

Abstract
Background
Nearly one-third of serous ovarian cancer (OVCA) patients will not respond to initial treatment with surgery and chemotherapy and die within one year of diagnosis. If patients who are unlikely to respond to current standard therapy can be identified up front, enhanced tumor analyses and treatment regimens could potentially be offered. Using the Cancer Genome Atlas (TCGA) serous OVCA database, we previously identified a robust molecular signature of 422-genes associated with chemo-response. Our objective was to test whether this signature is an accurate and sensitive predictor of chemo-response in serous OVCA.
Methods
We first constructed prediction models to predict chemo-response using our previously described 422-gene signature that was associated with response to treatment in serous OVCA. Performance of all prediction models were measured with area under the curves (AUCs, a measure of the model’s accuracy) and their respective confidence intervals (CIs). To optimize the prediction process, we determined which elements of the signature most contributed to chemo-response prediction. All prediction models were replicated and validated using six publicly available independent gene expression datasets.
Results
The 422-gene signature prediction models predicted chemo-response with AUCs of ~70 %. Optimization of prediction models identified the 34 most important genes in chemo-response prediction. These 34-gene models had improved performance, with AUCs approaching 80 %. Both 422-gene and 34-gene prediction models were replicated and validated in six independent datasets.
Conclusions
These prediction models serve as the foundation for the future development and implementation of a diagnostic tool to predict response to chemotherapy for serous OVCA patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-016-0548-9) contains supplementary material, which is available to authorized users.
Keywords: Ovarian cancer, Chemo-response, Prediction model, Data integration, Individualized treatment
Background
Epithelial ovarian cancer (OVCA) has the highest mortality rate of all gynecologic cancers [1]. The most common histological subtype of OVCA is serous [2]. The majority of patients present with advanced disease at diagnosis and, while some benefit from a treatment combining cytoreductive surgery and chemotherapy [3], nearly a third of patients with serous OVCA will not respond to this initial treatment and die from disease within one year after diagnosis [1, 4]. Despite significant research directed at understanding the biology of OVCA [5, 6], outcomes remain poor for a majority of patients, particularly those who do not respond to initial chemotherapy. A major limitation is the lack of validated biomarkers that can effectively predict response to chemotherapy [7, 8].
Previous attempts to define predictors of response to treatment have been limited by number of patients included, mixture of histological types and stages, and lack of validation in independent sets [9, 10]. In contrast, breast cancer gene signatures have been identified that can accurately predict recurrence [11] and chemotherapeutic response [12, 13]. These signatures were subsequently validated in independent clinical studies [13–15]. For example, one of these signatures, OncotypeDx, used 600 cases to create an association model and validated it in an additional 400 cases [11, 12]. Currently, there is no similar clinically available test for OVCA to identify which patients will respond to initial treatment [16].
In recently published studies using the Cancer Genome Atlas (TCGA) serous OVCA database [17], we identified a robust molecular signature associated with chemo-response by integrating publicly available biological and clinical data from 450 serous OVCA patients. This yielded a 422-gene molecular signature that was replicated in five independent gene expression experiments [18]. The contributing data used to identify this signature included gene expression, gene copy number alteration, gene mutations, DNA methylation, and miRNA profiles, all of which are available in TCGA dataset for serous OVCA. The presence of a strong association between the 422-gene signature and chemo-response from our previous work, though, does not imply that the signature also is predictive of chemo-response [9].
Therefore, the main objective of the present study was to determine the performance of the 422-gene signature as a predictor of chemo-response in serous OVCA. We also optimized and determined which of the elements of the signature contributed more to all prediction models. In this process, we identified a smaller set of 34 genes (the “optimized” set) from the original 422 signature that are predictive of response and that replicated the area under the curve (AUC) of the original complete gene set. Our data demonstrate that both the complete and the optimized models are predictive of outcome and are now replicated and validated in independent datasets.
Methods
Patients and data collection for prediction model
All data collection and processing, including the consenting process, were performed after approval by all local institutional review boards and in accord with the TCGA Human Subjects Protection and Data Access Policies, adopted by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI).
Patients with serous OVCA in TCGA were utilized to create a prediction model in the testing dataset, and were divided into two categories: complete responders (CR) and incomplete responders (IR). Clinical complete response (CR) was defined as progression-free survival 6 months after the first platinum-based treatment. In patients with incomplete response (IR), the disease either not did respond or progressed during treatment (refractory), or recurred within 6 months of treatment completion (resistant) [4, 19]. Patients defined as IR in our study are also clinically referred to as ‘platinum-resistant’ [20], with direct implications for treatment and prognosis. In the TCGA dataset, there were 292 patients classified as CR and 158 classified as IR. Table 1 describes the clinical characteristics of these patients. Chemo-response was the most significant prognostic factor for survival in multivariable analysis by Cox proportional hazards regression (p-value < 10−14), and patients with IR had a significantly decreased median survival compared to CR patients (Fig. 1) [18].
Table 1.
Clinical data from TCGA patients
| CR | IR | p-value* | |
|---|---|---|---|
| Number of Patients | 292 | 158 | |
| Age (Avg.) | 60 | 59.6 | N.S. |
| Grade | N.S. | ||
| Grade 1 | 4 | 1 | |
| Grade 2 | 35 | 18 | |
| Grade 3 | 246 | 135 | |
| Stage | p < 0.01 | ||
| Stage I | 10 | 3 | |
| Stage II | 19 | 1 | |
| Stage III | 224 | 123 | |
| Stage IV | 39 | 29 | |
| Surgical outcome | N.S. | ||
| Optimal (<1 cm residual) | 207 | 92 | |
| Suboptimal (>1 cm residual) | 52 | 57 | |
| Optimal Treatment | p < 0.001 | ||
| Optimal (Surgery + 6 cycles) | 179 | 66 | |
| Suboptimal | 113 | 92 |
*Multivariable analysis of TCGA clinical variables: Only FIGO stage and optimal treatment (including optimal surgery AND 6 cycles of platinum-based chemotherapy) were independently associated with chemo-response in serous OVCA
Fig. 1.
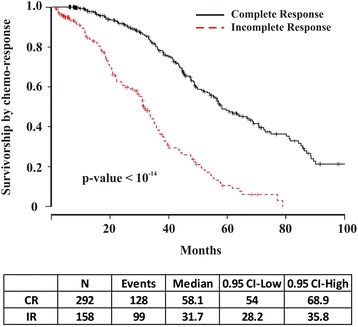
Survivorship by chemo-response in serous OVCA TCGA data. Chemo-response was the most significant factor in the multivariable analysis for survival. Complete responders (CR) have a median survival 2 years greater than IR
Gene signature and prediction analysis
We previously identified a 422-gene signature that is robustly associated with chemo-response [18]. To assess predictive performance of this signature, we applied the ‘Classification for MicroArrays’ (CMA) to TCGA serous OVCA data. CMA is a statistical tool designed to construct and evaluate classifiers (or prediction models) derived from microarray experiments using a large number of standard methods [21] and the R environment for statistical computing (www.r-project.org) [22].
Of the different methods available in the CMA package [21] to perform the analysis, nine methods consistently handle missing values, lower number of samples, and compute AUCs without reporting any errors: random forest [23], least absolute shrinkage and selection operator (Lasso) [24], Elastic Net [24], prediction analysis for microarrays (PAM) [25], diagonal discriminant analysis [26], partial least squares (PLS) [27], PLS - random forest [27], penalized logistic regression [28], and PLS - logistic regression [27]. We used these nine methods for the rest of the study to compare the predictive performance of all of the different datasets and for both the complete and optimized models. Two other available methods, linear and quadratic schrinkage, could not compute AUC. Fisher’s discriminant analysis could not handle more variables than subjects; neural networks was unstable/difficult to tune and interpret; k-nearest neighbors and support vector machines could not tune and evaluate AUCs.
Initially, all 422 genes associated with chemo-response in serous ovarian cancer [18] were utilized to construct prediction models, termed 422-gene prediction models. To assess how accurately the groups (CR and IR) were predicted, and to avoid over-fitting, cross-validation was used (internal validation of the classifier) [29]. The predictive performance was computed with corrections for TCGA batch-effect and to account for two other variables independently associated with chemo-response in serous OVCA (FIGO stage classification and optimal treatment, Table 1) [10]. Sensitivity, specificity and AUC of the predictor/classifier were also calculated. For each of the AUC measurements, we also computed a 95 % confidence interval (CI) to compare different models and different methods of classification. To illustrate the performance of the predictor in classifying chemo-response, a receiver operating characteristic (ROC) curve was generated. These analyses also facilitated comparison of the performance of the predictor models across independent serous OVCA datasets and assessed how consistently the models predicted chemo-response in OVCA patients based on sensitivity, specificity, misclassification rate, and AUC. Finally, we identified which patients were more likely to be misclassified and the clinical characteristics that were associated with misclassification.
Selection of most informative genes of prediction models
We focused on the selection of informative genes, because the composition of prediction models is paramount for their performance [9]. The selection process was performed with all available methods in the software package: two-sample t-test; Welch modification of the t-test; Wilcoxon rank sum test; F-test; Kruskal-Wallis test; “moderated” t and F test, respectively, using the package ‘limma’ in R statistics; one-step Recursive Feature Elimination (RFE) in combination with the linear support vector machines (SVM); random forest variable importance measure; least absolute shrinkage and selection operator (or Lasso); the regularized regression method or elastic net; component-wise boosting; and ad-hoc “Golub” criterion [21]. Using the gene selection tool, each gene was ranked depending on its relative importance in prediction models. These genes were ordered based on their rank and their relative ‘weight’ in the prediction process, and the prediction model analysis was applied by including only those genes that had been ranked at least once (one ‘hit’) by each method. These models, containing only the 34 selected and more informative genes, were termed 34-gene prediction models and comprised the optimized gene set as compared to the complete gene set.
Data retrieval for replication and validation analyses
Validation and replication of the prediction models was performed using datasets in the Gene Expression Omnibus (GEO) and the European Bioinformatics Institute, part of the European Molecular Biology Laboratory (EMBL-EBI), that contain gene expression paired with treatment response data (Table 2). Databases were downloaded in their raw state to maximize platform and annotation information, and then data were normalized. Response to therapy variables were coded to make outcomes comparable with TCGA: CR and IR. Also, patients that underwent optimal debulking (with largest residual disease of <1 cm) and completed six cycles of platinum-based therapy were considered to have ‘optimal treatment’. Lesser treatments were considered suboptimal. This analysis was performed because optimal treatment and FIGO stage of disease were also significantly and independently associated with chemo-response in TCGA (Table 1). Both clinical variables were collected, when available, and assessed for association to chemo-response in these new datasets in order to account for them in the prediction analysis. Also, batch-effect, if available, was accounted for to correct for any bias, as was also performed in the initial prediction model using the TCGA dataset.
Table 2.
Publicly available GEO datasets of patients with serous OVCA used for validation/replication of prediction models
| Repositories | Number of patients | Study Names | References | |
|---|---|---|---|---|
| CR | IR | |||
| GEO accession number | ||||
|
GSE23554 GSE3149 |
90 | 37 | MCC | Marchion, 2011 [32] Bild, 2006 [33] |
| GSE9891 | 185 | 55 | Australia | Tothill, 2008 [34] |
| GSE28739 | 20 | 30 | Trinh | Trinh, 2011 [36] |
| GSE17260 | 93 | 17 | Yoshihara | Yoshihara, 2010 [37] |
| GSE30161 | 32 | 23 | Ferriss | Ferriss, 2012 [35] |
| EMBL-EBI accession number | ||||
| E-MTAB-386 | 64 | 41 | Bentink | Bentink, 2012 [38] |
Replication and validation analyses
Initially, we replicated the prediction analysis of all 422 genes associated with chemo-response, or the complete prediction model, in all independent databases to assess how accurately chemo-response was predicted. Cross-validation was used for internal validation of the classifier, and predictive performance was computed with the same methods described for the TCGA dataset. Sensitivity, specificity, and AUC were calculated. Databases that contained information on all variables were later used for validation of the predictor/classifier of both the 422-gene and the 34-gene prediction models.
Validation of the 422-gene prediction models from TCGA data (the training set) and independent datasets (testing sets) was performed using the ‘prediction’ tool of the CMA package. Only independent datasets with information on all variables were used as testing sets. Sensitivity, specificity and AUC were used to measure the performance of the prediction model. Validation of the optimized 34-gene prediction models, including only the 34 most informative genes, was also performed in those independent datasets with information on all variables, and the same measures were used to assess the performance of the classifier.
Software
Apart from the ‘Classification for MicroArrays’ (CMA) utilized for the prediction analysis and already described, other analyses (i.e., logistic regression, Cox regression, Kaplan-Meier survival estimation), were performed using R software for statistical computing and graphics and utilizing Bioconductor packages as open source software for bioinformatics (bioconductor.org). Differential gene expression analysis was performed using Biometric Research Branch (BRB) ArrayTools, an integrated package for visualization and statistical analysis that utilizes Excel (Microsoft, Redmond, WA) as a front end, and with tools developed in the R statistical system. BRB-ArrayTools were developed by Dr. Richard Simon and the BRB-ArrayTools development team. Associations of the 34 most informative genes with survival were estimated with a multivariate Cox regression model and reported as hazard-ratios along with 95 % confidence intervals, and likelihood ratio test was used to compare models of survival, also within R environment.
To identify biological processes and pathways over-represented in the selected group of genes, we performed pathway enrichment analyses with MetaCore 6.0 [30] (GeneGo Inc., MI), an integrated knowledge-based platform for pathway analysis of OMICs data and gene lists, and other R-based tools, such as clusterProfiler [31], which mines the KEGG database (Kyoto Encyclopedia of Genes and Genomes, www.genome.jp/kegg).
Results
422-gene prediction model with all signature genes
Initially, we performed the prediction analysis of chemo-response including all 422 genes of the signature, which we refer to herein as the 422-gene prediction models. Figure 2 summarizes the predictive performance of all 422-gene prediction models. Their performance resulted in AUCs ranging from 53 to 73 %. Predictive performances of the 422-gene prediction models were not explained by prediction models constructed only with FIGO stage classification and optimal treatment, the clinical variables that have been independently associated with chemo-response (mean AUC around 58 %, Table 3).
Fig. 2.
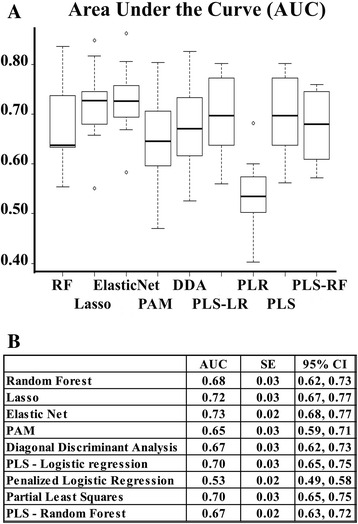
Area under the ROC curve (AUC) for the 422-gene prediction models. a Box plot representations of the AUC for the complete model by different methods. RF: Random forest; Lasso (least absolute shrinkage and selection operator); Elastic Net; PAM: Prediction analysis for microarrays; DDA: Diagonal discriminant analysis; PLS-LR: Partial least squares - Logistic regression; PLR: Penalized logistic regression; PLS: Partial least squares; PLS-RF: Partial least squares - Random forest. b Prediction performance measured in AUC, with their respective standard error, and confidence intervals (CI) by different methods using all 422 genes. PAM: Prediction analysis for microarrays; PLS: Partial least squares; Lasso: least absolute shrinkage and selection operator
Table 3.
AUCs and their CI comparing the 422-gene prediction model and clinical prediction models
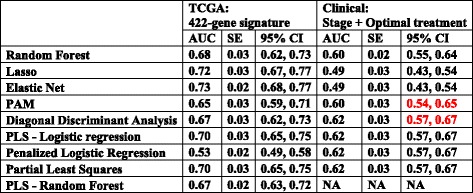
PAM: Prediction analysis for microarrays; PLS: Partial least squares; Lasso: least absolute shrinkage and selection operator; NA: not available (not computable)
CI of clinical prediction model WITH significant overlap with TCGA 422-gene prediction model CI: in red
Selection of the most informative genes and predictive performance of the 34-gene prediction model
The variable selection process identified 105 different genes to be relevant (i.e., at least one ‘hit’) in the prediction model. Of those, only 34 genes had more than 10 hits with all methods of gene selection (at least one hit per method): RHOT1, MYO7A, ZBTB10, MATK, ST18, RPS23, GCNT1, DROSHA, NUAK1, CCPG1, PDGFD, KLRAP1, MTAP, RNF13, THBS1, MLX, FAP, TIMP3, PRSS1, SLC7A11, PRSS2, OLFML3, RPS20, MCM5, POLE, STEAP4, LRRC8D, C10orf26 (WBP1L), ENTPD5, SYNE1, DPT, COPZ2, TRIO, and PDPR. These were considered to be the most relevant genes for the construction of the model. A new prediction for chemo-response was performed with these 34 genes, termed the optimized 34-gene prediction models, with corrections for batch-effect and clinical variables and using cross-validation (Fig. 3). By selecting only those genes that were most informative in prediction models and removing those that had little or no influence, the performance of the signature in terms of AUC increased across the board by 6–10 % regardless of the specific method.
Fig. 3.
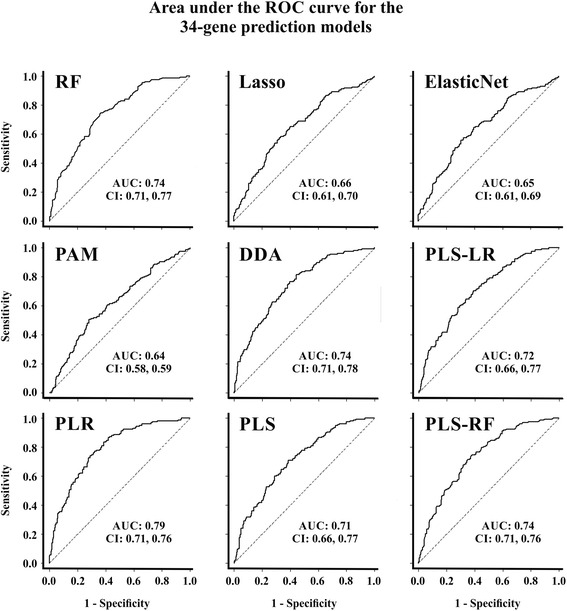
AUC for 34-gene predictions models. AUCs and CIs for the predicting methods using the most relevant genes in the prediction model/classifier: RF: Random forest; Lasso (least absolute shrinkage and selection operator); Elastic Net; PAM: Prediction analysis for microarrays; DDA: Diagonal discriminant analysis; PLS-LR: Partial least squares - Logistic regression; PLR: Penalized logistic regression; PLS: Partial least squares; PLS-RF: Partial least squares - Random forest
The 34 selected genes for the optimized prediction model were included in the initial 422-gene signature through different modes: 27 presented differential gene expression between CR and IR, 20 related to copy number variation between CR and IR (14 of them also had differential gene expression), five were correlated with genes with differential DNA methylation between CR and IR, and seven were correlated with miRNAs with different expression between CR and IR (Fig. 4). The relative chromosomal position of the 34 selected genes and the spatial distribution are displayed in Fig. 5 in a circular layout with matrix depiction of their relative expression (Table 4 has detailed information about these 34 genes). Multivariate analysis of survival using Cox proportional hazards regression identified that six of the 34 genes were independently associated with survival (Fig. 6).
Fig. 4.
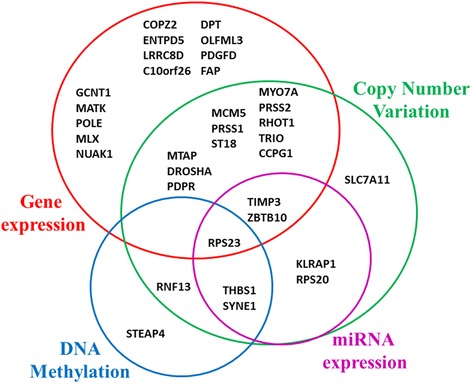
Origin of 34 genes selected in the optimized prediction model. Initially, genes were included in the 422-gene signature because of their differential gene expression (red), miRNA expression (pink), DNA methylation (blue), or copy number variation (green) between CR and IR. Some genes had more than one biological difference
Fig. 5.
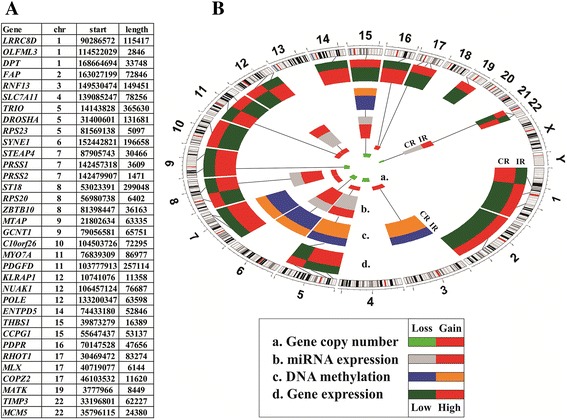
Genomic position of 34 genes selected for the optimized prediction model. a The 34 most informative genes from the prediction model and their chromosomal location: chr: number of the chromosome were the gene is located; start: of the gene position; length: of the gene in base-pairs (bp). The human genome version was hg19. b Circular layout with matrix depiction of different biological variables. From external to internal: Chromosome bands: circular representation of all chromosomes (centromere is in red); d Differential gene expression between incomplete and complete responders (CR/IR; red is over-expressed, green is under-expressed); c Differential DNA methylation between CR and IR (CR/IR; blue is hypomethylated, orange is hyper methylated); b Differential miRNA expression between CR and IR (CR/IR; red is over-expressed, grey is under-expressed); a Gene copy number variation between CR and IR (copy gain is red, green is copy loss). The order of genes in a is the same as in b. Lines represent correlations between different biological variables (for more details see Table 4)
Table 4.
Genomic information and reason for inclusion in the original 422-gene signature for the 34 genes selected in the prediction model
| Annotation | Expression | Copy number | DNA Methylation | miRNA expression | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Symbol | Name | Entrez-ID | chr | Start | Length | Fold-change (CR/IR) | Presence | cytoband | Methylated Genes | Methylation Status (CR/IR) | miRNA | CR/IR expression |
| LRRC8D | leucine rich repeat containing 8 family, member D | 55144 | 1 | 90286572 | 115417 | 1.13 | ||||||
| OLFML3 | olfactomedin-like 3 | 56944 | 1 | 114522029 | 2846 | 0.76 | ||||||
| DPT | dermatopontin | 1805 | 1 | 168664694 | 33748 | 0.9 | ||||||
| FAP | fibroblast activation protein, alpha | 2191 | 2 | 163027199 | 72846 | 0.7 | ||||||
| RNF13 | ring finger protein 13 | 11342 | 3 | 149530474 | 149451 | Gain | 3q22.1-q29 | CRKRS | 1.19 | |||
| SLC7A11 | solute carrier family 7, (cationic amino acid transporter, y + system) member 11 | 23657 | 4 | 139085247 | 78256 | Loss | 4q13.3-q35.2 | |||||
| TRIO | triple functional domain (PTPRF interacting) | 7204 | 5 | 14143828 | 365630 | 1.18 | Gain | 5p15.33-p13.1 | ||||
| DROSHA | drosha, ribonuclease type III | 29102 | 5 | 31400601 | 131681 | 1.16 | Gain | 5p15.33-p13.1 | ||||
| RPS23 | ribosomal protein S23 | 6228 | 5 | 81569138 | 5097 | 0.96 | Loss | 5q11.2-q21.1 | UNQ9217 | 1.15 | miR-22 | 0.8 |
| SYNE1 | spectrin repeat containing, nuclear envelope 1 | 23345 | 6 | 152442821 | 196658 | Loss | 6q15-q27 | LAD1, NFATC2, SLC1A2, STEAP4 | 0.77 | miR-22, miR-200b | 1.22 | |
| STEAP4 | STEAP family member 4 | 79689 | 7 | 87905743 | 30466 | SYNE1 | 0.82 | |||||
| PRSS1 | protease, serine, 1 (trypsin 1) | 5644 | 7 | 142457318 | 3609 | 1.32 | Gain | 7q32.1-q36.3 | ||||
| PRSS2 | protease, serine, 2 (trypsin 2) | 5645 | 7 | 142479907 | 1471 | 1.34 | Gain | 7q32.1-q36.3 | ||||
| ST18 | suppression of tumorigenicity 18 (breast carcinoma) (zinc finger protein) | 9705 | 8 | 53023391 | 299048 | 0.97 | Gain | 8p11.21-q24.3 | ||||
| RPS20 | ribosomal protein S20 | 6224 | 8 | 56980738 | 6402 | Gain | 8p11.21-q24.3 | miR-135b | 1.22 | |||
| ZBTB10 | zinc finger and BTB domain containing 10 | 65986 | 8 | 81398447 | 36163 | 1.2 | Gain | 8p11.21-q24.3 | miR-708 | 1.2 | ||
| MTAP | methylthioadenosine phosphorylase | 4507 | 9 | 21802634 | 63335 | 1.12 | Loss | 9p21.3-p21.2 | ||||
| GCNT1 | glucosaminyl (N-acetyl) transferase 1, core 2 | 2650 | 9 | 79056581 | 65751 | 1.17 | ||||||
| C10orf26 | WBP1L - chromosome 10 open reading frame 26 | 54838 | 10 | 104503726 | 72295 | 0.87 | No | |||||
| MYO7A | myosin VIIA | 4647 | 11 | 76839309 | 86977 | 1.08 | Gain | 11q13.5-q14.1 | ||||
| PDGFD | platelet derived growth factor D | 80310 | 11 | 103777913 | 257114 | 0.76 | No | |||||
| KLRAP1 | killer cell lectin-like receptor subfamily A pseudogene 1 | 10748 | 12 | 10741076 | 11358 | Gain | 12p13.33-p11.21 | miR-22 | 0.8 | |||
| NUAK1 | NUAK family, SNF1-like kinase, 1 | 9891 | 12 | 106457124 | 76687 | 0.75 | No | |||||
| POLE | polymerase (DNA directed), epsilon | 5426 | 12 | 133200347 | 63598 | 1.14 | No | |||||
| ENTPD5 | ectonucleoside triphosphate diphosphohydrolase 5 | 957 | 14 | 74433180 | 52846 | 1.13 | No | |||||
| THBS1 | thrombospondin 1 | 7057 | 15 | 39873279 | 16389 | Loss | 15q11.1-q21.1 | CORO6, LAD1, NFATC2, SLC1A2, SNAI1 | 0.74 | miR-22, miR-641, miR-200b | 1.22 | |
| CCPG1 | cell cycle progression 1 | 9236 | 15 | 55647437 | 53137 | 0.84 | Loss | 15q21.3 | ||||
| PDPR | pyruvate dehydrogenase phosphatase regulatory subunit | 55066 | 16 | 70147528 | 47656 | 1.04 | Loss | 16q12.2-q24.3 | ||||
| RHOT1 | ras homolog gene family, member T1 | 55288 | 17 | 30469472 | 83274 | 0.87 | Gain | 17p13.3-q21.2 | ||||
| MLX | MAX-like protein X | 6945 | 17 | 40719077 | 6144 | 0.88 | No | |||||
| COPZ2 | coatomer protein complex, subunit zeta 2 | 51226 | 17 | 46103532 | 11620 | 0.78 | No | |||||
| MATK | megakaryocyte-associated tyrosine kinase | 4145 | 19 | 3777966 | 8449 | 0.93 | No | |||||
| TIMP3 | TIMP metallopeptidase inhibitor 3 | 7078 | 22 | 33196801 | 62227 | 0.7 | Loss | 22q11.22 | miR-22 | 0.8 | ||
| MCM5 | minichromosome maintenance complex component 5 | 4174 | 22 | 35796115 | 24380 | 1.19 | Loss | 22q11.22-q13.33 | ||||
In the Expression, Copy number, DNA methylation, and miRNA expression, only those with significant differential values between CR and IR were represented. Some genes had more than one biological difference
Copy number shows the chromosomal region (cytoband) that was significantly correlated with gene expression in the 422-gene signature. DNA Methylation and miRNA expression shows the initial variables that were significantly correlated to gene expression in the 422-gene signature
Fig. 6.
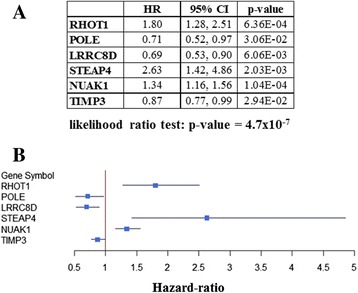
Multivariate survival analysis of the 34 selected genes. a Table with hazard-ratio (HR) or risk of death, with 95 % CIs and p-values for each of the genes independently associated with survival. b Forest plot of independently significant genes for survival. Blue boxes represent hazard-ratios (HR), and lines are their CIs. HR = 1 is non-significant. HR < 1 has decreased risk of death; HR > 1 has increased risk of death. Overall p-value of the survival model: p = 5.7×10−7
Pathway enrichment analysis using both GeneGo and clusterProfiler revealed that the 34 genes are particularly relevant to protein absorption and metabolism and DNA repair and replication (Table 5). Also, cytoskeletal remodeling and cell adhesion functions were significantly represented in GeneGo (Table 5), consistent with pathways that are frequently implicated in response to therapy and disease progression.
Table 5.
Pathway enrichment analysis of the selected 34 genes constituting the simplified prediction model
| Cluster Profiler pathway enrichment analysis | ||||
| KEGG ID | Description | Adjusted p-value |
FDR q-value |
Gene IDs |
| hsa03030 | DNA replication | 2.07E-02 | 7.28E-03 | POLE/MCM5 |
| hsa04974 | Protein digestion and absorption | 3.09E-02 | 1.08E-02 | PRSS1/PRSS2 |
| hsa03010 | Ribosome | 3.09E-02 | 1.08E-02 | RPS23/RPS20 |
| hsa00240 | Pyrimidine metabolism | 3.09E-02 | 1.08E-02 | POLE/ENTPD5 |
| hsa04972 | Pancreatic secretion | 3.09E-02 | 1.08E-02 | PRSS1/PRSS2 |
| GeneGO pathway enrichment analysis | ||||
| # | Description | Adjusted p-value |
FDR q-value |
Gene IDs |
| 1 | Cell adhesion_Chemokines and adhesion | 8.45E-03 | 9.91E-02 | Thrombospondin 1, TRIO |
| 2 | Immune response_IL-12 signaling pathway | 3.21E-02 | 9.91E-02 | G6NT |
| 3 | Cytoskeleton remodeling_Role of PDGFs in cell migration | 3.34E-02 | 9.91E-02 | PDGF-D |
| 4 | Triacylglycerol metabolism p.2 | 3.89E-02 | 9.91E-02 | CEL |
| 5 | Development_Thrombospondin-1 signaling | 3.89E-02 | 9.91E-02 | Thrombospondin 1 |
| 6 | Cell cycle_Start of DNA replication in early S phase | 4.43E-02 | 9.91E-02 | MCM5 |
| 7 | Role of Tissue factor in cancer independent of coagulation protease signaling | 4.84E-02 | 9.91E-02 | Thrombospondin 1 |
Replication of prediction models in independent datasets
After downloading the databases detailed in Table 2, only cases of serous OVCA were selected, and the outcome of interest (chemo-response) was coded to make outcomes comparable with the initial TCGA data set: CR and IR. Initially, we replicated the 422-gene prediction models with cross-validation to avoid over-fitting. Performance of prediction models were measured with AUC (Table 6). AUCs of the prediction models in the independent validation datasets were comparable with the testing set from the TCGA, with CIs overlapping the testing set for almost all methods that contained information from the full 422-gene signature (marked red in Table 6) [32–35].
Table 6.
Replication of 422-gene prediction models with different methods
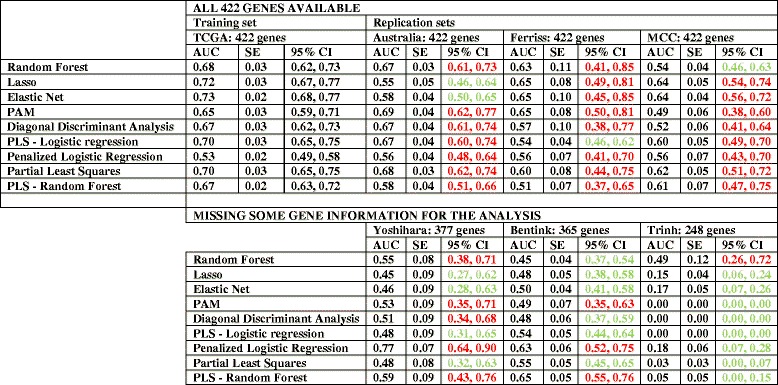
The table presents the results taking in consideration: 1) whether they were part of the training or replication set; 2) the number of genes available for analysis: on top analyses including all 422 genes; on the bottom analyses where all genes were not available (and the number included)
CI of replication datasets with WIDE overlap with TCGA testing set CI: in red. CI of replication datasets with NO overlap with TCGA testing set CI: in green. AUC: area under the ROC curve. CI: confidence intervals. SE: Standard Error. PAM: Prediction analysis for microarrays; PLS: Partial least squares; Lasso: least absolute shrinkage and selection operator
Unfortunately, all genes were not available in all platforms from the independent datasets, and three independent datasets had incomplete gene information [36–38]. The performance of these incomplete datasets was inferior to the TCGA classifier, with AUCs closer to 50 % (marked green in Table 6; see also Additional file 1: Table S1 for more details about classifier performance in relation to sensitivity, specificity, and standard errors in all available databases). Thus, the incomplete information in some of the replication sets provided insight into how well prediction models performed when some of the genes were not available.
Validation of prediction models in independent datasets
Validation of 422-gene prediction models were performed using TCGA data as the training set and independent datasets that contained complete information for all 422-genes as the testing sets. AUC and their CIs were used to validate the performance of the TCGA classifier in the three independent datasets with all gene expression information (Table 7). CIs of the AUC in the three different validation sets overlapped with the majority of the CIs of TCGA testing set (marked red in Table 7), validating the use of the 422-gene signature to predict chemo-response.
Table 7.
Validation of 422-gene prediction models in independent databases
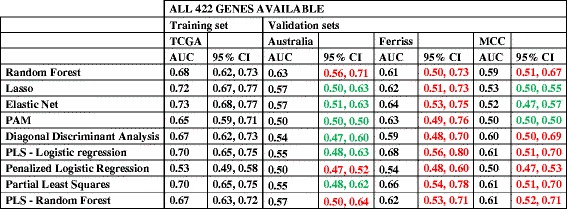
The table presents the results taking into consideration whether they were part of the training or validation set
CI of validation datasets WITH overlap with TCGA testing set CI: in red. CI of validation datasets with NO overlap with TCGA testing set CI: in green. AUC: area under the ROC curve. CI: confidence intervals. SE: Standard Error. PAM: Prediction analysis for microarrays; PLS: Partial least squares; Lasso: least absolute shrinkage and selection operator
Next, we validated TCGA 34-gene prediction models in the same three independent datasets with all gene information, that also included all the most informative 34 genes selected from the training set. Analysis of AUCs and CIs demonstrated that the optimized model performed very well in these validations sets, with AUC values ranging from 56 to 73 % and overlap of the majority of the CIs with the testing test (in red in Table 8). However, the CIs were wider in the validation set than in the testing set. For details about prediction models performance in relation to sensitivity, specificity, and standard deviation in all available databases see Additional file 2: Table S2.
Table 8.
Validation of optimized 34-gene prediction models
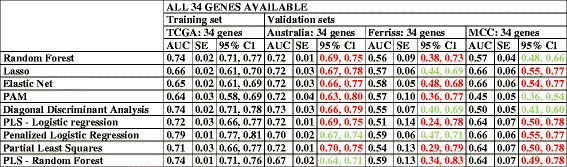
The table presents the results taking into consideration whether they were part of the training or validation set
CI of validation datasets WITH overlap with TCGA testing set CI: in red. CI of validation datasets with NO overlap with TCGA testing set CI: in green. AUC: area under the ROC curve. CI: confidence intervals. SE: Standard Error. PLS: Partial least squares
Clinical characteristics associated with misclassification in prediction models
Additional file 1: Tables S1 and Additional file 2: Table S2 list the misclassification rate for each of the prediction models in all analyzed datasets. To assess whether the prediction models could be further improved, we excluded those patients at high risk of misclassification and the clinical characteristics that are associated with this misclassification. First, we identified those samples that missed the true classification (either CR or IR) more than 20 % of the time on average: 158 of 450 patients in TCGA dataset. Next, we examined which baseline clinic characteristics (e.g., grade, stage, optimal debulking) were independently associated with misclassification using a multivariable regression analysis. The only independent variable associated with misclassification was ‘optimal treatment’ (p < 0.001), defined as treatment with optimal cytoreductive surgery and 6 cycles of platinum-based chemotherapy. Unfortunately, TCGA was not designed to study chemo-response, and other variables that may have affected the delivery of an optimal treatment were not collected and thus could not be included in the prediction models.
Discussion
Initial response to chemotherapy remains one of the most significant prognostic markers for serous OVCA patients. Identifying patients at high risk of chemoresistance early in the course of treatment has the potential to significantly alter clinical management, such as performing in depth tumor sequencing, more frequent disease monitoring or exploration of additional therapeutic options. Previous studies have focused in prediction models that classify ovarian cancer patients in prognostic groups [9, 10, 17]. While this is very valuable to clinicians and patients to assess the severity of the disease, prognosticators have limited immediate clinical application and they may not translate into effective therapeutic strategies. The design of our study was aimed to have immediate effect on treatment decisions. Using data available in TCGA for serous OVCA, we previously identified a 422-gene signature that is associated with chemo-response [18]. However, in order to develop this signature into a clinically useful test, it was paramount to establish its predictive performance. Herein we report the performance and validation of the 422-gene and optimized 34-gene models using TCGA and six other datasets. Specifically, 422-gene prediction models in the testing dataset (TCGA) presented AUCs around 70 %, and 34-gene predictive models performed slightly better, with AUCs above 70 %. This represents a significant improvement over current clinical estimations of complete chemo-response, which are between 40 and 60 % [3], setting the stage for the development of the 34-gene predictive model as a test to identify patients with serous OVCA at high risk for treatment failure.
The bulk of OVCA research is directed at understanding the biology of the disease [5, 6] and defining subsequent treatment for those who do not respond or recur after initial chemotherapy [16]. By comparison, we have a very poor understanding of which patients are at risk of failing initial treatment, due in large part to a lack of validated biomarkers or molecular signatures that can effectively predict chemo-response [7, 8]. Previous attempts have been limited by the number of patients included in the study, analysis of a mixture of histological types and stages, and lack of validation in independent datasets [9, 10, 16]. Some of these prediction models included serum levels of CA125, a tumor-associated glycoprotein of unknown function that is used clinically to assess disease burden, though it has limited value in predicting chemo-response [9, 10, 39]. In the present study, we have created prediction models of chemo-response from a 422-gene signature comprising only serous cases, the most common type of OVCA. These prediction models not only had AUCs over 70 %, but were replicated and validated in the largest collection of independent serous OVCA databases available to date.
Conclusions
One of the major strengths of this study is the replication and validation of prediction models in large independent datasets containing gene expression specific to serous OVCA and information about response to chemotherapy. However, it is important to recognize the limitations inherent in the retrospective design of the databases used to create and validate the prediction models, especially since these databases were not originally intended to study chemo-response as a primary outcome variable. Although this clinical parameter was recorded in all databases, it might contain biases. To minimize this, we used a strict definition of chemo-response to segregate patients into CR and IR. Regardless of these limitations, we demonstrate replication and validation of both the 422-gene and the 34-gene models across multiple databases, with overlapping CIs. Of particular significance is the validation of the optimized 34-gene models given the variability in gene expression analysis among different datasets. This highlights the utility of this gene signature as a bona fide indicator of chemo-response.
Identification of characteristics independently associated with misclassification could be very helpful in improving prediction models. For example, if we identify that patients with BMI > 50 are more likely to be misclassified, we might exclude those patients from the prediction process to enhance the classification of the other patients. Also, based on the observations made with patients with optimal treatment, fine-tuning of the prediction models may be possible, and perhaps necessary. Researchers in cardiovascular disease have been at the forefront of developing risk prediction equations utilizing clinical risk factors for the prediction of cardiovascular events, as in the Framingham Study [40]. Recently, new biomarkers and nonclinical measures have been studied to improve these risk prediction tools in the population [41]. Adding new biomarkers to the classic clinical risk factors may improve the model and offer better prediction of the 15–20 % of patients with cardiovascular events that have no clinical risks before the episode [41]. In OVCA, we found that there was one variable associated with misclassification, ‘optimal treatment’. Variables associated with the adequate delivery of optimal treatment in OVCA (surgery and chemotherapy) have the potential to improve prediction models significantly [42]. Quantification and different metrics for optimal treatment delivery could be determined in a prospective set of patients with comprehensive clinical data collection in order to better understand which factors influence treatment outcome.
A caveat to our study is that datasets with incomplete data performed poorly, which was accentuated in models with 34 genes. For example, in the Yoshihara dataset [37], we only lost expression information for one of the seven more influential genes in the model. However, this gene (MATK) was ranked second in importance, so the model underperformed. The Bentink database [38] lost fewer genes that Yoshihara (12 versus 14) but two of them were seven of the most influential genes, and the performance was inferior to the Yoshihara database. Trihn databse [36] lost four of the seven most influential genes, including the first two (RHOT1 and MATK), with serious consequences for its performance. Although this loss of performance could be seen as a problem for the construction of our prediction models, it also underscores that the importance of the genes included in the model is proportional to its performance, thereby supporting the robust performance of the 34-gene prediction models. Therefore, we propose that these prediction models are very promising and are robust across different databases and classifiers. Nevertheless, additional validation analyses are necessary to test their utility as a clinical test for response to chemotherapy in OVCA.
As to the pathways associated with the 422-gene signature, the majority of the 34 selected genes included in the optimized prediction model are drawn from cellular functions previously associated with response to chemotherapy [8, 18]. Specifically, the pathway enrichment analysis identified pathways involved in DNA damage repair, replication, protein metabolism, cell cycle and apoptosis, as well as cytoskeletal remodeling and cell adhesion functions, all of which have been associated with cancer transformation and proliferation [43]. Several genes in our optimized model have potential implications relative to the etiology or treatment of serous OVCA. For example, mutations in DNA polymerase epsilon (POLE) have been reported in endometrial and colon cancers with elevated numbers of somatic mutations [44, 45], which appears to correlate with a robust intratumoral T-cell response and better prognosis [46]. To date, however, there have been no reports of POLE differential gene expression and its association with prognosis in serous OVCA. Decreased expression of GCNT1 [47], DROSHA [48] and increased expression of TIMP3 [49] have been associated with decreased PFS and OS in independent analyses of TCGA. Of note, we found that POLE, LRRC3D, and TIMP3 mutations were all associated with improved survival.
Other genes, like FAP, have been associated with clinical resistance to chemotherapy in patients with serous OVCA, which carries worse prognosis and survival [50]. Although there is no direct evidence of clinical association with progression of disease in serous ovarian cancer, there are in vitro studies that implicate NUAK1 (ARK5) with invasion and progression in ovarian cancer cell lines [51]. ARK5 expression has been found to be elevated in serous OVCA, and its expression has been associated with invasion and metastasis in cancer in general [51]. Supporting the role of NUAK1/ARK5 in the aggressive cancer phenotype, this gene was independently associated with decreased survival (HR = 1.34, 95 % CI 1.16–1.56).
We also identified two genes, thrombospondin I (THBS1) and PDGFD, which participate in a wide range of signaling pathways that regulate cellular processes involved in cancer genesis and progression [52]. Thrombospondin I is a secreted protein that associates with the extracellular matrix and possesses a variety of biologic functions, including having potent antiangiogenic activity. In OVCA, decreased production of thrombospondin I results in increased expression of vascular endothelial growth factor (VEGF) and other angiogenic factors such as P1GF, FGF-2 and PDGFB [53]. PDGFD, also a member of the platelet-derived growth factor (PDGF) family, participates in cytoskeletal remodeling and, through its role in cell migration, contributes to vascular integrity and function mediated by perycite coverage of vessels. This action could be disrupted in tumors [43]. Also, platelet levels during treatment and recruitment to tumors have been associated with chemo-response [54]. Taken together, these data may provide a molecular basis for the efficacy of bevacizumab, a humanized antibody against VEGF, which has been shown to improve progression-free survival and even overall survival in a subset of serous OVCA patients [55, 56], or the use of multitarget tyrosine kinase inhibitors to simultaneously block VEGFR, PDGFR and FGFR pathways [57].
Up to a third of the patients undergoing treatment for advanced serous OVCA do not respond to treatment and have a very poor prognosis. Indeed, more than 40 % of the patients in this poor prognosis group have had optimal therapy, with optimal cytoreductive surgery and at least six cycles of platinum-based chemotherapy, indicating that lack of response is not due to incomplete clinical treatment. Hence, for many such patients, the inherit characteristics of the tumor appear to play a major role in lack of responsiveness. We have created 34-gene prediction models/classifiers that consistently predict these patients approximately 80 % of the time. This prediction model is specific for patients diagnosed with serous OVCA. Our ultimate goal is to create a diagnostic tool that will predict each patient’s response to chemotherapy. This is important, because, based on the potential molecular targets identified in the 34-gene prediction models, we could design clinical trials that compare standard therapy versus standard therapy in combination with molecular agents targeting resistance pathways for the women who are unlikely to respond to the usual upfront regimens.
Acknowledgements
We would like to thank ‘TCGA Research Network’ for generating, curating and providing high quality biological and clinical data.
Availability of data and materials
This study was performed in silico, with de-identified publicly available data. All data from TCGA is available at their website: https://tcga-data.nci.nih.gov/tcga/. Data for GEO sets are also available at NCBI website: http://www.ncbi.nlm.nih.gov/gds/. Datasets can be browsed by their accession number: GSE23554, GSE3149, GSE9891, GSE28739, GSE17260, and GSE30161. Software utilized by this study is also publicly available at Bioconductor website: http://bioconductor.org/.
Authors’ contributions
JGB, KWT, KKL, TG, and BJC conceived of the project and contributed to study design; JGB, AMN, RCK and MJK performed data collection; JGB, TG, and BJS performed computational analyses; JGB, AMN, RKC, MJG, KWT, KKL, TG, and BJS analyzed and interpreted the data; JBG, KWT, KKL, TGS, and BJS wrote the manuscript with input from all authors; all authors read and approved the final version of the manuscript.
Competing interests
KWT is a co-owner of Immortagen, Inc. All other authors declare that they have no competing interests.
Consent for publication
All authors have reviewed and approved the manuscript for submission.
Ethics approval and consent to participate
All data collection and processing, including the consenting process, were performed after approval by all local institutional review boards and in accord with the TCGA Human Subjects Protection and Data Access Policies, adopted by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI).
Financial support
The research was supported in part by the National Institutes of Health (NIH R01 CA99908 to KKL and BJS), the “St. Louis Ovarian Cancer Awareness Research Grant” from the 2012–2013 Foundation for Women’s Cancer grants.
Abbreviations
- AUC
Area under the curves
- BRB
Biometric research branch
- CI
Confidence interval
- CMA
Classification for MicroArrays
- CR
Complete responders
- EMBL-EBI
European Bioinformatics Institute, part of the European Molecular Biology Laboratory
- FDR
False discovery rate
- FIGO
International Federation of Obstetrics and Gynecology
- IR
Incomplete responders
- GEO
Gene expression omnibus
- HR
Hazard-ratio
- KEGG database
Kyoto encyclopedia of genes and genomes
- Lasso
Least absolute shrinkage and selection operator
- miRNA
micro RNA
- NCI
National Cancer Institute
- NHGRI
National Human Genome Research Institute
- OVCA
Serous ovarian cancer
- PAM
Prediction analysis for microarrays
- PLS
Partial least squares
- RFE
Recursive feature elimination
- ROC
Receiver operating characteristic
- SVM
Support vector machines
- TCGA
The Cancer Genome Atlas
Additional files
AUCs, CI, sensitivity, specificity and misclassification rate of testing and validation of 422-gene prediction models. (DOCX 32 kb)
AUCs, CI, sensitivity, specificity and misclassification rate of testing and validation for 34-gene prediction models. (DOCX 25 kb)
References
- 1.American Cancer Society . Cancer Facts & Figures 2014. Atlanta: American Cancer Society; 2014. [Google Scholar]
- 2.Gilks CB, Prat J. Ovarian carcinoma pathology and genetics: recent advances. Hum Pathol. 2009;40:1213–23. doi: 10.1016/j.humpath.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 3.Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–9. [DOI] [PubMed]
- 4.Friedlander ML, Stockler MR, Butow P, King MT, McAlpine J, Tinker A, Ledermann JA. Clinical trials of palliative chemotherapy in platinum-resistant or -refractory ovarian cancer: time to think differently? J Clin Oncol. 2013;31:2362. doi: 10.1200/JCO.2012.47.7927. [DOI] [PubMed] [Google Scholar]
- 5.Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997;57:850–6. [PubMed] [Google Scholar]
- 6.Johnson SW, Swiggard PA, Handel LM, Brennan JM, Godwin AK, Ozols RF, Hamilton TC. Relationship between platinum-DNA adduct formation and removal and cisplatin cytotoxicity in cisplatin-sensitive and -resistant human ovarian cancer cells. Cancer Res. 1994;54:5911–6. [PubMed] [Google Scholar]
- 7.Colombo PE, Fabbro M, Theillet C, Bibeau F, Rouanet P, Ray-Coquard I. Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer. Crit Rev Oncol Hematol. 2013. [DOI] [PubMed]
- 8.Hiss D. Optimizing molecular-targeted therapies in ovarian cancer: the renewed surge of interest in ovarian cancer biomarkers and cell signaling pathways. J Oncol. 2012;2012:737981. doi: 10.1155/2012/737981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oikonomopoulou K, Li L, Zheng Y, Simon I, Wolfert RL, Valik D, Nekulova M, Simickova M, Frgala T, Diamandis EP. Prediction of ovarian cancer prognosis and response to chemotherapy by a serum-based multiparametric biomarker panel. Br J Cancer. 2008;99:1103–13. doi: 10.1038/sj.bjc.6604630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y, Katsaros D, Shan SJ, de la Longrais IR, Porpiglia M, Scorilas A, Kim NW, Wolfert RL, Simon I, Li L, et al. A multiparametric panel for ovarian cancer diagnosis, prognosis, and response to chemotherapy. Clin Cancer Res. 2007;13:6984–92. doi: 10.1158/1078-0432.CCR-07-1409. [DOI] [PubMed] [Google Scholar]
- 11.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 12.Wan YW, Qian Y, Rathnagiriswaran S, Castranova V, Guo NL. A breast cancer prognostic signature predicts clinical outcomes in multiple tumor types. Oncol Rep. 2010;24:489–94. doi: 10.3892/or_00000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rathnagiriswaran S, Wan YW, Abraham J, Castranova V, Qian Y, Guo NL. A population-based gene signature is predictive of breast cancer survival and chemoresponse. Int J Oncol. 2010;36:607–16. doi: 10.3892/ijo_00000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 15.van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 16.Lloyd KL, Cree IA, Savage RS. Prediction of resistance to chemotherapy in ovarian cancer: a systematic review. BMC Cancer. 2015;15:117. doi: 10.1186/s12885-015-1101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez Bosquet J, Marchion DC, Chon H, Lancaster JM, Chanock S. Analysis of chemotherapeutic response in ovarian cancers using publically available high-throughput data. Cancer Res. 2014. [DOI] [PubMed]
- 19.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 20.Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133:624–31. doi: 10.1016/j.ygyno.2014.02.038. [DOI] [PubMed] [Google Scholar]
- 21.Slawski M, Daumer M, Boulesteix AL. CMA: a comprehensive Bioconductor package for supervised classification with high dimensional data. BMC Bioinformatics. 2008;9:439. doi: 10.1186/1471-2105-9-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R: A Language and Environment for Statistical Computing [www.r-project.org/].
- 23.Chen X, Ishwaran H. Random forests for genomic data analysis. Genomics. 2012;99:323–9. [DOI] [PMC free article] [PubMed]
- 24.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed]
- 25.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–72. [DOI] [PMC free article] [PubMed]
- 26.McLachlan GJ. Confidence intervals for the conditional probability of misallocation in discriminant analysis. Biometrics. 1975;31:161–7. [PubMed]
- 27.Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst. 2007;99:147–57. [DOI] [PubMed]
- 28.Zhu J, Hastie T. Classification of gene microarrays by penalized logistic regression. Biostatistics. 2004;5:427–43. [DOI] [PubMed]
- 29.Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J Clin Oncol. 2005;23:7332–41. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- 30.Piruzian E, Bruskin S, Ishkin A, Abdeev R, Moshkovskii S, Melnik S, Nikolsky Y, Nikolskaya T. Integrated network analysis of transcriptomic and proteomic data in psoriasis. BMC Syst Biol. 2010;4:41. doi: 10.1186/1752-0509-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchion DC, Cottrill HM, Xiong Y, Chen N, Bicaku E, Fulp WJ, Bansal N, Chon HS, Stickles XB, Kamath SG, et al. BAD phosphorylation determines ovarian cancer chemosensitivity and patient survival. Clin Cancer Res. 2011;17:6356–66. doi: 10.1158/1078-0432.CCR-11-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 34.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 35.Ferriss JS, Kim Y, Duska L, Birrer M, Levine DA, Moskaluk C, Theodorescu D, Lee JK. Multi-gene expression predictors of single drug responses to adjuvant chemotherapy in ovarian carcinoma: predicting platinum resistance. PLoS One. 2012;7:e30550. doi: 10.1371/journal.pone.0030550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trinh XB, Tjalma WA, Dirix LY, Vermeulen PB, Peeters DJ, Bachvarov D, Plante M, Berns EM, Helleman J, Van Laere SJ, van Dam PA. Microarray-based oncogenic pathway profiling in advanced serous papillary ovarian carcinoma. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshihara K, Tajima A, Yahata T, Kodama S, Fujiwara H, Suzuki M, Onishi Y, Hatae M, Sueyoshi K, Fujiwara H, et al. Gene expression profile for predicting survival in advanced-stage serous ovarian cancer across two independent datasets. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bentink S, Haibe-Kains B, Risch T, Fan JB, Hirsch MS, Holton K, Rubio R, April C, Chen J, Wickham-Garcia E, et al. Angiogenic mRNA and microRNA gene expression signature predicts a novel subtype of serous ovarian cancer. PLoS One. 2012;7 doi: 10.1371/journal.pone.0030269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felder M, Kapur A, Gonzalez-Bosquet J, Horibata S, Heintz J, Albrecht R, Fass L, Kaur J, Hu K, Shojaei H, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. doi: 10.1186/1476-4598-13-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D'Agostino RB Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–53. [DOI] [PubMed]
- 41.Ruwanpathirana T, Owen A, Reid CM. Review on cardiovascular risk prediction. Cardiovasc Ther. 2015;33:62–70. [DOI] [PubMed]
- 42.Aletti GD, Gostout BS, Podratz KC, Cliby WA. Ovarian cancer surgical resectability: relative impact of disease, patient status, and surgeon. Gynecol Oncol. 2006;100:33–7. [DOI] [PubMed]
- 43.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research N. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. [DOI] [PMC free article] [PubMed]
- 46.van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, de Bruyn M, Palles C, Nout RA, de Kroon CD, Osse EM, et al. POLE Proofreading Mutations Elicit an Antitumor Immune Response in Endometrial Cancer. Clin Cancer Res. 2015;21:3347–55. [DOI] [PMC free article] [PubMed]
- 47.Seagle BL, Eng KH, Yeh JY, Dandapani M, Schiller E, Samuelson R, Odunsi K, Shahabi S. Discovery of candidate tumor biomarkers for treatment with intraperitoneal chemotherapy for ovarian cancer. Sci Rep. 2016;6:21591. doi: 10.1038/srep21591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han C, Liu Y, Wan G, Choi HJ, Zhao L, Ivan C, He X, Sood AK, Zhang X, Lu X. The RNA-binding protein DDX1 promotes primary microRNA maturation and inhibits ovarian tumor progression. Cell Rep. 2014;8:1447–60. doi: 10.1016/j.celrep.2014.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheon DJ, Tong Y, Sim MS, Dering J, Berel D, Cui X, Lester J, Beach JA, Tighiouart M, Walts AE, et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin Cancer Res. 2014;20:711–23. doi: 10.1158/1078-0432.CCR-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryner L, Guan Y, Firestein R, Xiao Y, Choi Y, Rabe C, Lu S, Fuentes E, Huw LY, Lackner MR, et al. Upregulation of periostin and reactive stroma is associated with primary chemoresistance and predicts clinical outcomes in epithelial ovarian cancer. Clin Cancer Res. 2015;21:2941–51. doi: 10.1158/1078-0432.CCR-14-3111. [DOI] [PubMed] [Google Scholar]
- 51.Zhang HY, Li JH, Li G, Wang SR. Activation of ARK5/miR-1181/HOXA10 axis promotes epithelial-mesenchymal transition in ovarian cancer. Oncol Rep. 2015;34:1193–202. doi: 10.3892/or.2015.4113. [DOI] [PubMed] [Google Scholar]
- 52.Wu Q, Hou X, Xia J, Qian X, Miele L, Sarkar FH, Wang Z. Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat Rev. 2013;39:640–6. [DOI] [PMC free article] [PubMed]
- 53.Vikhanskaya F, Bani MR, Borsotti P, Ghilardi C, Ceruti R, Ghisleni G, Marabese M, Giavazzi R, Broggini M, Taraboletti G. p73 Overexpression increases VEGF and reduces thrombospondin-1 production: implications for tumor angiogenesis. Oncogene. 2001;20:7293–300. [DOI] [PubMed]
- 54.Bottsford-Miller J, Choi HJ, Dalton HJ, Stone RL, Cho MS, Haemmerle M, Nick AM, Pradeep S, Zand B, Previs RA, et al. Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin Cancer Res. 2015;21:602–10. [DOI] [PMC free article] [PubMed]
- 55.Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, Mannel RS, Homesley HD, Fowler J, Greer BE, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–83. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 56.Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, Carey MS, Beale P, Cervantes A, Kurzeder C, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–96. [DOI] [PubMed]
- 57.Antoniu SA, Kolb MR. Intedanib, a triple kinase inhibitor of VEGFR, FGFR and PDGFR for the treatment of cancer and idiopathic pulmonary fibrosis. IDrugs. 2010;13:332–45. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study was performed in silico, with de-identified publicly available data. All data from TCGA is available at their website: https://tcga-data.nci.nih.gov/tcga/. Data for GEO sets are also available at NCBI website: http://www.ncbi.nlm.nih.gov/gds/. Datasets can be browsed by their accession number: GSE23554, GSE3149, GSE9891, GSE28739, GSE17260, and GSE30161. Software utilized by this study is also publicly available at Bioconductor website: http://bioconductor.org/.