

Abstract
Chronic alterations in calcium (Ca2+) signalling in podocytes have been shown to cause proteinuria and progressive glomerular diseases. However, it is unclear whether short Ca2+ peaks influence glomerular biology and cause podocyte injury. Here we generated a DREADD (Designer Receptor Exclusively Activated by a Designer Drug) knock-in mouse line to manipulate intracellular Ca2+ levels. By mating to a podocyte-specific Cre driver we are able to investigate the impact of Ca2+ peaks on podocyte biology in living animals. Activation of the engineered G-protein coupled receptor with the synthetic compound clozapine-N-oxide (CNO) evoked a short and transient Ca2+ peak in podocytes immediately after CNO administration in vivo. Interestingly, this Ca2+ peak did neither affect glomerular perfusion nor filtration in the animals. Moreover, no obvious alterations in the glomerular morphology could be observed. Taken together, these in vivo findings suggest that chronic alterations and calcium overload rather than an induction of transient Ca2+ peaks contribute to podocyte disease.
Diseases of the glomerular filter are a leading cause of end stage renal failure. Podocyte injury is the common final pathway of any glomerular disease. Podocytes are highly specialized epithelial cells and part of the three-layered kidney filtration barrier as they enwrap the glomerular capillaries with their primary and secondary foot processes completely1. This barrier consists of the glomerular fenestrated endothelium, the glomerular basement membrane and the podocytes1. The latter form an interdigitating pattern of foot processes of adjacent cells. The only cell-cell contact of adjacent podocytes is the slit diaphragm, which is located in between the foot processes1. The slit diaphragm does not only serve as a filter to prevent loss of macromolecules from blood into primary urine, but also acts as a signalling platform2,3,4. In the last two decades several genes have been identified to be crucial to mediate signal transduction in podocytes5,6,7,8,9. In particular, the regulation of the actin-based cytoskeleton via several actin-binding proteins like alpha-actinin-4 and CD2AP7,10,11 appears of specific importance as cytoskeletal changes precede the development of foot process effacement and subsequently the onset of proteinuria as observed in patients with podocyte diseases12,13,14. Calcium was identified as an important second messenger in states of podocyte health and disease15. As early as 1978 Kerjaschki et al. reported that foot process effacement might be Ca2+-dependent16. The impact of Ca2+ for podocyte biology was further validated in the following years. Overexpression of the G-protein-coupled angiotensin II type 1 receptor, which results in an increased Ca2+ flux, causes glomerulosclerosis in rats17. Moreover, gain-of-function mutations in the non-selective cation channel TRPC6 were identified to cause familial FSGS8,18. In addition, TRPC6 was found to be upregulated in different glomerular diseases, e.g. minimal change disease or membranous glomerulonephritis19. TRPC channels themselves are tightly regulated20 and activated by various stimuli (such as e.g. angiotensin II AT1 receptor). Some reports see a role for TRPC channels to regulate the contractile and motile phenotype of podocytes, as TRPC5 activates Rac1 and TRPC6 RhoA through Ca2+ signalling20. If this balance is disturbed, podocytes develop either an increased motility through overactivation of Rac1 or a contractile phenotype through overactivation of RhoA. In vivo, expression of either constitutively active Rac1 or RhoA both caused albuminuria and foot process effacement21,22. Additional findings on the Ca2+ dependent degradation of the actin-binding protein synaptopodin via the calcineurin/cathepsin-L pathway further emphasise the importance of Ca2+ in podocytes23,24.
Taken together, numerous reports highlight the role of Ca2+ dependent signalling in podocytes. However, most studies only provide indirect evidence that indeed Ca2+ signalling is required to regulate podocyte function in vivo as they focus on e.g. receptor function and do not study Ca2+ levels in vivo. Due to technical limitations most experiments performed to investigate the role of Ca2+ have been performed on cultured podocytes ex vivo. To further study the role of Ca2+ dependent signalling events in podocytes in vivo and its implication on podocyte function we applied the DREADD (Designer Receptor Exclusively Activated by a Designer Drug) concept and generated a novel podocyte-specific transgenic mouse model in which we were able to induce increased intracellular Ca2+ levels by administration of a specific chemical compound. To this end, we use a previously well characterized mutant human muscarinic type 3 receptor (hM3D), which leads to an exclusive activation by binding of the inert compound Clozapine-N-Oxide (CNO)25,26. With this in vivo approach we demonstrate that single transient Ca2+ peaks do not affect glomerular function.
Results
Conditional expression of a functional hM3D in murine immortalized podocytes
We first validated the hM3D construct in murine immortalized mouse podocytes and generated cell lines either expressing FLAG-tagged hM3D or GFP as control. Immunofluorescence stainings revealed a clear membrane localization of the receptor (Fig. 1A). To verify functionality of the receptor we administered CNO and studied phosphorylation of MAP kinases as a readout for intracellular Ca2+ signalling. CNO led to a significantly increased phosphorylation of the MAP-Kinases MEK1/2 (Fig. 1B). In addition, we performed Ca2+-imaging studies with the Ca2+ indicator dye Fluo-8. Treatment with 2 μM CNO increased intracellular calcium levels in the hM3D-expressing cells, but not in control cells (Fig. 1C,D). Depicted are the Ca2+ signals of 12 different hM3D-expressing cells (Fig. 1D). Of note, some cells showed a single peak upon stimulation with CNO (Fig. 1D upper panel) while others revealed an oscillating response only (Fig. 1D middle panel) or did not react at all (Fig. 1D lower panel). This is most likely a technical artefact due to the lentiviral system used to express the hM3D transgene in the podocyte cell line. While the use of this retroviral gene transfer system allows expression of transgenes even in non-proliferating podocytes, which are hardly to transfect, it does not allow to control for expression levels as integration is random and can occur only once or several times. Experiments were performed in three biological replicates and a total of 45 cells were included in the statistical analysis shown in Fig. 1E. We analysed the fluorescence signal after CNO stimulation in comparison to the basal level prior to CNO stimulation. We observed a significant increase in intracellular Ca2+ upon CNO stimulation in hM3D expressing cells (p < 0.0001). We also excluded receptor desensitization or internalization by repetitive Ca2+ imaging experiments at 0 hrs and 24 hrs after the initial stimulation (Fig. 1F). This data clearly demonstrates the functionality of the hM3D-receptor and the ability to induce an increase of intracellular calcium levels in vitro.
Figure 1. Conditional expression of a functional hM3D in murine immortalized podocytes.

(A) Murine immortalized podocytes stably expressing the FLAG.hM3D were stained with a FLAG antibody to confirm expression and membrane localisation (scale bar = 50 μm). (B) Immortalized mouse podocytes stably expressing FLAG.hM3D or GFP were treated with 1 μM CNO for 10 min. Western Blot analysis revealed a significant increase of the phosphorylation of the MAP-Kinase MEK1/2 after CNO treatment in the receptor expressing cells. (t-test: *p < 0.05). (C,D) Representative images of Ca2+ imaging with Fluo-8 (scale bar = 20 μm). FLAG hM3D expressing podocytes were used for Ca2+ imaging with Fluo-8. Administration of 2 μM CNO leads to an immediate increase of intracellular Ca2+ levels. The time point of CNO administration is indicated with a red tick in panel one. Ionomycin was used as positive control (indicated with a green tick in panel one). 12 cells are depicted from which some reached saturation of the fluorescence signal. Fluorescence is depicted as F/F1–5, where the mean value of the first five measurements (F1–5) prior to CNO stimulation was used for normalisation. Experiments were performed in three biological replicates. (E) Statistical analysis revealed a significant increase of intracellular Ca2+ after CNO stimulation. 45 cells from three biological replicates were included in the statistics. (t-test: p < 0.0001). (F) FLAG.hM3D expressing podocytes were used for repetitive Ca2+ imaging with Fluo-8. Podocytes were treated with CNO at 0 and 24 hrs. At both time points an immediate increase of intracellular Ca2+ levels could be observed. Fluorescence is depicted as F/F1–5, where the mean value of the first five measurements prior to CNO stimulation was used for normalisation.
Increased intracellular Ca2+ levels after CNO stimulation cause Rac1 activation
Small GTPases are known to be crucial for podocyte function and maintenance21,22 and are under tight control of intracellular Ca2+ levels27. Moreover, GTPases play a crucial role for the organisation of the actin cytoskeleton and cell migration28. To investigate the effects of increased intracellular Ca2+ levels after CNO stimulation on small GTPases in podocytes, we performed GTPase pull-down assays for active Rac1 and RhoA. Podocytes were treated with 1 μM CNO for 15 min and the GTP-bound active Rac1 and RhoA were pulled down (Fig. 2A). Comparing the GFP-expressing control cells with the FLAG.hM3D-expressing podocytes revealed no statistically significant differences of active Rac1 or RhoA levels in the receptor expressing cells in comparison to control cells, although a trend towards increased active Rac1 and decreased RhoA levels could be observed (Fig. 2A).
Figure 2. Increased intracellular Ca2+-levels after CNO treatment cause Rac1 activation.

(A) GTPase pull-down assays were performed for active RhoA and Rac1. FLAG.hM3D expressing podocytes and GFP expressing control podocytes were stimulated with 1 μM CNO for 15 min. Pull down of active Rac1 and RhoA revealed no significant changes of active Rac1 or RhoA in FLAG.hM3D-expressing cells in comparison to control cells. Active Rac1 and RhoA levels were normalized to tubulin. (two-tailed t-test: p > 0.05).
Generation of a transgenic podocyte-specific hM3D mouse model
Next, we generated a knock-in mouse model to manipulate Ca2+ levels in podocytes specifically in vivo. To this end, a ROSA26 targeting vector was generated in which expression of the C-terminal FLAG-tagged hM3D construct is driven by a CAGS promotor as published previously29. To ascertain cell type specific expression of the receptor transgene a loxP-flanked Neo.Stop cassette was inserted upstream of the receptor sequence. In addition, we included an IRES.GFP to visualize transgene expression (Fig. 3A). C57/BL6 ES cells of the Bruce 4 lineage were transfected with the targeting vector and positive clones were identified via selection and southern blot analysis (Fig. 3B). Positive clones were used to generate chimeras that were intercrossed with C57/BL6 mice to establish the ROSA26 LoxP-STOP-LoxP hM3D mouse strain. Mice carrying the hM3D construct in the ROSA26 locus were mated with Nphs2. Cre mice to achieve a podocyte specific receptor expression after Cre mediated recombination and deletion of the STOP codon (Fig. 3A)30. We further validated expression of the receptor by isolating and culturing primary glomerular cells in vitro. Podocytes from hM3D-transgenic mice also expressed GFP and could easily been identified (Fig. 3C). To validate the functionality of the hM3D receptor in primary podocytes we labelled the cells with the Ca2+ indicator dye Fluo-8 and stimulated the cells with 100 μM CNO. hM3D/GFP-positive cells showed a strong increase in the intracellular Ca2+-levels upon CNO treatment, while hM3D-negative cells did not respond to CNO treatment (Fig. 3D). Experiments were performed in three biological replicates. Analyses of the signal intensities revealed an immediate increase of intracellular Ca2+ with a constant decline of the signal (Fig. 3E). Of note, the strong oscillatory phenotype observed in some virus transduced cultured podocytes was not seen in primary cells. Other glomerular cells, like endothelial or mesangial cells, which were co-cultured in this mixed primary cell culture system, served as internal controls and did not respond to CNO nor did the outgrowing glomerular cells from hM3D negative control animals. Thus, our data clearly shows the podocyte-specific expression of the hM3D receptor and its functionality in isolated primary podocytes.
Figure 3. Generation of a transgenic podocyte-specific hM3D mouse model.

(A) Schematic of the ROSA26 locus with the FLAG.hM3D sequence before and after Cre recombination. Triangles indicate loxP sites and rectangles indicate exons. (B) Southern Blot analyses were performed with a ROSA26 and a neomycin probe to screen for positive ES-cell clones. (C) Isolated primary glomerular cells were cultured and GFP expression was visualized by confocal microscopy. Glomerular cells from ROSA26hM3D/wt; PodCretg/wt show cytoplasmic GFP expression. ROSA26wt/wt; PodCretg/wt control cells were GFP negative. (scale bar = 50 μm). (D,E) Representative images of Ca2+ imaging with Fluo-8 (scale bar = 20 μm). Primary glomerular cells were used for Ca2+ imaging with Fluo-8. Cells expressing the receptor showed an instant increase of intracellular Ca2+ levels after administration of 100 μM CNO. Control cells did not react to CNO (scale bar = 20 μm). Experiments were performed in three biological replicates. Fluorescence is depicted as F/F1–5, where the mean value of the first five measurements prior to CNO stimulation was used for normalisation.
Administration of CNO leads to increased Ca2+-levels in podocytes in vivo
To assess the functionality of the receptor in vivo we mated our ROSAhM3D/wt; Nphs2.Cretg/wt mice with the Ca2+-reporter strain GCaMP3tg/wt 31. Using Multi-Photon-microscopy we were able to study the effects of CNO administration on podocytes in vivo. After i.a. injection of 5 mg/kg bodyweight CNO an immediate elevation of the intracellular Ca2+ levels was observed in podocytes (Fig. 4A,B). All podocytes responded simultaneously and showed a significant single transient Ca2+ peak (Fig. 4B,C).
Figure 4. Administration of CNO leads to increased Ca2+-levels in podocytes in vivo.

(A) Glomeruli of ROSA26hM3D/wt; NPHS2.Cretg/wt; GCaMP3tg/wt mice were visualised in the intact kidney in vivo using a Multi-Photon-microscope. CNO (5 mg/kg) was injected i.a. after 10 sec and caused an immediate increase in intracellular Ca2+ levels (scale bar = 25 μm). (B) Fluorescence is depicted as F/F1–10, where the mean value of the first ten measurements (F1–10) prior to CNO stimulation was used for normalisation. Experiments were performed in three biological replicates. (C) Statistical analysis performed for Multi-Photon-imaging experiments revealed a significant increase of the intracellular Ca2+ after CNO stimulation. N = 18 cells were included in the statistical analysis (t-test p < 0.0001).
hM3D-mediated Ca2+-peaks do not effect glomerular filtration or ex vivo kidney perfusion
Having shown the feasibility of our transgenic mouse model to transiently increase Ca2+ levels in podocytes we set out to study the effects of CNO and a subsequent increase of intracellular Ca2+-levels in podocytes on glomerular filtration and glomerular perfusion. First, we performed transcutaneous glomerular filtration rate measurements to study the glomerular filtration rate (GFR) as published by Schreiber et al.32. Using this method, we were able to record the glomerular filtration rate after CNO administration in freely moving mice over 60 minutes. We did not observe any significant differences in GFR between hM3D-expressing animals in comparison to their littermate controls (Fig. 5A).
Figure 5. Increased intracellular Ca2+levels do not affect glomerular filtration or ex vivo kidney perfusion.
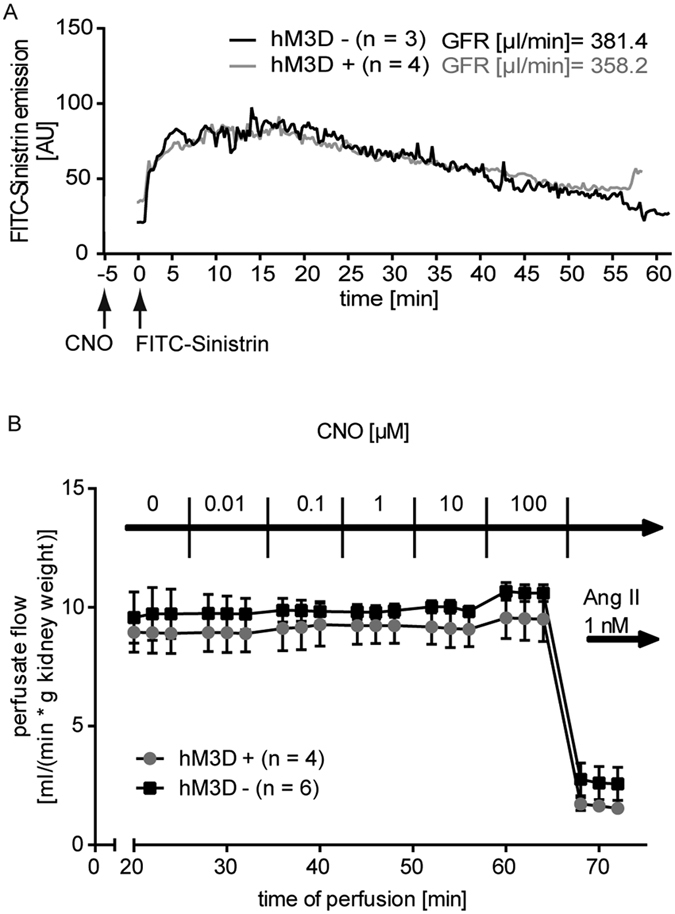
(A) Transcutaneous GFR measurement with hM3D-expressing animals in comparison to their littermate controls does not reveal changes after CNO (5 mg/kg bodyweight) administration. Displayed are means (n = 4: hM3D positive, n = 3: hM3D negative). (B) Ex vivo kidney perfusion was performed with increasing CNO concentrations. Comparing hM3D-expressing mice with their control littermates does not show differences in the vascular resistance. Angiotensin II was used as positive control. Displayed are means with standard error of the mean (SEM).
Second, we investigated the impact of increased intracellular Ca2+-levels on glomerular perfusion. As podocytes harbour a contractile actin-myosin-based machinery33 we hypothesized, that podocytes adapt their contractile behaviour due to the changes in Ca2+ concentration, which in turn might alter vascular resistance. To this end, we performed ex vivo perfusion experiments as published previously34. Kidneys of hM3D-expressing mice and their littermate controls were subjected with increasing CNO concentrations, starting at 0.01 μM up to 100 μM. However, we could not observe a change in the vascular resistance after CNO administration (Fig. 5B). Administration of angiotensin II, which leads to constriction of the vas afferens, resulted in an immediate and strong decrease of the perfusion rate and served as positive control. Taken together, these results show that administration of CNO and a subsequent increase in intracellular Ca2+ does not change the glomerular filtration rate nor renal vascular resistance.
Increased intracellular Ca2+-levels do not cause glomerular disease
After investigating short-term effects of increased intracellular Ca2+ levels on podocyte function, we next investigated effects after repetitive administration of CNO. Animals expressing the hM3D construct and their littermate controls received CNO for 5 consecutive days (5 mg/kg bodyweight i.p.). However, we could not observe any signs of glomerular disease based on histological analyses as well as urinary albumin-to-creatinine ratios (Fig. 6B–D). Immunofluorescence stainings for the actin-binding protein synaptopodin as well as the slit diaphragm protein podocin did also not reveal differences between the hM3D- expressing and control mice (Fig. 6C). Next, to investigate if a prolonged application of CNO causes glomerular disease, we administered CNO (5 mg/kg bodyweight i.p.) to mice expressing the hM3D construct and their control hM3D-negative littermates for 18 consecutive days. Again, urinary and histological analyses did not reveal any signs of glomerular disease (Fig. 7B–D). In conclusion, even a repetitive administration of CNO for 18 days failed to induce glomerular disease.
Figure 6. Increased intracellular Ca2+-levels do not cause glomerular disease.

(A) Scheme of CNO administration. 5 mg/kg bodyweight CNO was injected i.p. on 5 consecutive days. (B) Urinary albumin to creatinine ratios did not show an increase after 5 days of CNO administration in both groups. Displayed are means with SEM; 2way ANOVA and Bonferroni’s multiple comparison test (p > 0.05). (C) Kidney sections of hM3D-expressing animals and their littermate controls after 5 days of CNO treatment were stained with a podocin and synaptopodin antibody to visualize the slit diaphragm and the actin cytoskeleton respectively (scale bar = 20 μm). (D) PAS (periodic acid Schiff stainings) were performed on day 5 of CNO administration and did not reveal differences in histology (scale bar = 50 μm).
Figure 7. Long-term CNO treatment does not cause glomerular disease.

(A) Scheme of CNO administration. 5 mg/kg bodyweight CNO was injected i.p. on 18 consecutive days. (B) Urinary albumin to creatinine ratios did not show an increase after 18 days of CNO administration in both groups. Displayed are means with standard error of the mean (SEM); 2 way ANOVA and Bonferroni’s multiple comparison test (p > 0.05). (C) Kidney sections of hM3D-expressing animals and their littermate controls after 18 days of CNO stimulation were stained with a podocin and synaptopodin antibody to visualize the slit diaphragm and the actin cytoskeleton respectively (scale bar = 20 μm). (D) PAS (periodic-acid Schiff stainings) were performed on day 18 of CNO administration and did not reveal differences in histology (scale bar = 50 μm).
Discussion
In this study, we present a novel transgenic mouse model to study Ca2+ signalling in podocytes in vivo. We used a modified DREADD, a Gq-coupled human muscarinic type 3 receptor (hM3D). Due to two point mutations (Y149C/A239G) this receptor can solely be activated by the chemical compound CNO but no longer by its native ligand acetylcholine25,26. Moreover, great advantages of the usage of CNO are its bioavailability for rodents35, the natural affinity of its parent compound clozapine to M3 receptors, which makes CNO a potent agonist of the mutated M3 receptor26 and its pharmacological inert behaviour25,36. The highly specific binding of CNO to the receptor causes activation of the Phospholipase C (PLC) pathway in the targeted cell type. This activation leads to binding of IP3 to the IP3-receptor at the ER resulting in a strong Ca2+ release from the ER into the cytoplasm. This allows studying effects of increasing intracellular Ca2+ levels in vivo in a novel, highly specific manner. As we inserted a LoxP-STOP-LoxP cassette in the ROSA26 locus upstream of the hM3D sequence, this mouse model may be used to study effects of Gq-coupled receptor activation in a variety of tissues and cell types. Here, we mated the ROSAhM3D/wt mice to a podocyte-specific Cre recombinase driver line (NPHS2.Cretg/wt)30 to achieve exclusive expression of the receptor transgene in podocytes. Doing so, the increase of intracellular Ca2+ after CNO administration solely occurred in podocytes.
Surprisingly, despite the proven functionality of the hM3D receptor in podocytes as demonstrated in vitro, ex vivo and in vivo, activation of the PLC pathway did not affect glomerular filtration or perfusion nor caused glomerular disease even after a prolonged stimulation for 18 consecutive days. Apparently, short and transient Ca2+ peaks as visualized by calcium imaging in vivo in this study are not sufficient to cause overt morphological alterations or functional changes at the renal filtration barrier. We did not observe any effect on glomerular perfusion, filtration or the onset of albuminuria. Our findings are in line with previously reported effects of G-protein coupled receptors in regulating glomerular biology. Expression of a constitutively active Gq-coupled receptor mutant in podocytes did also not result in any glomerular dysfunction under physiological conditions37. Only when a second hit was present, e.g. the PAN nephrosis model or diabetic conditions, glomerular malfunction was aggravated to a greater extent as compared to wildtype controls37.
These findings do not allow the assumption that Ca2+ signalling events are dispensable for podocyte function. As we activated the PLC pathway, increased Ca2+ levels primarily derive from the ER. This might be of particular importance in podocytes with their delicate cytoskeletal architecture. Increased Ca2+ levels close to the ER, which is primarily found in perinuclear regions in the podocyte cell body, might not resemble the intracellular Ca2+ concentration as observed in patients with TRPC6 gain-of-function mutations8. Moreover, podocytes rely on a stringent control of their intracellular Ca2+ levels as they harbor several homeostasis mechanisms including the Na+-Ca2+-exchanger, the ATP-dependent plasma membrane Ca2+ pump (PMCA) as well as Ca2+-buffers like calbindin and parvalbumin38. It is well conceivable that increasing intracellular Ca2+ levels due to the CNO administration might be compensated by these homeostasis mechanisms. In addition, the slit diaphragm might serve as a Ca2+ microdomain orchestrating Ca2+-dependent proteins like the TRPC channels, GTPases and synaptopodin. Furthermore, administration of CNO did only result in a transient Ca2+ peak in podocytes.
One might also speculate, whether the hM3D-transgene gets rapidly internalized or undergoes desensitization upon stimulation. However, studies performed in hippocampal neurons expressing a similar hM3D-transgene showed repetitive activation of the receptor 24 hrs after the initial CNO treatment25, suggesting that receptor internalisation and desensitization does not occur. Similarly, we could observe a repetitive activation of the receptor and subsequent increase of intracellular Ca2+ levels by stimulating podocytes with CNO at 0 and 24 hrs. It is therefore easily conceivable, that a sustained Ca2+ influx rather than a single Ca2+ peak is required to induce cytoskeletal changes to cause foot process effacement in podocytes.
In conclusion, we present a novel DREADD mouse model to study Ca2+ signalling events in vivo. As the DREADD sequence is integrated into the ROSA26 locus it can be easily applied for all murine tissues by mating the loxP-STOP-loxP hM3D mice with tissue specific Cre recombinase mouse lines. In podocytes, a transient Ca2+ peak alone is not sufficient to impair podocyte function challenging the current view of transient Ca2+ peak as regulators of glomerular biology.
Material and Methods
Cell culture
Immortalized mouse podocytes were cultured with RPMI-1640 medium (Sigma-Aldrich, St. Louis, USA) with 10% fetal bovine serum, 5% sodium pyruvate solution 100 mM (Sigma-Aldrich, St. Louis, USA) and 5% HEPES buffer solution 1 M (Life Technologies, Carlsbad, USA) as previously reported39. The undifferentiated and proliferating cells were cultured at 33 °C in the presence of 2.5 μl murine IFN-gamma (Provitro, Berlin, Germany). To induce podocyte differentiation the cells were shifted to 37 °C for 14 days in the absence of IFN-gamma. To generate podocyte cell lines with stable expression of FLAG-tagged hM3D or GFP we used the commercially available pLenti6/V5-dest vector (Thermo Fisher, Waltham, USA) as previously published40. To this end, podocytes were grown at 33 °C and transduced with a Lentivirus containing the hM3D or GFP construct respectively.
Immunofluorescence on cells
Immortalized mouse podocytes were grown at 37 °C for 14 days. The cells were fixed with 4% paraformaldehyde, further blocking was performed with 5% normal donkey serum for 30 min. The cells were washed with 1 x PBS (phosphate buffered saline: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4) containing 1 mM CaCl2 and 0.5 mM MgCl2 for three times. The cells were stained with an anti-FLAG antibody over night at 4 °C. The primary antibody was diluted in 1 x PBS with 1 mM CaCl2, 0.5 mM MgCl2 and 0.1% Triton-X and with 5% NDS. Prior to incubation with the secondary antibody, cells were washed again three times with 1 x PBS with 1 mM CaCl2 and 0.5 mM MgCl2. The respective secondary antibody was diluted in 1 x PBS with 1 mM CaCl2, 0.5 mM MgCl2 and 0.1% Triton-X. Mounting was performed with Prolong Gold with DAPI (Thermo Fisher, Waltham, USA). Antibody dilution and commercial source are listed in Table 1. All images were taken with an Axiovert 200 M microscope/C-Apochromat 63x/1.20 W water immersion objective (all from Carl Zeiss MicroImaging GmbH). Images were further processed with ImageJ/Fiji Software and Photoshop CS4 version 11.0.
Table 1. Antibodies.
| Name | Company | Catalog No. | host species | Dilution IF | Dilution WB |
|---|---|---|---|---|---|
| Anti-FLAG | Sigma | F7425 | Rabbit | 1:100 | |
| Anti-Podocin | Sigma | P0372 | Rabbit | 1:100 | |
| Anti-pMEK1/2 | Cell signaling | 2338S | Rabbit | 1:1000 | |
| Anti-MEK1/2 | Cell signaling | 9126 | Rabbit | 1:1000 | |
| Anti-Synaptopodin | PROGEN Biotechnik | 65294 | mouse | 1:10 | |
| goat anti-rabbit IgG HRP conjugated | Jackson Immuno Research | 111-035-003 | Goat | 1:30.000 | |
| donkey anti-rabbit IgG (H + L)-Alexa Fluo 488 | Jackson Immuno Research | 711-545-152 | Donkey | 1:250 | |
| donkey anti-rabbit IgG (H + L)-Cy3, MinX | Jackson Immuno Research | 711-165-152 | Donkey | 1:250 |
Western Blot
Immortalized mouse podocytes were stimulated with 1 μM CNO (Enzo Life Sciences, Farmingdale, USA) diluted in medium for 10 min. Afterwards the cells were harvested using a cell scraper and lysed in 100 μl 1% Trition-X 100 buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 50 mM NaF, 15 mM Na4P2O7) and 1 mM of the serine protease inhibitor PMSF Phenylmethanesulfonyl fluoride (Sigma-Aldrich, St. Louis, USA) and 2 mM of the phosphatase inhibitor Na3VO4 (Sigma-Aldrich, St. Louis, USA). Following incubation on ice for 15 min and subsequent centrifugation (14.000 × g, 4 °C, 15 min) the supernatant was used for further analyses. The diluted lysates (with 2x SDS-PAGE buffer) were separated by SDS-PAGE. For gel electrophoresis the XCell SureLockTM Mini-Cell System was used. Electrophoresis was performed with 70 V for 30 min, followed by 25 mA/gel for 1:45 h. Blotting was performed on PVDF membranes (Carl Roth, Karlsruhe, Germany) for 1 h at 12 V. After the transfer, membranes were blocked with 5% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, USA) for 30 min, followed by three washing steps. The primary antibody was diluted in washing buffer (30 mM Tris, 300 mM NaCl and 0.3% (v/v) Tween20, pH 7.5) and incubation was performed over night at 4 °C. The respective secondary antibody was diluted in washing buffer and incubated 45 min at room temperature, followed by visualization with chemiluminescence with a Fusion Solo S (Vilber Lourmat, Eberhardzell, Germany). Antibody dilution and commercial source are listed in Table 1.
Ca2+-imaging
Immortalized mouse podocytes were grown at 33 °C and cultured on 8 chamber slides (ibidi GmbH, Planegg/Martinsried, Germany). The cells were incubated with 5 μM Fluo-8 (Sigma-Aldrich, St. Louis, USA) in imaging buffer (125 mM NaCl, 5 mM KCl, 1.2 mM MgSO4*7H2O, 25 mM HEPES, 6 mM Glucose) for 30 min at 37 °C. Afterwards they were kept in imaging buffer for 20 min at room temperature. Imaging was performed with a Confocal microscope LSM710/Axiobserver Z1(Carl Zeiss Microimaging, Jena, Germany) using an 40x objective. Pictures were taken every 0.78 secs. 2 μM CNO was added after 10 pictures for the immortalized mouse podocytes and 100 μM after 20 pictures for the primary glomerular cells. 3 μM Ionomycin was used as positive control and added to the immortalized cells after 280 pictures and after 130 pictures to the primary cells.
GTPase pull down assay
Immortalized mouse podocytes were grown at 33 °C. Five confluent dishes of each cell line were stimulated with 1 μM CNO. After 15 min the cells were pooled and lysed according to the according to manufacturer’s instructions. Rho and Rac1 pull down assays (Thermo Fisher, Waltham, USA) were performed according to manufacturer’s instructions.
Transgenic mouse models
Generation of the ROSA26hM3D/wt mice is described in the results section in detail. Briefly, to generate mice expressing the hM3D transgene in podocytes, we mated ROSA26hM3D/wt mice to NPHS2.Cretg/wt mice30. Multi-Photon-imaging was performed using the Ca2+ reporter mouse GCaMP3tg/wt 31. ROSAhM3D/wt; NPHS2.Cretg/wt mice were mated with GCaMP3tg/wt to achieve a simultaneous hM3D and Ca2+ reporter expression in podocytes exclusively. All animals were on a pure C57/Bl6 background. The mouse holding was done in the University of Cologne animal facility according to standardized specific pathogen-free conditions. All animal experiments were performed in accordance with the guidelines provided by the LANUV NRW (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen/State Agency for Nature, Environment and Consumer Protection North Rhine-Westphalia). The experimental protocol was examined and approved by the LANUV NRW.
Isolation of primary cells
To isolate primary cells we perfused the kidneys via the renal arteries with magnetic beads (Invitrogen, Carlsbad, USA) and isolated glomeruli as previously published41,42. The isolated glomeruli were transferred in 12-well dishes with RPMI-1640 medium (Sigma-Aldrich, St. Louis, USA) with 10% fetal bovine serum, 5% sodium pyruvate solution 100 mM (Sigma-Aldrich, St. Louis, USA) and 5% HEPES buffer solution 1 M (Life Technologies, Carlsbad, USA). After three days glomeruli were detached from the surface with ice cold PBS. All cells grown on the dish remained attached and were used for further analysis.
Administration of CNO in vivo
Mice were injected intra-peritoneally with 5 mg/kg bodyweight CNO (Enzo Life Sciences, Farmingdale, USA) dissolved in NaCl either 5 or 18 consecutive days as published previously25. Spot urine was collected on day 1 and day 5 and day 1, day 11 and day 18 respectively. Tissue samples were harvested at day of sacrifice.
Urinary analysis
Quantification of the urinary albumin levels was performed with a specific mouse albumin ELISA kit according to the manufactor’s instructions (ICL/Dunn Labortechnik GmbH, Asbach, Germany). Creatinine levels were quantified with a urinary creatinine kit according to the manufactor’s instructions (Biomol, Hamburg, Germany).
Histological analysis on mouse tissue
Mice were perfused with phosphate-buffered saline via the heart and kidneys were either fixed in 4% paraformaldehyde overnight or frozen in Tissue-Tek® O.C.T.™ compound (Sakura, Leiden, Netherlands). Paraformaldehyde fixed kidneys were embedded in paraffin and used for cutting 2 μm thick sections. To study morphological changes we performed a periodic-acid Schiff staining. To this end, sections were stained with 0.9% periodic acid (Carl Roth, Karlsruhe, Germany) for 10 min, followed by incubation in Schiff’s reagent for 10 min (VWR, Radnor, USA). After washing with tap water for 2 min sections were incubated for 10 min in Mayer’s hematoxylin solution (Sigma-Aldrich, St. Louis, USA). Mounting was performed with histomount. For immunofluorescence analysis frozen kidney sections (5 μm) were used. After fixation with paraformaldehyde sections were blocked with 5% normal donkey serum in phosphate buffered saline with 0, 1% Triton-X (PBS-T) and subsequently incubated with primary antibody diluted in PBS-T with 5% NDS overnight at 4 °C. The respective secondary antibody was diluted in PBS-T and incubation was performed for 45 min at room temperature. Mounting was done with Prolong Gold antifade with DAPI (Thermo Fisher, Waltham, USA). All images were taken with an Axiovert 200 M microscope/C-Apochromat 63x/1.20 W (all from Carl Zeiss MicroImaging GmbH, Jena, Germany). Images were further processed with ImageJ/Fiji Software and Photoshop CS4 version 11.0. Antibody dilution and commercial source are listed in Table 1.
Multi-Photon – Calcium imaging
4 weeks old ROSAhM3D/wt; NPHS2.Cretg/wt; GCaMP3tg/wt mice were anaesthetized using isoflurane and buprenorphine (0.1 mg/kg). A tube was placed into the trachea to facilitate breathing, and the right carotid artery was cannulated for dye infusion and injection of CNO. 70 kDa Texas red dextrane was injected i.a. to label the vasculature. The left kidney was exteriorized via a small incision at the left flank of the animal. The mouse was placed onto a heated mouse holder and the kidney was stabilized and covered with a coverslip for imaging. Body temperature was maintained during the imaging time. Calcium imaging was performed with a TSC SP8 upright Multi-Photon microscope (Leica) using an IR Apo L25x/0.95 W objective and a Coherent Chameleon Vision II laser at a wavelength of 940 nm. For acquisition two external hybrid detectors were used. The time courses of calcium signals were acquired at 1 frame per second over 90 seconds. After 10 seconds of baseline recording, 5 mg/kg bodyweight CNO was injected. The images were further processed with Leica Application Suite X, Version 1.1.0.
Transcutaneous glomerular filtration rate measurement
To study the glomerular filtration rate we made use of a transcutaneous measurement approach32. Therefore ROSAhM3D/wt; NPHS2.Cretg/wt mice were anaesthetized with isofluran and a sensor was taped directly on their skin. After injecting 5 mg/kg CNO i.p., we injected 15 mg/100 g FITC-Sinistrin i.v. and measured the GFR for 60 min in the freely moving mice. Calculation of the GFR was performed according to Schreiber et al.32.
Isolated perfused mouse kidney
Kidneys from ROSAhM3D/wt; NPHS2.Cretg/wt mice and their littermate controls were perfused ex-situ at a constant perfusion pressure (100 mmHg) as described in detail previously34. Perfusion medium consisted of a modified Krebs-Henseleit buffer supplemented with bovine serum albumin (6 g/100 ml) and human erythrocytes (10% hematocrit). The renal vein was cannulated and samples of the venous perfusate were collected and weighed every 2 minutes for the determination of renal blood flow. Three samples were taken during each experimental period and the last two values were averaged for statistical analysis.
Statistical analysis
All results are expressed as means ± SEM, except for the GFR measurement, which is depicted as mean. Statistical significance was evaluated using GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, CA). For two groups t-test was applied and a P-value < 0.05 was considered significant. For two groups with two independent variables 2 way ANOVA combined with Bonferroni’s multiple comparison test was applied and a P-value < 0.05 was considered significant.
Additional Information
How to cite this article: Koehler, S. et al. Single and Transient Ca2+ Peaks in Podocytes do not induce Changes in Glomerular Filtration and Perfusion. Sci. Rep. 6, 35400; doi: 10.1038/srep35400 (2016).
Acknowledgments
We thank S. Keller, S. Kunath and N. Urban and the CECAD mouse facility for excellent technical support. We thank S. Shankland (University of Washington, Seattle, WA) for providing us with the immortalized mouse podocyte cell line. P. T. Brinkkoetter received funding from the Deutsche Forschungsgemeinschaft (BR2955/4-1 and BR2955/6–1) and Köln Fortune Program of the University of Cologne, Germany. B. Schermer obtained funding from the Deutsche Forschungsgemeinschaft (SCHE1562/2). T. Benzing received funding from the Deutsche Forschungsgemeinschaft (SFB635 and 829). F. Schweda received funding from the Deutsche Forschungsgemeinschaft (SFB699). M.J.H. was supported by Deutsche Forschungsgemeinschaft grant HA-6212/2.
Footnotes
Author Contributions S.K., S.B., B.S., T.B. and P.T.B. conceived the study; S.K., S.B., A.K., J.B., M.J.H., H.H., M.C.V., C.M.W., F.T.W. and F.S. carried out the experiments; S.K., S.B., F.T.W., F.S., B.S., T.B. and P.T.B. designed experiments and analyzed the data; S.K., S.B., B.S., T.B. and P.T.B. wrote the manuscript with input from all other authors. All authors approved the manuscript.
References
- Pavenstädt H., Kriz W. & Kretzler M. Cell biology of the glomerular podocyte. Physiol. Rev. 83, 253–307 (2003). [DOI] [PubMed] [Google Scholar]
- Benzing T. Signaling at the Slit Diaphragm. J. Am. Soc. Nephrol. 15, 1382–1391 (2004). [DOI] [PubMed] [Google Scholar]
- Garg P., Verma R. & Holzman L. B. Slit diaphragm junctional complex and regulation of the cytoskeleton. Nephron Exp. Nephrol. 106, e67–e72 (2007). [DOI] [PubMed] [Google Scholar]
- Huber T. B. & Benzing T. The slit diaphragm: a signaling platform to regulate podocyte function. Curr. Opin. Nephrol. Hypertens. 14, 211–216 (2005). [DOI] [PubMed] [Google Scholar]
- Boute N. et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 24, 349–354 (2000). [DOI] [PubMed] [Google Scholar]
- Kestilä M. et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol. Cell 1, 575–582 (1998). [DOI] [PubMed] [Google Scholar]
- Shih N. Y. et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286, 312–315 (1999). [DOI] [PubMed] [Google Scholar]
- Winn M. P. et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308, 1801–1804 (2005). [DOI] [PubMed] [Google Scholar]
- Kaplan J. M. et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 (2000). [DOI] [PubMed] [Google Scholar]
- Lehtonen S., Zhao F. & Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. Am. J. Physiol. Renal Physiol. 283, F734–F743 (2002). [DOI] [PubMed] [Google Scholar]
- Lehtonen S. et al. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc. Natl. Acad. Sci. USA 102, 9814–9819 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W. & LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 67, 404–419 (2005). [DOI] [PubMed] [Google Scholar]
- Kerjaschki D. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J. Clin. Invest. 108, 1583–1587 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda T., McQuistan T., Orlando R. A. & Farquhar M. G. Loss of glomerular foot processes is associated with uncoupling of podocalyxin from the actin cytoskeleton. J. Clin. Invest. 108, 289–301 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieder N. & Greka A. Calcium, TRPC channels, and regulation of the actin cytoskeleton in podocytes: towards a future of targeted therapies. Pediatr. Nephrol. Berl. Ger. 10.1007/s00467-015-3224-1 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerjaschki D. Polycation-induced dislocation of slit diaphragms and formation of cell junctions in rat kidney glomeruli: the effects of low temperature, divalent cations, colchicine, and cytochalasin B. Lab. Investig. J. Tech. Methods Pathol. 39, 430–440 (1978). [PubMed] [Google Scholar]
- Hoffmann S., Podlich D., Hähnel B., Kriz W. & Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J. Am. Soc. Nephrol. Jasn 15, 1475–1487 (2004). [DOI] [PubMed] [Google Scholar]
- Reiser J. et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 37, 739–744 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller C. C. et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J. Am. Soc. Nephrol. Jasn 18, 29–36 (2007). [DOI] [PubMed] [Google Scholar]
- Tian D. et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 3, ra77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. et al. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol. Cell. Biol. 33, 4755–4764 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Jiang R., Aoudjit L., Jones N. & Takano T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. JASN 22, 1621–1630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul C. et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 14, 931–938 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundel P. et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol. 139, 193–204 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander G. M. et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster B. N., Li X., Pausch M. H., Herlitze S. & Roth B. L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 104, 5163–5168 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price L. S. et al. Calcium signaling regulates translocation and activation of Rac. J. Biol. Chem. 278, 39413–39421 (2003). [DOI] [PubMed] [Google Scholar]
- Ridley A. J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgardt B. F. et al. PDK1 Deficiency in POMC-Expressing Cells Reveals FOXO1-Dependent and -Independent Pathways in Control of Energy Homeostasis and Stress Response. Cell Metab. 7, 291–301 (2008). [DOI] [PubMed] [Google Scholar]
- Moeller M. J., Sanden S. K., Soofi A., Wiggins R. C. & Holzman L. B. Podocyte-specific expression of cre recombinase in transgenic mice. Genes. N. Y. N 2000 35, 39–42 (2003). [DOI] [PubMed] [Google Scholar]
- Zariwala H. A. et al. A Cre-dependent GCaMP3 reporter mouse for neuronal imaging in vivo. J. Neurosci. Off. J. Soc. Neurosci. 32, 3131–3141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber A. et al. Transcutaneous measurement of renal function in conscious mice. Am. J. Physiol. - Ren. Physiol. 303, F783–F788 (2012). [DOI] [PubMed] [Google Scholar]
- Drenckhahn D. & Franke R. P. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab. Investig. J. Tech. Methods Pathol. 59, 673–682 (1988). [PubMed] [Google Scholar]
- Schweda F., Wagner C., Krämer B. K., Schnermann J. & Kurtz A. Preserved macula densa-dependent renin secretion in A1 adenosine receptor knockout mice. Am. J. Physiol. Renal Physiol. 284, F770–F777 (2003). [DOI] [PubMed] [Google Scholar]
- Bender D., Holschbach M. & Stöcklin G. Synthesis of n.c.a. carbon-11 labelled clozapine and its major metabolite clozapine-N-oxide and comparison of their biodistribution in mice. Nucl. Med. Biol. 21, 921–925 (1994). [DOI] [PubMed] [Google Scholar]
- Weiner D. M. et al. The role of M1 muscarinic receptor agonism of N-desmethylclozapine in the unique clinical effects of clozapine. Psychopharmacology (Berl.) 177, 207–216 (2004). [DOI] [PubMed] [Google Scholar]
- Wang L. et al. Gq signaling causes glomerular injury by activating TRPC6. J. Clin. Invest. 125, 1913–1926 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2, a004051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankland S. J., Pippin J. W., Reiser J. & Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 72, 26–36 (2007). [DOI] [PubMed] [Google Scholar]
- Brinkkoetter P. T. et al. p35, the non-cyclin activator of Cdk5, protects podocytes against apoptosis in vitro and in vivo. Kidney Int. 77, 690–699 (2010). [DOI] [PubMed] [Google Scholar]
- Boerries M. et al. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 83, 1052–1064 (2013). [DOI] [PubMed] [Google Scholar]
- Takemoto M. et al. Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J. 25, 1160–1174 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]