

An estimated 50 gigatons of carbon is annually fixed within marine systems, of which heterotrophic microbial populations process nearly half. These communities vary in composition and activity across spatial and temporal scales, so understanding how these changes affect global processes requires the delineation of functional roles for individual members. In a step toward ascertaining these roles, we applied proteomic stable isotope probing to quantify the assimilation of organic carbon from DFAAs into microbial protein biomass, since the turnover of DFAAs accounts for a substantial fraction of marine microbial carbon metabolism that is directed into biomass production. We conducted experiments at two coastal North Pacific locations and found taxonomically distinct responses. This approach allowed us to compare amino acid assimilation by specific bacterioplankton populations and characterize their allocation of this substrate among cellular functions.
KEYWORDS: environmental microbiology, marine microbiology, microbial communities, microbial ecology, proteomics
ABSTRACT
Heterotrophic marine bacterioplankton are a critical component of the carbon cycle, processing nearly a quarter of annual primary production, yet defining how substrate utilization preferences and resource partitioning structure microbial communities remains a challenge. In this study, proteomic stable isotope probing (proteomic SIP) was used to characterize population-specific assimilation of dissolved free amino acids (DFAAs), a major source of dissolved organic carbon for bacterial secondary production in aquatic environments. Microcosms of seawater collected from Newport, Oregon, and Monterey Bay, California, were incubated with 1 µM 13C-labeled amino acids for 15 and 32 h. The taxonomic compositions of microcosm metaproteomes were highly similar to those of the sampled natural communities, with Rhodobacteriales, SAR11, and Flavobacteriales representing the dominant taxa. Analysis of 13C incorporation into protein biomass allowed for quantification of the isotopic enrichment of identified proteins and subsequent determination of differential amino acid assimilation patterns between specific bacterioplankton populations. Proteins associated with Rhodobacterales tended to have a significantly high frequency of 13C-enriched peptides, opposite the trend for Flavobacteriales and SAR11 proteins. Rhodobacterales proteins associated with amino acid transport and metabolism had an increased frequency of 13C-enriched spectra at time point 2. Alteromonadales proteins also had a significantly high frequency of 13C-enriched peptides, particularly within ribosomal proteins, demonstrating their rapid growth during incubations. Overall, proteomic SIP facilitated quantitative comparisons of DFAA assimilation by specific taxa, both between sympatric populations and between protein functional groups within discrete populations, allowing an unprecedented examination of population level metabolic responses to resource acquisition in complex microbial communities.
IMPORTANCE An estimated 50 gigatons of carbon is annually fixed within marine systems, of which heterotrophic microbial populations process nearly half. These communities vary in composition and activity across spatial and temporal scales, so understanding how these changes affect global processes requires the delineation of functional roles for individual members. In a step toward ascertaining these roles, we applied proteomic stable isotope probing to quantify the assimilation of organic carbon from DFAAs into microbial protein biomass, since the turnover of DFAAs accounts for a substantial fraction of marine microbial carbon metabolism that is directed into biomass production. We conducted experiments at two coastal North Pacific locations and found taxonomically distinct responses. This approach allowed us to compare amino acid assimilation by specific bacterioplankton populations and characterize their allocation of this substrate among cellular functions.
INTRODUCTION
Marine microbial communities are an essential component of global carbon cycles (1–4), but problems of scale and cultivability (5) have hindered progress in defining the functional roles of individual taxa within ecosystems. The development of culture-independent tools has revealed the phylogenetic and functional diversity of marine microbial communities and allowed for detailed observations of temporal and spatial population dynamics in natural environments (6–11). However, understanding how community diversity is maintained in a competitive environment, and relating the dynamics of specific populations to ecosystem processes remains a challenge.
Our current understanding of the biogeochemical significance of marine bacterioplankton relies on decades of research on marine chemistry and measurements of bulk microbial community processes (12–17), with many studies focused on defining the role of dissolved free amino acids (DFAAs) as a substrate for heterotrophic bacteria (18–20). Turnover of bulk DFAAs can account for between 5 and 55% of total dissolved organic carbon (DOC) turnover in marine systems. Approximately 50% of the carbon from DFAAs utilized by microbes is assimilated (21, 22). While bulk measurements have illustrated the importance of heterotrophic bacterioplankton to the rapid utilization of this labile substrate, the specific contribution of individual populations toward DFAA turnover, as well as other DOC components, remains an area of active research.
Experiments have examined the roles of individual bacterioplankton taxa in DOC turnover through indirect assessments of gene expression (11, 23–28) and direct observations of isotopically labeled substrate incorporation into cellular biomass. Several experimental techniques have been developed to determine taxonomically resolved carbon assimilation patterns for natural microbial populations. A combination of microautoradiography and fluorescence in situ hybridization (MAR-FISH) has been used to show amino acid and other types of DOC utilization by single marine bacterioplankton cells that are taxonomically identified with FISH probes (29–32). Another technique, DNA stable isotope probing (DNA-SIP), applies metagenomic approaches to whole-community DNA that is separated based on differences in density due to isotopic label incorporation into newly synthesized DNA (33–36). A third option, Chip-SIP, is a recently developed high-throughput method that quantifies isotopic label incorporation into RNA molecules that are hybridized to a microarray (37–39).
A fourth technique measures stable isotope incorporation into protein biomass through the use of high-accuracy mass spectrometry (MS). Currently, there are two analytical approaches that differ in how isotopic labeled proteins are identified and how isotope incorporation is quantified. The protein-SIP approach produces detailed quantification of label incorporation into a defined set of identified proteins. First, unlabeled peptides are identified by tandem mass spectrometry (MS/MS), and then isotope incorporation into a predefined set of peptides is characterized through manual examination of mass spectra from coeluted isotopologues (40–44). The proteomic stable isotope probing (proteomic SIP) technique (45) used in this study expands on current metaproteomic approaches (46–48) by providing detailed information on stable isotope enrichment patterns for a wide diversity of peptides, in addition to identifying and quantifying thousands of unlabeled microbial proteins. Labeled peptide detection is made possible with the Sipros program (45, 49, 50), which finds peptide spectral matches (PSM) across a defined range and interval of percent 13C enrichments (e.g., 0 to 100% 13C at 1% increments in this study). Proteomic SIP has been applied with success to acid mine drainage biofilms to track activity related to 15N and 2H assimilation (45, 51). Here we report the application of proteomic SIP to quantify assimilation of 13C-labeled amino acids over two time points spanning 32 h by specific populations of bacterioplankton within samples of two coastal marine microbial communities.
The nature of the data generated from these experiments allowed us to establish two metrics for quantifying labeled substrate incorporation into detected proteins. The first metric, “label frequency,” is the relative frequency or proportion of PSM that are labeled (13C enrichment greater than naturally occurring levels) within the total set of PSM for an individual protein or for a taxonomic or functional group of proteins. Label frequency quantifies the breadth of de novo protein synthesis, but it does not discriminate between mechanisms of substrate incorporation, such as direct uptake and assimilation of a substrate, indirect incorporation from recycling of labeled biomass, or label incorporation from cross-feeding. The second metric, “average enrichment,” does provide a quantitative measure of labeled substrate incorporation into newly synthesized proteins. Average enrichment is the average percent 13C atom enrichment of the labeled PSM assigned to a given protein or group of proteins. Accordingly, high “average enrichment” denotes the direct assimilation of 13C-labeled substrate. Using these metrics, we quantified taxonomic differences in amino acid assimilation, showing general conservation among genera within the same taxonomic order. We also propose physiological mechanisms for these taxonomic differences based on differences in label frequency between groups of proteins associated with specific clusters of orthologous groups of proteins (COG) functional categories.
RESULTS
General metaproteomic results.
An overview of our experimental approach is illustrated in Fig. 1 and briefly described here. Eight microcosm incubations were performed on samples from two locations. Two microcosms that contained surface water sampled from the Oregon coast, OR1 and OR2, were incubated with 13C-amino acids for 15 h and 32 h, respectively (time point 1 and time point 2, respectively). Six microcosms contained Monterey Bay (MB) surface water that was also incubated with the same substrate over the same two time points (MB1a, MB1b, and MB1c [each incubated for 15 h] and MB2a, MB2b, and MB2c [each incubated for 32 h]). At each time point, microcosm microbial communities were collected, and then proteins were extracted and analyzed using high-resolution tandem mass spectrometry. Sipros searches of mass spectra obtained for each microcosm were performed against a peptide matching database consisting of isolate marine microbial protein sequences and predicted coding sequences (CDS) from assembled metagenomes (52) from the Monterey Bay samples. The taxonomic affiliations of predicted CDS from the metagenome assembly and the peptide matching database are indicated in Fig. S1A in the supplemental material. Total PSM, peptide identifications, protein identifications, and statistics concerning 13C enrichment of PSM are detailed in Table 1.
FIG 1 .
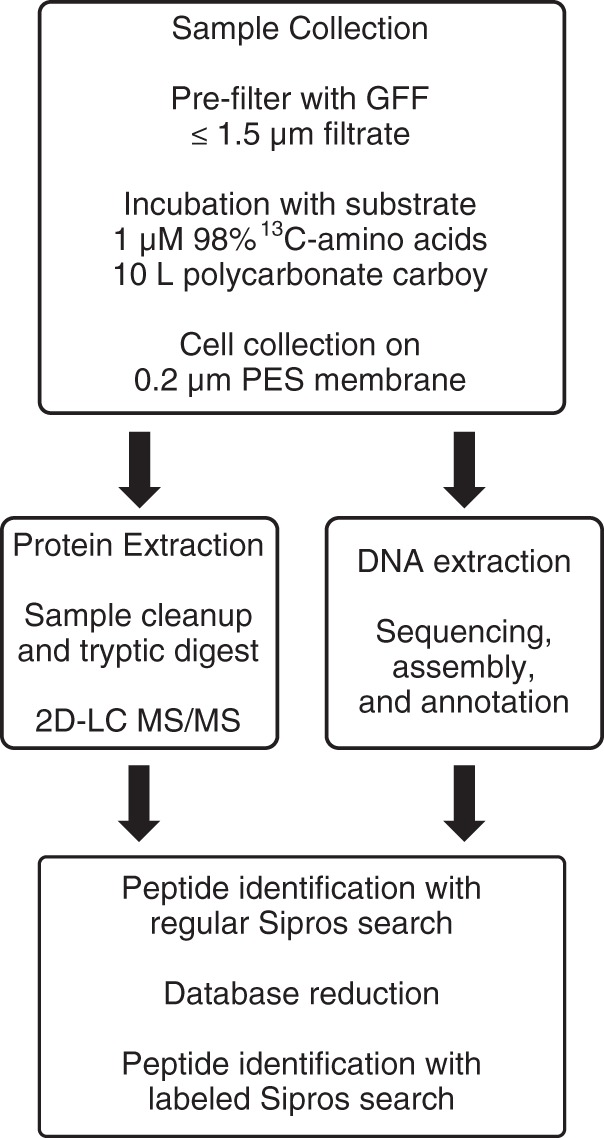
Overview of proteomic SIP experiment. GFF, glass fiber filters; PES, polyether sulfone.
TABLE 1 .
Summary of metaproteomics results
| Sample name |
Sample location | Incubation time (h) |
Peptide spectral matches |
No. of peptides identified |
No. of proteins identified |
Label frequency (%) |
Avg enrichment (%) |
|---|---|---|---|---|---|---|---|
| MB1a | Monterey Bay, CA | 15 | 22,208 | 18,631 | 3,378 | 2.43 | 14.96 |
| MB1b | Monterey Bay, CA | 15 | 22,669 | 19,800 | 3,613 | 2.72 | 16.01 |
| MB1c | Monterey Bay, CA | 15 | 25,157 | 19,668 | 3,513 | 2.14 | 15.57 |
| MB2a | Monterey Bay, CA | 32 | 15,959 | 15,170 | 2,885 | 3.95 | 20.17 |
| MB2b | Monterey Bay, CA | 32 | 17,928 | 14,992 | 2,772 | 4.51 | 24.77 |
| MB2c | Monterey Bay, CA | 32 | 17,510 | 15,675 | 2,860 | 5.72 | 39.93 |
| OR1 | Newport, OR | 15 | 6,554 | 6,441 | 1,190 | 9.22 | 33.04 |
| OR2 | Newport, OR | 32 | 6,374 | 6,180 | 1,147 | 10.34 | 20.76 |
Taxonomic distribution of predicted open reading frames (ORFs), metagenome reads, and 16S rDNA amplicons. (A) Proportions of predicted ORFs for taxonomic groups in the assembled MB metagenomes and in the final database used for peptide spectral matching. (B) Distribution of mapped metagenomic reads among the assembled contigs for the initial MB0 sample and MB1. (C) Relative proportions of 16S rDNA amplicon taxonomic assignments for MB0, MB1, and MB2. Download Figure S1, EPS file, 0.3 MB (313.8KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Substantially more peptide identifications came from metagenome-derived protein sequences than from isolate genome sequences (Fig. 2), even when the metagenome was not derived from the same sample as the metaproteome. Most of the peptides identified from isolate genome sequences were also identified in metagenomic protein sequences. The metagenome database performed better with the matched MB metaproteome samples than for the unmatched OR samples, for which no metagenome was available, yet nearly 60% of the proteins identified in the OR coast metaproteomes were from the MB metagenome sequences and not isolate genome sequences, many of which were collected from the Oregon coast (5).
FIG 2 .
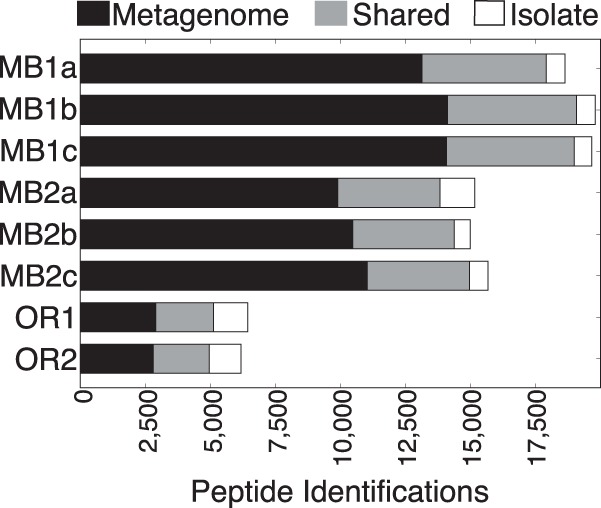
Peptide identifications by sequence source. “Metagenome” refers to peptides identified in predicted protein coding sequences (CDS) from an assembled metagenome from the Monterey Bay (MB) samples. “Isolate” refers to peptides identified in CDS from reference genomes. “Shared” refers to peptides found in CDS from both sources.
Taxonomic and functional composition of metaproteomes and metagenomes.
The replicate proteomes from the Monterey Bay experiment were highly correlated at both time points, as evidenced by pairwise determination of the concordance correlation coefficient between the relative abundances of identified proteins defined by normalized balanced spectral counts (see Table S2 in the supplemental material). Within-replicate pairwise comparisons of MB1 and MB2 samples had average correlation scores of 0.82 and 0.71, respectively. The average correlation between all pairwise comparisons between time point 1 and time point 2 replicates was 0.66, indicating greater differences in measured protein abundances between time points than between replicates within a time point. The correlation between time points is comparable to the correlation coefficient for a comparison between OR1 and OR2 (ρ = 0.65).
Shotgun metagenome sequencing (for time points 0 and 1, MB0 and MB1) and 16S rDNA sequencing (for MB0, MB1, and MB2) of the Monterey Bay sample indicated substantial population shifts that occurred between sample collection and subsequent time points. The relative abundances of taxonomic groups as determined by metagenomic read recruitment (see Fig. S1B in the supplemental material) and operational taxonomic unit (OTU) assignment of 16S rDNA amplicons (Fig. S1C) revealed a relative increase in Alteromonadales populations compared to a relative decline in the abundance of other initially low-abundance taxa during the first 15 h of incubation. Both sequencing data sets showed a relative increase in Flavobacteriales populations over the course of the incubations; however, this trend was not evident in the proteomic data where the relative abundance of mass spectra declined between MB1 and MB2 (Fig. S2). Overall, the taxonomic distribution of mass spectra in the metaproteomes resembled the composition of the sampled MB community as determined by DNA sequencing.
Comparison of metagenomes and metaproteomes. (A to C) Relative abundance of mapped reads from the MB0 and MB1 metagenomes (A), average relative spectral counts in MB1 and MB2 (B), and relative spectral counts in OR1 and OR2 (C). Download Figure S2, EPS file, 0.1 MB (129.1KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
When proteins were assigned by taxonomy, observed differences in relative spectral counts (see Fig. S2 in the supplemental material) and total protein identifications (Fig. 3A) were greater between sampling locations than between time points. Proteins associated with the order Flavobacteriales represented a substantial proportion of total protein identifications in all samples, but relative counts declined from the first time point to the second time point in all of the samples. Total protein identifications for SAR11 were substantially lower in the OR samples (8 and 9 in OR1 and OR2) than in the MB samples (average numbers of 231 and 234 in MB1 and MB2); in both locations, most of these proteins were assigned to the coastal Oregon isolate HTCC1062 (53). The relative contribution of Rhodobacterales protein identifications was similar between the two locations, but absolute counts were lower in the OR samples (91 and 161 in OR1 and OR2 and average numbers of 231 and 234 in MB1 and MB2). Alteromonadales proteins contributed a substantially greater fraction of the community proteome in the two OR samples, but absolute numbers of these protein identifications were similar between the locations. Other Gammaproteobacteria clades, such as members of the OMG, SAR86, and SAR92 clades, had relatively higher numbers of protein identifications in the MB proteomes. Conversely, Oceanospirillales protein assignments accounted for less than 1% of protein identifications in the six MB samples versus 8.7% and 16.7% of identified proteins in OR1 and OR2.
FIG 3 .

Relative abundance of protein identifications by taxonomy and COG functional category. (A) Stacked bar chart of proportion of total protein identifications in each sample for abundant orders of Bacteria. “Other” refers to low abundance and unclassified proteins, including identified eukaryote and Archaea proteins. (B) Heat map of proportion of protein identifications in each sample for general COG categories represented in the metaproteomes.
We determined the proportion of identified proteins assigned to COG categories (Fig. 3B) and found negligible differences between the functional profiles of the bacterioplankton communities at both locations. The most frequently detected proteins were assigned to COG category J (translation and ribosomal structure and biogenesis), followed by proteins belonging to categories C (energy production and conversion), P (inorganic ion transport and metabolism), E (amino acid transport and metabolism), G (carbohydrate transport and metabolism), and S (unknown function). Compared to the six MB proteomes, a higher proportion of protein identifications in the two OR proteomes were assigned to COG categories J and C, while a lower portion of proteins were assigned to categories P and S.
Community level assimilation of amino acids determined by 13C labeling of proteomes.
We analyzed the 13C enrichment of all samples in order to assess community level amino acid assimilation with respect to both location and incubation time. We observed significant changes in both the label frequency (P = 0.01 by t test) and average enrichment (P = 0.05 by t test) in the MB samples between time points 1 and 2, while there was little change between OR1 and OR2 (Fig. 4 and Table 1). At the first time point, the three MB1 samples exhibited similar label frequency (average frequency of 2.4% ± 0.29% standard deviation [SD]) and average enrichment (average frequency of 15.5% ± 0.53% SD), but the three MB2 proteomes exhibited greater variability in label frequency (average frequency of 4.7% ± 0.90% SD) and average enrichment (average frequency of 28.3% ± 10% SD). The patterns of 13C enrichment in the MB2 proteomes were more similar to the OR1 and OR2 proteomes than the MB1 proteomes, as reflected by the histogram of labeled spectra (Fig. 4). The three MB1 samples had unimodal distributions centered around 35% 13C enrichment, whereas the distributions of labeled spectra from the three MB2 samples and the two OR samples were more representative of a bimodal distribution, with a lower peak at 16 to 18% and the second peak above 50% 13C enrichment (Fig. 4), suggesting that incubation time increased the heterogeneity in label incorporation across populations.
FIG 4 .
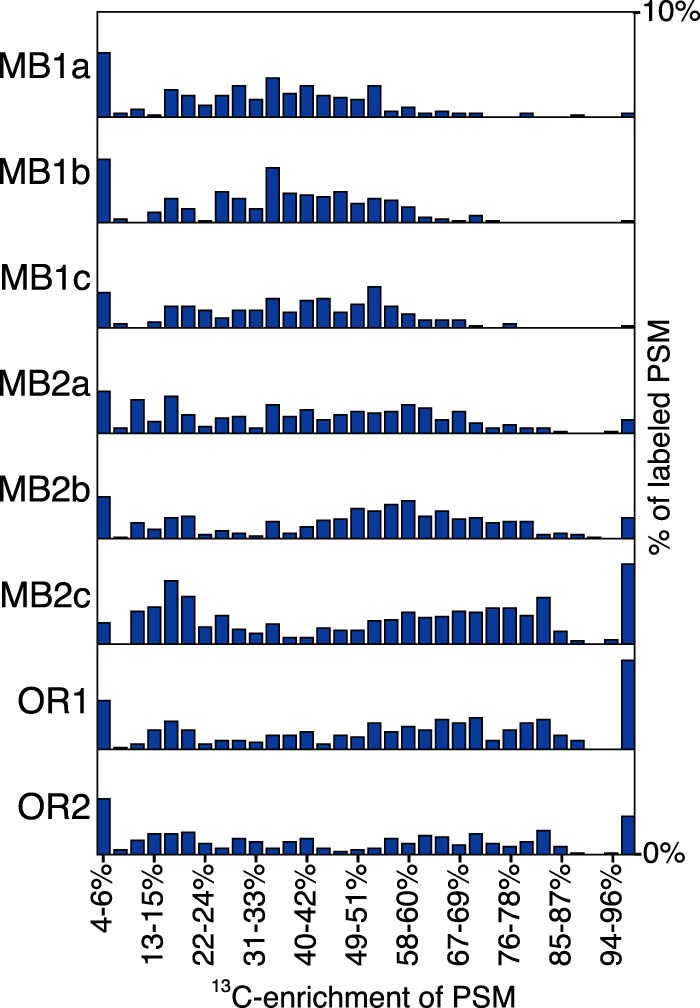
Histogram of highly labeled peptide spectral matches (PSM). The histogram depicts the proportion of labeled spectra (13C enrichment of ≥2%) from 4% to 99% 13C enrichment within 3% enrichment bins. Each distribution depicts bars on a relative scale of 0 to 10% of total labeled spectra. Label frequency and average enrichment values for the samples are presented in Table 1.
The change in overall label frequency at both locations was positively and significantly correlated to the change in overall cell counts (Pearson’s r = 0.89). Average cell counts from the OR samples indicated that the microbial community increased nearly 3-fold during the first 15 h but increased by only 32% in the subsequent 17 h (counts of 0.496 × 106, 1.48 × 106, and 1.96 × 106 cells ml−1, respectively). Average cell counts from the samples from Monterey Bay, California, suggest a higher initial population size (2.16 × 106 cells ml−1), which increased by 8% in the first 15 h and again by 26% in the following 17 h (2.33 × 106 and 2.94 × 106 cells ml−1, respectively). Unlike label frequency, average enrichment was not significantly correlated with change in cell counts (Pearson’s r = 0.10); in the OR samples, average enrichment actually decreased between time points.
Taxonomic differences in amino acid assimilation.
Incorporation of amino acids into proteins was not limited to specific taxa; labeled PSM were detected for all taxonomic groups with greater than 12 protein identifications in any one metaproteome. To test the null hypothesis that all taxa could equally assimilate 13C-amino acids, we compared each taxon’s observed label frequency and average enrichment values calculated from each taxon’s proteome against a null distribution consisting of label frequency and average enrichment values for 1,000 randomly selected subsets with equivalent numbers of balanced spectral counts (Fig. 5). This null distribution, created independently for each test, represented the expected distribution of label frequency and average enrichment values if taxonomic labels were randomly assigned to PSM. Significant values were defined as the values more extreme than the inner 95% of expected values. Z-scores for each value presented in Fig. 5 are derived from each null distribution. For brevity, we focus on the most abundant taxa, namely, those within the Proteobacteria and Bacteroidetes phyla.
FIG 5 .

Label frequency and average enrichment of taxa. (A and B) more-represented (A) and less-represented (B) orders in the metaproteomes. The z-scores of label frequency (x axis) and average enrichment (y axis) based on comparisons of observed values with distributions under the null model are shown. The shaded area represents the inner 95% of values under a standard normal distribution. Significance testing is described in Materials and Methods.
Flavobacteriales and SAR11 showed label frequency and average enrichment values that were significantly low compared to the null distribution in nearly all of the samples from both locations (Fig. 5A). Genus level assignment of proteins revealed a consistent trend in labeling for Flavobacteriales, with the notable exception of the genus Flavobacterium which had high label frequency in the OR and MB2 samples (see Fig. S3C in the supplemental material), while also supplanting Polaribacter as the Flavobacteriales genus with the most protein identifications in the MB2 samples. Average enrichment of Alteromonadales proteomes were significantly high in all samples except MB2b, and label frequency was significantly high in the three MB1 samples and in MB2c, but not in the OR samples (Fig. 5A). This trend was consistent at the genus level where label frequency was generally higher at time point 1 and average enrichment was generally high at both time points (Fig. S3B). In all eight metaproteomes, Rhodobacterales proteins had significantly high label frequency that increased between time points in the MB samples, with average enrichment significantly high in the MB samples, but not in the two OR samples (Fig. 5A). The labeling of genera within the Rhodobacterales was generally consistent, but with a wide range of label frequency values from insignificant to significantly high (Fig. S3A).
Label frequency and average enrichment for genera of three abundant orders, Rhodobacterales (A), Alteromonadales (B), and Flavobacteriales (C). The figure depicts Z-scores of label frequency (x axis) and average enrichment (y axis) based on comparisons of observed values with distributions under the null model. The shaded area represents the inner 95% of values under a standard normal distribution. Significance testing is described in Materials and Methods. Download Figure S3, EPS file, 0.7 MB (742.8KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oceanospirillales proteins had high average enrichment at both OR time points, whereas label frequency varied between samples (Fig. 5B). Proteins from SAR92 bacteria were insignificantly labeled in all but one sample, MB1b, where both label frequency and average enrichment values were significantly low (Fig. 5B). Gammaproteobacteria proteins from the SAR86 and OMG clades had labeling that was generally insignificant or low across all samples (Fig. 5B). The SAR116 proteome was significantly high in label frequency in MB1b, MB1c, and MB2b, but its average enrichment was significantly high only in MB2c (Fig. 5B). Average enrichment values for the Methylophilales proteome were significantly low in five of the samples, but label frequency was significantly high in two MB2 samples and in OR2 (Fig. 5B). Chroococcales proteins were significantly low in labeling in most samples (Fig. 5B).
In addition to assessing differential substrate assimilation between taxa, our experiment permitted the assessment of label frequency for proteins assigned to different COG functional categories within specific taxonomic groups. Significance testing was conducted in a similar manner as the previously described tests for differential 13C enrichment of taxonomic groups. Here the null distribution assumes that 13C enrichment of proteins within a taxonomic group is independent of COG category label, i.e., all types of proteins should have equal label frequency. By comparing the observed label frequency for each COG category to the values expected under the null distribution, we can assess the physiological basis for observed taxonomic differences in 13C enrichment. The results of this analysis show that the Alteromonadales proteomes had significantly high label frequency in COG category J (translation) in all six MB samples (see Fig. S4B in the supplemental material). This is in contrast to the Flavobacteriales and Rhodobacterales proteomes in which label frequencies were significantly high for COG category J at MB1, but significantly lower than expected at MB2 (Fig. S4A and S4C). Flavobacteriales proteins in COG category P (inorganic ion transport and metabolism) had significantly high labeling in all time point 2 proteomes. The Rhodobacterales proteome had increased labeling in COG category E (amino acid transport and metabolism) in the MB2 samples and both OR samples. Additionally, this taxonomic order had significantly low labeling of carbohydrate metabolism proteins (COG category G) in five of the six MB proteomes. Both SAR11 and Rhodobacterales had consistently high labeling of proteins associated with protein modification, turnover, and chaperones (COG category O).
Relative labeling of COG categories. The panels depict biclustered heat maps of label frequency (Z-scores) of COG functional categories for each sample within three dominant orders: Rhodobacterales (A), Alteromonadales (B), and Flavobacteriales (C). Download Figure S4, EPS file, 0.3 MB (317.9KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Microbial community activity varies between time and location.
Variability among replicates and geographic location allows us to speculate on the physiological responses of different populations that could be driving the observed patterns of 13C enrichment. In this study, we found that the MB2 samples and the OR1 sample had similar label frequency and average enrichment values, suggesting more rapid DFAA assimilation in the OR samples than in the MB samples. This observation could result from a substantially greater fraction of cells being in an active state and ready to assimilate the amended amino acids in the OR samples; total cell numbers increased relatively more in the OR samples than the MB samples within the time frame of the experiment. Alternatively, the OR samples could have included greater initial populations of taxa with the capacity for rapid amino acid assimilation, as evidenced by the greater relative numbers of protein identifications and mass spectra associated with two highly labeled Gammaproteobacteria populations, Alteromonadales and Oceanospirillales. The OR2 samples had increased label frequency and decreased average enrichment relative to the OR1 samples, suggesting that the pool of labeled proteins had undergone greater turnover and the signal had been diluted among greater numbers of proteins. The apparent bimodal distribution of labeled spectra in MB2 and OR1 samples can indicate heterogeneity among community members with respect to amino acid assimilation representing both moderately and highly 13C-enriched populations. By testing and defining assimilation patterns for distinct taxonomic groups, amino acid utilization preferences were determined for the abundant microbial community members. Assessment of substrate assimilation for less abundant populations was also possible, including the findings of low DFAA assimilation by SAR86, which was also found in a previous MAR-FISH study (32).
Distinct populations differ in amino acid assimilation.
The relative dominance of Flavobacteriales proteins in the coastal samples is consistent with the results of other studies (29, 54–57). The two OR samples in this study exhibited higher relative abundances of Flavobacteriales mass spectra compared to a previous OR coast metaproteome (27); this could be due in part to better representation of proteins owing to inclusion of metagenomic data in the database. The significantly low relative assimilation of DFAAs by Flavobacteriales populations is similar to findings in a previous MAR-FISH study (29). Substrate concentration could have affected the overall activity of this population with regard to the low label frequency observed compared to other taxa. A Chip-SIP study that tracked 15N incorporation of DFAAs in San Francisco Bay found that members of the Bacteroidetes incorporated relatively more substrate with increasing substrate concentration (39), suggesting that this population might have been more highly labeled in our experiments if the amended substrate concentration had been greater. Experimental conditions removed particulate matter of ≥1.5 µm in size, potentially removing an important resource for Flavobacteriales populations, which are often identified as particulate colonizers and degraders responding initially to the presence of increased particulate matter occurring during phytoplankton blooms (11, 58). The observed relative decrease in protein identifications over the course of the experiment would seem to confirm this hypothesis; however, metagenomic sequence libraries (see Fig. S1B in the supplemental material) and 16S sequence libraries (Fig. S1C) provide evidence for an actively growing population, suggesting that alternative unlabeled substrates were utilized. The observed high label frequency but low average enrichment for proteins assigned to the genus Flavobacterium in the MB2 replicates (Fig. S3C) could be interpreted as a cross-feeding event in which this population utilized labeled by-products generated from DFAA metabolism of other community members.
In both the OR and MB proteomes, SAR11 proteins had significantly low label frequency that was even lower relative to expected values at time point 2, indicating relatively lower DFAA assimilation compared to the general community. This relative decline was also observed in the metagenome sequence libraries for the first 15 h of incubation but was not observed in the 16S amplicon data, as the primers used here have been reported to miss this clade (59). The observed low label frequency in this experiment differs from a prior MAR-FISH study that found SAR11 cells were highly represented in the fraction of cells actively assimilating DFAAs (31). However, substrate concentrations were much lower in that study (0.5 nM), suggesting that differences in observations may be based on an advantage of SAR11 cells in competing for substrates at low concentrations, and potentially being inhibited at high concentrations. The observed high label frequency in SAR11 proteins assigned to COG category O is congruent with previous findings of highly expressed protein maintenance and recycling genes in Sargasso Sea samples (28). The significant investment of resources into proteome maintenance provides insight into the observed low growth rate of SAR11 (53) which would accompany an overall low relative label frequency. Alternatively, increased expression of chaperones and heat shock proteins could represent a general stress response. In a previous study of transcriptional responses to bottle incubations, SAR11 was found to have increased expression of stress response genes and decreased expression of transporters (60). Although a previous metaproteome study of coastal Oregon (27) identified substantially more SAR11 proteins than these two OR proteomes, samples in this study were taken from waters adjoining an estuary. The relative abundance of SAR11 has been observed to increase with distance from near-shore bloom conditions, opposite the trend observed for populations of Flavobacteriales and Rhodobacterales (61).
Populations of the metabolically versatile Rhodobacterales order have been shown to play important roles in the processing of carbon in coastal environments (26, 57, 62–65). This lineage has also been associated with the utilization of low-molecular-weight DOC, specifically DFAAs (30, 39). Our assimilation data support these hypotheses, as Rhodobacterales proteins were highly labeled in samples from both coastal locations and generally increased in both labeling metrics between time points. Increased relative abundance in both the metagenomes (see Fig. S1B in the supplemental material) and 16S rDNA amplicon libraries during the first 15 h of incubation also indicates an active population. The proteomic SIP results are similar to findings that 15N incorporation from DFAAs by this taxon was similar at both high (5 µM) and medium (500 nM) concentrations (39), as depletion of the labeled substrate would be expected over the course of the incubation. Indeed, the observed high label frequency for Rhodobacterales amino acid metabolism proteins in the time point 2 samples suggests that this population responded to depleted DFAAs by building more machinery to acquire this substrate. The moderate percent 13C enrichment values observed also fit with previous Chip-SIP observations of 25% higher N-use efficiency than C from DFAAs (38), suggesting the role of DFAAs as a significant source of N. Additionally, Rhodobacterales represented a substantial fraction of the initial sampled MB community (Fig. S1B and S1C), which if active would assimilate labeled substrate across a greater number of cells, resulting in low to moderate average enrichment values compared to a population of new cells with newly synthesized proteins composed of a greater proportion of newly acquired labeled substrate.
Previous work examining marine community responses to dissolved organic matter and deep seawater amendment have characterized members of the Alteromonadales order as fast-growing copiotrophic organisms (66, 67). In this study, label frequency and average enrichment values indicated that this population assimilated a higher percentage of 13C from labeled DFAAs into protein biomass than other taxa, suggesting rapid initial growth in response to the incubation conditions that was confirmed by sequencing data (see Fig. S1B and S1C in the supplemental material). At time point 2, there was greater variability in the labeling of this taxon and in the relative abundance of 16S OTUs assigned to this taxonomic order. In most samples, their label frequency declined while average enrichment remained high, concurrent with the decline in relative abundance in 16S libraries. This could be due to continued, less rapid growth on unlabeled substrates with limited turnover of early synthesized proteins, specifically ribosomes and translational machinery, where these bacteria are possibly less competitive for amino acid uptake at lower concentrations. Consistent with this hypothesis, Chip-SIP experiments found that Alteromonadales increased labeling with increased substrate concentration (39). Unlike the Flavobacteriales and Rhodobacterales proteomes, there was not a distinction between time points for labeling of COG categories. Instead, this population consistently devoted labeled substrate to translational machinery and energy production proteins, with little apparent turnover or dilution of the label signal.
MATERIALS AND METHODS
Sample collection, processing, and substrate incubation experiments.
Oregon coast samples (OR) were collected in Newport, OR (44°37.069′N, 124°3.455′W, ~0 km from shore), on 16 July 2013 at 5:00 p.m. during incoming high tide. Monterey Bay (MB) samples were collected from surface waters (36°53.387′N, 121°57.257′W, ~10 km from shore) while on board the RV Rachel Carson on 14 October 2013. All samples were prefiltered using a Geotech polycarbonate filter holder through 142-mm Whatman 934-AH glass fiber filters (GFF) (autoclaved and rinsed with distilled water) with a nominal retention size of 1.5 µm. Microcosm incubations of filtrate were carried out in acid-washed and rinsed 10-liter polycarbonate carboys, amended with a final concentration of ~1 µM 98 atom% 13C-labeled algal amino acids (catalog no. 426199-1G; Sigma-Aldrich). Incubations were performed at constant temperatures close to the temperature of the ambient water column (16°C for OR samples and 19°C for MB samples) for 15 and 32 h. Upon harvest, cells from replicate microcosms were concentrated in parallel on 0.2-µm polyethersulfone (PES) membrane filters (42 mm; Pall) using a peristaltic pump, Pall 42-mm polycarbonate in-line filter holders, and 42-mm GFF support filters. Filters were changed as needed for maintaining a consistent flow of ~100 ml min−1. Upon completion, membrane filters were transferred to sterile 15-ml tubes, immediately frozen on dry ice, and subsequently transferred to a −80°C freezer for long-term storage. For all MB samples, 1 liter of retentate was reserved for DNA extraction and subsequent metagenome sequencing as described by Mueller et al. (52). Formaldehyde-fixed and SYBR green-stained cells were counted with a Guava Technologies flow cytometer as previously described (68, 69).
Protein extraction, purification, digestion, and mass spectrometry.
Protein was extracted from cells concentrated on membrane filters as follows. First, membrane filters were cut into thin strips and placed in sterile 15-ml tubes. Next, 3 ml of MoBio solution ST1B was added, and tubes were vortexed. Three milliliters of sodium dodecyl sulfate (SDS) lysis buffer plus dithiothreitol (DTT) was then added to each sample, and the tubes were vortexed and subsequently incubated for 15 min at 90°C. Cell lysate was transferred to new 2-ml Eppendorf Protein LoBind tubes, and cellular debris was removed by centrifugation. Supernatant from the tubes was transferred to new 2-ml Protein Lo Bind tubes, and protein was precipitated overnight with trichloroacetic acid (TCA). The concentration of protein in extracts was quantified using the Qubit protein assay kit (Invitrogen). Extractions from microbial cells collected from 9 liters of seawater typically yielded ~200 µg of purified protein, which was sufficient for full mass spectrometry runs requiring 50 to 100-µg protein digest.
Protein pellets were resolubilized in 6 M guanidine and 10 mM DTT. Then, 50 µg of protein was further cleaned up and digested by modified trypsin on centrifugal filters with a 30,000-molecular-weight cutoff (70, 71). Each sample was first digested overnight at an enzyme/substrate ratio of 1:100 (weight/weight) at room temperature with gentle shaking, followed by a secondary digestion for 4 h. For each sample, 25 µg of peptides was loaded offline into a 150-µm-inner-diameter (ID) two-dimensional (2D) back column (Polymicro Technologies) packed with 3 cm of C18 reverse-phase (RP) resin (Luna; Phenomenex) and 3 cm of strong-cation-exchange (SCX) resin (Luna; Phenomenex). The back column loaded with peptides was desalted offline with 100% solvent A (95% H2O, 5% acetonitrile, and 0.1% formic acid) and washed with a 1-h gradient from 100% solvent A to 100% solvent B (30% H2O, 70% acetonitrile, and 0.1% formic acid) to move peptides from RP resin to SCX resin. The back column was then connected to a 100-µm-ID front column (New Objective) packed in-house with 15 cm of C18 RP resin and placed in-line with a U3000 quaternary high-performance liquid chromatography (HPLC) pump (Dionex). Multidimensional protein identification technology (MudPIT) was used for the liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) measurements (72). Each mass spectrometric run was configured with 11 SCX fractionations using 5%, 7%, 10%, 12%, 15%, 17%, 20%, 25%, 35%, 50%, and 100% of solvent D (500 mM ammonium acetate dissolved in solvent A). Each SCX fraction was separated by a 110-min RP gradient from 100% solvent A to 50% solvent B. The LC eluent was directly nanosprayed (Proxeon) into an LTQ Orbitrap Elite mass spectrometer (Thermo Scientific). Both MS scans and collision-induced dissociation (CID)MS/MS scans were acquired in Orbitrap with a resolution of 30,000 and 15,000, respectively. After each MS scan, the eight most abundant precursor ions were selected under automated data-dependent acquisition for MS/MS analysis by CID.
Metaproteome database construction, spectra searches, and label quantification.
Given that sample-specific metagenomes were not available for OR samples, multiple databases comprised of protein sequences from isolate marine microbes and metagenomes generated for the MB samples were constructed for confident identification of peptide spectral matches (PSM) with the Sipros program (45, 49, 50). All searches considered tryptic peptides in the range of 6 to 60 amino acids with a maximum of two missed cuts. Parent ion and fragment ion mass tolerances were 0.05 Da and 0.02 Da, respectively. Regular searches considered only natural 13C abundance (1.109%). SIP searches considered 13C enrichments of 0% to 100% in 1% increments. A peptide level 1% false-discovery rate (FDR) using decoy sequences was applied in both regular and SIP searches. The two-peptide rule, one unique plus one shared peptide identification, was required for protein identifications. All Sipros searches were performed on the Titan supercomputer.
In order to determine whether peptide mass spectra from a known unlabeled proteome were falsely identified as being enriched with 13C label, the spectra from two unlabeled coastal marine samples were searched with Sipros in SIP enrichment-searching mode. The FDR for spectra that were incorrectly assigned enrichment values above the natural abundance of 13C (≥2%) averaged 2.1%. No spectra were falsely assigned 13C enrichment greater than 7% (see Table S1 in the supplemental material).
False detection rate of labeled spectra. The table lists numbers of and proportion of total PSM from two unlabeled metaproteomes that were identified at the corresponding level of percent 13C enrichment. Searches were conducted against the same sequence database used for SIP searches in this study. Download Table S1, PDF file, 0.04 MB (43.1KB, pdf) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pairwise concordance correlation coefficients. Results are from pairwise tests between all metaproteomes using relative balanced spectral counts for each identified protein. Download Table S2, PDF file, 0.03 MB (35.3KB, pdf) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparisons between individual search outputs of multiple database configurations were made to evaluate the influence of database design on protein identification results. Regular searches of the OR samples were performed against an initial database of protein coding sequences (CDS) from 133 available marine bacteria and archaea genomes. Subsequent searches queried a smaller database of CDS from 36 of these genomes with the most protein identifications. A separate database of CDS was also created from an assembly of metagenomes from MB samples (52), which included sequences from pooled DNA extracted from the three MB1 replicates in this study. Reads from MB metagenomes were assembled using MetaRay, version 2.3.1 (73). The resulting contigs were clustered at 99% sequence identity with cd-hit-est (74) and merged with Minimus2 (75). Then, CDS were predicted using Prodigal (76). The taxon of each CDS was assigned based on the respective best hit against GenBank RefSeq (downloaded 14 June 2014) protein sequences using the Diamond search algorithm in “blastp” mode with default settings (77). Functional assignments of individual coding sequences were made against the eggNOG database, v.4.0 (78), based on respective best scoring matches against hidden Markov model (hmm) profiles of bacterial cluster alignment (bactNOG) protein families using HMMSCAN (79). Metagenome abundance abundances (see Fig. S1B in the supplemental material) were determined by mapping sequencing reads back to the assembled contigs using Bowtie 2 (80).
Each database, isolate genome CDS or assembled metagenome CDS database, was used for regular PSM searches of all eight samples considered in this study. In preliminary database search tests, no SIP searches identified labeled peptides (13C enrichment of ≥2%) that were not also identified in regular searches (data not shown). This allowed us to reduce the protein database for SIP searches to include only proteins with peptide identifications in any of the eight regular searches (search results in Table 1; taxonomic distribution of CDS in the metagenome and the final database used for searches in Fig. S1A in the supplemental material).
16S rDNA amplicon library preparation and analysis.
For all MB1 and MB2 replicates plus three time point 0 (MB0) replicates, the extracted whole-community DNA (52) was subjected to PCR amplification of the v4 rRNA gene region using previously described dual indexed primers (81). Two sets of PCRs (technical replicates using two sets of barcodes) were carried out in triplicate using 25-µl volume, ~1-ng template, 20 cycles, and the Thermo Phusion high-fidelity PCR kit. Triplicate reaction mixtures were pooled, size selected, excised from 1% agarose gel, purified with the PureLink Quick gel extraction kit (Invitrogen), and then quantified with Qubit dsDNA (double-stranded DNA) BR assay kit (Thermo Fisher). Libraries were sequenced on the Illumina MiSeq platform at the Oregon State University Center for Genome Research and Biocomputing using the MiSeq Reagent kit v2 generating two 250-bp reads. Reads were quality filtered using Sickle (version 1.33; N. A. Joshi and J. N. Fass, 2011; available at https://github.com/najoshi/sickle), a minimum quality score of 25, and a minimum length of 150. Subsequent assembly, quality filtering, unsupervised OTU picking at 97% identity, taxonomic assignment with alignment to the Silva database (82), and relative abundance profiles were conducted in the mothur software package (83).
Metaproteome data processing and statistical analyses.
Label incorporation into the peptides and proteomes of distinct taxonomic groups from each community were defined using two separate metrics: “label frequency” (the proportion of PSM with 13C content of ≥2%) and “average enrichment” (the average percentage of 13C in labeled PSM). Both metrics were applied to define labeling for the PSM associated with a set of proteins from a specific sample, from a specific taxon within a sample, or from a COG functional category within a specific taxonomic grouping. Significance testing for label frequency and average enrichment was conducted using a nonparametric approach in order to overcome differences in total detected label between samples and total PSM for proteins associated with taxonomic groups within a sample or COG functional categories within a taxon. For each subset of proteins that was tested, 1,000 permutations of equivalent numbers of spectra were randomly selected in order to define the distribution of expected values for label frequency and average enrichment. The null distribution for tests comparing taxonomic groups within a sample and tests comparing COG functional categories within a taxonomic group were drawn from each sample’s total spectra and from each taxon’s associated spectra within a sample, respectively. We defined significant observations as those that were as extreme or more extreme than the critical values delineating the inner 95% of values obtained by random permutations. The mean and standard deviation of each random distribution were used to define Z-scores for illustration purposes.
Conclusions.
In this study of two North Pacific coastal microbial communities, we used proteomic SIP to identify thousands of proteins within each sample, providing access to the expressed genome of many of the diverse populations of planktonic cells (≤1.5 µm) within these communities. While we identified more than 1,000 proteins for each of the OR samples without the use of a matched metagenome, comparable to numbers found in previous marine metaproteomic experiments (47), the use of a matched metagenome database with the MB samples resulted in an approximately 2- to 3-fold increase in protein identifications. By extending metaproteomics with the application of 13C-based proteomic SIP, we quantified DFAA assimilation patterns among discrete populations and compared de novo protein synthesis between protein functional groups.
Using proteomic SIP data along with comparisons of shifts in relative abundance from 16S rDNA amplicon sequencing and metagenomics provided additional insight into community dynamics that would remain obscured if only one technique was utilized. Proteomic SIP data indicated that Flavobacteriales populations had low average enrichment over the course of the incubations but did exhibit increases in both label frequency and relative abundance in sequence libraries; taken together, these provide compelling evidence for an active community potentially obtaining label through cross-feeding. Sequencing data also helped differentiate two populations with high label frequency. Rhodobacterales were an abundant component of the initial sampled community; while their proteomes consistently demonstrated high label frequency across samples and time points, their relatively moderate average enrichment suggested 13C-amino acid assimilation across an extant active population. Conversely, the high average enrichment of Alteromonadales populations indicated rapid 13C-amino acid conversion into newly synthesized cells, which was corroborated by increased relative abundance in the sequencing data.
Analysis of DFAA assimilation across protein functional categories by Rhodobacterales and Alteromonadales during the second half of the incubation provided insight into these observed differences in population level responses. Natural communities experience substrate availability that is affected by rates of production and uptake, interpopulation competition, and substrate lability, all of which interact to shape the community. Observed increases in label frequency and relative abundance for Rhodobacterales were accompanied by significant synthesis of amino acid uptake and metabolism proteins, potentially conferring a competitive advantage for continued uptake of this substrate. Conversely, the initial rapid increase in the relative abundance of Alteromonadales, concurrent with ribosomal protein biosynthesis, did not continue during the second time period during which time available DFAAs were likely depleted.
ACKNOWLEDGMENTS
This research used resources of the Oak Ridge Leadership Computing Facility, which is a DOE Office of Science User Facility supported under contract DE-AC05-00OR22725. Oak Ridge National Laboratory is managed by the University of Tennessee-Battelle, L.L.C. for the U.S. Department of Energy (DOE). Work at Lawrence Livermore National Laboratory (LLNL) was conducted under the auspices of DOE contract DE-AC52-07NA27344.
We thank Francisco Chavez, the crew of the RV Rachel Carson, the Monterey Bay Research Institute, the lab of Stephen Giovannoni, and the Oregon State University Center for Genomic Research and Biocomputing.
This work was funded by the Gordon and Betty Moore Foundation Marine Microbiology Initiative (grant GBMF3302).
The funding organization had no role in the design, data collection and interpretation, or the decision to submit this work for publication.
REFERENCES
- 1.Pomeroy LR. 1974. The ocean’s food web, a changing paradigm. BioScience 24:499–504. [Google Scholar]
- 2.Williams PJLB. 1981. Incorporation of microheterotrophic processes into the classical paradigm of the plankton food web. Kiel Meeresforsch Sonderh 5:1–28. [Google Scholar]
- 3.Azam F, Fenchel T, Field J, Gray J, Meyer-Reil L, Thingstad F. 1983. The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 10:257–263. doi: 10.3354/meps010257. [DOI] [Google Scholar]
- 4.Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connon SA, Giovannoni SJ. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol 68:3878–3885. doi: 10.1128/AEM.68.8.3878-3885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt TM, DeLong EF, Pace NR. 1991. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol 173:4371–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giovannoni SJ, Britschgi TB, Moyer CL, Field KG. 1990. Genetic diversity in Sargasso Sea bacterioplankton. Nature 345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- 8.Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, Jaroszewski L, Cieplak P, Miller CS, Li H, Mashiyama ST, Joachimiak MP, van Belle C, Chandonia J-M, Soergel DA, Zhai Y, Natarajan K, Lee S, Raphael BJ, Bafna V, Friedman R, Brenner SE, Godzik A, Eisenberg D, Dixon JE, Taylor SS, Strausberg RL, Frazier M, Venter JC. 2007. The Sorcerer II Global Ocean Sampling Expedition: expanding the universe of protein families. PLoS Biol 5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P, Boss E, Bowler C, Follows M, Karp-Boss L, Krzic U, Reynaud EG, Sardet C, Sieracki M, Velayoudon D. 2015. Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 10.Giovannoni SJ, Rappé MS. 2000. Evolution, diversity, and molecular ecology of marine prokaryotes, p 47–84. In Kirchman DL. (ed), Microbial ecology of the oceans. Wiley, New York, NY. [Google Scholar]
- 11.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Weber M, Klindworth A, Otto A, Lange J, Bernhardt J, Reinsch C, Hecker M, Peplies J, Bockelmann FD, Callies U, Gerdts G, Wichels A, Wiltshire KH, Glöckner FO, Schweder T, Amann R. 2012. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 12.Williams PJLB. 1970. Heterotrophic utilization of dissolved organic compounds in the sea I. Size distribution of population and relationship between respiration and incorporation of growth substrates. J Mar Biol Assoc U K 50:859–870. doi: 10.1017/S0025315400005841. [DOI] [Google Scholar]
- 13.Williams PJLB, Gray RW. 1970. Heterotrophic utilization of dissolved organic compounds in the sea II. Observations on the responses of heterotrophic marine populations to abrupt increases in amino acid concentration. J Mar Biol Assoc U K 50:871–881. doi: 10.1017/S0025315400005853. [DOI] [Google Scholar]
- 14.Andrews P, Williams PJL. 1971. Heterotrophic utilization of dissolved organic compounds in the sea III. Measurement of the oxidation rates and concentrations of glucose and amino acids in sea water. J Mar Biol Assoc U K 51:111–125. doi: 10.1017/S0025315400006500. [DOI] [Google Scholar]
- 15.Pomeroy LR, Johannes RE. 1968. Occurrence and respiration of ultraplankton in the upper 500 meters of the ocean. Deep Sea Res Oceanogr Abstr 15:381–391. doi: 10.1016/0011-7471(68)90014-4. [DOI] [Google Scholar]
- 16.Pocklington R. 1972. Determination of nanomolar quantities of free amino acids dissolved in North Atlantic ocean waters. Anal Biochem 45:403–421. doi: 10.1016/0003-2697(72)90202-3. [DOI] [PubMed] [Google Scholar]
- 17.Siegel A, Degens ET. 1966. Concentration of dissolved amino acids from saline waters by ligand-exchange chromatography. Science 151:1098–1101. doi: 10.1126/science.151.3714.1098. [DOI] [PubMed] [Google Scholar]
- 18.Kirchman DL. 2003. The contribution of monomers and other low-molecular weight compounds to the flux of dissolved organic material in aquatic ecosystems, p 217–241. In Findlay SEG, Sinsabaugh RL (ed), Aquatic ecosystems: interactivity of dissolved organic matter. Academic Press, San Diego, CA. [Google Scholar]
- 19.Crawford CC, Hobbie JE, Webb KL. 1974. The utilization of dissolved free amino acids by estuarine microorganisms. Ecology 55:551–563. doi: 10.2307/1935146. [DOI] [Google Scholar]
- 20.Hobbie JE, Crawford CC, Webb KL. 1968. Amino acid flux in an estuary. Science 159:1463–1464. doi: 10.1126/science.159.3822.1463. [DOI] [PubMed] [Google Scholar]
- 21.Keil RG, Kirchman DL. 1991. Contribution of dissolved free amino acids and ammonium to the nitrogen requirements of heterotrophic bacterioplankton. Mar Ecol Prog Ser 73:1–10. doi: 10.3354/meps073001. [DOI] [Google Scholar]
- 22.Suttle CA, Chan AM, Fuhrman JA. 1991. Dissolved free amino acids in the Sargasso Sea: uptake and respiration rates, turnover times, and concentrations. Mar Ecol Prog Ser 70:189–199. doi: 10.3354/meps070189. [DOI] [Google Scholar]
- 23.Ottesen EA, Marin R, Preston CM, Young CR, Ryan JP, Scholin CA, DeLong EF. 2011. Metatranscriptomic analysis of autonomously collected and preserved marine bacterioplankton. ISME J 5:1881–1895. doi: 10.1038/ismej.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gifford SM, Sharma S, Moran MA. 2014. Linking activity and function to ecosystem dynamics in a coastal bacterioplankton community. Front Microbiol 5:185. doi: 10.3389/fmicb.2014.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gifford SM, Sharma S, Booth M, Moran MA. 2013. Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J 7:281–298. doi: 10.1038/ismej.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gómez-Consarnau L, Lindh MV, Gasol JM, Pinhassi J. 2012. Structuring of bacterioplankton communities by specific dissolved organic carbon compounds. Environ Microbiol 14:2361–2378. doi: 10.1111/j.1462-2920.2012.02804.x. [DOI] [PubMed] [Google Scholar]
- 27.Sowell SM, Abraham PE, Shah M, Verberkmoes NC, Smith DP, Barofsky DF, Giovannoni SJ. 2011. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J 5:856–865. doi: 10.1038/ismej.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sowell SM, Wilhelm LJ, Norbeck AD, Lipton MS, Nicora CD, Barofsky DF, Carlson CA, Smith RD, Giovanonni SJ. 2009. Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J 3:93–105. doi: 10.1038/ismej.2008.83. [DOI] [PubMed] [Google Scholar]
- 29.Cottrell MT, Kirchman DL. 2000. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol 66:1692–1697. doi: 10.1128/AEM.66.4.1692-1697.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alonso-Sáez L, Gasol JM. 2007. Seasonal variations in the contributions of different bacterial groups to the uptake of low-molecular-weight compounds in northwestern Mediterranean coastal waters. Appl Environ Microbiol 73:3528–3535. doi: 10.1128/AEM.02627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malmstrom RR, Kiene RP, Cottrell MT, Kirchman DL. 2004. Contribution of SAR11 bacteria to dissolved dimethylsulfoniopropionate and amino acid uptake in the North Atlantic Ocean. Appl Environ Microbiol 70:4129–4135. doi: 10.1128/AEM.70.7.4129-4135.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikrad MP, Cottrell MT, Kirchman DL. 2014. Uptake of dissolved organic carbon by gammaproteobacterial subgroups in coastal waters of the West Antarctic Peninsula. Appl Environ Microbiol 80:3362–3368. doi: 10.1128/AEM.00121-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson CE, Carlson CA. 2012. Tracking differential incorporation of dissolved organic carbon types among diverse lineages of Sargasso Sea bacterioplankton. Environ Microbiol 14:1500–1516. doi: 10.1111/j.1462-2920.2012.02738.x. [DOI] [PubMed] [Google Scholar]
- 34.Dumont MG, Murrell JC. 2005. Stable isotope probing — linking microbial identity to function. Nat Rev Microbiol 3:499–504. doi: 10.1038/nrmicro1162. [DOI] [PubMed] [Google Scholar]
- 35.Neufeld JD, Dumont MG, Vohra J, Murrell JC. 2007. Methodological considerations for the use of stable isotope probing in microbial ecology. Microb Ecol 53:435–442. doi: 10.1007/s00248-006-9125-x. [DOI] [PubMed] [Google Scholar]
- 36.DeLorenzo S, Bräuer SL, Edgmont CA, Herfort L, Tebo BM, Zuber P. 2012. Ubiquitous dissolved inorganic carbon assimilation by marine bacteria in the Pacific Northwest coastal ocean as determined by stable isotope probing. PLoS One 7:e46695. doi: 10.1371/journal.pone.0046695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayali X, Weber PK, Brodie EL, Mabery S, Hoeprich PD, Pett-Ridge J. 2012. High-throughput isotopic analysis of RNA microarrays to quantify microbial resource use. ISME J 6:1210–1221. doi: 10.1038/ismej.2011.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayali X, Weber PK, Pett-Ridge J. 2013. Taxon-specific C/N relative use efficiency for amino acids in an estuarine community. FEMS Microbiol Ecol 83:402–412. doi: 10.1111/j.1574-6941.12000.x. [DOI] [PubMed] [Google Scholar]
- 39.Mayali X, Weber PK, Mabery S, Pett-Ridge J. 2014. Phylogenetic patterns in the microbial response to resource availability: amino acid incorporation in San Francisco Bay. PLoS One 9:e95842. doi: 10.1371/journal.pone.0095842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jehmlich N, Schmidt F, Taubert M, Seifert J, Bastida F, von Bergen M, Richnow H-H, Vogt C. 2010. Protein-based stable isotope probing. Nat Protoc 5:1957–1966. doi: 10.1038/nprot.2010.166. [DOI] [PubMed] [Google Scholar]
- 41.Seifert J, Taubert M, Jehmlich N, Schmidt F, Völker U, Vogt C, Richnow H-H, von Bergen M. 2012. Protein-based stable isotope probing (protein-SIP) in functional metaproteomics. Mass Spectrom Rev 31:683–697. doi: 10.1002/mas.21346. [DOI] [PubMed] [Google Scholar]
- 42.Jehmlich N, Schmidt F, von Bergen M, Richnow H-H, Vogt C. 2008. Protein-based stable isotope probing (protein-SIP) reveals active species within anoxic mixed cultures. ISME J 2:1122–1133. doi: 10.1038/ismej.2008.64. [DOI] [PubMed] [Google Scholar]
- 43.Taubert M, Vogt C, Wubet T, Kleinsteuber S, Tarkka MT, Harms H, Buscot F, Richnow H-H, von Bergen M, Seifert J. 2012. Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J 6:2291–2301. doi: 10.1038/ismej.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slysz GW, Steinke L, Ward DM, Klatt CG, Clauss TRW, Purvine SO, Payne SH, Anderson GA, Smith RD, Lipton MS. 2014. Automated data extraction from in situ protein-stable isotope probing studies. J Proteome Res 13:1200–1210. doi: 10.1021/pr400633j. [DOI] [PubMed] [Google Scholar]
- 45.Pan C, Fischer CR, Hyatt D, Bowen BP, Hettich RL, Banfield JF. 2011. Quantitative tracking of isotope flows in proteomes of microbial communities. Mol Cell Proteomics 10:M110.006049. doi: 10.1074/mcp.M110.006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hettich RL, Sharma R, Chourey K, Giannone RJ. 2012. Microbial metaproteomics: identifying the repertoire of proteins that microorganisms use to compete and cooperate in complex environmental communities. Curr Opin Microbiol 15:373–380. doi: 10.1016/j.mib.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Wang D-Z, Xie Z-X, Zhang S-F. 2014. Marine metaproteomics: current status and future directions. J Proteomics 97:27–35. doi: 10.1016/j.jprot.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 48.Williams TJ, Cavicchioli R. 2014. Marine metaproteomics: deciphering the microbial metabolic food web. Trends Microbiol 22:248–260. doi: 10.1016/j.tim.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Ahn T-H, Li Z, Pan C. 2013. Sipros/ProRata: a versatile informatics system for quantitative community proteomics. Bioinformatics 29:2064–2065. doi: 10.1093/bioinformatics/btt329. [DOI] [PubMed] [Google Scholar]
- 50.Hyatt D, Pan C. 2012. Exhaustive database searching for amino acid mutations in proteomes. Bioinformatics 28:1895–1901. doi: 10.1093/bioinformatics/bts274. [DOI] [PubMed] [Google Scholar]
- 51.Justice NB, Li Z, Wang Y, Spaudling SE, Mosier AC, Hettich RL, Pan C, Banfield JF. 2014. 15N- and 2H proteomic stable isotope probing links nitrogen flow to archaeal heterotrophic activity. Environ Microbiol 16:3224–3237. doi: 10.1111/1462-2920.12488. [DOI] [PubMed] [Google Scholar]
- 52.Mueller RS, Bryson S, Kieft B, Li Z, Pett-Ridge J, Chavez F, Hettich RL, Pan C, Mayali X. 2015. Metagenome sequencing of a coastal marine microbial community from Monterey Bay, California. Genome Announc 3(2):e00341-15. doi: 10.1128/genomeA.00341-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630–633. doi: 10.1038/nature00917. [DOI] [PubMed] [Google Scholar]
- 54.Eilers H, Pernthaler J, Glöckner FO, Amann R. 2000. Culturability and in situ abundance of pelagic bacteria from the North Sea. Appl Environ Microbiol 66:3044–3051. doi: 10.1128/AEM.66.7.3044-3051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alonso C, Warnecke F, Amann R, Pernthaler J. 2007. High local and global diversity of Flavobacteria in marine plankton. Environ Microbiol 9:1253–1266. doi: 10.1111/j.1462-2920.2007.01244.x. [DOI] [PubMed] [Google Scholar]
- 56.Gómez-Pereira PR, Fuchs BM, Alonso C, Oliver MJ, van Beusekom JEE, Amann R. 2010. Distinct flavobacterial communities in contrasting water masses of the North Atlantic Ocean. ISME J 4:472–487. doi: 10.1038/ismej.2009.142. [DOI] [PubMed] [Google Scholar]
- 57.Tada Y, Taniguchi A, Nagao I, Miki T, Uematsu M, Tsuda A, Hamasaki K. 2011. Differing growth responses of major phylogenetic groups of marine bacteria to natural phytoplankton blooms in the western North Pacific Ocean. Appl Environ Microbiol 77:4055–4065. doi: 10.1128/AEM.02952-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abell GCJ, Bowman JP. 2005. Colonization and community dynamics of class Flavobacteria on diatom detritus in experimental mesocosms based on Southern Ocean seawater. FEMS Microbiol Ecol 53:379–391. doi: 10.1016/j.femsec.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 59.Walters W, Hyde ER, Berg-Lyons D, Ackermann G, Humphrey G, Parada A, Gilbert JA, Jansson JK, Caporaso JG, Fuhrman JA, Apprill A, Knight R. 2016. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1:e00009-15. doi: 10.1128/mSystems.00009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stewart FJ, Dalsgaard T, Young CR, Thamdrup B, Revsbech NP, Ulloa O, Canfield DE, DeLong EF. 2012. Experimental incubations elicit profound changes in community transcription in OMZ bacterioplankton. PLoS One 7:e37118. doi: 10.1371/journal.pone.0037118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morris RM, Longnecker K, Giovannoni SJ. 2006. Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environ Microbiol 8:1361–1370. doi: 10.1111/j.1462-2920.2006.01029.x. [DOI] [PubMed] [Google Scholar]
- 62.Mou X, Hodson RE, Moran MA. 2007. Bacterioplankton assemblages transforming dissolved organic compounds in coastal seawater. Environ Microbiol 9:2025–2037. doi: 10.1111/j.1462-2920.2007.01318.x. [DOI] [PubMed] [Google Scholar]
- 63.Alonso C, Pernthaler J. 2006. Roseobacter and SAR11 dominate microbial glucose uptake in coastal North Sea waters. Environ Microbiol 8:2022–2030. doi: 10.1111/j.1462-2920.2006.01082.x. [DOI] [PubMed] [Google Scholar]
- 64.Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, Howard EC, King E, Oakley CA, Reisch CR, Rinta-Kanto JM, Sharma S, Sun S, Varaljay V, Vila-Costa M, Westrich JR, Moran MA. 2010. Genome characteristics of a generalist marine bacterial lineage. ISME J 4:784–798. doi: 10.1038/ismej.2009.150. [DOI] [PubMed] [Google Scholar]
- 65.Voget S, Wemheuer B, Brinkhoff T, Vollmers J, Dietrich S, Giebel H-A, Beardsley C, Sardemann C, Bakenhus I, Billerbeck S, Daniel R, Simon M. 2015. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J 9:371–384. doi: 10.1038/ismej.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR, Chisholm SW, DeLong EF. 2010. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci U S A 107:16420–16427. doi: 10.1073/pnas.1010732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi Y, McCarren J, DeLong EF. 2012. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment. Environ Microbiol 14:191–206. doi: 10.1111/j.1462-2920.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- 68.Stingl U, Tripp HJ, Giovannoni SJ. 2007. Improvements of high-throughput culturing yielded novel SAR11 strains and other abundant marine bacteria from the Oregon coast and the Bermuda Atlantic Time series study site. ISME J 1:361–371. doi: 10.1038/ismej.2007.49. [DOI] [PubMed] [Google Scholar]
- 69.Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ. 2008. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452:741–744. doi: 10.1038/nature06776. [DOI] [PubMed] [Google Scholar]
- 70.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 71.Li Z, Wang Y, Yao Q, Justice NB, Ahn T-H, Xu D, Hettich RL, Banfield JF, Pan C. 2014. Diverse and divergent protein post-translational modifications in two growth stages of a natural microbial community. Nat Commun 5:4405. doi: 10.1038/ncomms5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Washburn MP, Wolters D, Yates JR. 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 73.Boisvert S, Raymond F, Godzaridis E, Laviolette F, Corbeil J. 2012. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol 13:R122. doi: 10.1186/gb-2012-13-12-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 75.Treangen TJ, Sommer DD, Angly FE, Koren S, Pop M. 2011. Next generation sequence assembly with AMOS. Curr Protoc Bioinformatics Chapter 1:Unit 11.8. doi: 10.1002/0471250953.bi1108s33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28:2223–2230. doi: 10.1093/bioinformatics/bts429. [DOI] [PubMed] [Google Scholar]
- 77.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 78.Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, Gabaldón T, Rattei T, Creevey C, Kuhn M, Jensen LJ, von Mering C, Bork P. 2014. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res 42:D231–D239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Taxonomic distribution of predicted open reading frames (ORFs), metagenome reads, and 16S rDNA amplicons. (A) Proportions of predicted ORFs for taxonomic groups in the assembled MB metagenomes and in the final database used for peptide spectral matching. (B) Distribution of mapped metagenomic reads among the assembled contigs for the initial MB0 sample and MB1. (C) Relative proportions of 16S rDNA amplicon taxonomic assignments for MB0, MB1, and MB2. Download Figure S1, EPS file, 0.3 MB (313.8KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of metagenomes and metaproteomes. (A to C) Relative abundance of mapped reads from the MB0 and MB1 metagenomes (A), average relative spectral counts in MB1 and MB2 (B), and relative spectral counts in OR1 and OR2 (C). Download Figure S2, EPS file, 0.1 MB (129.1KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Label frequency and average enrichment for genera of three abundant orders, Rhodobacterales (A), Alteromonadales (B), and Flavobacteriales (C). The figure depicts Z-scores of label frequency (x axis) and average enrichment (y axis) based on comparisons of observed values with distributions under the null model. The shaded area represents the inner 95% of values under a standard normal distribution. Significance testing is described in Materials and Methods. Download Figure S3, EPS file, 0.7 MB (742.8KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relative labeling of COG categories. The panels depict biclustered heat maps of label frequency (Z-scores) of COG functional categories for each sample within three dominant orders: Rhodobacterales (A), Alteromonadales (B), and Flavobacteriales (C). Download Figure S4, EPS file, 0.3 MB (317.9KB, eps) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
False detection rate of labeled spectra. The table lists numbers of and proportion of total PSM from two unlabeled metaproteomes that were identified at the corresponding level of percent 13C enrichment. Searches were conducted against the same sequence database used for SIP searches in this study. Download Table S1, PDF file, 0.04 MB (43.1KB, pdf) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pairwise concordance correlation coefficients. Results are from pairwise tests between all metaproteomes using relative balanced spectral counts for each identified protein. Download Table S2, PDF file, 0.03 MB (35.3KB, pdf) .
Copyright © 2016 Bryson et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.