resolution, respectively, in relatively short scan times, thus providing a time resolution in the order of minutes. On the other hand, experiments using synchrotron radiation are limited by their total duration, because it is not common to be granted more than a couple of days of beamtime at a time. This limitation necessitates important compromises between number of samples and time‐on‐stream that can be measured. In contrast, the deactivation of cobalt‐based FTS catalysts is typically quite slow. For instance, we have shown in our previous work that the deactivation of the Co/TiO2 FTS catalyst under study takes approximately 10 days.7 This is one of the reasons that the problem of cobalt FTS catalyst deactivation has not yet been fully solved despite numerous studies using for example, combined XRD/EXAFS under reaction conditions.12, 13, 14, 16, 20, 21, 22, 23, 24, 25, 26
Catalyst deactivation is one of the main problems facing the industrial application of the FTS process, which converts synthesis gas (CO and H2) into long‐chain hydrocarbons. Given that the synthesis gas can be produced from alternative energy sources such as natural gas, coal, and biomass, FTS is a promising way to deal with depleting oil reserves. Furthermore, FTS fuels are naturally very clean, because they are low in sulfur and aromatics.27, 28, 29, 30, 31, 32
It is for these reasons that our operando system was designed to use a laboratory‐based MoKα X‐ray source. More specifically, we will show here that the constructed combined operando XRD/Raman spectroscopy setup can be used for extended periods of time, for example, up to 10 days. However, that is certainly not a hard limit, and even longer‐term experiments should be possible.
Results and Discussion
The design and construction of the novel operando setup are described first, then the XRD and Raman spectroscopy results are presented for the Co/TiO2 catalyst during reduction and FTS at atmospheric pressure and elevated pressure (10 bar). Finally, the catalytic activity results are given.
Design and construction of the operando setup
A schematic of the setup is shown in Figure 1 A, and a photograph of the complete setup is shown in Figure 1 B. The setup is based on a horizontally mounted capillary (outer diameter: 1 mm, wall thickness: 0.01 mm) that holds the catalyst material, perpendicular to the incoming X‐ray beam.33, 34 The sample is held in place with two plugs of quartz wool on both sides of the catalyst bed. The use of a laboratory X‐ray source requires that the height of the sample capillary can be adjusted to be in the center of rotation of the X‐ray source and detector. A Raman spectroscopy probe is installed on a x,y,z movable stage so that the Raman excitation laser can be focused on the sample. Two photographs of details of the setup are shown in Figure 1 C and 1 D.
Figure 1.
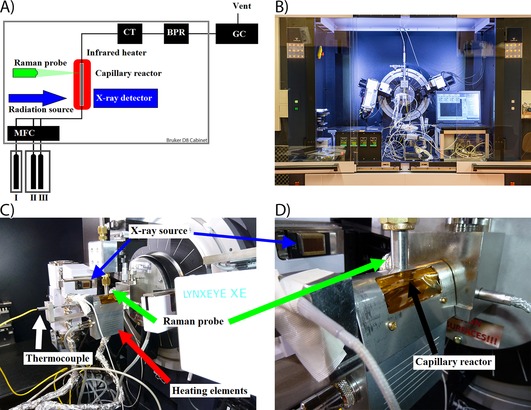
A) Schematic of the combined operando XRD/Raman/GC setup. I) He; II) CO; III) H2; MFC=mass flow controllers; CT=cold trap; BPR=back pressure regulator. B–D) Photographs of the setup as installed inside the XRD apparatus.
During preliminary testing of the setup using a conventional CuKα X‐ray source, the measurements were hampered by a low signal‐to‐noise ratio and a relatively high background. The consequence of this is poor data quality even if using long scan times. To improve the X‐ray penetration into the capillary, MoKα radiation with a photon energy of 17 479 eV and a corresponding wavelength of 0.7093 Å was used. This resulted in an improved signal‐to‐noise ratio and a lower background signal, as shown in the comparison in Figure 2. Another benefit of using the MoKα radiation is that the diffraction peaks are shifted to lower angles and the 2θ range is compressed compared to the CuKα radiation. This results in shorter scan times (hence increased time resolution). Simultaneously, the maximum 2θ value is lower, which leaves more physical space in the X‐ray diffractometer setup for the operando Raman spectroscopy probe.
Figure 2.
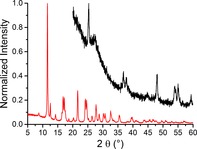
Comparison of the X‐ray diffractograms of the 10 wt % Co/TiO2 FTS catalyst as measured with CuKα (black) and MoKα (red) radiation. The position of the diffraction peaks changes because of the difference in X‐ray wavelength. Both scans took approximately 20 min.
Reduction behavior of Co/TiO2
The XRD results of the reduction of the 15 wt % Co/TiO2 catalyst are shown in Figure 3. The diffraction patterns (Figure 3 A) change during the reduction of the catalyst. First, the peak at 2θ of approximately 7° disappears; this peak is caused by Co3O4.35 Simultaneously, a peak at approximately 18° is formed, which is assigned to CoO.36 Finally a peak appears at approximately 20°. This peak is caused by metallic Co in the cubic closed‐packed (CCP) crystal structure.36 The same changes can be seen in Figure 3 B, where four selected diffractograms are plotted separately. The assignment of the different peaks is indicated by the vertical lines in Figure 3 B.
Figure 3.
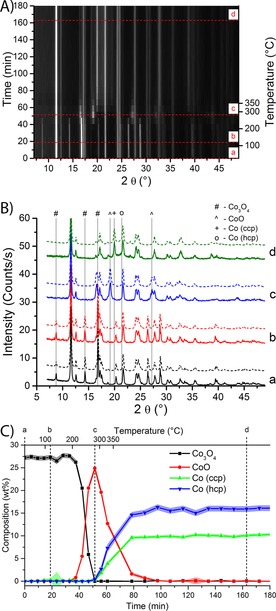
XRD patterns of a 15 wt % Co/TiO2 FTS catalyst during H2 reduction. A) XRD patterns as a function of time (and temperature on the right axis). Black indicates low diffracted intensity, and white means high intensity. The peak at 2θ angle of 12° is not shown completely to increase the visibility of the other diffraction peaks. B) Selected diffraction patterns (full lines) and corresponding Rietveld refinements (dashed lines) of time points as indicated in A). The diffractograms are offset for clarity. The main diffraction peaks of the Co species are indicated by symbols. Peaks caused by the support and minor Co peaks are not indicated. C) Results of the Rietveld refinements. The weight fraction of the different Co oxidation states is plotted as a function of time and reduction temperature. Standard deviations of the fits are shown as the shaded area around the lines.
The composition of the catalyst (as quantified by Rietveld refinement and shown in Figure 3 C) indicates that the reduction behavior of the 15 wt % Co/TiO2 catalyst follows a two‐step process. The first step (reduction from Co3O4 to CoO) starts after approximately 40 min or 200 °C. The amount of CoO reaches a maximum at approximately 50 min (250 °C). The second step is the reduction from CoO to metallic Co. The amount of metallic Co increases steadily until a plateau is reached. From this point on (≈90 min), no oxidized Co species are detected anymore. Both the CCP and the hexagonal closed‐packed (HCP) crystal structures of Co are formed, approximately 10 wt % of CCP Co and approximately 15 wt % of HCP Co. The peaks attributable to HCP Co are so severely broadened that they are difficult to recognize in the diffractograms (Figure 3 B). The crystallite sizes of HCP Co and CCP Co will be discussed in more detail later.
The discrepancy between the calculated weight loading (15 wt % Co) and the XRD quantification (25 wt % Co in total) could be caused by a combination of inaccuracies during impregnation and artifacts of the quantification procedure. Multiple analysis of the catalyst sample to determine the chemical content gave very similar results, indicating that the deviations should be related to the XRD quantification procedure. A potential source of inaccuracy could be the presence of amorphous TiO2 that would decrease the detected wt % of TiO2, in turn leading to an overestimation of the Co content in the quantification. In addition, the amount of amorphous Ti species might change as Ti4+ might be (partially) reduced into Ti3+ during the FTS experiment. Taking this point further, one could imagine using the difference between the “real” (externally verified) Co wt % and the detected Co wt % as an indirect way to detect the formation of amorphous Ti species. Further experiments would be needed to confirm the validity this methodology.
As an alternative approach to solve the discrepancy, we have tried to use anatase as an internal standard during the Rietveld refinement, effectively fixing the anatase contribution to 63.75 wt % (=75 wt % (the amount of anatase in titania) of 85 wt % (the amount of titania in the catalyst according to the impregnation for a 15 wt % Co/TiO2 catalyst)). This approach resulted in significant contributions of amorphous material, approximately 10 wt %. Simultaneously, the detected amount of Co decreased to approximately 24 wt %, still significantly higher than the calculated weight loading from the impregnation. Hence, we decided not to use an internal standard for the rest of the Rietveld refinements, because it did not solve the discrepancy between the calculated and the detected weight loadings. Instead, we are forced to accept that the quantification cannot be used to determine the precise composition of the catalyst material in an absolute way. However, we can still see important trends in the relative change in composition, and draw relevant conclusions from that.
The two‐step reduction behavior of supported Co/TiO2 catalysts that we have seen for this catalyst has previously been demonstrated by using temperature‐programmed reduction (TPR)7, 22 and XANES spectroscopy.16, 18, 24 Also the mixture of the HCP and CCP crystal structures upon reduction of supported Co catalysts has been observed before.16, 37, 38
Fischer–Tropsch synthesis of Co/TiO2
In a first series of experiments, the 15 wt % Co/TiO2 FTS catalyst was studied under FTS conditions at 250 °C and atmospheric pressure. The results of these experiments are shown in Figure 4. As can be seen in Figure 4 A, the diffraction patterns do not change significantly over the course of the experiment. The 15 wt % Co/TiO2 FTS catalyst consists of approximately 12 wt % CCP Co and approximately 17 wt % of HCP Co, as shown in Figure 4 B. Again, the amount of Co from XRD analysis is higher than the calculated weight loading. The contributions of the two metallic Co crystal structures are stable for the duration of the experiment (in light of the experimental error). No contributions for CoO or Co2C are detected.
Figure 4.

XRD results of a 15 wt % Co/TiO2 FTS catalyst during FTS at atmospheric pressure and at 250 °C. A) XRD patterns as a function of time under FTS conditions. Black indicates low diffracted intensity, and white means high intensity. The peak at 2θ of 12° is not shown completely to increase the visibility of the other diffraction peaks. B) The composition of the catalyst as a function of time on stream. The contributions of different Co species are plotted; standard deviations from the fit are indicated as the shaded area around the lines. C) Particle sizes of three of the Co species as a function of time on stream. Standard deviations are shown as the shaded area. D) Selected experimental diffractograms (full lines) and the Rietveld refinements (dashed lines) at the time points as indicated in Figure B and C. Peak positions for the Co species are indicated by vertical lines. Diffraction peaks attributable to TiO2 support peaks and minor diffraction lines are not indicated.
The crystallite sizes of the 15 wt % FTS Co/TiO2 catalyst under reaction conditions (as shown in Figure 4 C) are approximately 20 nm for the CCP phase and approximately 2.5 nm for the HCP phase. The particles sizes do not change significantly during the reaction. If anything, there seems to be a downward trend in the particle size of the CCP crystallites but this can hardly be called significant in light of the experimental error. Hence, no clear signs of sintering of metal nanoparticles are observed. The stability of the catalyst is also illustrated in Figure 4 D, which shows four X‐ray diffractograms, along with their respective Rietveld refinements. There is an excellent agreement between the refinements and the experimental data.
In a second series of experiments, the 15 wt % Co/TiO2 FTS catalyst was studied during FTS at 250 °C and 10 bar. The XRD results of these experiments are shown in Figure 5 A. The data at the start of the experiment are relatively noisy, because the sample moved out of the center of rotation of the XRD apparatus, which has negatively affected the signal quality. After approximately 50 h the results stabilize; from that moment the composition of the catalyst (as shown in Figure 5 B) is in approximate agreement with that of the same sample at atmospheric pressure. The catalyst then consists of approximately 18 wt % of HCP Co and about 10 wt % of CCP Co.
Figure 5.
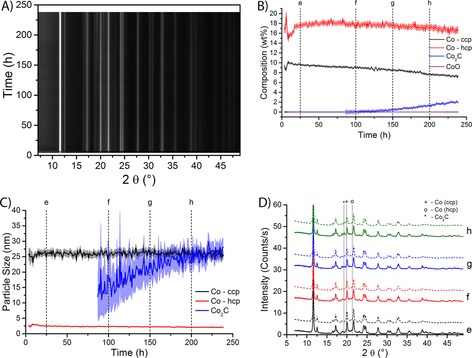
XRD results of the 15 wt % Co/TiO2 FTS catalyst during FTS at 10 bar and at 250 °C. A) XRD patterns as a function of time under FTS conditions. Black indicates low diffracted intensity, and white means high intensity. The peak at 2θ of 12° is not shown completely to increase the visibility of the other diffraction peaks. B) The composition of the catalyst as a function of time on stream. The contributions of different Co species are plotted; standard deviations from the fit are indicated as the shaded area around the lines. C) Particle sizes of three of the Co species as a function of time on stream. Standard deviations are shown as the shaded area. D) Selected experimental diffractograms (full lines) and the Rietveld refinements (dashed lines) at the time points as indicated in Figure B and C. Peak positions for the Co species are indicated by vertical lines. Diffraction peaks attributable to TiO2 support peaks and minor diffraction lines are not indicated.
Remarkably, after about 150 h there is a slow but steady increase in the contribution of Co2C. After 250 h of reaction, the catalyst contains approximately 2 wt % of Co2C. Along with the increase in the detection of Co2C, a small but significant decrease in the contribution of CCP Co is detected. The contribution of HCP Co decreases also, but not significantly. This suggests that primarily CCP Co is converted into Co2C. In Figure 5 C the particle sizes of CCP Co are 25 nm, those of HCP Co 2.5 nm. The particle size is higher than during the experiment at atmospheric pressure (about 20 nm). The reason for this is currently unknown. Sintering is not a likely explanation, because the particle size is high from the start of the experiment (i.e., there is no observed growth of the particles), however, the data at the start of the experiment is extremely noisy. As in the experiments at atmospheric pressure, the particle sizes of both Co crystal structures do not change significantly during the experiment. The HCP particles are approximately 2.5 nm and the CCP particles are approximately 25 nm. The particle size of Co2C increases from approximately 20 nm after 150 h of reaction to about 30 nm at the end of the experiment. However, the experimental error of the particle size is rather large (≈20 nm) because the contribution of Co2C is small, especially during the first few hours after 150 h. As the amount of Co2C increases, the uncertainty in the particle size decreases to approximately 4 nm.
The formation of Co2C is clearly seen in the selected diffractograms that are shown in Figure 5 D. A small peak at the indicated position of Co2C is visible in the diffractograms after 150 h and 200 h. This peak is not visible in the diffractograms after 25 h and 100 h, which is consistent with the results of the Rietveld refinement.
The XRD results suggest that the FTS catalyst contains both small HCP particles (≈2.5 nm) and larger CCP particles (≈25 nm) during the FTS reaction. This finding is in contrast to the results from XRD experiments on the fresh catalyst, which indicate that the catalyst consists of Co3O4 particles of approximately 18 nm.7 It is unlikely that reducing 18 nm particles somehow results in metallic particles of 2.5 nm. A solution to this contradiction is that metallic cobalt particles are not completely CCP, but small HCP domains exist within the particles.39 The transition from CCP to HCP is a stacking fault. The existence of stacking faults in metallic cobalt has also been reported after reduction of bulk Co3O4 40 and for alumina‐supported cobalt nanoparticles.39, 41
Remarkably, during FTS at elevated pressure (10 bar) part of the metallic Co is converted into cobalt carbide (Co2C). Remember that we did not find any Co2C during FTS at low pressure. This suggests that the formation of Co2C is dependent on a high partial pressure of CO. Furthermore, the conversion starts only after approximately 125 h of reaction. The formation of Co2C is accompanied by a decrease of CCP Co, whereas the contribution of HCP Co does not change. Hence, it is probable that the CCP crystal structure is converted into the carbide, and not the HCP phase. This is corroborated by the particle size of Co2C (≈20 nm, increasing to ≈25–30 nm), which is much closer to the particle size of the CCP phase (about 25–30 nm) than to the size of the HCP domains (≈2.5 nm).
Cobalt carbide is generally accepted not to be active in FTS catalysis, thus the conversion of metallic Co to Co2C is a possible cause for catalyst deactivation.27, 42 Although not very common, Co2C has been found in Co‐based FTS catalysts before. The presence of Co2C in spent Al2O3‐supported FTS catalysts was reported by Jacobs et al.43 Using synchrotron XRD, Karaca et al. observed a small amount of Co2C in their Co/Al2O3 catalyst after approximately 8 h of FTS conditions at a pressure of 20 bar.14 Co2C has also been detected in catalysts supported on activated carbon.44 According to Lynch, the formation of Co2C could be directly correlated to the loss of catalytic activity.45 On the other hand, Claeys et al. reported that the formation of Co2C is not a significant cause of catalyst deactivation.33 With the aid of a combination of spectroscopy and microscopy we have recently reported results that suggest that the formation of Co2C at the surface of metallic Co nanoparticles might be related to catalyst deactivation.8
The presence of Co2C was reported to increase the selectivity towards alcohols in carbon‐supported catalysts for the synthesis of higher alcohols at pressures of 30 bar.46, 47 This was explained by calculations that show that absorption of molecular CO is easier on a Co2C surface than on a metallic Co surface. However, hydrocarbon chain growth happened mostly on metallic surfaces.46
Carburization of Co/TiO2
To study the formation of cobalt carbide (Co2C) in more detail, the 15 wt % Co/TiO2 FTS catalyst was exposed to pure CO at atmospheric pressure and 250 °C, after reduction in H2 under the same condition as used earlier. The results of the carburization experiment are shown in Figure 6.
Figure 6.
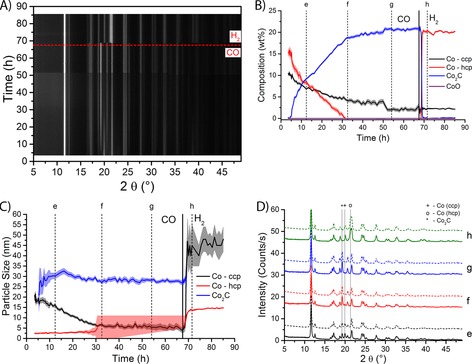
XRD results of a 15 wt % Co/TiO2 FTS catalyst during carburization and subsequent hydrogenation (both at 250 °C and atmospheric pressure). A) XRD patterns as a function of time. The horizontal dashed line indicates the switch from CO to H2. Black indicates low diffracted intensity, and white means high intensity. The peak at 2θ of 12° is not shown completely to increase the visibility of the other diffraction peaks. B) The composition of the catalyst as a function of time on stream. The contributions of different Co species are plotted; standard deviations from the fit are indicated as the shaded area around the lines. The vertical full line at 67 h indicates the switch from CO to H2. C) Particle sizes of three of the Co species as a function of time on stream. Standard deviations are shown as the shaded area. D) Selected experimental diffractograms (full lines) and the Rietveld refinements (dashed lines) at the time points as indicated in Figure B) and C). Peak positions for the Co species are indicated by vertical lines. Diffraction peaks attributable to TiO2 support peaks and minor diffraction lines are not indicated.
At the start of the experiment the catalyst is composed of about 15 wt % of HCP Co and about 10 wt % of CCP Co, as shown in Figure 6 B. Almost immediately upon exposure to CO, there is a sharp increase in the contribution of Co2C. However, the complete conversion of metallic Co into the carbide takes quite long, approximately 50 h. Even after 65 h under CO there is still approximately 2.5 wt % of CCP Co in the catalyst. Remarkably, the conversion of the HCP crystal structure is quicker than of the CCP phase, in contrast to the experiments under FTS conditions, where primarily the CCP phase was converted into Co2C. Although conversion starts at a higher value, the contribution of HCP Co is decreased to 0 wt % after approximately 32 h under CO.
After approximately 67 h of exposure to CO, the gas flow was switched to 1 mL min−1 of H2; this moment is indicated by the black vertical line in Figure 6 B. There is an immediate conversion of Co2C into metallic Co. However, only the HCP crystal structure is formed, whereas the contribution of CCP Co stays constant at the value before the switch from CO to H2.
The selective formation of HCP Co from Co2C is well accepted in literature.33, 39, 41, 48, 49, 50, 51 Carburization followed by hydrogenation of Co FTS catalysts was even suggested as a way to convert the CCP structure into the more active HCP structure.52 However, it has been reported that Co/Al2O3 FTS catalysts that were activated in CO or in syngas have lower activity and selectivity than catalysts that were reduced in H2.53 The findings were explained by the increased methane production of Co2C compared with metallic Co. Co/TiO2 FTS catalysts that were pretreated in CO were carbidic at first, but they slowly became more metallic during FTS, as shown by EXAFS and XANES spectroscopy.54 However, the catalytic activity and selectivity were still lower than for the same catalyst that was reduced in H2.
The particle sizes of the different Co species are shown in Figure 6 C. Initially, the CCP Co nanoparticles are approximately 20 nm and HCP Co nanoparticles are approximately 2.5 nm in size. This is in line with the results under FTS conditions. However, upon exposure to CO, the particle size of the CCP crystal structure decreases and stabilizes after about 32 h at approximately 5 nm. Meanwhile, the HCP Co particle size is more or less stable at approximately 2.5 nm. The particle size of Co2C starts off at approximately 25 nm, and this increases to approximately 33 nm after approximately 15 h. The particle size then decreases slightly to become stable at approximately 28 nm. Upon exposure to H2, the HCP Co phase (HCP) consists of bigger particles (≈15 nm) than the CCP phase that disappeared under CO atmosphere (≈2.5 nm). At the same time, there is a sharp increase in the particle size of the minority species (CCP Co), but the experimental error is relatively large owing to the small amount of CCP Co that is detected in these fits. In the diffractograms (Figure 6 D) we can clearly see the increase in the Co2C peaks (marked with ′*′) with increasing reaction time (diffractograms e–g), along with a corresponding decrease in both the CCP and HCP peaks.
Raman spectroscopy of Co/TiO2
Simultaneously to the XRD experiments, operando Raman spectra were acquired. Bands attributable to Co species were not detected because their bands are located below 750 cm−1, too low for the Raman spectrometer used.55, 56, 57, 58 The spectra in the region of 1000–2000 cm−1 for the 15 wt % Co/TiO2 FTS catalyst during FTS at atmospheric pressure are shown in Figure 7 A and B. Two Raman bands are visible in the spectra, at approximately 1350 cm−1 and 1600 cm−1; these bands are characteristic of coke formation.19 The band at 1350 cm−1 is assigned to disordered‐type graphitic carbonaceous species, and it is commonly called the D band. The band at approximately 1600 cm−1 is assigned to the graphite lattice and is called the G band.19, 59, 60
Figure 7.
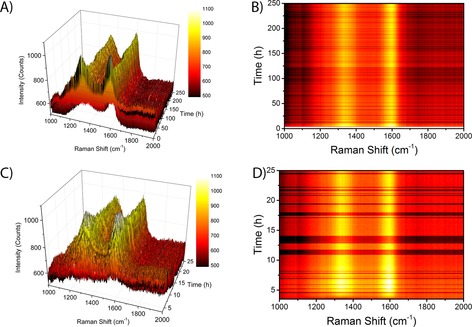
Operando Raman spectroscopy of a 15 wt % Co/TiO2 FTS catalyst during FTS at atmospheric pressure and 250 °C. A) 3 D plot of the Raman spectra as a function of time during the first 250 h of FTS reaction. B) Contour plot of the Raman spectra during the first 250 h of FTS reaction, using the same color map as in A). C) 3D plot of the Raman spectra as a function of time during the first 25 h of FTS reaction. D) Contour plot of the Raman spectra during the first 25 h of FTS reaction, using the same color map as C).
There are large differences in the intensity of the Raman bands over time. However, upon closer examination, we see that there are also large fluctuations in the background signal of the Raman spectra (especially visible in Figure 7 B as darker horizontal lines in the spectra). The fluctuations in the Raman signal are possibly caused by (small) changes in the sample position (moving in and out of the focal point of the Raman probe). In any case, we cannot assign any chemical significance to the changes in absolute intensity. Taking into account the fluctuations in the background, the intensity of the Raman bands looks quite constant during the 250 h of FTS reaction. In Figure 7 C and D we show a zoom‐in of the Raman spectra during the first 25 h of FTS reaction. The formation of the 1350 cm−1 and the 1600 cm−1 Raman bands takes approximately 5 h. The averaged Raman spectra at the beginning (10–20 h) and at the end (230–240 h) of the FTS reaction are shown in Figure 8 A. The spectra are essentially the same, and we can clearly see the two Raman bands at 1350 and 1600 cm−1. Based on these spectra we can define three spectral regions to look at the intensity in a more quantitative way. The two Raman bands are indicated by the grey vertical lines in the spectra, and the background is defined as the region of 1800–2000 cm−1. We can now normalize the two Raman bands by dividing the average intensity in the peak regions by the intensity in the background region.
Figure 8.
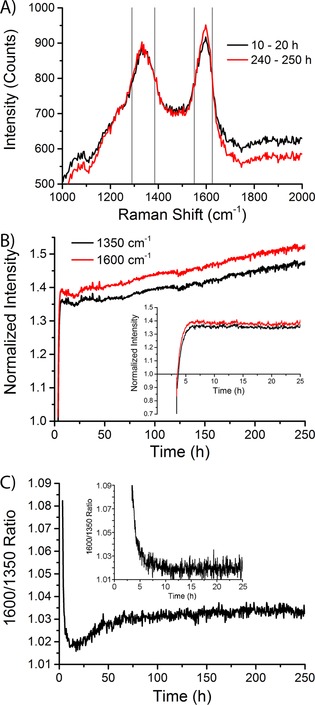
Analysis of the Raman spectroscopy data of a 15 wt % Co/TiO2 FTS catalyst during FTS at atmospheric pressure and 250 °C. A) Averaged Raman spectra as measured from 10–20 h of FTS reaction (black spectrum) and from 240–250 h of FTS reaction (red spectrum). Gray vertical lines define the spectral regions of the 1350 cm−1 and the 1600 cm−1 Raman peaks. B) Intensity of the 1350 cm−1 (black) and the 1600 cm−1 (red) Raman peaks, normalized to the background (defined as the averaged intensity in the featureless region from 1800–2000 cm−1), as a function of time during the first 250 h of FTS reaction. Inset: during the first 25 h. C) Ratio of the intensities of the 1600 cm−1 to the 1350 cm−1 Raman peaks during the first 250 h of reaction. Inset: during the first 25 h.
The normalized intensity of the two Raman bands as a function of time is shown in Figure 8 B. The 1600 cm−1 band (G band) has a higher intensity than the 1350 cm−1 band (D band) for the whole duration of the experiment. There is a rapid increase in the intensity of both Raman bands. The inset graph shows that the majority of the increase in the intensity happens in the first ≈6 h of reaction. After that moment, there is a further, more or less linear increase in the normalized intensity of the Raman bands. A continuous increase in the amount of coke is observed.
The ratio of the intensity of the 1600 cm−1 to the 1350 cm−1 band (I 1600/I 1350) is shown in Figure 8 C. These values were calculated by dividing the average intensity in the 1600 cm−1 by the average intensity in the 1350 cm−1 band. There is an initial decrease from a relatively high value of approximately 1.09 to a minimum of approximately 1.02. After the initial decrease, a slightly higher plateau at a ratio of approximately 1.03 is reached after about 75 h.
In addition to the quantitative analysis described above, we have also performed a deconvolution of the Raman spectra following the approach from Sadezky et al.,61 which has also been used in our group for the analysis of coke formed on propane dehydrogenation catalysts.59, 60 First, the background of the Raman spectra was subtracted. Then, a set of five Gaussian and Lorentzian functions was used to find the best fit to the experimental data. The set of functions is given in Table 1, with the nomenclature of the bands as proposed by Sadezky et al.61 A Gaussian was used instead of a Lorentzian for the G band; this resulted in visually better fits. Furthermore, we found only one Q band, at 1297 cm−1, which is probably caused by the capillary. The D2 band at 1620 cm−1 that was mentioned by Sadezky et al.61 was not detected in our fits.58, 59 The inability to resolve the D2 band indicates the poorly ordered structure of the graphite that is formed.62
Table 1.
Characteristics of the Raman bands that are used in the deconvolution of the Raman spectra.
| Band | Initial position [cm−1] | Shape | Assignment |
|---|---|---|---|
| Q | 1297 | Gaussian | capillary |
| G | 1587 | Gaussian | graphite |
| D1 | 1320 | Lorentzian | defects in graphite |
| D3 | 1500 | Gaussian | amorphous carbon |
| D4 | 1220 | Gaussian | disordered graphite |
The deconvoluted Raman spectra during the experiment are given in Figure 9 A. The spectra were averaged over a period of 10 h to increase the signal to noise ratio. There is a close agreement between the experimental data (black curves) and the spectral fits (red curves). The ratio of the G band intensity to the D1 band intensity is shown in Figure 9 B. The G/D1 ratio is seen to slightly increase during the first 100 h for the 15 wt % Co/TiO2 FTS catalyst (Figure 9 B). This is in broad agreement with the results from the quantitative treatment of the Raman spectra. This finding suggests that some of the defects in the graphite sheets disappear during the first 100 h of FTS reaction, or that the amount of graphitic carbon increases. However, the increase in the G/D1 ratio is rather small and hardly significant.
Figure 9.
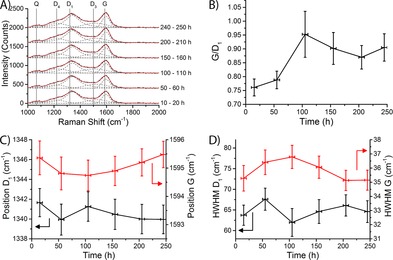
Deconvolution of the Raman spectra of the 15 wt % Co/TiO2 FTS catalyst during FTS at atmospheric pressure and at 250 °C. A) Deconvoluted Raman spectra averaged over different times under FTS conditions. Experimental data are indicated by black curves, fits are indicated by red curves; gray dashed curves indicate the contribution of the different Raman bands; vertical gray lines indicate the idealized positions of the Raman peaks. B) Ratio of the intensity of the G and the D1 bands as a function of time under FTS conditions. Horizontal error bars indicate the time the spectra were averaged over; vertical error bars indicate the standard error. C) Spectral position of the D1 band (black, on the left axis) and the G band (red, on the right axis). D) HWHM of the D1 band (black, on the left axis) and the G band (red, on the right axis).
The position of the G band is an indication for the size of graphite crystallites, with larger crystals causing the G band to shift to lower wavenumbers.62 On the other hand, a decreased half width half maximum (HWHM) of both the D1 and the G band is a sign of increased graphitization of the coke species.60 However, the positions (Figure 9 C) and the HWHMs (Figure 9 D) of both the D1 and the G bands stay constant for the entire duration of the experiment, within experimental error. From this, we conclude that the coke species formed on this catalyst are stable during FTS.
No Raman bands are discernible in the spectra that were acquired during FTS at elevated pressure (10 bar) using the 15 wt % Co/TiO2 FTS catalysts, as the spectra (not shown in the interest of brevity) are dominated by a large background of fluorescence. The presence of the fluorescence in the spectra makes interpretation of the Raman spectra impossible. The contribution of the coke bands to the Raman spectra is overshadowed by the fluorescent background. However, this does not mean that coke is not present during this experiment, or even that the amount of coke is small. Only a small amount of a fluorescent species is enough to cause a very high background. It is even possible that the coke species themselves are fluorescent.
There is a strong indication that coke formation could be (partly) responsible for catalyst deactivation, either by physically blocking the pores of the support, poisoning the Co surface, or by physically blocking access to the Co surface.27 The deposition of carbon has been suggested to be a relatively slow process, with the initial deactivation attributed to sintering of Co.64, 65 These findings were based on modeling approaches to catalyst deactivation. In contrast, we have seen that the formation of Raman bands belonging to coke is rather quick.
Catalytic activity during Fischer–Tropsch synthesis
The catalytic activity was measured using a GC during the operando XRD/Raman experiments. However, because an internal standard for the GC was not used during the experiments, accurate activity and selectivity data could not be calculated. However, we can calculate the production of light FTS products (C1–4), which we will use as an approximation for the “real” catalytic activity. We will express the catalytic productivity as 10−5 mol of product per gram of Co per second (10−5 mol gCo −1 s−1).
The catalytic productivity during FTS at atmospheric pressure using the 15 wt % Co/TiO2 FTS catalyst is shown in Figure 10 A. The production of CH4 decreases rather quickly during the first ≈50 h of reaction. The production of C2–4 species decreases at a similar rate. At the end of the reaction the production of methane seems to increase a little again. This is probably caused by hydrogenation of coke species, and not by an increased activity of the catalyst.
Figure 10.

Catalytic productivity of the 15 wt % Co/TiO2 FTS catalyst during FTS at A) atmospheric pressure and B) a pressure of 10 bar, both at 250 °C. Black curves indicate the CH4 production; red curves indicate the production of C2–4 species.
The production of light products during FTS at a pressure of 10 bar of the 15 wt % Co/TiO2 FTS catalyst is shown in Figure 10 B. The production starts out by increasing to a maximum. The further decline in the production of FTS products is now less pronounced than in the experiments at atmospheric pressure. However, there is still a significant deactivation. Slower deactivation at higher pressure was observed by de la Peña O'Shea et al.,66 who explained it by increased segregation of the surface of the catalyst at high CO partial pressure. This roughening of the surface was said to increase the Co surface area and lead to higher activity.67 Second, the production of both methane and the higher products is higher than at atmospheric pressure for the duration of the entire experiment.
The ratios of the production of CH4 to the production of C2–4 for the 15 wt % Co/TiO2 FTS catalyst at atmospheric pressure and at 10 bar are shown as blue lines in Figure 10 A and B, respectively. As the methane selectivity of Co FTS catalysts increases if Co2C is present in the catalyst,53, 54 an increase in the CH4/C2–4 ratio during FTS could be an indication for the formation of Co2C. Indeed, the CH4/C2–4 ratio (blue lines in Figure 10) is higher during the experiment at elevated pressure (at which Co2C is formed) than during the experiment at atmospheric pressure (at which Co2C is not observed in XRD analysis). Furthermore, during the experiment at elevated pressure (Figure 10 B), there is a continuous increase in the CH4/C2–4 ratio, after an initial steep increase. There are two artifacts after approximately 190 h and approximately 230 h, which are probably caused by pressure drops in the GC. The increase in the CH4/C2–4 ratio suggests that the carburization of the catalyst starts immediately upon exposure to high partial pressures of CO. The Co2C is not seen in the XRD analysis until about 150 h into the experiment, but this is to be expected if the initial Co2C phase is amorphous or consists of small particles, only to becoming crystalline after approximately 150 h of FTS reaction.
On the other hand, the CH4/C2–4 ratio during FTS at atmospheric pressure stays almost constant (after the initial steep decrease) during the experiment. This is in line with the XRD results, which did not show the formation of Co2C. However, careful examination of the CH4/C2–4 ratio reveals that there is a small increase in the ratio after approximately 200 h of FTS reaction. This could suggest that an amorphous (hence not visible in XRD) carbide phase is formed from this moment. Even longer experiments can confirm or deny this hypothesis.
Conclusions
This paper discusses the design and application of an operando setup for combined XRD and Raman spectroscopy, with which the product stream is simultaneously analyzed with an online GC. Many different reactions and catalyst materials can be tested with the described instrument. A major improvement over previous operando XRD/Raman setups12, 13, 14, 15, 16, 17, 18, 19, 24, 25 is the possibility of using of a laboratory X‐ray source, instead of a synchrotron. This allows us to monitor catalytic reactions for an extended time, without limitation of the amount of beamtime that is granted. The use of MoKα radiation resulted in improved data quality to that of conventional CuKα radiation.
We have applied the setup to study the long‐term deactivation of a 15 wt % Co/TiO2 FTS catalyst. From the operando XRD experiments it was found that sintering or bulk oxidation do not play a significant role in the deactivation of the catalysts. The particle size of the Co nanoparticles was remarkably stable during the experiments, and the nanoparticles stayed completely metallic. Furthermore, there are many stacking faults in the Co nanoparticles, resulting in small HCP domains (≈3 nm) in otherwise CCP particles (≈25 nm). The metallic nanoparticles were converted into Co2C, but this only happened during FTS at elevated pressure (10 bar). No Co2C was found during FTS at atmospheric pressure. This is probably because the formation of Co2C is only feasible if the partial pressure of CO is high enough.
The focus of the operando Raman spectroscopy experiments was to study the formation of coke deposits during FTS. Unfortunately, the Raman experiments at 10 bar were hampered by fluorescence, which overshadowed the Raman bands. At atmospheric pressure, there was clear evidence for coke formation in the form of the formation of Raman bands at about 1350 cm−1 (D band) and at 1600 cm−1 (G band). Quantitative analysis and deconvolution of the Raman bands revealed that the coke species were quite stable, in terms of graphite crystallite sizes and number of defects in the graphite structure. The catalyst saw a quick initial increase in the amount of coke, with most of the coke band intensities forming in the first ≈15 h of FTS reaction. After the initial increase there was a slower increase in the coke band intensities. The intensity of the coke bands of the catalyst kept increasing for the duration of the experiment.
Experimental Section
As was reported earlier, the fresh catalyst sample was prepared by impregnating samples of ≈2 g of vacuum‐dried TiO2 (P25) powder with a saturated Co(NO3)2 solution (Sigma–Aldrich, >99 %) until the pores of the support were filled.7, 8 The powder was then dried at 60 °C and calcined at 350 °C. This resulted in a catalyst with a Co loading of 10 wt %. The process was repeated to reach a final Co loading of 15 wt %.7, 8
The operando reactor that was used in this study was designed by iKey, South Africa.33, 34 Inert and reactant gases (He and CO and H2, respectively (all from Linde)) were delivered to the reactor at elevated pressures (up to 20 bar) to the reactor by using Bronckhorst EL‐FLOW select mass flow controllers that are calibrated for flow rates between 0 and 1 mL min−1. All gas lines were heat‐traced to preheat the reactant gases and to prevent condensation of liquid products. A RT trap was inserted in the gas line downstream of the reactor to collect the liquid products. The pressure in the capillary reactor was controlled by a Bronckhorst pressure sensor coupled to a control valve.
Samples of the product stream were analyzed with an on‐line Interscience TraceGC 1300 gas chromatograph (GC). The different gases in the product stream were analyzed on three separate channels, one flame ionization Detector (FID) for the analysis of C1–C6 hydrocarbons and two thermal conductivity detectors (TCD) for the analysis of He and H2 and the permanent gases (CO, CH4, O2, N2, CO2 and ethane), respectively. The separation of the FID channel was performed using an Rtx‐1 precolumn and an Al2O3/Na2SO4 main column. A backflush was performed on the precolumn after the elution of C6 hydrocarbons. The permanent gases are separated on a Molsieve 5 A column.
Preliminary XRD experiments were performed by using a Bruker D8 Discover using a CuKα X‐ray source and a MoKα X‐ray source. A similar XRD apparatus with a MoKα X‐ray source was used during the operando experiments. The XRD data were obtained in parallel‐beam geometry. X‐ray diffractograms were measured from 2θ angles of 5° to 49°. Step sizes between 0.02° and 0.05° were used. Scan times were varied as a compromise between data quality and acquisition speed. Shorter scan times were used when relatively quick changes in composition and/or crystallite sizes were expected (i.e., during the reduction of the catalyst). Longer scan times (with better data quality) were used when changes in composition and/or crystallite sizes were expected to be slower, such as during the FTS reaction.
XRD data analysis was performed by using the fundamental parameters approach68, 69 as implemented in the Bruker Topas software, which allows semiautomated fitting of multiple XRD patterns at the same time. Full‐profile fitting (Rietveld refinement) was performed to extract the composition of the sample and the crystallite sizes of the relevant species. Background subtraction was performed by fitting 3rd or 4th order Chebychev polynomials to the data. The diffraction patterns of the support (i.e., anatase and rutile) and several cobalt species (i.e., Co3O4, CoO, Co2C, and metallic cobalt (CCP and HCP crystal structures)) were taken from the PDF‐4+ 2015 database by the International Centre for Diffraction Data and used in the Rietveld refinement, without further refinement of the unit cell parameters. Instrument parameters (most notably the instrumental line broadening and the X‐ray emission profile of the source) were obtained from a previously measured, highly crystalline Si sample. R‐values were approximately 8–9 for the high quality data and 10–11 for the lower quality (faster) scans, which is comparable to previous experiments by Rayner et al. on a similar catalyst system.70 Crystallite sizes for the relevant Co species were estimated using the Scherrer equation, assuming k=0.89, and after correcting for the instrumental line broadening.
Sample amounts of approximately 10–20 mg of catalyst material (10 wt % or 15 wt % Co/TiO2 FTS catalyst) were loaded into a boro‐silicate glass capillary (length: 8 cm, outer diameter: 1 mm, wall thickness: 0.01 mm). The catalyst was held in place by two plugs of quartz wool. A thermocouple was mounted inside of the capillary, immediately against the quartz wool. The catalyst bed length was chosen such that all catalyst material was within the heating zone of the infrared furnace. The capillary was attached to the holder with rubber ferrules. The setup was then checked for leaks at 10 bar with He.
The catalyst was first reduced in H2 (flow rate: 1.00 mL min−1) by heating from RT to 350 °C with a ramp rate of 5 °C min−1. Quick XRD scans (5 min per scan) were measured during this period. The temperature was then held constant for 2 h, while slightly longer XRD scans (10 min per scan) were measured. The catalyst was then cooled down to 250 °C. We have selected this reaction temperature as we anticipated that sintering would be faster at 250 °C than at lower reaction temperatures. The catalyst was then exposed to FTS conditions: 1.00 mL min−1 H2 and 0.50 mL min−1 CO. For the tests at high pressure, the pressure was slowly increased to 10 bar, this took approximately 3 h. After this, the flows were reduced to 0.66 mL min−1 of H2 and 0.33 mL min−1 of CO. The pressure was kept constant at 10 bar. The pressure was kept constant at 0 bar (relative to atmospheric pressure) for the experiments that were performed at low pressure. XRD scans of 1 h each were measured continuously. A sample of the product stream was analyzed approximately every 23 min. Raman spectra from ≈750 cm−1 to ≈3500 cm−1 were measured continuously with an Avantes Ava‐Raman‐532TEC spectrometer fitted with a 532 nm laser light source (10 s integration time per spectrum, 24 spectra averaged).
Acknowledgements
Michael Claeys and Nico Fischer (University of Cape Town, South Africa) are acknowledged for the design of the operando reactor. We are very grateful to Marjan Versluijs‐Helder (Utrecht University, UU) for her help with the XRD measurements and data analysis. We thank Bruker AXS for their help with the selection of the XRD apparatus. Jan Falkenhagen, Joris Goetze and Pasi Paalanen (UU) have helped with the design of the setup and the selection of the GC. We thank Oscar Kerkenaar, Ad Mens, Jan Willem de Rijk, and Rien van Zwienen (UU) for their help with building the setup. Fouad Soulimani (UU) is acknowledged for helping with Raman spectroscopy.
K. H. Cats, B. M. Weckhuysen, ChemCatChem 2016, 8, 1531.
References
- 1. Handbook of Heterogeneous Catalysis (Eds.: G. Ertl, H. Knözinger, F. Schüth, J. Weitkamp), Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 2. Guerrero-Pérez M. O., Bañares M. A., Chem. Commun. 2002, 1292–1293. [DOI] [PubMed] [Google Scholar]
- 3. Bañares M. A., Guerrero-Pérez M. O., Fierro J. L. G., Cortez G. G., J. Mater. Chem. 2002, 12, 3337–3342. [Google Scholar]
- 4. Weckhuysen B. M., Phys. Chem. Chem. Phys. 2003, 5, 4351–4360. [Google Scholar]
- 5. Weckhuysen B. M., Chem. Commun. 2002, 97–110. [DOI] [PubMed] [Google Scholar]
- 6. Weckhuysen B. M., Natl. Sci. Rev. 2015, 2, 147–149. [Google Scholar]
- 7. Cats K. H., Gonzalez-Jimenez I. D., Liu Y., Nelson J., van Campen D., Meirer F., van der Eerden A. M. J., de Groot F. M. F., Andrews J. C., Weckhuysen B. M., Chem. Commun. 2013, 49, 4622–4624. [DOI] [PubMed] [Google Scholar]
- 8.K. H. Cats, J. C. Andrews, O. Stéphan, K. March, C. Karunakaran, F. Meirer, F. M. F. de Groot, B. M. Weckhuysen, Catal. Sci. Technol 2016, DOI: 10.1039/c5cy01524c.
- 9. Beale A. M., O'Brien M. G., Weckhuysen B. M. in Characterization of Solid Materials and Heterogeneous Catalysts: from Structure to Surface Reactivity, Vol. 1&2 (Eds.: M. Che, J. C. Védrine), Wiley-VCH, Weinheim, 2012, pp. 1075–1117. [Google Scholar]
- 10. Tinnemans S. J., Mesu J. G., Kervinen K., Visser T., Nijhuis T. A., Beale A. M., Keller D. E., van der Eerden A. M. J., Weckhuysen B. M., Catal. Today 2006, 113, 3–15. [Google Scholar]
- 11. Bentrup U., Chem. Soc. Rev. 2010, 39, 4718–4730. [DOI] [PubMed] [Google Scholar]
- 12. Tsakoumis N. E., Voronov A., Rønning M., van Beek W., Borg Ø., Rytter E., Holmen A., J. Catal. 2012, 291, 138–148. [Google Scholar]
- 13. Rochet A., Moizan V., Pichon C., Diehl F., Berliet A., Briois V., Catal. Today 2011, 171, 186–191. [Google Scholar]
- 14. Karaca H., Hong J., Fongarland P., Roussel P., Griboval-Constant A., Lacroix M., Hortmann K., Safonova O. V., Khodakov A. Y., Chem. Commun. 2010, 46, 788–790. [DOI] [PubMed] [Google Scholar]
- 15. Scalbert J., Legens C., Clémençon I., Taleb A.-L., Sorbier L., Diehl F., Chem. Commun. 2014, 50, 7866–7869. [DOI] [PubMed] [Google Scholar]
- 16. Kumar N., Payzant E. A., Jothimurugesan K., Spivey J. J., Phys. Chem. Chem. Phys. 2011, 13, 14735–14741. [DOI] [PubMed] [Google Scholar]
- 17. Sadeqzadeh M., Karaca H., Safonova O. V., Fongarland P., Chambrey S., Roussel P., Griboval-Constant A., Lacroix M., Curulla-Ferré D., Luck F. et al., Catal. Today 2011, 164, 62–67. [Google Scholar]
- 18. Rønning M., Tsakoumis N. E., Voronov A., Johnsen R. E., Norby P., van Beek W., Borg Ø., Rytter E., Holmen A., Catal. Today 2010, 155, 289–295. [Google Scholar]
- 19. de Smit E., Cinquini F., Beale A. M., Safonova O. V., van Beek W., Sautet P., Weckhuysen B. M., J. Am. Chem. Soc. 2010, 132, 14928–14941. [DOI] [PubMed] [Google Scholar]
- 20. Thüne P. C., Weststrate C. J., Moodley P., Saib A. M., van de Loosdrecht J., Miller J. T., Niemantsverdriet J. W., Catal. Sci. Technol. 2011, 1, 689–697. [Google Scholar]
- 21. Boldrin P., Gallagher J. R., Combes G. B., Enache D. I., James D., Ellis P. R., Kelly G., Claridge J. B., Rosseinsky M. J., Chem. Sci. 2015, 6, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morales F., de Groot F. M. F., Glatzel P., Kleimenov E., Bluhm H., Hävecker M., Knop-Gericke A., Weckhuysen B. M., J. Phys. Chem. B 2004, 108, 16201–16207. [Google Scholar]
- 23. Morales F., Grandjean D., Mens A., de Groot F. M. F., Weckhuysen B. M., J. Phys. Chem. B 2006, 110, 8626–8639. [DOI] [PubMed] [Google Scholar]
- 24. Jacobs G., Ji Y., Davis B. H., Cronauer D., Kropf A. J., Marshall C. L., Appl. Catal. A 2007, 333, 177–191. [Google Scholar]
- 25. Hong J., Marceau E., Khodakov A. Y., Griboval-Constant A., La Fontaine C., Briois V., Chem. Eur. J. 2012, 18, 2802–2805. [DOI] [PubMed] [Google Scholar]
- 26. Grunwaldt J.-D., Schroer C. G., Chem. Soc. Rev. 2010, 39, 4741–4753. [DOI] [PubMed] [Google Scholar]
- 27. Tsakoumis N. E., Rønning M., Borg Ø., Rytter E., Holmen A., Catal. Today 2010, 154, 162–182. [Google Scholar]
- 28. van der Laan G. P., Beenackers A. A. C. M., Catal. Rev. 1999, 41, 255–318. [Google Scholar]
- 29. Khodakov A. Y., Chu W., Fongarland P., Chem. Rev. 2007, 107, 1692–1744. [DOI] [PubMed] [Google Scholar]
- 30. James O. O., Chowdhury B., Mesubi M. A., Maity S., RSC Adv. 2012, 2, 7347. [Google Scholar]
- 31. Zhang Q., Deng W., Wang Y., J. Energy Chem. 2013, 22, 27–38. [Google Scholar]
- 32. Dry M. E., Catal. Today 2002, 71, 227–241. [Google Scholar]
- 33. Fischer N., Clapham B., Feltes T., Claeys M., ACS Catal. 2015, 5, 113–121. [Google Scholar]
- 34. Fischer N., Clapham B., Feltes T., van Steen E., Claeys M., Angew. Chem. Int. Ed. 2014, 53, 1342–1345; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1366–1369. [Google Scholar]
- 35. Roth W. L., J. Phys. Chem. Solids 1964, 25, 1–10. [Google Scholar]
- 36. Wyckoff R. W. G., Crystal Structures, Interscience Publishers, New York, 1963. [Google Scholar]
- 37. Enache D. I., Rebours B., Roy-Auberger M., Revel R., J. Catal. 2002, 205, 346–353. [Google Scholar]
- 38. Srinivasan R., De Angelis R. J., Reucroft P. J., Dhere A. G., Bentley J., J. Catal. 1989, 116, 144–163. [Google Scholar]
- 39. Ducreux O., Rebours B., Lynch J., Roy-Auberger M., Bazin D., Oil Gas Sci. Technol. 2009, 64, 49–62. [Google Scholar]
- 40. Cherepanova S. V., Bulavchenko O. A., Tsybulya S. V., J. Struct. Chem. 2008, 49, 512–516. [Google Scholar]
- 41. Karaca H., Safonova O. V., Chambrey S., Fongarland P., Roussel P., Griboval-Constant A., Lacroix M., Khodakov A. Y., J. Catal. 2011, 277, 14–26. [Google Scholar]
- 42. Weller S., Hofer L. J. E., Anderson R. B., J. Am. Chem. Soc. 1948, 70, 799–801. [Google Scholar]
- 43. Jacobs G., Patterson P. M., Zhang Y., Das T., Li J., Davis B. H., Appl. Catal. A 2002, 233, 215–226. [Google Scholar]
- 44. Xiong J., Ding Y., Wang T., Yan L., Chen W., Zhu H., Lu Y., Catal. Lett. 2005, 102, 265–269. [Google Scholar]
- 45. Lynch J., Oil Gas Sci. Technol. 2002, 57, 281–305. [Google Scholar]
- 46. Pei Y.-P., Liu J.-X., Zhao Y.-H., Ding Y.-J., Liu T., Dong W.-D., Zhu H.-J., Su H.-Y., Yan L., Li J.-L. et al., ACS Catal. 2015, 5, 3620–3624. [Google Scholar]
- 47. Pei Y., Ding Y., Zang J., Song X., Dong W., Zhu H., Wang T., Chen W., Chin. J. Catal. 2015, 36, 252–259. [Google Scholar]
- 48. Peña D., Griboval-Constant A., Lecocq V., Diehl F., Khodakov A. Y., Catal. Today 2013, 215, 43–51. [Google Scholar]
- 49. Mohandas J. C., Gnanamani M. K., Jacobs G., Ma W., Ji Y., Khalid S., Davis B. H., ACS Catal. 2011, 1, 1581–1588. [Google Scholar]
- 50. Liu J.-X., Su H.-Y., Sun D.-P., Zhang B.-Y., Li W.-X., J. Am. Chem. Soc. 2013, 135, 16284–16287. [DOI] [PubMed] [Google Scholar]
- 51. Gnanamani M. K., Jacobs G., Shafer W. D., Davis B. H., Catal. Today 2013, 215, 13–17. [Google Scholar]
- 52. Kwak G., Woo M. H., Kang S. C., Park H.-G., Lee Y.-J., Jun K.-W., Ha K.-S., J. Catal. 2013, 307, 27–36. [Google Scholar]
- 53. Pan Z., Parvari M., Bukur D. B., Top. Catal. 2014, 57, 470–478. [Google Scholar]
- 54. Yang J., Jacobs G., Jermwongratanachai T., Pendyala V. R. R., Ma W., Chen D., Holmen A., Davis B. H., Catal. Lett. 2014, 144, 123–132. [Google Scholar]
- 55. Ai L.-H., Jiang J., Powder Technol. 2009, 195, 11–14. [Google Scholar]
- 56. Jiang J., Li L., Mater. Lett. 2007, 61, 4894–4896. [Google Scholar]
- 57. Choi H. C., Jung Y. M., Noda I., Bin Kim S., J. Phys. Chem. B 2003, 107, 5806–5811. [Google Scholar]
- 58. Vuurman M. A., Stufkens D. J., Oskam A., Deo G., Wachs I. E., J. Chem. Soc. Faraday Trans. 1996, 92, 3259–3265. [Google Scholar]
- 59. Sattler J. J. H. B., Beale A. M., Weckhuysen B. M., Phys. Chem. Chem. Phys. 2013, 15, 12095–12103. [DOI] [PubMed] [Google Scholar]
- 60. Sattler J. J. H. B., Mens A. M., Weckhuysen B. M., ChemCatChem 2014, 6, 3139–3145. [Google Scholar]
- 61. Sadezky A., Muckenhuber H., Grothe H., Niessner R., Pöschl U., Carbon 2005, 43, 1731–1742. [Google Scholar]
- 62. Guedes A., Valentim B., Prieto A. C., Rodrigues S., Noronha F., Int. J. Coal Geol. 2010, 83, 415–422. [Google Scholar]
- 63. Tuinstra F., Koenig J. L., J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar]
- 64. Argyle M. D., Frost T. S., Bartholomew C. H., Top. Catal. 2014, 57, 415–429. [Google Scholar]
- 65. Sadeqzadeh M., Chambrey S., Hong J., Fongarland P., Luck F., Curulla-Ferré D., Schweich D., Bousquet J., Khodakov A. Y., Ind. Eng. Chem. Res. 2014, 53, 6913–6922. [Google Scholar]
- 66. de La Peña O'Shea V. A., Alvarez-Galvan M. C., Campos-Martin J. M., Fierro J. L. G., Catal. Lett. 2005, 100, 105–116. [Google Scholar]
- 67. Schulz H., Nie Z., Ousmanov F., Catal. Today 2002, 71, 351–360. [Google Scholar]
- 68. Cheary R. W., Coelho A., J. Appl. Crystallogr. 1992, 25, 109–121. [Google Scholar]
- 69. Cheary R. W., Coelho A. A., Cline J. P., J. Res. Natl. Inst. Stand. Technol. 2004, 109, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rayner M. K., Billing D. G., Coville N. J., Acta Crystallogr. Sect. B 2014, 70, 498–509. [DOI] [PubMed] [Google Scholar]