

Abstract
BACKGROUND
The definition of hereditary prostate cancer (HPC) is based on family history and age at onset. Intuitively, HPC is a serious subtype of prostate cancer but there are only limited data on the clinical phenotype of HPC. Here, we aimed to compare the prognosis of HPC to the sporadic form of prostate cancer (SPC).
METHODS
HPC patients were identified through a national registry of HPC families in the Netherlands, selecting patients diagnosed from the year 2000 onward (n = 324). SPC patients were identified from the Netherlands Cancer Registry (NCR) between 2003 and 2006 for a population‐based study into the genetic susceptibility of PC (n = 1,664). Detailed clinical data were collected by NCR‐registrars, using a standardized registration form. Follow‐up extended up to the end of 2013. Differences between the groups were evaluated by cross‐tabulations and tested for statistical significance while accounting for familial dependency of observations by GEE. Differences in progression‐free and overall survival were evaluated using χ2 testing with GEE in a proportional‐hazards model.
RESULTS
HPC patients were on average 3 years younger at diagnosis, had lower PSA values, lower Gleason scores, and more often locally confined disease. Of the HPC patients, 35% had high‐risk disease (NICE‐criteria) versus 51% of the SPC patients. HPC patients were less often treated with active surveillance. Kaplan–Meier 5‐year progression‐free survival after radical prostatectomy was comparable for HPC (78%) and SPC (74%; P = 0.30). The 5‐year overall survival was 85% (95%CI 81–89%) for HPC versus 80% (95%CI 78–82%) for SPC (P = 0.03).
CONCLUSIONS
HPC has a favorable clinical phenotype but patients more often underwent radical treatment. The major limitation of HPC is the absence of a genetics‐based definition of HPC, which may lead to over‐diagnosis of PC in men with a family history of prostate cancer. The HPC definition should, therefore, be re‐evaluated, aiming at a reduction of over‐diagnosis and overtreatment among men with multiple relatives diagnosed with PC. Prostate 76:897–904, 2016. © 2016 The Authors. The Prostate published by Wiley Periodicals, Inc.
Keywords: prostate cancer, hereditary prostate cancer, clinical characteristics, phenotype, family history
INTRODUCTION
Prostate cancer (PC) poses a major burden to society. It is the most frequently diagnosed cancer among Western males 1. If detected at an early stage, PC can be treated curatively. Serum prostate‐specific antigen (PSA), available since 1987, is the only commonly used test for early detection of PC 2. In the Netherlands, the general practitioners’ guidelines on prostate cancer testing require detailed counseling about its pros and cons. As opposed to the general population (in which predominantly sporadic PC–SPC–occurs), where overtreatment due to PSA testing is assumed to outweigh the proven mortality benefit, PSA testing is advised to men in families with hereditary PC (HPC) 3, 4. It is not entirely clear, however, what the basis is for this advice.
In the Netherlands, the only genetic tests when a high‐penetrance genetic predisposition for PC is suspected, are tests for BRCA2 mutations and, in some centers, a HOXB13‐variant. Because these and other HPC‐related mutations are rare and explain only a small proportion of HPC families, the tests are not widely used. As a result, the definition of HPC is still based on family history and age at onset only 5. HPC is defined as PC detected in at least (i) three first‐degree relatives; (ii) two first‐ or second‐degree relatives diagnosed before the age of 55; or (iii) three consecutive generations. Since the introduction of this definition in 1993, the increase in opportunistic PSA testing, is even more marked among family members of PC patients 6. This may have led to an increase in the detection of relatively many “low‐risk” PCs within families. As a consequence, an increasing part of the HPC families might represent clusters of PC due to increased PSA testing instead of high‐penetrance genetic predisposition. This is supported by the findings of two recent studies that showed that the same low‐penetrance genetic risk variants are found in SPC and HPC 7, 8. Even more so, screening studies among unaffected men in HPC families did not find an increased risk of PC 9, 10. These observations might also lead to the conclusion that HPC patients have a relatively favorable prognosis, while intuitively it is the more “serious phenotype” of PC. Only a few studies, all comprising series from the pre‐PSA era, have specifically reported on the clinical and prognostic characteristics of HPC. They did not identify any differences in staging, grading, or prognosis 11, 12. If the clinical phenotype of PC in HPC‐families is indeed similar or, given the increase in prostate cancer case finding since the introduction of PSA, even favorable as compared to SPC, the current management of HPC families might need to be revised. It would indicate a need for focusing on prevention of over‐diagnosis in men with a positive family history. In this study, we aimed to evaluate whether clinical phenotype differences exist between HPC and SPC diagnosed in the PSA era.
MATERIALS AND METHODS
HPC Patients
The Netherlands Foundation for the Detection of Hereditary Tumours (NFDHT) holds a nation‐wide registry of Dutch HPC‐families. The NFDHT‐registry is clinician‐driven, that is, all probands of families have been referred by a physician who suspects HPC based on family history taking. After referral, a genetic registrar contacts the family and collects pedigree and medical data for all PC patients. All affected men are asked to provide written informed consent for access to their medical files to confirm the diagnoses. For men who are already deceased before registration, their next of kin are requested to provide informed consent.
For this study (conducted as part of the European Union 7th Framework Programme “ProMark: genetic PC variants as markers of disease progression”), all HPC‐families registered until June 2011 were contacted (196 families, reporting 869 HPC patients, 676 of whom were confirmed by medical file review). The NFDHT data were expanded by extracting standard diagnostic data from the Netherlands Cancer Registry (NCR) and by medical file review. The medical file review was performed by the registrars of the NCR held by the Netherlands Comprehensive Cancer Organisation (IKNL) using a standardized registration form.
SPC Patients
From 2003 to 2006, all men up to 75 years of age who were newly diagnosed with PC in the catchment area of the Netherlands Cancer Registry (NCR), location Nijmegen, were selected from the NCR to participate in the European Union 6th Framework Programme‐funded Polygene study, which has been described in more detail before 13. In short, for this study into the genetic susceptibility of PC, detailed clinical information was collected from medical files from a population‐based series of PC patients (n = 1,664). In 2013, additional data were gathered for all these men by the registrars of the NCR, using an identical form as in the aforementioned data collection for HPC patients.
The completed data set for both groups comprised PC diagnostics, clinical, and postsurgical TNM‐stage and Gleason grade (or WHO grade if Gleason score was not reported), primary and salvage therapies, disease progression, and vital status. The patients were stratified by PC aggressiveness, according to the 2014 NICE‐classification, which is similar to the d'Amico classification: high‐risk = all PC with lymph node or distant metastasis and PC with the following characteristics: ≥cT2c, PSA > 20, or biopsy Gleason score ≥ 8; intermediate risk = PC without high‐risk features and with cT2b, PSA 10–20 ng/ml, or biopsy Gleason score = 7; low‐risk = PC without high‐risk or intermediate‐risk features 14, 15. For patients with an unknown Gleason score, WHO‐grade ≥ 3 was considered as a criterion for high‐risk, WHO‐grade 2 for intermediate‐risk, and WHO‐grade 1 for low‐risk PC. The study protocols were approved by the Institutional Review Board of the Radboud University Medical Center.
Statistical Analysis
To improve the comparability of the HPC and SPC patients, only the HPC patients who were diagnosed in the year 2000 or later (i.e., the PSA era) and who were 75 years of age or younger at diagnosis were selected. A total of 324 out of the 676 confirmed HPC patients met these criteria. For three patients, no clinical data could be retrieved (two because they were diagnosed abroad, one because the medical file was already destroyed), leaving 321 HPC patients available for analysis. All 1,664 SPC patients were included in the analysis. Differences between the two groups were evaluated by cross‐tabulations. To account for familial dependence within the HPC data, χ2 tests and Mixed Models using Generalized Estimating Equations (GEE) were performed to test for differences in categorical and continuous variables, respectively. Progression after radical prostatectomy (RP) was defined as two separate serum PSA measurements ≥ 0.2 ng/ml, histologically confirmed recurrence, initiation of salvage treatment (started in the absence of “formal” recurrence) or metastasis. To evaluate differences in Kaplan–Meier progression‐free and overall survival, we performed GEE analyses with a proportional hazards‐model. Relative survival as an alternative for PC‐specific survival was calculated as the ratio of the survival per patient group to the survival in the age‐ and gender‐matched Dutch population. To examine the possible effect of age on this analysis, relative survival was also calculated stratified by age (age at diagnosis <55, 55–65, and 65–75 years of age). Where appropriate, 95% confidence intervals (95%CI) were calculated. Data analyses were performed using SAS software (SAS system 9.3, SAS Institute, Cary, NC).
RESULTS
The mean age of the HPC patients was 62.8 years versus 65.6 years for SPC (mean difference 2.7 years, 95%CI 1.8–3.7 years) (Table I). HPC patients had lower pre‐diagnostic serum PSA values, for example, 18% of the HPC patients had a serum PSA >20 ng/ml versus 29% of SPC (P = 0.02). The HPC patients less often had locally extended disease (cT‐stage ≥ T3: 12.8% vs. 23.1% of the SPC patients, P < 0.01) and less often presented with lymph node (5.0% vs. 8.3%, P = 0.04) or distant metastases (6.2% vs. 9.1%, P = 0.09). Gleason scores were more often unknown for HPC patients (21% vs. 8%). When recoding the WHO‐grades into Gleason scores, HPC patients more often had low‐risk disease according to the NICE‐classification: 34% versus 21%. Also, HPC patients less often had high‐risk disease: 38% versus 51% (P < 0.01).
Table I.
Clinical Characteristics of the HPC Patients and SPC Patients
| HPC patients (N = 321) | SPC patients (N = 1664) | ||||
|---|---|---|---|---|---|
| N | % | N | % | Difference | |
| Age at diagnosis a | P < 0.01 b | ||||
| 40–45 | 1 | 0.3 | 3 | 0.2 | |
| 45–50 | 9 | 3.1 | 15 | 0.9 | |
| 50–55 | 30 | 9.3 | 70 | 4.2 | |
| 55–60 | 69 | 21.5 | 245 | 14.7 | |
| 60–65 | 88 | 27.4 | 336 | 20.2 | |
| 65–70 | 66 | 20.6 | 471 | 28.3 | |
| 70–75 | 58 | 18.1 | 524 | 31.5 | |
| Period of diagnosis | – | ||||
| 2000–2002 | 86 | 26.8 | – | ||
| 2003–2005 | 132 | 41.1 | 1,524 | 91.6 | |
| 2006–2008 | 79 | 24.6 | 140 | 8.4 | |
| 2009–2011 | 24 | 7.5 | – | – | |
| Mean age at diagnosis in years a | 62.8 | 65.6 | Mean difference 2.7 years (95%CI 1.8–3.7 years) c | ||
| Method of diagnosis | P < 0.01 b | ||||
| Needle biopsy | 297 | 92.5 | 1,503 | 90.4 | |
| TURP | 8 | 2.5 | 111 | 6.7 | |
| (Cysto) prostatectomy | 1 | 0.3 | 23 | 1.4 | |
| Unknown/other | 15 | 4.7 | 27 | 1.6 | |
| Serum PSA at diagnosis (ng/ml) | P = 0.02 b | ||||
| <4 | 48 | 14.9 | 170 | 10.2 | |
| 4–10 | 134 | 41.7 | 653 | 39.2 | |
| 10–20 | 62 | 19.3 | 342 | 20.6 | |
| >20 | 59 | 18.4 | 475 | 28.5 | |
| Unknown | 18 | 5.6 | 24 | 1.4 | |
| cTNM‐stage | |||||
| cT1 | 120 | 37.4 | 622 | 37.4 | P = 0.02 b |
| cT2 | 138 | 43.0 | 601 | 36.1 | |
| cT3 | 34 | 10.6 | 333 | 20.0 | |
| cT4 | 7 | 2.2 | 52 | 3.1 | |
| cT0/Tx | 22 | 6.9 | 56 | 3.4 | |
| cN0/Nx | 305 | 95.0 | 1,526 | 91.7 | P < 0.01 b |
| cN+ | 16 | 5.0 | 138 | 8.3 | |
| cM0/Mx | 301 | 93.8 | 1,513 | 90.9 | P = 0.09 b |
| cM1 | 20 | 6.2 | 151 | 9.1 | |
| Gleason score biopsy | P = 0.30 b | ||||
| 2–6 | 187 | 58.3 | 979 | 58.8 | |
| 7 | 42 | 13.1 | 325 | 19.5 | |
| 8–10 | 24 | 7.5 | 222 | 13.3 | |
| Gleason score unknown: | 68 | 21.2 | 138 | 8.3 | |
| (WHO grade 1) | 22 | 6.9 | 51 | 3.1 | |
| (WHO grade 2) | 16 | 5.0 | 21 | 1.3 | |
| (WHO grade 3/4) | 4 | 1.2 | 5 | 0.3 | |
| (WHO grade unknown) | 26 | 8.1 | 61 | 3.7 | |
| NICE‐risk stratification d | P < 0.01 b | ||||
| Low‐risk PC | 109 | 34.0 | 349 | 21.0 | |
| Intermediate risk PC | 90 | 28.0 | 462 | 27.8 | |
| High‐risk PC | 122 | 38.0 | 853 | 51.3 | |
95%CI, 95% confidence interval; HPC, hereditary prostate cancer; PC, prostate cancer; PSA, prostate‐specific antigen; SPC, sporadic prostate cancer; TURP, trans‐urethral resection of the prostate.
The maximum age for inclusion in this study was set at 75 years of age, according to the Polygene study.
χ2 test using generalized estimating equations to test for differences between categorical variables.
Mixed model using generalized estimating equations used to test for differences between normally distributed continuous variables.
PC risk stratification based on the 2014 NICE‐guidelines. High‐risk PC = all PC with lymph node or distant metastasis and localized PC with any or more of the following characteristics: cT ≥ T2c, PSA > 20 or biopsy Gleason score ≥ 8; intermediate risk PC = localized PC without any of the high‐risk features and with cT = T2b, PSA 10–20 ng/ml or biopsy Gleason score = 7; low‐risk PC = localized PC without any high‐risk or intermediate‐risk features, that is, cT1‐T2a, PSA < 10 ng/ml and biopsy Gleason score ≤ 6. If the Gleason score of the biopsy was not reported/unknown, a WHO grade ≥ 3 was considered as a criterion for high‐risk PC, WHO grade 2 was considered a criterion for intermediate‐risk PC and WHO grade 1 as considered a criterion for low‐risk PC, if available.
Despite the favorable stage and grade distribution, HPC patients were less often treated with active surveillance: 7% versus 14% (Table II). Most often, they underwent localized radical treatment: 41% underwent a RP and 21% received RT versus 36% and 11%, respectively, of the SPC patients. HPC patients less often received hormonal therapy (HT) as monotherapy.
Table II.
Treatment Characteristics and Prognosis of the HPC and SPC Patients
| HPC patients (N = 321) | SPC patients (N = 1,664) | Difference | |||
|---|---|---|---|---|---|
| Primary treatment | |||||
| Active surveillance | 22 | 6.9 | 227 | 13.7 | |
| Localized therapy | |||||
| RP | 130 | 40.5 | 600 | 36.1 | |
| Cryotherapy | – | 3 | 0.2 | ||
| Radiation therapy | 66 | 20.6 | 183 | 11.0 | |
| EBRT | 34 | 10.6 | 123 | 7.4 | |
| Brachytherapy | 32 | 10.0 | 60 | 3.6 | |
| RP + RT | 2 | 0.6 | 5 | 0.3 | |
| Systemic therapy | |||||
| RP + HT | 5 | 1.6 | 16 | 1.0 | |
| EBRT + HT | 50 | 15.6 | 288 | 17.3 | |
| HT monotherapy | 37 | 11.5 | 323 | 19.4 | |
| Chemotherapy | – | – | 1 | 0.1 | |
| Other therapy | – | – | 3 | 0.2 | |
| Unknown | 8 | 2.5 | 15 | 0.9 | |
| Progression after RP a | |||||
| 5‐year progression‐free survival | 78% | (95%CI 71–86%) | 74% | (95%CI 69–77%) | P = 0.30 b |
| Survival | |||||
| 5‐year overall survival | 85% | (95%CI 81–89%) | 80% | (95%CI 78–82%) | P = 0.03 b |
| 5‐year relative survival | 98% | (95%CI 94–100%) | 90% | (95%CI 88–92%) | |
EBRT, external‐beam radiation therapy; HT, hormonal therapy; RP, radical prostatectomy; RT, radiation therapy.
Progression after RP was defined as the occurrence of any of the following events: biochemical recurrence, that is, two serum PSA measurements ≥ 0.2 ng/ml; histological evidence of local recurrence; initiation of salvage treatment (e.g., radiation therapy) without documented evidence of biochemical recurrence; detection of metastatic disease.
χ2 testing with generalized estimating equations in a proportional‐hazards model was used to test for differences in survival.
The median follow‐up was 66 months for HPC patients and 94 months for SPC patients. Biochemical recurrence rates after RP were comparable (Table II), with a 5‐year progression‐free survival of 78% for HPC versus 74% for SPC (P = 0.30). The 5‐year overall survival was 85% (95%CI 81–89%) for HPC and 80% (95%CI 78–82%) for SPC. The 5‐year relative survival was 98% (95%CI 94–100%) for HPC and 90% (95%CI 88–92%) for SPC (P < 0.01). When stratifying by age, this advantage for HPC was present only in men diagnosed under 65 years of age (Fig. 1).
Figure 1.
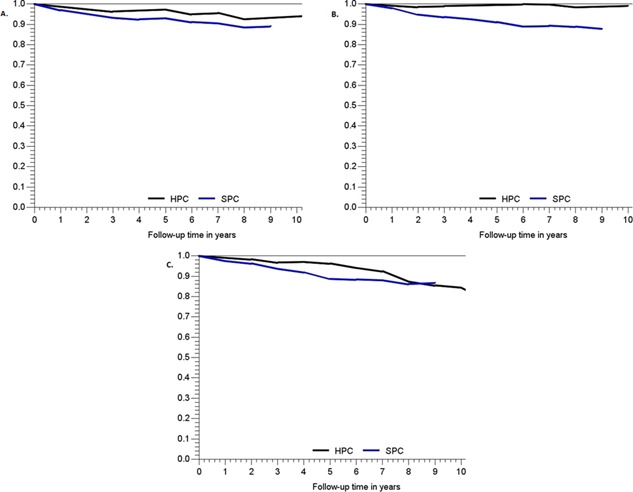
Relative survival per patient group, stratified by age at diagnosis. A: Age at diagnosis <55 years of age. B: Age at diagnosis 55–64 years of age. C: Age at diagnosis ≥65 years of age.
DISCUSSION
We found that in the Dutch population, HPC patients were diagnosed at a younger age. This is logical considering one of the criteria for HPC is age‐related and it is consistent with previous reports 11, 12. As men in HPC families are intuitively expected to be at risk for early onset, aggressive PC, men in “HPC families” are advised to undergo annual PSA screening between 50 and 75 years of age in the Netherlands 3. This means that the screening advice itself can lower the average age at PC diagnosis. A similar phenomenon could occur if the first PC patient in a family is one of the older brothers. His younger siblings might be more aware of PC, which could lead to lower ages at diagnosis. It may also lead to the detection of more non‐aggressive PCs and eventually even a HPC “diagnosis” 16. The younger age at diagnosis was accompanied by a beneficial clinical phenotype of HPC, as can be illustrated by the lower percentage of high‐risk disease (38% vs. 51%). One should bear in mind that, if the Gleason score was not available, the WHO‐grade was used for NICE‐risk stratification. This may have led to small differences, as a higher percentage of the HPC patients had an unknown Gleason score (21% vs. 8%). This was probably due to the fact that part of the HPC patients was diagnosed between 2000 and 2003, when WHO‐grading was still the standard. Because a central pathology revision was not performed, we preferred to use the WHO‐grade as a surrogate for the Gleason score, instead of leaving Gleason score “missing.”
HPC patients were less often treated with active surveillance. This is remarkable, as more HPC patients were eligible for this option according to the risk classification (34% low‐risk disease vs. 21% for SPC). One explanation for this could be that active surveillance was not yet commonly advised as a therapy option in the beginning of the 21st century (2000–2002). Another explanation may be that men in HPC families were more inclined to undergo radical treatment, that is, they might have been more afraid of dying from PC or they have close relatives with good experiences of a specific treatment. The role of the urologist could also be of importance, although we are not aware of any specific data on this topic. Interestingly, brachytherapy was chosen more often by HPC patients (10% vs. 4%). It could be that this relatively low‐impact treatment (as compared to RP) was chosen as an alternative for active surveillance for low‐risk HPC patients.
No differences were seen in progression‐free survival after RP. Unfortunately, the absolute numbers were too small to evaluate progression after other local therapies (including RT). Remarkably, for HPC, the relative survival in patients diagnosed under 65 years of age was superior to SPC. This might be the result of extensive and early PSA testing, which is known to happen particularly within families 6.
Unfortunately, we were not able to stratify the SPC cohort into men with and without a family history of PC, that is, “familial PC” (FPC) and “true” SPC, as the questionnaire covering this item was only completed by 956 of the 1664 Polygene‐participants at a later stage of the study in 2008. In an attempt to deal with this, family history was included as an item in the clinical data collection, but it appeared not to be reported in the medical files in a remarkably high 75% of the cases. An exploratory analysis of the 956 SPC patients who completed the questionnaire identified a beneficial stage distribution for the 210 men with FPC, which would be in line with the overall results in this study (FPC vs. SPC: cT ≥ T2c 28% vs. 37% [P = 0.06]; cN+ 4% vs. 8% [P = 0.06]; cM+ 3% vs. 5% [P = 0.22]). The differences were not statistically significant though, and the Gleason score distribution was similar (data not shown).
Another limitation of this study is the relatively short median follow‐up of 5 years for HPC patients and 8 years for SPC patients. Although the survival analyses take the differences in length of follow‐up into account, obviously, longer follow‐up is required to see whether the suggested observations for the patients with longer follow‐up will also hold for the rest of the group.
A possible further limitation of this study is that the HPC cases and SPC cases came from different catchment populations: the whole country and the Nijmegen region, respectively. We can, therefore, not exclude that regional differences in treatment preference have caused differences in treatment choice between the two groups. These treatment preferences can be either urologist‐based, but also family‐based. Particularly if the proband has had a positive treatment outcome with his treatment of choice, one might expect to see similar treatment choices within a HPC family. This could be one of the possible explanations for the relatively low number of HPC patients treated with “active surveillance.”
Recent studies showed that SPC and HPC were largely similar with respect to the prevalence and associated risks for known low‐penetrance genetic variants 7, 17, 18. Most of these studies focused on the potential of these single nucleotide polymorphisms (SNPs) to aid in the identification of men at increased risk of PC, concluding that the SNPs also explain part of the PC risk in familial PC (FPC) or HPC. Much fewer studies have reported in detail on the clinical phenotype of HPC. A literature search (November 2015) in PubMed identified only two original (non‐review) articles on prognosis of FPC and HPC 11, 12. Grönberg et al. evaluated the prognosis for 241 familial PC cases as compared to 304 non‐familial cases, whereas Bratt et al. evaluated 201 HPC patients and 402 age‐matched controls with non‐hereditary PC. Both found no differences with respect to tumor grade, initial therapy, or overall and prostate‐cancer specific survival. To our knowledge, the current analysis is the largest and only PSA era study into the clinical characteristics of HPC. Given our results and the results of the literature search, we conclude that there is no convincing evidence that HPC is more aggressive than SPC.
Obviously, in a certain proportion of HPC families, aggressive PC is highly prevalent. In these families, it is important to provide timely PC counseling and screening. Even more so, in families with many high‐risk PCs, testing for BRCA2‐ and HOXB13‐mutations is more and more becoming the standard of care. Low‐penetrance markers have been discovered that also appear to play a role in HPC, although the proportion of HPC that is attributable to known variants in these genes is small. Because of this fact and the diagnostic dilemma's as discussed before, HPC appears to be a multifactorial disease more than anything else 10. In families in which mainly asymptomatic low‐risk PCs are diagnosed, it is doubtful whether family members benefit from increased PC awareness, diagnostics, and treatment. Therefore, in the absence of a genetic test for HPC, we suggest that HPC might be redefined. The focus should move from the number of affected relatives to the phenotype of the PCs within a family. One suggestion could be to ignore all patients with low‐risk PC (i.e., <cT2b stage and Gleason score ≤ 6 and PSA < 10) as well as patients with localized prostate cancer with an estimated life‐expectancy of less than 10 years. On the other hand, in families with only one or two male siblings, the minimum number of PC cases to consider PC testing for offspring might have to be ignored if the PC phenotype is very aggressive. In the future, this phenotypic definition of HPC may be replaced by a genetic definition, if a comprehensive genetic PC‐test including all known high‐risk mutations as well as the PC susceptibility SNPs would become available and proves to be useful in clinical practice 19, 20. Although the currently performed research cannot be used to provide direct evidence for these suggestions, we feel that the results of this study into the phenotypic differences between SPC and HPC warrants a critical review of the usefulness of the current HPC definition.
CONCLUSIONS
HPC in the PSA era has a beneficial clinical phenotype as compared to SPC, emphasizing the need for an incorporation of the clinical phenotype into the HPC definition. This might reduce over‐diagnosis and overtreatment among men with multiple relatives diagnosed with PC.
ACKNOWLEDGMENTS
Dr. Cremers was supported by Contract Number 202059 (ProMark: genetic prostate cancer variants as biomarkers of disease progression; www.promark-fp7.eu) from the Seventh Framework Program from the European Union and by an Agiko stipend (number 92003573) of the Netherlands Organisation of Health Research and Development. We are indebted to all HPC patients and their relatives for their registration at the Netherlands Foundation for the Detection of Hereditary Tumours and to the Netherlands Cancer Registry for participating in our study.
The copyright line for this article was changed on 18 April 2016 after original online publication.
Conflicts of interest: The authors have no conflict of interest to disclose.
REFERENCES
- 1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin 2014; 64(1):9–29. [DOI] [PubMed] [Google Scholar]
- 2. Stamey TA, Yang N, Hay AR, McNeal JE, Freiha FS, Redwine E. Prostate‐specific antigen as a serum marker for adenocarcinoma of the prostate. N Engl J Med 1987; 317(15):909–916. [DOI] [PubMed] [Google Scholar]
- 3.Stichting Opsporing Erfelijke Tumoren en de Vereniging Klinische Genetica Nederland Werkgroep Klinische Oncogenetica. Erfelijke tumoren: Richtlijnen voor diagnostiek en preventie: Stichting Opsporing Erfelijke Tumoren en de Vereniging Klinische Genetica Nederland, Werkgroep Klinische Oncogenetica; 2010.
- 4. Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Zappa M, Nelen V, Kwiatkowski M, Lujan M, Maattanen L, Lilja H, Denis LJ, Recker F, Paez A, Bangma CH, Carlsson S, Puliti D, Villers A, Rebillard X, Hakama M, Stenman UH, Kujala P, Taari K, Aus G, Huber A, van der Kwast TH, van Schaik RH, de Koning HJ, Moss SM, Auvinen A, Investigators E. Screening and prostate cancer mortality: Results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow‐up. Lancet 2014; 384(9959):2027–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carter BS, Bova GS, Beaty TH, Steinberg GD, Childs B, Isaacs WB, Walsh PC. Hereditary prostate cancer: Epidemiologic and clinical features. J Urol 1993; 150(3):797–802. [DOI] [PubMed] [Google Scholar]
- 6. Brandt A, Bermejo JL, Sundquist J, Hemminki K. Age‐specific risk of incident prostate cancer and risk of death from prostate cancer defined by the number of affected family members. Eur Urol 2010; 58(2):275–280. [DOI] [PubMed] [Google Scholar]
- 7. Teerlink CC, Thibodeau SN, McDonnell SK, Schaid DJ, Rinckleb A, Maier C, Vogel W, Cancel‐Tassin G, Egrot C, Cussenot O, Foulkes WD, Giles GG, Hopper JL, Severi G, Eeles R, Easton D, Kote‐Jarai Z, Guy M, Cooney KA, Ray AM, Zuhlke KA, Lange EM, Fitzgerald LM, Stanford JL, Ostrander EA, Wiley KE, Isaacs SD, Walsh PC, Isaacs WB, Wahlfors T, Tammela T, Schleutker J, Wiklund F, Gronberg H, Emanuelsson M, Carpten J, Bailey‐Wilson J, Whittemore AS, Oakley‐Girvan I, Hsieh CL, Catalona WJ, Zheng SL, Jin G, Lu L, Xu J, International Consortium for Prostate Cancer G, Camp NJ, Cannon‐Albright LA. Association analysis of 9,560 prostate cancer cases from the International Consortium of Prostate Cancer Genetics confirms the role of reported prostate cancer associated SNPs for familial disease. Hum Genet 2014; 133(3):347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cremers RG, Galesloot TE, Aben KK, van Oort IM, Vasen HF, Vermeulen SH, Kiemeney LA. Known susceptibility SNPs for sporadic prostate cancer show a similar association with “hereditary” prostate cancer. Prostate 2015; 75(5):474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiemeney LA, Broeders MJ, Pelger M, Kil PJ, Schroder FH, Witjes JA, Vasen HF. Screening for prostate cancer in Dutch hereditary prostate cancer families. Int J Cancer 2008; 122(4):871–876. [DOI] [PubMed] [Google Scholar]
- 10. Lynch HT, Kosoko‐Lasaki O, Leslie SW, Rendell M, Shaw T, Snyder C, D'Amico AV, Buxbaum S, Isaacs WB, Loeb S, Moul JW, Powell I. Screening for familial and hereditary prostate cancer. Int J Cancer 2015. doi: 10.1002/ijc.29949. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 11. Gronberg H, Damber L, Tavelin B, Damber JE. No difference in survival between sporadic, familial and hereditary prostate cancer. Br J Urol 1998; 82(4):564–567. [DOI] [PubMed] [Google Scholar]
- 12. Bratt O, Damber JE, Emanuelsson M, Gronberg H. Hereditary prostate cancer: Clinical characteristics and survival. J Urol 2002; 167(6):2423–2426. [PubMed] [Google Scholar]
- 13. Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, Benediktsdottir KR, Magnusdottir DN, Orlygsdottir G, Jakobsdottir M, Stacey SN, Sigurdsson A, Wahlfors T, Tammela T, Breyer JP, McReynolds KM, Bradley KM, Saez B, Godino J, Navarrete S, Fuertes F, Murillo L, Polo E, Aben KK, van Oort IM, Suarez BK, Helfand BT, Kan D, Zanon C, Frigge ML, Kristjansson K, Gulcher JR, Einarsson GV, Jonsson E, Catalona WJ, Mayordomo JI, Kiemeney LA, Smith JR, Schleutker J, Barkardottir RB, Kong A, Thorsteinsdottir U, Rafnar T, Stefansson K. Genome‐wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet 2009; 41(10):1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Institute for Health and Care Excellence. CG175: Prostate cancer: Diagnosis and treatment. NICE Clinical Guidelines. London: National Institute for Health and Care Excellence; 2014.
- 15. D'Amico AV, Whittington R, Malkowicz SB, Cote K, Loffredo M, Schultz D, Chen MH, Tomaszewski JE, Renshaw AA, Wein A, Richie JP. Biochemical outcome after radical prostatectomy or external beam radiation therapy for patients with clinically localized prostate carcinoma in the prostate specific antigen era. Cancer 2002; 95(2):281–286. [DOI] [PubMed] [Google Scholar]
- 16. Hemminki K, Rawal R, Bermejo JL. Prostate cancer screening, changing age‐specific incidence trends and implications on familial risk. Int J Cancer 2005; 113(2):312–315. [DOI] [PubMed] [Google Scholar]
- 17. Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Ghoussaini M, Luccarini C, Dennis J, Jugurnauth‐Little S, Dadaev T, Neal DE, Hamdy FC, Donovan JL, Muir K, Giles GG, Severi G, Wiklund F, Gronberg H, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Lindstrom S, Kraft P, Hunter DJ, Gapstur S, Chanock SJ, Berndt SI, Albanes D, Andriole G, Schleutker J, Weischer M, Canzian F, Riboli E, Key TJ, Travis RC, Campa D, Ingles SA, John EM, Hayes RB, Pharoah PD, Pashayan N, Khaw KT, Stanford JL, Ostrander EA, Signorello LB, Thibodeau SN, Schaid D, Maier C, Vogel W, Kibel AS, Cybulski C, Lubinski J, Cannon‐Albright L, Brenner H, Park JY, Kaneva R, Batra J, Spurdle AB, Clements JA, Teixeira MR, Dicks E, Lee A, Dunning AM, Baynes C, Conroy D, Maranian MJ, Ahmed S, Govindasami K, Guy M, Wilkinson RA, Sawyer EJ, Morgan A, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As NJ, Woodhouse CJ, Thompson A, Dudderidge T, Ogden C, Cooper CS, Lophatananon A, Cox A, Southey MC, Hopper JL, English DR, Aly M, Adolfsson J, Xu J, Zheng SL, Yeager M, Kaaks R, Diver WR, Gaudet MM, Stern MC, Corral R, Joshi AD, Shahabi A, Wahlfors T, Tammela TL, Auvinen A, Virtamo J, Klarskov P, Nordestgaard BG, Roder MA, Nielsen SF, Bojesen SE, Siddiq A, Fitzgerald LM, Kolb S, Kwon EM, Karyadi DM, Blot WJ, Zheng W, Cai Q, McDonnell SK, Rinckleb AE, Drake B, Colditz G, Wokolorczyk D, Stephenson RA, Teerlink C, Muller H, Rothenbacher D, Sellers TA, Lin HY, Slavov C, Mitev V, Lose F, Srinivasan S, Maia S, Paulo P, Lange E, Cooney KA, Antoniou AC, Vincent D, Bacot F, Tessier DC, Initiative CO‐CRUG‐E, Australian Prostate Cancer B, Oncology UKGPCSCBAoUSSo, Collaborators UKPS, Consortium P, Kote‐Jarai Z, Easton DF. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet 2013; 45(4):385–391, 391e381–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cremers RG, Galesloot TE, Aben KK, van Oort IM, Vasen HF, Vermeulen SH, Kiemeney LA. Known susceptibility SNPs for sporadic prostate cancer show a similar association with “hereditary” prostate cancer. Prostate 2015; 75(5):474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al Olama AA, Kote‐Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, Benlloch S, Hazelett DJ, Wang Z, Saunders E, Leongamornlert D, Lindstrom S, Jugurnauth‐Little S, Dadaev T, Tymrakiewicz M, Stram DO, Rand K, Wan P, Stram A, Sheng X, Pooler LC, Park K, Xia L, Tyrer J, Kolonel LN, Le Marchand L, Hoover RN, Machiela MJ, Yeager M, Burdette L, Chung CC, Hutchinson A, Yu K, Goh C, Ahmed M, Govindasami K, Guy M, Tammela TL, Auvinen A, Wahlfors T, Schleutker J, Visakorpi T, Leinonen KA, Xu J, Aly M, Donovan J, Travis RC, Key TJ, Siddiq A, Canzian F, Khaw KT, Takahashi A, Kubo M, Pharoah P, Pashayan N, Weischer M, Nordestgaard BG, Nielsen SF, Klarskov P, Røder MA, Iversen P, Thibodeau SN, McDonnell SK, Schaid DJ, Stanford JL, Kolb S, Holt S, Knudsen B, Coll AH, Gapstur SM, Diver WR, Stevens VL, Maier C, Luedeke M, Herkommer K, Rinckleb AE, Strom SS, Pettaway C, Yeboah ED, Tettey Y, Biritwum RB, Adjei AA, Tay E, Truelove A, Niwa S, Chokkalingam AP, Cannon‐Albright L, Cybulski C, Wokołorczyk D, Kluźniak W, Park J, Sellers T, Lin HY, Isaacs WB, Partin AW, Brenner H, Dieffenbach AK, Stegmaier C, Chen C, Giovannucci EL, Ma J, Stampfer M, Penney KL, Mucci L, John EM, Ingles SA, Kittles RA, Murphy AB, Pandha H, Michael A, Kierzek AM, Blot W, Signorello LB, Zheng W, Albanes D, Virtamo J, Weinstein S, Nemesure B, Carpten J, Leske C, Wu SY, Hennis A, Kibel AS, Rybicki BA, Neslund‐Dudas C, Hsing AW, Chu L, Goodman PJ, Klein EA, Zheng SL, Batra J, Clements J, Spurdle A, Teixeira MR, Paulo P, Maia S, Slavov C, Kaneva R, Mitev V, Witte JS, Casey G, Gillanders EM, Seminara D, Riboli E, Hamdy FC, Coetzee GA, Li Q, Freedman ML, Hunter DJ, Muir K, Gronberg H, Neal DE, Southey M, Giles GG, Severi G; Breast and Prostate Cancer Cohort Consortium (BPC3); PRACTICAL (Prostate Cancer Association Group to Investigate Cancer‐Associated Alterations in the Genome) Consortium; COGS (Collaborative Oncological Gene‐environment Study) Consortium; GAME‐ON/ELLIPSE Consortium, Cook MB, Nakagawa H, Wiklund F, Kraft P, Chanock SJ, Henderson BE, Easton DF, Eeles RA, Haiman CA. A meta‐analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 2014; 46(10):1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langeberg WJ, Isaacs WB, Stanford JL. Genetic etiology of hereditary prostate cancer. Front Biosci 2007; 12:4101–4110. [DOI] [PubMed] [Google Scholar]