

Abstract
Trinucleotide repeats (TNRs) undergo frequent mutations in families afflicted with certain neurodegenerative disorders and in model organisms. TNR instability is modulated both by the repeat tract itself and by cellular proteins. Here we identified the Saccharomyces cerevisiae DNA helicase Srs2 as a potent and selective inhibitor of expansions. srs2 mutants had up to 40-fold increased expansion rates of CTG, CAG, and CGG repeats. The expansion phenotype was specific, as mutation rates at dinucleotide repeats, at unique sequences, or for TNR contractions in srs2 mutants were not altered. Srs2 is known to suppress inappropriate genetic recombination; however, the TNR expansion phenotype of srs2 mutants was largely independent of RAD51 and RAD52. Instead, Srs2 mainly functioned with DNA polymerase delta to block expansions. The helicase activity of Srs2 was important, because a point mutant lacking ATPase function was defective in blocking expansions. Purified Srs2 was substantially better than bacterial UvrD helicase at in vitro unwinding of a DNA substrate that mimicked a TNR hairpin. Disruption of the related helicase gene SGS1 did not lead to excess expansions, nor did wild-type SGS1 suppress the expansion phenotype of an srs2 strain. We conclude that Srs2 selectively blocks triplet repeat expansions through its helicase activity and primarily in conjunction with polymerase delta.
Trinucleotide repeats (TNRs) undergo frequent expansions in families afflicted with Huntington's disease, fragile X syndrome, and at least 13 other neurodegenerative diseases (7, 8, 35). In addition to this biomedical relevance, the unusual genetic mechanisms underlying TNR instability have sparked substantial scientific interest. Studies in the field of human genetics and with model organisms indicate that the TNR DNA itself plays a major role in its own mutability (7, 8, 17, 35, 45). For example, the risk of expansion is closely tied to cis-acting features, such as the sequence and length of the TNR tract, and whether the repeat is perfect or imperfect. While these “rules” can usually predict expansion risk, there are clear exceptions. For example, the risk of an expansion can vary greatly depending on whether the gene is inherited maternally or paternally (35, 45). In addition, genetic background and other factors have been suggested to explain, for example, the variations in germ line instability among individuals with similar allele lengths at the Huntington's disease locus (22).
These observations provide some of the evidence suggesting that cellular proteins modulate the likelihood of TNR instability. What proteins might be involved? Important clues come from models of expansion (7, 12, 17, 45). While expansions result from more than one genetic mechanism, a central feature of essentially all expansion models is that single-stranded TNR sequences fold into aberrant DNA structures, usually hairpins (11), which are crucial intermediates in the mutation process (12, 17, 45). If a hairpin cannot be prevented from forming or is not removed quickly enough, an expansion will ensue. Thus, hairpins are thought to be direct precursors of expansions. Proteins that either prevent hairpin formation or accelerate hairpin removal would help reduce expansion rates. One well-characterized protein that helps block hairpin formation is the flap endonuclease FEN-1 (24), which removes some single-stranded TNR flaps (10, 24, 25, 40, 43, 44). However, it is likely that other proteins also help avoid expansions.
In this study, the Srs2 DNA helicase was identified in a genetic screen for inhibitors of expansion in Saccharomyces cerevisiae. Characterization of srs2 mutants indicates that the helicase is a potent and selective inhibitor of TNR expansions.
MATERIALS AND METHODS
Strains.
Most of the S. cerevisiae strains used were derived from MW3317-21A (18) (MATα Δtrp1 ura3-52 ade2Δ ade8 hom3-10 his3-Kpn1 met4 met13). Isogenic derivatives containing disruptions of msh2, rad27, rad52, or rad51 were constructed by standard techniques (26, 39) and confirmed by Southern blotting and phenotypic analysis. The strain harboring the srs2::TRP1 mutation, HKY723-5D (MATa hpr5::TRP1 leu2-3,112 his3-11, 15 ade2-2 ura3-1 trp1-1 can1-100 RAD5+), was kindly provided by Hannah Klein, New York University School of Medicine. A strain harboring sgs1::kanMX4 was obtained from Open Biosystems. BL 492, a pol32Δ derivative of BL490 (MATa leu2Δ1 trpΔ63 ura3-52 his3-200) was a generous gift from Sergei Mirkin (University of Illinois, Chicago). The chromosomal integration of TNR-containing plasmids and confirmation of correct single integrants were done as described previously (33, 37).
Plasmids.
All triplet repeat-containing plasmids were constructed with the pBL94 vector as described previously (38). Plasmids pHK209 (BamHI-SalI insert of SRS2 in YCp50), pHK282 (EcoRI-SalI insert of srs2K41A in pRS314) and pFP56 (srs2::TRP1) were generous gifts from Hannah Klein. To create low-copy-number pSRS2 plasmids, the SRS2 coding fragment was recovered as a BamHI-SalI fragment from pHK209 and subcloned into pRS314 (marked with TRP1) or pRS315 (marked with LEU2) (42). When low-copy-number pSRS2 was used for complementation studies of the srs2::LEU2 and srs2::TRP1 alleles, the appropriate pSRS2 plasmid was always added to the strain already containing the triplet repeat reporter. The high-copy-number pSRS2 plasmid used for overexpression studies was created by transfer of the SRS2 gene as an EcoRI-SalI fragment from pHK209 into pRS424 (6). The reporter plasmid pSH44, containing 16.5 repeats of the dinucleotide repeat GT (14), was kindly provided by Tom Petes, University of North Carolina. The 2μm plasmid YEplac195-SGS1 was a generous gift from Alan Morgan, University of Liverpool. For this study, the SGS1 gene was moved as a SalI fragment into the corresponding site of the high-copy-number plasmid pRS424.
Genetic assays and molecular analysis of mutated TNR alleles.
Expansion and contraction rates were measured by fluctuation analysis as described previously (33, 37). Briefly, TNR tracts were cloned into a yeast promoter-reporter to create spacing-sensitive expression of a downstream URA3 gene. Tracts containing up to 25 TNRs or a subset of repeats plus randomized sequence DNA equivalent to 25 repeats were used to score expansions of +5 repeats or more (33, 37). To measure contraction rates, the assay is performed with longer starting tracts (here we used 25 repeats plus 24 random-order nucleotides, equivalent to eight repeats), referred to as CTG25+8 (37) Contractions of −5 or more repeats are identified as Ura+ colonies. Dinucleotide repeat mutation rates were measured as described previously (14). Forward mutation rates for the CAN1 gene were determined by selection for canavanine resistance. Three to five independent clones were tested for each of the above assays to ensure reproducibility. Single-colony PCR analysis of TNR expansions and contractions was performed by a published method (33). The percent bona fide expansions or contractions, determined by PCR analysis, was multiplied by the apparent expansion and contraction rates derived from fluctuation analysis. All rates reported here reflect this correction factor.
Western blot analysis.
The expression of Srs2 was analyzed by Western blotting. Briefly, 107 cells/ml from a 5-ml overnight culture were harvested and lysed in sample buffer with glass beads; 10 μl of this lysate (containing 5 μg of total protein) was loaded onto a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) 7.5% polyacrylamide gel and separated by electrophoresis, the proteins were transferred to a nitrocellulose membrane (Amersham), and the membrane was blocked overnight at 4°C in Odyssey blocking buffer (Li-Cor). Immunodetection was done with rabbit anti-Srs2 antiserum (from Patrick Sung, Yale University) or with anti-Rfc3 antiserum (from Peter Burgers, Washington University), followed by probing with Alexa Fluor 680 goat anti-rabbit immunoglobulin G (Molecular Probes) and fluorescence detection and quantitation with the Odyssey system, per the manufacturer's protocol.
Helicase substrate analysis and unwinding assays.
End labeling with 32P by T4 polynucleotide kinase (New England Biolabs) was performed according to the manufacturer's protocol. Oligonucleotide strands (one radiolabeled) were mixed at equimolar concentrations with the unlabeled complement, heated at 95°C for 5 min, and then slowly cooled to room temperature. The location of the radiolabel on either the longer or shorter strand is stipulated in the legends to Fig. 3 and 4. Mung bean nuclease (New England Biolabs) digestions (total volume, 20 μl) were performed with 1 unit of enzyme in the manufacturer's reaction buffer at 37°C for 40 min. Reactions were quenched by addition of SDS to 0.01% final concentration, followed by ethanol precipitation, resuspension in formamide loading buffer, and analysis on a 15% sequencing gel.
FIG. 3.

Srs2 unwinds CTG repeat-containing double-stranded DNA. (A) Schematic diagram of putative Srs2 action on a CTG hairpin in vivo. In the diagram, a newly synthesized CTG tract has folded onto itself to form a hairpin (the template strand has been omitted for clarity). Srs2 (diamond) loads onto the 3′ DNA end and unwinds the structure. The single-stranded product might then reanneal properly to the template strand or might be subject to nuclease digestion, followed by resynthesis of the tract. Either outcome would help eliminate hairpins and thereby reduce the appearance of expansions. (B) Partial DNA duplexes containing CTG repeats can be tested in vitro as potential Srs2 helicase substrates. Srs2 action is envisioned to work as in A. The products of unwinding are single-stranded DNAs which can be readily assayed by gel electrophoresis. (C) Predicted structure of the partially double-stranded substrate. The 3′ tail of the longer strand provides a loading site for the helicase. The duplex contains five CTG repeats to mimic a TNR hairpin. The nine complementary base pairs add thermodynamic stability to the duplex and help ensure that the CTG repeats align as predicted. The arrow shows the predicted junction between single- and double-stranded DNA; 24 nucleotides of the bottom strand should be protected from mung bean nuclease, an enzyme that cleaves single-stranded DNA but not double-stranded regions. (D) Nuclease analysis of DNA structure. The annealed substrate, 5′-end labeled on the longer strand with 32P, was incubated with mung bean nuclease. The products were analyzed on a denaturing 15% polyacrylamide gel. Lanes 1, 2, and 3 contain molecular size markers of 34, 24, and 9 nucleotides, respectively. Lane 4 contains the undigested helicase substrate, and lane 5 contains the product after nuclease digestion. (E) Srs2 helicase activity. The DNA substrate, 5′-end labeled on the shorter strand with 32P, was incubated under standard helicase assay conditions (Materials and Methods) with 50 nM Srs2 for the indicated times. Δ, heat-denatured control.
FIG. 4.

Srs2 unwinds a TNR substrate substantially faster than does UvrD. DNA substrates radiolabeled on the shorter strand were incubated with either Srs2 (35 nM) or UvrD (200 nM) under standard conditions (19, 30) for the indicated times, and the reaction products were separated by PAGE and visualized by phosphorimaging. Both DNA substrates contained a 14-nucleotide-long 3′ tail, which is usable by either Srs2 or UvrD. (A) Schematic diagram of the (CTG)10-containing substrate. Along with the 9 bp of nonrepeating DNA (identical to Fig. 3C), the duplex region is 39 bp in total length. (B) Control substrate (19) without triplet repeats. The duplex region measures 40 bp.
For helicase assays, the radiolabeled partial duplex DNA substrate was incubated with 25 to 35 nM purified Srs2 for 0 to 15 min under standard helicase assay conditions; 30°C in the presence of 300 nM (nucleotides) DNA substrate, 25 mM Tris-Cl (pH 7.5), 2.5 mM MgCl2, 1 mM dithiothreitol, 100 μg of bovine serum albumin per ml, and 2 mM ATP (19). UvrD was used at 200 nM for 0 to 20 min as follows (30); the reaction mixture contained 25 mM Tris-Cl (pH 7.5), 3 mM MgCl2, 20 mM NaCl, 50 mM 2-mercaptoethanol, 50 μg of bovine serum albumin per ml, and 10 nM DNA substrate. UvrD was diluted in helicase II storage buffer and then incubated for 5 min at 37°C with the reaction mix lacking ATP. The unwinding reaction was initiated by addition of ATP to a final concentration of 3 mM. For all helicase reactions, the DNA products were subsequently analyzed by nondenaturing 15% PAGE.
RESULTS
Identification of SRS2 as a gene affecting triplet repeat expansions.
To find candidate genes that help protect the cell from expansions, random transposon mutants of S. cerevisiae were screened for increased expansions of a (CTG)13 reporter (4). A CTG sequence was used because CTG•CAG tracts are unstable in several human genetic diseases. The length of 13 is relevant in S. cerevisiae because it falls close to a crucial threshold length (37) at which expansions become increasingly more frequent. We reasoned that yeast mutations that alter (CTG)13 expansion rates might be specific for TNRs, since thresholds are unique to TNRs. Thresholds are well known in human TNR instability, where lengths of ≈35 repeats are associated with increased risk of expansion (35).
DNA sequencing (4) identified srs2::LEU2 as a disrupted gene that gave increased expansion rates. Additional genetic assays were performed to verify the srs2 mutation. Both the srs2::LEU2 mutant and an srs2::TRP1 mutant showed moderate UV sensitivity, consistent with published results (1). Nonhomologous end joining, measured by a plasmid repair assay, was reduced 2.5-fold in the srs2::LEU2 background, in good agreement with published values (13). Identical expansion phenotypes were observed with both the srs2::LEU2 and the srs2::TRP1 strains, and a low-copy-number number pSRS2 plasmid reversed the hyperexpansion phenotype of the srs2::LEU2 mutation (see below). Together with the sequencing data, these results verified SRS2 as the mutated gene.
The hyperexpansion phenotype of the srs2 mutant was surprising because neither spontaneous nor damage-induced mutagenesis, measured as forward mutations at CAN1, is increased in srs2 strains (2). To evaluate the hyperexpansion phenotype more thoroughly, quantitative mutation rates were determined. Expansions of the (CTG)13 tract (37) were elevated 40- to 44-fold over that in the wild type for our srs2::LEU2 allele and for the srs2::TRP1 strain obtained from another laboratory (Table 1). The wild-type SRS2 gene on a low-copy-number plasmid (pSRS2) reduced the expansion rate to wild-type levels. The expansion sizes of CTG tracts in srs2::LEU2 were also measured (33). Starting from an initial (CTG)13 tract, the expanded alleles ranged from +5 to +10 repeats, with a median of +8, similar to expansions in the wild-type strain (Fig. 1). As the two mutational spectra overlap, we conclude that the number of expansions, not their size, accounts for the dramatic increase in the expansion rate of the srs2 strain. There was a single case of a large expansion (+31 repeats) in the srs2::LEU2 mutant (Fig. 1). However 98% of CTG expansions in the srs2 strain were less than twice the original tract size, consistent with a replicational model of TNR repeat instability (12).
TABLE 1.
Mutation rates in srs2 mutant strainsa
| Mutation (exponent) and genotype | Mean no. of mutations/cell generation, 10−n (± SD) | Ratio (fold over wild type) |
|---|---|---|
| Expansions of (CTG)13 (10−7) | ||
| SRS2 | 1.4 (± 0.2) | 1.0 |
| srs2::LEU2 | 56 (± 11) | 40 |
| srs2::TRP1 | 62 (± 11) | 44 |
| srs2::LEU2 + pSRS2 | 1.8 (± 0.8) | 1.3 |
| rad51::kanMX | 1.3 (± 0.5) | 0.9 |
| srs2::TRP1 rad51::kanMX | 16 (± 0.6) | 11 |
| rad52::LEU2 | ≈1 | ≈0.7 |
| srs2::TRP1 rad52::LEU2 | 46 (± 5) | 33 |
| pol32Δ | 41 (± 3) | 29 |
| srs2::LEU2 pol32Δ | 28 (± 12) | 20 |
| pol32Δ + high-copy SRS2 | 2.0 (± 0.5) | 1.4 |
| srs2::LEU2 + psrs2K41A | 24 (± 9) | 17 |
| sgs1::kanMX4 | 2.4 (± 0.5) | 1.7 |
| srs2::LEU2 + high-copy SGS1 | 60 (± 11) | 43 |
| Expansions of (CTG)25 (10−5) | ||
| SRS2 | 1.0 (± 0.3) | 1.0 |
| srs2::LEU2 | 4.9 (± 0.8) | 4.9 |
| srs2::TRP1 | 5.1 (± 1.1) | 5.1 |
| srs2::LEU2 + low-copy pSRS2 | 1.4 (± 0.2) | 1.4 |
| Expansions of (CAG)25 (10−7) | ||
| SRS2 | <5 | 1.0 |
| srs2::LEU2 | 23 (± 0.7) | 4.6 |
| Expansions of (CGG)25 (10−5) | ||
| SRS2 | 6.8 (± 0.7) | 1.0 |
| srs2::LEU2 | 23 (± 5) | 3.4 |
| Dinucleotide repeat mutations (10−5) | ||
| SRS2 | 3.8 (± 0.2) | 1.0 |
| srs2::LEU2 | 4.3 (± 0.3) | 1.1 |
| msh2Δ | 1,400 (± 10) | 370 |
| CAN1 forward mutations (10−7) | ||
| SRS2 | 3.0 (± 0.4) | 1.0 |
| srs2::LEU2 | 3.7 (± 0.3) | 1.2 |
| rad27Δ | 180 (± 10) | 60 |
| Contractions of (CTG)25+8 (10−5) | ||
| SRS2 | 2.7 (± 0.2) | 1.0 |
| srs2::LEU2 | 2.5 (± 0.3) | 0.9 |
| srs2::TRP1 | 2.2 (± 0.4) | 0.8 |
Rates were determined by the method of the median(20). Rates are the averages of three to six determinations for genetically independent clones. The exponent is shown separately for each section. The ratio value is the mutation rate for the indicated genotype divided by the wild-type rate.
FIG. 1.
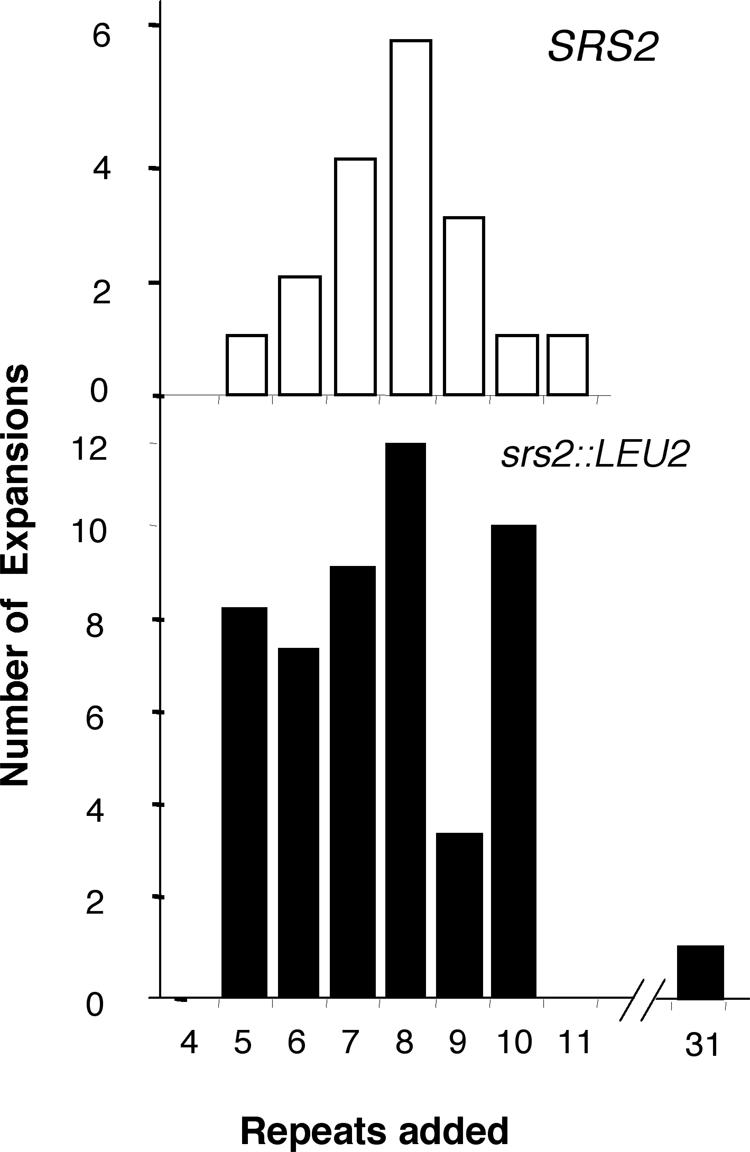
(CTG)13 expansion sizes in wild-type and srs2::LEU2 strains. A subset of the 5-fluoroorotic acid-resistant colonies obtained from fluctuation analysis were subjected to single-colony PCR analysis and sizing on sequencing gels to an accuracy of ±1 to 2 repeats (33). The x axis denotes the number of repeats added to the original tract of 13 repeats.
Hyperexpansion phenotype of an srs2 mutant is mainly associated with DNA polymerase delta, not with genetic recombination.
Genetic and biochemical studies (1, 3, 19, 47) indicate that Srs2 functions as an antirecombinase, and many srs2 phenotypes are linked to recombination (16). Therefore, it was important to know whether the hyperexpansion phenotype of srs2 was also a result of hyperrecombination. If so, expansion rates should be reduced to near wild-type levels when recombination is inactivated. In contrast to this prediction, the results in Table 1 show that double mutants (rad51 srs2 and rad52 srs2) still retained much of the instability (11- to 33-fold) seen in an srs2 single mutant (40- to 44-fold). Thus, loss of recombination reduced the srs2 effect on CTG expansions by only 1.3- to 4-fold. While a portion of the srs2 expansion phenotype might be due to recombinational misprocessing of TNRs, some other process is responsible for the majority of the phenotype. Expansion rates were also unaffected when either rad51 or rad52 was mutated singly (Table 1), arguing against unequal sister chromatid exchange (9) as a mechanism of expansion.
An alternative possibility is that Srs2 functions with DNA polymerase delta to block expansions. This idea is based on the finding that Srs2 interacts in two-hybrid experiments with the Pol32 subunit of polymerase delta (15). If Srs2 and polymerase delta are both needed for inhibiting TNR expansions, then a mutant lacking POL32 should show a high expansion rate. A (CTG)13 tract was 29-fold more unstable in a pol32Δ strain than its wild-type counterpart (Table 1). Similarly, the repeat tract was 20-fold destabilized in an srs2 pol32 double mutant. This epistasis analysis is consistent with the idea that the majority of expansions prevented by Srs2 occur in conjunction with Pol32, and hence polymerase delta. Statistical analysis by Student's t test showed no significant differences between the expansion rates for srs2 compared to pol32Δ (P = 0.08) or for pol32Δ compared to srs2 pol32Δ (P = 0.14); however, there was a significant difference between srs2 and srs2 pol32Δ (P = 0.04).
The epistasis relationship between Srs2 and polymerase delta may be somewhat complex. To examine this idea further, we tested the effect of Srs2 overexpression. If a direct physical interaction between Srs2 and Pol32 is important for preventing expansions (for example, to recruit Srs2 to a replication fork), overexpressing Srs2 in a pol32Δ background should not alleviate the hyperexpansion phenotype. In contrast to the prediction, we found that the expansion rate for Srs2 overexpression in a pol32Δ strain was indistinguishable from wild-type levels (Table 1). Immunoblotting (Fig. 2B) showed that Srs2 levels were elevated three- to fourfold when cells harbored SRS2 on a multicopy plasmid. We conclude that Srs2, when overexpressed, does not require physical interaction with Pol32 to block expansions. Caution is warranted when interpreting this result, however, because the interaction may be more important when Srs2 is present at normal levels.
FIG. 2.

Immunoblot analysis of Srs2 expression. Whole-cell extracts were separated by SDS-7.5% PAGE, transferred to a nitrocellulose filter, blotted with polyclonal Srs2 antiserum, and detected by fluorescence. (A) Lane 1, purified Srs2. Lane 2, extract from srs2::LEU2 cells. Lane 3, extract from srs2::LEU2 cells containing low-copy-number pSRS2 plasmid. Lane 4, extract from srs2::LEU2 cells containing the low-copy-number mutant plasmid psrs2K41A. Bottom strip, loading control immunoblot for Rfc3. Similar results were seen in two other repetitions of this experiment. (B) Lane 1, extract from wild-type cells. Lane 2, extract from wild-type cells containing a high-copy-number pSRS2 plasmid. Lane 3, extract from pol32Δ cells. Lane 4, extract from pol32Δ cells with a high-copy-number pSRS2 plasmid. A similar result was seen in a repetition of this experiment.
Srs2 selectively blocks TNR expansions.
Expansion rates were also examined for a longer TNR, (CTG)25. In a previous study (37), we showed that (CTG)25 is above the apparent threshold length of 13 repeats in S. cerevisiae. As a reference, wild-type cells undergo expansions of (CTG)25 70-fold more often than for (CTG)13 (37). It was therefore of interest to know whether Srs2 can block expansions of a TNR above the threshold, where expansions occur at a relatively high rate even in the wild type. We found that the absence of Srs2 further increased instability for (CTG)25. Expansion rates (Table 1) were approximately fivefold higher in the srs2::LEU2 and srs2::TRP1 mutants compared to the wild type, and the low-copy-number pSRS2 plasmid complemented the srs2::LEU2 mutation. Thus, Srs2 still provides protection against expansions for longer CTG tracts. Two other TNRs capable of forming hairpins, (CAG)25 and (CGG)25, showed 3.4- to 4.6-fold enhancement of expansion rates in an srs2 mutant, respectively (Table 1), suggesting that Srs2 suppresses expansions for at least three different TNR sequences. In contrast, srs2 had no effect on the non-hairpin-forming and genetically stable repeat (CTA)25 (see supplemental material at http://www.unmc.edu/Eppley/publications/chart_lah3.html). Similarly, there was no detectable increase in mutation rates for poly(GT) dinucleotide repeats (Table 1) or for forward mutations that inactivate the CAN1 gene (Table 1). Together, these results indicate a high degree of specificity for Srs2 on TNR sequences capable of hairpin formation.
TNRs also undergo contractions, in which repeating units are deleted. It is thought that contractions also occur via a hairpin-dependent mechanism, although a hairpin on the template strand is predicted to lead to contractions, not on the daughter strand, as for expansions. There was no significant effect of srs2 on contractions of a (CTG)25 reporter (37) (Table 1). Contractions of a shorter tract, (CTG)15 (37), were also essentially unaffected by the srs2 mutation (3.9 × 10−6 ± 1.2 × 10−6 versus 2.4 × 10−6 ± 0.6 × 10−6 for the wild type). These results suggest that Srs2 acts primarily on the daughter DNA strand, not the template strand, to block triplet repeat instability. Alternatively, the mechanisms of expansion and contraction might be somewhat different and therefore differentially sensitive to the loss of Srs2, even though hairpins likely mediate both types of mutation.
Srs2 helicase activity is important for preventing expansions.
Disease-causing TNRs form hairpins (11) that are likely precursors of expansions (12, 17, 45). Since Srs2 has helicase activity (2, 16, 19, 47), we hypothesized that Srs2 helps prevent expansions by unwinding hairpins. If so, the ATPase activity of Srs2 should be important for blocking expansions in vivo. We found that an srs2 point mutant lacking ATPase function, srs2K41A (19), gave a (CTG)13 expansion rate within about twofold of that of the null allele (Table 1). The expression levels of the wild-type and Srs2K41A proteins were similar (Fig. 2A), so the expansion defects in the K41A mutant cannot be attributed to low expression.
If Srs2 acts in vivo to unwind triplet repeat hairpins and prevent expansion, then perhaps the activity can be mimicked in vitro with double-stranded DNA containing CTG repeats within the duplex region (Fig. 3A and B). A DNA substrate was designed to test this idea (Fig. 3C), based on the following rationale. A single-stranded (CTG)13 repeat tract (the presumed intermediate during expansion in vivo) can fold into a hairpin with maximally five to six repeats on one side of the stem, a short loop, and then five to six repeats on the other side of the stem. Thus, a helicase substrate with five CTG repeats (Fig. 3C) is reasonable. Control experiments indicated that the molecule adopted the predicted structure (Fig. 3D). Srs2 helicase readily unwound the DNA substrate (Fig. 3E) in an ATP-dependent manner (data not shown). The fact that Srs2 was able to unwind duplex DNA containing CTG repeats supports the idea that Srs2 prevents expansions by unwinding TNR hairpins.
To address the specificity of helicase action, we compared Srs2 to UvrD from Escherichia coli. UvrD protein, also called DNA helicase II, was chosen for comparison because it has been well studied (29, 31, 32) and has the same 3′-to-5′ polarity (28) as Srs2. If Srs2 is especially active at unwinding TNR-containing substrates, then perhaps there would be a difference in helicase activity compared to UvrD. Figure 4A demonstrates the activity of both enzymes on a (CTG)10 partial duplex. Srs2 completely unwound the substrate by the 10-min time point, approximately twice the time required to unwind (CTG)5 (compare Fig. 4A and 3E). In contrast, bacterial UvrD proceeded slowly through the (CTG)10 substrate (Fig. 4A), requiring 20 min for full unwinding despite being assayed at a higher enzyme concentration (200 nM for UvrD versus 35 nM for Srs2). Both enzymes were active on a control substrate without repeats (Fig. 4B). The difference in UvrD's activity cannot be attributed to different duplex lengths, since the total duplex region in the two substrates was 39 and 40 bp long. The findings in Fig. 4 suggest that either the sequence or the structure or both of the (CTG)10 substrate presents a barrier to unwinding that can be overcome by Srs2 faster than by UvrD, another 3′-to-5′ helicase.
We also asked whether a related yeast helicase, Sgs1, could substitute for Srs2 in vivo. Sgs1 is a member of the RecQ family of DNA helicases with 3′-5′ polarity (16). SRS2 and SGS1 mutants share several phenotypes, but single mutants also show differences (16). Although sgs1 mutants show increased gross chromosomal rearrangements (34), an sgs1::kanMX4 strain in our assay gave a low expansion rate that was very close to the wild-type value (Table 1). Thus, sgs1 and srs2 mutants have different phenotypes for TNRs. In some circumstances, SGS1 in high copy number can suppress srs2 phenotypes (27). However, a multicopy plasmid containing SGS1 (27) failed to suppress the expansion defect conferred by srs2::LEU2 (Table 1). Our results that SRS2 and SGS1 behave differently at TNRs are in general agreement with those of White et al. (48), who found that an sgs1 mutation caused stabilization (not destabilization) of CGG arrays. These results suggest that Srs2 plays a unique role in stabilizing triplet repeat tracts that cannot be replaced by the functionally similar protein Sgs1.
DISCUSSION
We identified srs2 in a blind screen for mutations that result in high expansion rates. Subsequent analysis showed that CTG, CAG, and CGG repeats are destabilized in srs2 mutants. The srs2 mutator phenotype is specific for TNR expansions, as there was no detectable increase in srs2 backgrounds for TNR contractions, dinucleotide repeat alterations, or mutations in unique DNA sequences. Epistasis analysis indicated that the srs2 expansion phenotype was mostly associated with DNA polymerase delta, with only a fraction of the expansion rate in srs2 mutants attributable to recombination. Expansion sizes in an srs2 mutant were also consistent with a replicational source. The helicase activity of Srs2 is important for preventing expansions in vivo (although the related helicase Sgs1 cannot compensate for loss of Srs2), and purified Srs2 is substantially faster than bacterial UvrD helicase at unwinding a DNA substrate that mimics a TNR hairpin. Together these results indicate a novel, specific, and direct role for Srs2 in preventing TNR expansions.
What molecular features might account for the selective inhibition of TNR expansions by Srs2? We suggest that Srs2 acts in vivo to unwind most TNR hairpins before they become fixed as expansions. The unwound hairpin might either reanneal in register with the template strand or become a substrate for nuclease cleavage and thereby remove the excess DNA prior to completion of the expansion process. This idea helps explain why srs2 strains are mutators only at TNRs, which are unique among microsatellites in using a hairpin-based mechanism to expand in large increments. Other repeating elements such as dinucleotides change length primarily one or two repeats at a time, due to replication slippage (41), and thus the small loop-out intermediates would not present a target for Srs2 helicase action. To explain the stabilization of TNR expansions but not contractions, it is possible that Srs2 may not have access to hairpins on the template strand or that loading of the helicase may be precluded. If true, contractions could occur regardless of Srs2 status.
The final issue is the TNR length dependence of the srs2 phenotype. While both 13-repeat and 25-repeat tracts are protected by Srs2, a greater protective effect is seen for the shorter allele. Perhaps Srs2 helicase is more active on shorter hairpins, but longer hairpins are not unwound as readily. Helicase assays showed that longer times are required for Srs2 unwinding of 10-repeat versus 5-repeat substrates. Alternatively, the protective function of Srs2 may be reduced for the (CTG)25 tract because it is already relatively unstable. These possibilities will be addressed in future work.
Our results also suggest that Srs2 helicase prevents expansions primarily in conjunction with DNA polymerase delta, since the srs2K41A mutant, the pol32Δ mutant, and the srs2 pol32Δ double mutant all gave high (CTG)13 expansion rates within twofold of the srs2 value. Strains bearing the srs2 or pol32Δ mutation also showed a similar dependence on repeat tract length; for (CTG)25, a smaller increase in expansion phenotype was seen for srs2 (Table 1) and no significant effect was seen in pol32Δ strains (36). Polymerase delta is essential for replication, but it also acts during recombination and in some repair pathways (5). Recombination is not a major source of TNR instability in our system, as the loss of RAD51 or RAD52 functions in srs2 backgrounds reduced the expansion rate by 3.5-fold or less compared to an srs2 single mutant. Srs2 and polymerase delta must work primarily through replication and/or repair to inhibit expansions. A role for Srs2 in DNA replication has been suggested previously (21), and expansion sizes in an srs2 mutant are consistent with replication-mediated events (12). We are not aware of any reports in the literature testing whether repeat replication is compromised in srs2 mutants, but this would be a very interesting question to address.
Several models consistent with our epistasis results can be envisioned for inhibiting expansions. The first is a direct protein-protein interaction of Srs2 and polymerase delta. For example, the polymerase might stall when it synthesizes a TNR tract that subsequently folds into a hairpin and then recruit Srs2 for unwinding. A putative Srs2-polymerase delta complex might help explain the circa twofold reduction in expansion rates for srs2K41A compared to the srs2 null mutant. This difference allows for the possibility that there is an ATPase-independent component for inhibiting expansions that can be conferred by the point mutant protein (perhaps via protein-protein contacts). Our viewpoint is that the large majority (17-fold) of the srs2 effect is ATPase dependent. If a direct physical interaction between Srs2 and polymerase delta is necessary, however, our results with overexpression of Srs2 in a pol32Δ strain indicate that Pol32 cannot be the only crucial protein-protein contact (keeping in mind the caveat regarding overexpression).
A second model is that Srs2 and Pol32 inhibit expansions via the same pathway but without direct contact. Model two is in closer agreement with the Srs2 overexpression study. A third possibility is that expansions arise by a more indirect mechanism, such as defects in checkpoint response associated with loss of Srs2 (23, 46). If true, perhaps Srs2 is involved in a specialized repair process that responds to replicational stress (23), which in the case of triplet repeats could be replication fork stalling. Model three does not seem to be mutually exclusive with the first two models.
In srs2 rad52 strains, the propensity to expand is similar to that of an srs2 single mutant. Also, rad51 and rad52 single mutants show no specific phenotype in our assay. Both findings are consistent with our interpretation that recombination plays only a minor role in expansions in our system. In contrast, the srs2 rad51 double mutant showed a larger (≈3.5-fold) reduction in expansion rates than the srs2 strains, suggesting some specialized effect of Rad51. Perhaps Rad51 sequesters or inhibits another protein that can partially substitute for Srs2. In rad51 srs2 mutants, this other factor might then act to avoid some expansions that would otherwise occur due to the absence of Srs2. The identity of this putative second factor is unknown. Alternatively, the absence of both Srs2 and Rad51 might result in alteration in repair pathway choice, leading to an effect on expansion rate. This alternate pathway remains hypothetical, however, until appropriate experiments can be done.
In summary, our results show that the Srs2 helicase acts through an important new mechanism to provide a selective and potent block to TNR expansions. These studies provide a paradigm for how one helicase, Srs2, can suppress expansions.
Acknowledgments
This work was supported by NIH grant GM61961, by a postdoctoral fellowship from the Huntington's Disease Society of America, and by NIH training grant T32 CA09746.
We thank Hannah Klein for providing strains and plasmids and for valuable comments on the manuscript, Patrick Sung for providing purified Srs2 protein, antibodies, and advice on helicase substrate design, Steve Matson for purified UvrD protein, Luis Marky for guidance on stabilizing the oligonucleotide partial duplex, Alan Morgan for providing an Sgs1-overexpressing plasmid, Sergei Mirkin for a pol32Δ strain, and Peter Burgers for anti-Rfc3 antiserum.
REFERENCES
- 1.Aboussekhra, A., R. Chanet, A. Adjiri, and F. Fabre. 1992. Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to prokaryotic RecA proteins. Mol. Cell. Biol. 12:3224-3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aboussekhra, A., R. Chanet, Z. Zgaga, C. Cassier-Chauvat, M. Heude, and F. Fabre. 1989. RADH, a gene of Saccharomyces cerevisiae encoding a putative DNA helicase involved in DNA repair. Characteristics of radH mutants and sequence of the gene. Nucleic Acids Res. 17:7211-7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilera, A., and H. L. Klein. 1989. Genetic and molecular analysis of recombination events in Saccharomyces cerevisiae occurring in the presence of the hyper-recombination mutation. hpr1. Genetics 122:503-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya, S., M. L. Rolfsmeier, M. J. Dixon, K. Wagoner, and R. S. Lahue. 2002. Identification of RTG2 as a modifier gene for CAG•CTG repeat instability in Saccharomyces cerevisiae. Genetics 162:579-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgers, P. M. J. 1998. Eukaryotic DNA polymerases in DNA replication and DNA repair. Chromosoma 107:218-227. [DOI] [PubMed] [Google Scholar]
- 6.Christianson, T. W., R. S. Sikorski, M. Dante, J. H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119-122. [DOI] [PubMed] [Google Scholar]
- 7.Cleary, J. D., and C. E. Pearson. 2003. The contribution of CIS-elements to disease-associated repeat instability: clinical and experimental evidence. Cytogenet. Genome Res. 100:25-55. [DOI] [PubMed] [Google Scholar]
- 8.Cummings, C. J., and H. Y. Zoghbi. 2000. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum. Mol. Genet. 9:909-916. [DOI] [PubMed] [Google Scholar]
- 9.Dong, Z., and M. Fasullo. 2003. Multiple recombination pathways for sister chromatid exchange in Saccharomyces cerevisiae: role of RAD1 and the RAD52 epistasis group genes. Nucleic Acids Res. 31:2576-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freudenreich, C. H., S. M. Kantrow, and V. A. Zakian. 1998. Expansion and length-dependent fragility of CTG repeats in yeast. Science 279:853-856. [DOI] [PubMed] [Google Scholar]
- 11.Gacy, A. M., G. Goellner, N. Juranic, S. Macura, and C. T. McMurray. 1995. Trinucleotide repeats that expand in human disease form hairpin structure in vitro. Cell 81:533-540. [DOI] [PubMed] [Google Scholar]
- 12.Gordenin, D. A., T. A. Kunkel, and M. A. Resnick. 1997. Repeat expansion—all in a flap? Nat. Genet. 16:116-118. [DOI] [PubMed] [Google Scholar]
- 13.Hegde, V., and H. Klein. 2000. Requirement for the SRS2 DNA helicase gene in non-homologous end joining in yeast. Nucleic Acids Res. 28:2779-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson, S. T., and T. D. Petes. 1992. Instability of simple sequence DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 12:2749-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang, M.-E., A. de Calignon, A. Nicolas, and F. Galibert. 2000. POL32, a subunit of the Saccharomyces cerevisiae DNA polymerase delta, defines a link between DNA replication and the mutagenic bypass repair pathway. Curr. Genet. 38:178-187. [DOI] [PubMed] [Google Scholar]
- 16.Klein, H. 2000. A radical solution to death. Nat. Genet. 25:132-134. [DOI] [PubMed] [Google Scholar]
- 17.Kovtun, I., G. Goellner, and C. T. McMurray. 2001. Structural features of trinucleotide repeats associated with DNA expansion. Biochem. Cell Biol. 79:325-336. [PubMed] [Google Scholar]
- 18.Kramer, B., W. Kramer, M. S. Williamson, and S. Fogel. 1989. Heteroduplex DNA correction in Saccharomyces cerevisiae is mismatch specific and requires functional PMS genes. Mol. Cell. Biol. 9:4432-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krejci, L., S. Van Komen, Y. Li, J. Villemain, M. S. Reddy, H. Klein, T. Ellenberger, and P. Sung. 2003. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423:305-309. [DOI] [PubMed] [Google Scholar]
- 20.Lea, D. E., and C. A. Coulson. 1948. The distribution of the number of mutants in bacterial populations. J. Genet. 49:264-284. [DOI] [PubMed] [Google Scholar]
- 21.Lee, S. K., R. E. Johnson, S. L. Yu, L. Prakash, and S. Prakash. 1999. Requirement of yeast SGS1 and SRS2 genes for replication and transcription. Science 286:2339-2342. [DOI] [PubMed] [Google Scholar]
- 22.Leeflang, E. P., S. Tavare, P. Marjoram, C. O. S. Neal, J. Srinidhi, M. E. MacDonald, M. de Young, N. S. Wexler, J. F. Gusella, and N. Arnheim. 1999. Analysis of germline mutation spectra at the Huntington's disease locus supports a mitotic mutation mechanism. Hum. Mol. Genet. 8:173-183. [DOI] [PubMed] [Google Scholar]
- 23.Liberi, G., I. Chiolo, A. Pellicioli, M. Lopes, P. Plevani, M. Muzi-Falconi, and M. Foiani. 2000. Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J. 19:5027-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, Y., and R. A. Bambara. 2003. Analysis of human flap endonuclease 1 mutants reveals a mechanism to prevent triplet repeat expansion. J. Biol. Chem. 278:3278-13739. [DOI] [PubMed] [Google Scholar]
- 25.Liu, Y., H. Zhang, J. Veeraraghavan, R. A. Bambara, and C. H. Freudenreich. 2004. Saccharomyces cerevisiae flap endonuclease I uses flap equilibration to maintain triplet repeat stability. Mol. Cell. Biol. 24:4049-4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manivasakam, P., S. C. Weber, J. McElver, and R. H. Schiestl. 1995. Micro-homology mediated PCR targeting in Saccharomyces cerevisiae. Nucleic Acids Res. 23:2799-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mankouri, H. W., T. J. Craig, and A. Morgan. 2002. SGS1 is a multicopy suppressor of srs2: functional overlap between DNA helicases. Nucleic Acids Res. 30:1103-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matson, S. W. 1986. Escherichia coli helicase II (uvrD gene product) translocates unidirectionally in a 3′ to 5′ direction. J. Biol. Chem. 261:10169-10175. [PubMed] [Google Scholar]
- 29.Matson, S. W., and K. A. Kaiser-Rogers. 1990. DNA helicases. Annu. Rev. Biochem. 59:289-329. [DOI] [PubMed] [Google Scholar]
- 30.Mechanic, L. E., Frankel, B. A., and Matson, S. W. 2000. Escherichia coli MutL loads DNA helicase II onto DNA. J. Biol. Chem. 275:38337-38346. [DOI] [PubMed] [Google Scholar]
- 31.Mechanic, L. E., Frankel, B. A., and Matson, S. W., M. C. Hall, and S. W. Matson. 1999. Escherichia coli DNA helicase II is active as a monomer. J. Biol. Chem. 274:12488-12498. [DOI] [PubMed] [Google Scholar]
- 32.Mechanic, L. E., M. E. Latta, and S. W. Matson. 1999. A region near the C-terminal end of Escherichia coli DNA helicase II is required for single-stranded DNA binding. J. Bacteriol. 181:2519-2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miret, J. J., L. Pessoa-Brandao, and R. S. Lahue. 1998. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 95:12438-12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myung, K., A. Datta, C. Chen, and R. D. Kolodner. 2001. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat. Genet. 27:113-116. [DOI] [PubMed] [Google Scholar]
- 35.Paulson, H. L., and K. H. Fischbeck. 1996. Trinucleotide repeats in neurogenetic disorders. Annu. Rev. Neurosci. 19:79-107. [DOI] [PubMed] [Google Scholar]
- 36.Pelletier, R., A. S. Krasilnikova, G. M. Samadishwily, R. S. Lahue, and S. M. Mirkin. 2003. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 23:1349-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rolfsmeier, M. L., M. J. Dixon, L. Pessoa-Brandao, R. Pelletier, J. J. Miret, and R. S. Lahue. 2001. Cis-elements governing trinucleotide repeat instability in Saccharomyces cerevisiae. Genetics 157:1569-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rolfsmeier, M. L., and R. S. Lahue. 2000. Stabilizing effects of interruptions on trinucleotide repeat expansions in Saccharomyces cerevisiae. Mol. Cell. Biol. 20:173-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose, M. D., F. Winston, and P. Hieter. 1988. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 40.Schweitzer, J. K., and D. M. Livingston. 1998. Expansions of CAG repeat tracts are frequent in a yeast mutant defective in Okazaki fragment maturation. Hum. Mol. Genet. 7:69-74. [DOI] [PubMed] [Google Scholar]
- 41.Sia, E. A., R. J. Kokoska, M. Dominska, P. Greenwell, and T. D. Petes. 1997. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol. 17:2851-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spiro, C., and C. T. McMurray. 2003. Nuclease-deficient FEN-1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability. Mol. Cell. Biol. 23:6063-6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spiro, C., R. Pelletier, M. L. Rolfsmeier, M. J. Dixon, R. S. Lahue, G. Gupta, M. S. Park, X. Chen, S. V. S. Mariappan, and C. T. McMurray. 1999. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 4:1079-1085. [DOI] [PubMed] [Google Scholar]
- 45.Usdin, K., and E. Grabczyk. 2000. DNA repeat expansions and human disease. Cell. Mol. Life Sci. 57:914-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaze, M. B., A. Pellicioli, S. E. Lee, G. Ira, G. Liberi, A. Arbel-Eden, M. Foiani, and J. E. Haber. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 10:373-385. [DOI] [PubMed] [Google Scholar]
- 47.Veaute, X., J. Jeusset, C. Soustelle, S. C. Kowalczykowski, E. Le Cam, and F. Fabre. 2003. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423:309-312. [DOI] [PubMed] [Google Scholar]
- 48.White, P. J., R. H. Borts, and M. C. Hirst. 1999. Stability of the human fragile X (CGG)n triplet repeat array in Saccharomyces cerevisiae deficient in aspects of DNA metabolism. Mol. Cell. Biol. 19:5675-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]