

Abstract
A combination of solution NMR, dynamic light scattering (DLS), and fluorescence quenching assays were employed to obtain insights into the dynamics and structural features of a polyplex system consisting of HIV-1 transactivation response element (TAR) and PEGylated generation 5 poly(amidoamine) dendrimer (G5-PEG). NMR chemical shift mapping and 13C spin relaxation based dynamics measurements depict the polyplex system as a highly dynamic assembly where the RNA, with its local structure and dynamics preserved, rapidly exchanges (< ms) between free and polyplex-bound forms, at least for the probed N:P ratios. Recovery of quenched fluorescence shows that RNA in the polyplex can be competitively exchanged over a wide range of amine to phosphate (N:P) charge ratios. The rapid exchange between free and bound forms may contribute to the mechanism by which RNA is released into the cell upon delivery while being protected by the polyplex environment from immediate degradation by nucleases.
Keywords: therapeutic RNA delivery, PAMAM, PEGylation, TAR, DLS, NMR spin relaxation
Graphical abstract
INTRODUCTION
Therapeutic RNA holds great promise for treating cancer and other genetic diseases.1–8 However, the development of vectors for effective transport of therapeutic RNA to target cellular locations remains a challenge. Polycationic polymer (POCPs) based vectors are attractive due to versatility in synthesis and functional group modifications available in order to meet requirements such as safety, stability in intra and extracellular fluids, and release of the nucleic acid cargo.9,10 The positive charge density of POCPs enables spontaneous formation of nanometer-sized particles (polyplexes) with polyanionic RNA. siRNA polyplex formulations using POCPs like linear poly(ethyleneimine) (L-PEI)11,12 and cyclodextrins1 have advanced to clinical trials. Despite these potential advantages, POCPs have displayed poor efficacy compared to their viral counterparts.11,13 Improved designs of the POCPs, particularly to balance specificity of transport, toxicity, and efficiency of action are hindered by poor understanding of the mechanistic pathway of polyplexes. Studies have shown correlations of knockdown/transfection efficiencies of therapeutic RNA polyplexes with modulations of polymer properties.14–16 However, there is lack of molecular level understanding of polyplex structure and dynamics and how these properties vary as a function of key vector properties such as molecular weight, hydrophobicity, and charge density.17 Polyplexes are often difficult to characterize in terms of molecular stoichiometry, thermodynamic constants, exchange kinetics, and dynamics. Polyplex morphology has been explored using electron microscopy (EM)18 and atomic force microscopy (AFM) imaging.19,20 Studies based on molecular dynamics (MD) simulations,21–24 circular diochroism (CD),25–27 Infrared (IR) spectroscopy,28 and small angle X-ray scattering studies (SAXS)29,30 have helped gain some molecular level information on polyplexes. Despite these efforts a complete atomic level picture, particularly dynamics of polyplexes are lacking in the field.
Using 1D 1H NMR spectroscopy, we recently provided evidence that a 20-bp DNA duplex rapidly exchanges (exchange times < ms) between free and bound forms of polyplexes formed with generation 5 poly(amidoamine) (G5 PAMAM) dendrimer with 10% of its primary amines functionalized with 5000 molecular weight poly(ethyleneglycol) (PEG), (G5-PEG).31 The G5 PAMAM scaffold was PEGylated to increase polyplex solubility and this form has been shown to be effective for in vivo knockdown of protein expression.32 Based on the 1H NMR chemical shifts, there was no evidence for changes in the DNA duplex structure or dynamics upon binding to the dendrimer. This study immediately raised the question of whether a similar exchange mechanism might apply for RNA/polymer polyplexes. Unlike plasmid DNA, RNA therapeutics are active in the cytosol and do not require nuclear delivery. In addition, DNA and RNA have different physiochemical, structural, and dynamic properties. DNA typically adopts a B-form helix whereas RNA can form alternative secondary structures that contain A-form helices as well as non-canonical motifs such as bulges and apical loops. RNA and DNA also have distinct dynamic properties. While DNA B-form helix is more locally flexible than RNA A-form helices, non-canonical motifs in RNA can give rise to complex dynamics, including local motions in junctions such as bulges and collective motions of helical domains across these junctions.33,34
We use 2D NMR spectroscopy to characterize the interaction between a 13C/15N isotopically enriched model RNA system, the 29-nucleotide transactivation response element (TAR) from the human immunodeficiency virus type 1 (HIV-1)35 and G5-PEG.31 TAR is one of the key regulatory RNA elements in the HIV-1 viral genome which plays essential roles in viral replication. Although there has been interest in studying TAR/PAMAM dendrimer interactions in the context of Tat-TAR inhibition,36,37 here we focus on using TAR as a model flexible RNA system38–41 that contains non-canonical secondary structural elements, including a 3-nucleotide bulge and 6-nucleotide apical loop. G5-PEG has several advantages for NMR-based studies of polyplex. The G5 PAMAM scaffold is structurally well-defined, has a fairly narrow molecular weight distribution, and is well characterized in terms of NMR spectral properties.42–47 The 10% PEGylation gives G5 PAMAM excellent biological properties.12,32,48–50 Moreover, it provides convenient aqueous solubility properties for NMR experiments.31
We report exchange and dynamic properties of HIV-1 TAR/G5-PEG polyplexes formed across a range of N:P ratios. Using a combination of solution state 1D and 2D NMR spectroscopy, including 13C spin relaxation based dynamics measurements, DLS, and fluorescence spectroscopy, we show that the free RNA exists in rapid exchange with polyplex particles in the smaller size range ~12–40 nm that are detectable by NMR and larger particles in the size range ~140–200 nm that are NMR ‘invisible’. Interestingly, we find that interactions with the dendrimer results in negligible changes in the structure and dynamics of RNA that is in rapid exchange with smaller polyplex particles, although we cannot rule out changes that might occur in larger particles not observed in the NMR experiments.
EXPERIMENTAL SECTION
Materials
Sodium chloride (NaCl), sodium dihydrogen phosphate (NaH2PO4), ethylenediamine tetraacetic acid (EDTA), 0.1 N hydrochloric acid (HCl), tris (hydroxymethyl)-aminomethane, Triton-X, and methoxypoly(ethyleneglycol) tresylate (5000 MW; PEG 5000) were purchased from Sigma, St. Louis, MO. 99.96% deuterium oxide (D2O) was purchased from Cambridge Isotope Laboratories, Inc.. G5 PAMAM dendrimer was purchased from Dendritech Inc. (Midland, MI) and purified using published protocols.46 HPLC purified TAR tagged with fluorescein at the 3′ end was purchased from Dharmacon (Lafayette, CO).
RNA Synthesis and Purification
Unlabeled and uniformly 13C/15N labeled TAR was prepared by in vitro transcription using a dsDNA (Integrated DNA Technologies) template containing T7 promoter at 5′-end. T7 RNA polymerase (Takara Mirus Bio, Inc.) was used to transcribe the dsDNA sequence in the presence of unlabeled (SILANTES, ISOTEC, Inc.) or 13C/15N labeled (Cambridge Isotope Laboratories, Inc.) ribonucleotide triphosphates. The RNA was purified using 20% (w/v) denaturing polyacrylamide gel electrophoresis (PAGE) in 8 M urea and 1X TBE buffer followed by electroelution in 20 mM Tris (pH 8) buffer and ethanol precipitation. The purified RNA pellet was redissolved in Tris buffer and exchanged into NMR buffer (15 mM sodium phosphate, 25 mM sodium chloride, 0.1 mM EDTA, pH 7.4) using a centricon ultracell YM-3 concentrator (Millipore Corp.). The concentration of RNA was measured by optical absorption using Nanodrop 2000 (Thermo Scientific) assuming an extinction coefficient of 268900 L/M.
PEGylation of G5 PAMAM Dendrimer
G5 PAMAM dendrimer (0.0624 g, 2.22 × 10−6 mol, 1 equiv.) was dissolved in 4.0 mL of 1X PBS in a 50 mL round bottom flask. In a vial, 5,000 MW PEG tresylate (0.128 g, 2.6×10−5 mol, 12 equiv.) was dissolved in 2.0 mL of 1X PBS. PEG solution was added to dendrimer solution dropwise and stirred for 4 days at room temperature. The resulting conjugate was purified by 8 rounds of dialysis (first 2 rounds 2 hours each, the rest 4 hours each) in 4 L of water using 30,000-MW cutoff dialysis tubing. Solvent was removed using lyophilization yielding a puffy white solid. The yield was 0.169 g. The purified and lyophilized product was analyzed by MALDI, which gave average molecular weight of 85239 g/mol corresponding to an average of 12 PEG-5000 per dendrimer. From potentiometric titration, the number of primary amines was determined to be 101 per dendrimer.
Dynamic Light Scattering
Polyplexes of TAR/G5-PEG were prepared at the same concentration, buffer, and salt conditions as NMR experiments at N:P ratios 0, 0.25, 1, and 5 (at TAR concentration of 200 μM) and incubated for at least 30 min before measurements were recorded. The hydrodynamic diameter was measured at room temperature using Malvern Zetasizer Nano ZS (Worchestershire, United Kingdom). Three rounds of measurements were performed for each sample, with the diameter obtained in each round being an average of at least 6–10 measurements with duration of 40 s. Standard deviation was obtained by averaging the width of each peak obtained from the three rounds of measurements. The dispersant viscosity and refractive index were assumed to be that of water (cP 0.8872 and 1.33, respectively) whereas the material refractive index was assumed to be that of a standard protein sample (1.59). Since the intensity distribution showed multiple peaks (polydisperse sample, polydispersity index (PDI) > 0.1), we report intensity average and number average diameters separately instead of the commonly used Z-average size.
NMR experiments
All NMR experiments were carried out on a 600 MHz Bruker instrument equipped with a 5 mm triple-resonance cryogenic probe at a temperature of 37°C. The buffer used was pH 7.4 NMR buffer (15 mM sodium phosphate, 25 mM NaCl, 0.01% EDTA). The spectra were processed and analyzed using NMRpipe software,51 unless mentioned otherwise.
1D 1H NMR Titrations
200 μM TAR (13C/15N labeled or unlabeled) in NMR buffer was lyophilized and suspended in D2O twice to minimize 1H-based signals, including those from exchangeable amide protons that overlap in chemical shift with aromatic RNA signals of interest. Increasing volumes of 4 mM G5-PEG (NMR buffer, lyophilized and resuspended in D2O twice) was added to the TAR sample resulting in increasing N:P ratios (0, 0.17, 0.25, 0.5, 1.0, and 5.0). 1D 1H NMR spectra were acquired with or without 13C/15N decoupling during acquisition for labeled or unlabeled RNA respectively following each G5-PEG addition. The spectra were collected using excitation sculpting water suppression schemes. For 1D 1H NMR titration of TAR into G5-PEG, 5 μM G5-PEG was titrated to decreasing N:P ratios (4, 3, 2, 0.5, and 0.25) by adding increasing volumes of 1mM TAR in NMR buffer.
1H CPMG Relaxation Dispersion
1H NMR titration of G5-PEG into 200 μM unlabeled TAR to increasing N:P ratios (0, 0.25, 0.5, and 1.0) was performed as mentioned above. For each titration point, effective “transverse” relaxation rates (R2,effective) were measured using 1D Carr-Purcell-Meiboom-Gill (CPMG) experiments31,52,53 at three CPMG field strengths, νCPMG (νCPMG = 1/2τcp in Hz, where τcp is the interval between two 180° proton pulses during the CPMG element); 625 Hz (τcp = 0.8 ms), 250 Hz (τcp = 2.0 ms), and 125 Hz (τcp = 4.0 ms). Relaxation data was collected using 14 delay series with two duplicate measurements for error estimation; [1, 10, 20 (x2), 30, 40, 60, 80, 100, 120, 140, 160 (x2), 200 ms] at 625 Hz, [1, 6, 12, 18, 24, 30 (x2), 42, 48, 54, 60, 70 (x2), 80 ms] at 250 Hz, [1, 2, 4 (x2), 6, 8, 12, 16, 20, 24, 28, 32 (x2), 40 ms] at 125 Hz for N:P 0; [1, 8 (x2), 16, 24, 32, 40, 60, 80, 100, 120, 140 (x2), 160 ms] at 625 Hz, [1, 6 (x2), 12, 18, 24, 30, 36, 42, 48, 54, 60 (x2), 64 ms] at 250 Hz, [1, 4 (x2), 6, 8, 10, 12, 14, 16, 18, 20, 26 (x2), 32 ms] at 125 Hz for N:P 0.25; [1, 6 (x2), 12, 18, 24, 30, 36, 40, 50, 60 (x2), 70, 80 ms] at 625 Hz, [1, 4 (x2), 6, 8, 10, 12, 14, 16, 18, 20, 26 (x2), 32 ms] at 250 Hz, [1, 2, 4 (x2), 5, 7, 6, 8, 9, 10, 12 (x2), 14, 16 ms] at 125 Hz for N:P 0.5; [1 (x2), 2, 4, 6, 8, 10, 12, 14, 16, 18 (x2), 24, 30 ms] at 625 Hz, [1, 2 (x2), 3, 4, 5, 6, 7, 8, 9, 10 (x2), 11, 12 ms] at 250 Hz, [1 (x2), 2 (x3), 3 (x2), 4 (x3), 5 (x2), 6 (x2) ms] at 125 Hz for N:P 1. Spectra were processed and analyzed using mNova software (Mestrelab research) to obtain integrated area. The regions 6.6–9.0 ppm (for H2/H6/H8) and 5.1–6.3 ppm (for H5/H1′) were integrated separately. The integrated area versus relaxation delay times were fitted to a monoexponential decay using Origin Software (Origin Corporation) to obtain R2,effective. To estimate R2,polyplex values, the R2,effective values measured at the highest field strength (νCPMG = 625 Hz) for different N:P ratios were globally fit to eq. 131 employing a nonlinear regression algorithm using an in house python script. The fits were obtained using random initial values of each parameter using the constraints 0 ≤ free ≤ 1; 0 ≤ R2,polyplex ≤ 500.
2D 1H-13C HSQC
2D 1H-13C spectra were obtained for the same samples used in 1H NMR titrations at N:P of 0, 0.17, 0.25, 0.5, and 1.0. Spectra were processed using NMRpipe51 and Sparky 3 NMR Assignment and Integration Software (University of California, San Francisco).
Measurement of 13C R1 and R2 Relaxation Rates
13C “longitudinal” relaxation rate constant (R1) and “transverse” relaxation rate constant (R2) were measured for the same samples used in 1H NMR and HSQC titrations at N:P of 0 and 0.25 using 2D R1 and R1ρ relaxation experiments39 for base C2, C6, and C8 and sugar C1′ carbons. To maintain adequate signal to noise ratio, we carried out 13C R2/R1 relaxation at low dendrimer concentration (N:P of 0.25). The R1ρ relaxation experiment was carried out using a 3.5 kHz spin-lock field strength and a spin-lock carrier centered at aromatic C6 (for C2, C6, and C8) or sugar C1′ resonances. The spin-lock power was sufficiently high to suppress undesired chemical exchange (Rex) contributions. For N:P 0, relaxation data were collected using the delay series [10, 55, 140, 250 (x3) ms] for R1 and [4, 16, 32, 48 (x3) ms] for R1ρ, with triplicate measurements for error estimation for aromatic (C2, C6, and C8) carbons and [20, 140 (x2), 280, and 580 (x2) ms] for R1 and [0.4, 8.4, 24.4 (x2), and 48.4 (x2) ms] for R1ρ, with two duplicate measurements for error estimation for sugar C1′ carbons. For N:P 0.25, the delay series [10, 140 (x2), 280, and 480 (x2) ms] for R1 and [0.4, 8.4 (x2), 16.4, and 28.4 (x2) ms] for R1ρ, with two duplicate measurements for error estimation, was used for both aromatic (C2,C6,C8) and sugar (C1′) carbons. The spectra were processed with nmrPipe51 and relaxation rate constants determined by fitting the resonance intensities to monoexponential decays using Mathematica 6.0 (Wolfram Research, Inc.). The R2 values were calculated from the relationship R1ρ = R1 cos2θ + R2 sin2θ where θ = arctan (spinlock power/ resonance offset).
Calculation of Observed Polyplex Hydrodynamic Diameter using 1H NMR Relaxation Data
1H R2,polyplex values were used to calculate the rotational correlation time (τm) of the polyplex. For a X-H group, R2 relaxation rates are dependent on a linear combination of spectral density functions evaluated at five frequencies, J(0), J(ωH), J(ωX), J(ωX + ωH), and J(ωX − ωH), where ωH and ωX are the frequencies of H and X nuclei respectively at a given spectrometer field.54 Assuming that dipole-dipole coupling dominates 1H R2 relaxation due to directly bonded carbon as well as neighboring protons within 4 Å, R2,polyplex was expressed as a sum of R2 relaxation rates31,39,55
where d is the dipolar coupling constant given by
μ0 is the permittivity of free space, γH and γC are the magnetogyric ratios of 1H and 13C respectively and h is the Plank’s constant. rHC is the distance of the aromatic and sugar protons to their directly bonded carbon (1.104 Å and 1.115 Å for base C–H bond and sugar C1′–H1′ bond respectively) and rHH are the distances to neighboring protons within 4 Å. These distances were obtained from the solution NMR structure of free TAR (protein structure database ID: 1ANR).56 J(ω) as defined by the “simplified” model-free formalism for isotropic overall tumbling.54
where S2 is the generalized order parameter. An average of residue specific S2 values (0.763 and 0.809 for H2/H6/H8 and H5/H1′ respectively) previously published39 for elongated UUCG-TAR based on 13C relaxation data was used. Assuming spherical shape, polyplex hydrodynamic diameter (D) was computed using Stokes-Einstein equation for the range of τm values obtained from 1H relaxation data.
where R = D/2 is the radius of a sphere. kB is the Boltzman’s constant, T is temperature, η is the viscosity of the solvent at temperature T.
Calculation of Observed Polyplex Hydrodynamic Diameter using 13C Relaxation Data
Residue specific 13C R2,polyplex were obtained using 13C R2 values in eq. 1 where pfree and pbound values were obtained from analysis of 1H relaxation at N:P of 0.25 (Table 1). Since chemical shift anisotropy (CSA) also significantly contributes to carbon relaxation, R2,polyplex was obtained as a sum of contribution due to dipole-dipole coupling and CSA as given by39,55
where d and J(ω) are as defined earlier and CSA = 134.3 ppm, 178.8 ppm, 149.9 ppm, 40 ppm for C8, C6, C2, and C1′ respectively.55,57 Residue specific S2 values (ranging from 0.43 to 0.93) were used in the J(ω) expression.39 Assuming spherical shape, polyplex hydrodynamic diameter was obtained using the Stokes-Einstein equation as mentioned above.
Table 1.
Exchange Parameters Obtained from Global Fitting Relaxation Data using eq. 1 (see main text) for H2/H6/H8 (6.6–9.0 ppm) and H5/H1′ (5.1–6.3 ppm). pfree = 1 − pbound where pbound is the fraction of RNA bound in the small-sized polyplexes in fast exchange with free RNA.
| N:P | pfree | R2,polyplex (s−1) (H2/H6/H8) | R2,polyplex (s−1) (H5/H1′) |
|---|---|---|---|
| 0 | 1 | 311 ± 111 | 236 ± 84 |
| 0.25 | 0.97 ± 0.02 | ||
| 0.50 | 0.89 ± 0.06 | ||
| 1.0 | 0.64 ± 0.21 |
Fluorescence Quenching Based Competition Assay
TAR with 3′ end tagged with fluorescein was employed for the fluorescence quenching based assay. The RNA was annealed by heating to 95°C for 5 min and then cooled on ice for an hour prior to use. The RNA was diluted in pH 7.4 fluorescence assay buffer (50 mM Tris-HCl, 50 mM KCl, 0.01% Trition-X). The G5-PEG was dissolved in the assay buffer and serially diluted to obtain aliquots of concentrations from 2 nM to 2000 nM. 100 nM TAR was incubated for 30 min in 1:1 volume with varying concentrations of the G5-PEG in a 384-well plate such that the final RNA concentration in each well was 50 nM and the N:P ratios varied from 0 to 28. Fluorescence intensities were measured in triplicates using an Omega star plate reader (BMG Labtech) with a 485 nm excitation and 520 nm detection. For the exchange assay, to selected N:P ratio sample wells, 0.3 μL of 500 μM untagged TAR was added and the resulting fluorescence intensity was measured in a similar fashion after 30 min incubation. The assay was also repeated for N:P points 1, 10, and 70 within 2 min of adding untagged TAR.
RESULTS AND DISCUSSION
DLS Provides Evidence for a Major (~12–40 nm) and Minor (~140–200 nm) Polyplex Particles
We initially used DLS to characterize the size of TAR/G5-PEG polyplex particles formed at different N:P ratios. The hydrodynamic diameter obtained using cumulants based intensity mean (Z-average diameter),58 which is the commonly used method to obtain particle size from light scattering data, yielded values of ~67–170 nm consistent with literature reported values for dendrimer based polyplex systems.20,31,32,59 However, the polydispersity index (PDI), which reports on the homogeneity of the sample with respect to particle sizes was high (0.27–0.37) and peaks in the range of ~12–40 nm were also observed (Figure S1). Since the Z-average diameter gives good description of size only for unimodal distribution with narrow polydispersity (PDI < 0.1),58 we also examined the intensity weighting by the number of particles (number average distribution) assuming that the conversion from intensity to number holds for our measurements (Figure 1A). The number average analysis indicates that particles of ~12–34 nm size range are the major species in the population. The number average diameter increases from N:P of 0.25 to 1 and only slightly decreases at N:P of 5. Interestingly, such small polyplex sizes haven’t been reported previously for similar polyplex systems using DLS. For example, DLS data previously reported for siRNA/G5-PEG polyplexes in tris-HCL buffer at N:P of 10 gave a diameter of ~100 nm based on Z-average analysis.32 This could be due to different sample conditions or due to differences in size estimates obtained from Z-average versus number average analysis. Furthermore, it is known that larger particles scatter more light compared to smaller particles causing the average diameter obtained by DLS to be biased towards larger particles present in the solution.58 Although DLS reported sizes are usually in the larger size range (> 50 nm), there have been reports of smaller sizes for unPEGylated PAMAM/ siRNA polyplexes using TEM and AFM (15–130 nm)20 and nanoparticle tracking analysis (NTA) (10–80 nm).60 Although the DLS experiments were carried out at room temperature compared to the NMR experiments at 37°C (described below) which can affect the equilibrium populations, the sizes obtained by DLS are consistent with size estimates (~7.5–15 nm) from our NMR results.
Figure 1.
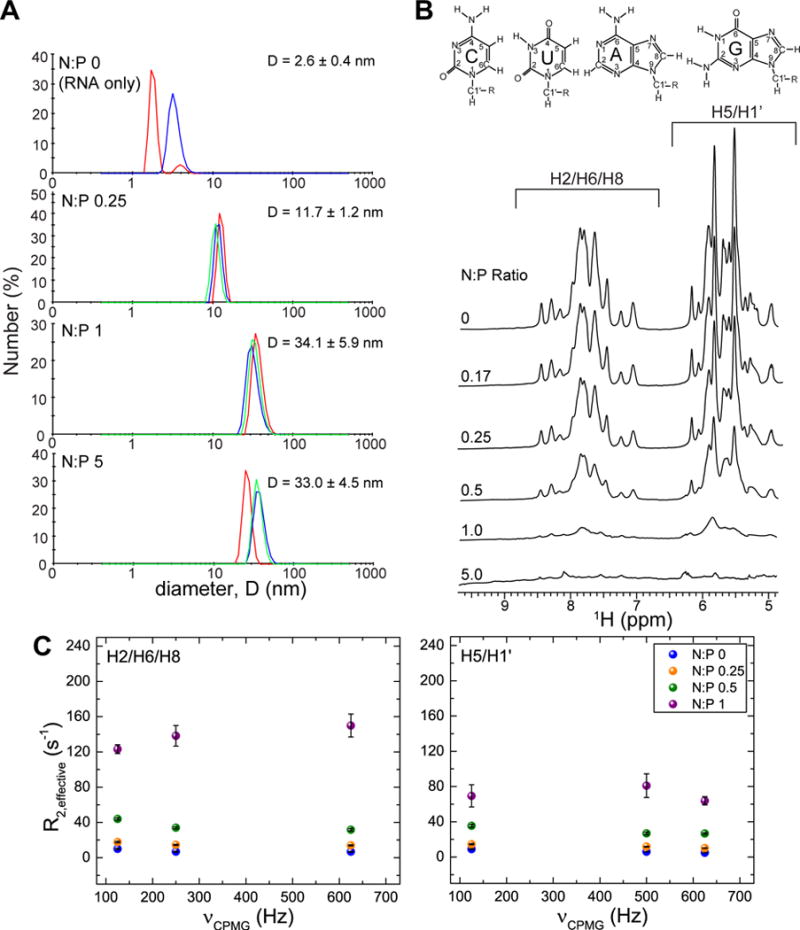
TAR exists in rapid exchange between free and polyplex-bound species. (A) DLS measurements of polyplex samples at N:P ratios 0 to 5 at NMR concentrations (200 μM TAR) showing presence of ~12–34 nm polyplexes. Data is presented as number average diameter. Overlay of three peaks in the same graph indicates triplicate measurements. Only two measurements were collected for N:P 0. The diameter of G5-PEG only is 5.9 nm.31 (B) Stacked overlay of 1H NMR spectra (aromatic H2/H6/H8, sugar H1′, and aromatic H5 region) of 200 μM TAR titrated to increasing N:P ratios with G5-PEG at 37°C in pH 7.4 NMR buffer (15mM sodium phosphate, 25mM NaCl, 0.01% EDTA) exchanged to 99.99% D2O (C) R2,effective as a function of different CPMG fields in Hz for N:P ratios 0, 0.25, 0.5, and 1 measured independently for H2/H6/H8 and H5/H1′ protons.
TAR Exists in Rapid Exchange (< ms timescale) between Free and Polyplex-Bound Particles
1D 1H NMR titrations of G5-PEG into a solution of unlabeled TAR showed gradual line broadening of TAR resonances without significant changes in the TAR chemical shifts (Figure 1B). The line broadening indicates that TAR is interacting with G5-PEG but the lack of significant chemical shift perturbations suggests that the interaction does not significantly affect the structural and dynamic properties of TAR. This is striking considering that TAR is a highly flexible molecule40,61–63 and that chemical shift perturbations are observed when varying salt concentrations,64 upon addition of DMSO,65 and a wide variety of ligands that bind to TAR non-specifically.38,66
Analysis of the spectra did not reveal evidence for free RNA with sharp narrow lines, as would be expected if small and large species with similar chemical shifts exchange at slow timescales. The line broadening could arise due to microsecond-to-millisecond chemical exchange, which results in an exchange contribution (Rex) to the observed effective “transverse” relaxation rate constant (R2,effective = R2 + Rex, where R2 is the intrinsic “transverse” relaxation rate constant). The exchange could represent transitions between free and bound RNA and/or changes in the RNA conformation in the bound state. Chemical exchange with a small particle can cause significant line broadening if there were significant differences in chemical shifts of free and bound state. For a low population of such a bound state, if the chemical shift difference is large enough, and on the slow to intermediate timescales, then the line broadening could be significant without inducing a change in the observed RNA chemical shift.
To assess Rex contributions, we carried out 1H Carr-Purcell-Meiboom-Gill (CPMG)52,53 relaxation dispersion experiments (Figure 1C) on polyplexes of unlabeled TAR and G5-PEG using an approach similar to that described previously for DNA.31 In this experiment, R2,effective is measured during a relaxation period as a function of a time interval (τcp) between successive 180° refocusing pulses which serve to suppress the Rex chemical exchange contribution. We relied on 1H CPMG experiment rather than 13C CPMG dispersion due to poor sensitivity in 2D 1H-13C HSQC spectra beyond N:P of 0.25. The regions of the 1H NMR spectra representing H2/H6/H8 aromatic protons (6.6–9.0 ppm) and aromatic H5 and sugar H1′ protons (5.1–6.3 ppm) were integrated separately and R2,effective values were obtained from monoexponential decay fits of the integrated area versus decay times (Figure S2). R2,effective values were measured for various τcp delays and for N:P ratios of 0, 0.25, 0.5, and 1. We observed an increase in measured R2,effective with increasing N:P ratios even when using a short τcp delay of 0.8 ms to maximally suppress Rex contributions. Increasing the N:P ratio from 0 to 1 resulted in an increase in R2,effective from 6.7 ± 0.4 s−1 to 150 ± 13 s−1 for H2/H6/H8 and 4.7 ± 0.1 s−1 at N:P of 0 to 63.8 ± 4.7 s−1 at N:P of 1 for H5/H1′. These values are comparable to that reported previously published for DNA/G5-PEG where R2,effective also increased by ~20 fold from 8.1 ± 0.2 s−1 at N:P 0 to 135.6 ± 14.5 s−1 at N:P of 1 for H2/H6/H8 and 8.7 ± 0.4 s−1 at N:P 0 to 162.7 ± 13.5 s−1 at N:P 1.33 The measured R2,effective values showed much smaller (< 10 Hz) variations when varying the τcp delay and carrying out the CPMG experiments at three field strengths (νCPMG = 625 Hz, 250 Hz, and 125 Hz) (Figure 1C). These data indicate that the increase in line broadening and R2,effective is not principally due to an exchange contribution in the intermediate time scale but rather due to rapid exchange of the RNA with large polyplex particles which results in an increase in the intrinsic R2 as reported previously for DNA polyplexes.31 Indeed, the R2,effective values increase as the amount of dendrimer increases suggesting rapid exchange with polyplex particles growing in size (at least up to the N:P ratios probed by the experiment). This is consistent with DLS data that also showed an increase in the number average size from N:P of 0.25 to 1. We were, however, unable to measure R2,effective for N:P of 5 due to severe line broadening.
To estimate the R2,effective values for the polyplex-bound form, the R2,effective values measured at the highest CPMG field strength for different N:P ratios were globally fit to eq. 1,31
| eq. 1 |
where, pbound describes the fraction of RNA bound in the small-sized polyplexes in fast exchange with free RNA, and pfree + pbound = 1. R2,free and R2,polyplex are the R2,effective rates for free RNA and RNA bound to small-sized polyplexes respectively. The large ~140–200 nm particles observed by DLS are likely in slow exchange which do not contribute to the observed R2, effective values. This equation expresses R2,effective as a population weighted average for free and polyplex-bound species assuming a two-state binding equilibrium for a fast exchange scenario where the chemical exchange rate constant (kex = k1 + k−1, where k1 and k−1 are forward and backward rates respectively) is much larger than 2π × Δω (where Δω = ωpolyplex – ωfree is the chemical shift difference between free and bound species). The fits were obtained using random initial values of each parameter using the constraints 0 ≤ pfree ≤ 1; 0 ≤ R2,polyplex ≤ 500. The R2,polyplex values (Table 1) obtained from the two-state fit are similar to those reported previously for DNA.31
Based on the estimated value of R2,polyplex, we can estimate the rate of exchange (kex) to be greater than R2,polyplex − R2,free ~304 s−1 for H2/H6/H8 and ~231 s−1 for H5/H1′. The largest Δω observed in the proton dimension observed in 2D 1H-13C HSQC spectra described below was ~0.08 ppm (~300 rad s−1 at 600 MHz spectrometer field). Therefore, for all chemical shift differences smaller than this the kex will be greater placing the system in fast exchange.
We employed the fitted R2,polyplex values to calculate approximate hydrodynamic diameter for the polyplex particles. Under the assumptions of isotropic tumbling with internal motions being much smaller than overall tumbling, and that dipole-dipole coupling dominates the “transverse” relaxation rate, the overall rotational correlation times obtained (see materials and method) were estimated to be ~36–240 ns. Assuming spherical shape, this corresponds to hydrodynamic diameter in the range 7.5–14 nm consistent with previously reported values for DNA polyplexes.31 Using the population of free and bound at N:P of 0.25 from the two state fit of 1H relaxation data shown in Table 1, 13C R2,polyplex values were also calculated using residue specific 13C relaxation data as a cross-validation (see materials and methods). Despite a number of approximations employed, these values are close to number average size of the polyplexes (~12–40 nm) as measured by DLS.
It should be noted that in addition to line broadening, we observe a significant reduction in the integrated area of the 1H NMR resonances with increasing N:P ratios. Even though this decrease in intensity could result due to relaxation during the experiment itself, we cannot rule out some degree of exchange with much larger particles such as the ~140–200 nm particles observed by DLS that are not directly observable by NMR.67 A similar decrease in resonance signals was reported in the previous study31 with 20-bp DNA duplex which was attributed to the small polyplexes in equilibrium with larger NMR ‘invisible’ particles.
Insights into RNA-Dendrimer Interactions from 2D NMR Chemical Shift Mapping Experiments
Previous studies relied primarily on 1H NMR experiments to examine the impact of dendrimer binding on DNA structure.31 These experiments did not allow for characterization of any structural and dynamic perturbations with site-specific resolution. Here, we used uniformly 13C/15N labeled TAR and 2D 1H-13C HSQC experiments to analyze TAR G5-PEG interactions and the effects on TAR structure and dynamics with site-specific resolution. The ability to measure 13C and 15N chemical shifts provides additional probes of structure and dynamics. Prior studies have shown that the TAR 2D 1H-13C HSQC spectra are very sensitive to changes in physicochemical conditions, including salt,64 pH,68 and presence of a wide range of added ligands.65,66,69 This chemical shift sensitivity is a result of TAR’s high degree of conformational flexibility and high susceptibity to structural and dynamic perturbations due to environmental cues. Remarkably, despite high susceptibility to chemical shift perturbations, we observed little change in the 2D 1H-13C HSQC spectra of TAR following the addition of dendrimer resulting in N:P ratio up to a value of 1. The only significant perturbations observed were for the flexible bulge residues U23 and C24, and the flexible apical loop residue A35 (Figure 2). These perturbations including other small chemical shift changes in both 1H and 13C dimensions are similar to those observed for TAR conformational changes induced by positively charged ions such as Na+ and Mg2+.64 At physiological pH, the terminal –CH2H2NH2 dendrimer surface is protonated yielding –CH2H2NH3+ end groups (up to ~101 per dendrimer molecule). These charged end groups of the dendrimer could be interacting with TAR in a manner similar to positively charged metal ions.
Figure 2.
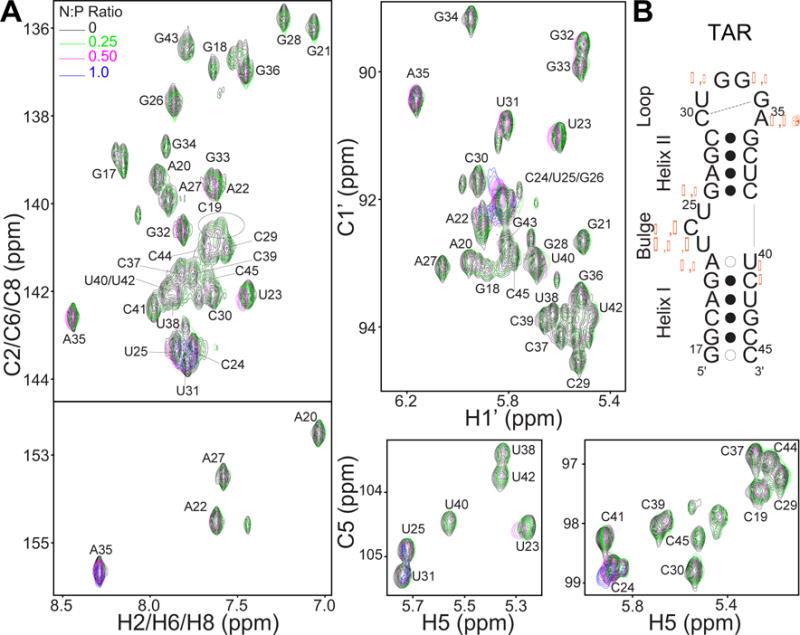
Effect of dendrimer interaction on RNA structure. (A) Overlay of 2D 13C–1H HSQC spectra of uniformly 13C/15N labeled 200 μM TAR titrated to increasing N:P ratios with G5-PEG at 37°C showing cross peaks for H2–C2/H6–C6/H8–C8, H1′–C1′, and H5/C5 correlations. Spectra for N:P of 0.17 has been omitted for clarity (B) Secondary structure of TAR. Residues that show chemical shift perturbations are indicated by orange diamond, circle, triangle, and square symbols representing H8–C8, H2–C2, H1′–C1′, and H5–C5 perturbations respectively. Larger symbol denotes greater magnitude of chemical shift perturbation.
Characterizing how Dendrimer Interactions affect RNA Dynamics
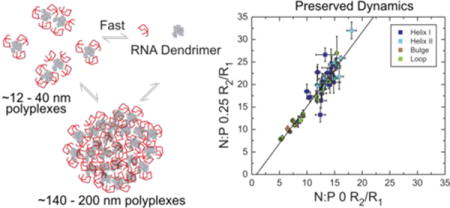
To more fully characterize the impact of dendrimer binding on the TAR dynamics we measured 13C “longitudinal” relaxation rate (R1) and “transverse” relaxation rates (R2) values on the aromatic (C2/C6/C8) and sugar (C1′) carbons. The measured 13C R2/R1 values for TAR in absence of dendrimer and at N:P ratio of 0.25 is shown in Figure 3A. The values obtained for free TAR are in agreement with previously published data.39,40 In the presence of dendrimer, nearly all TAR residues show an increase in R2/R1, consistent with a slow-down in overall tumbling due to formation of small sized polyplex particles as indicated by 1H NMR relaxation and DLS data. Although the overall tumbling of the RNA is slowed down, the bulge residues: U23, C24, U25 and the apical loop residues: U31, G32, G33, A35 retain lower R2/R1 values indicating that the RNA retains internal flexibility when bound to the small sized polyplexes. The correlation plot (Figure 3B) of R2/R1 values at N:P of 0 vs N:P of 0.25 shows good aggrement indicating that the dynamic properties of TAR are not significantly affected upon interacting with dendrimers at this N:P ratio. The outlier points correspond to the terminal residue (C45) or near terminal residue (G18), which could be the most affected due to dendrimer interaction. Overall these results further support that despite interactions with the dendrimer that slow down overall tumbling, at this N:P ratio the RNA largely retains key elements of flexibility observed in the free state, including high flexibility at the bulge and apical loop.
Figure 3.
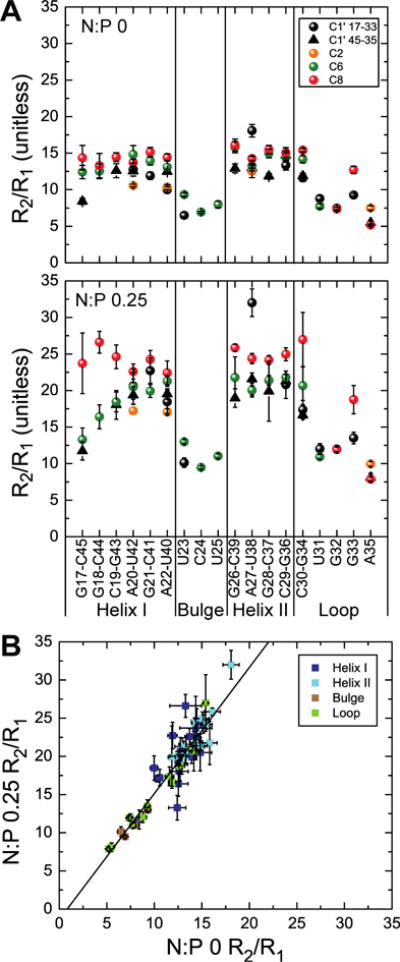
Examining impact of dendrimer interaction on TAR dynamics by 13C spin relaxation (A) Ratio of 13C “transverse” relaxation (R2) to “longitudinal” relaxation (R1) values (R2/R1) measured on TAR at N:P 0 and 0.25 plotted as a function of the nucleotide residue (as base-paired in the helices or unpaired in the bulge and apical loop). Values for different nuclei are denoted using different symbols and colors (B) Correlation of measured R2/R1 values at N:P 0 and 0.25.
To cross validate 1H NMR relaxation based overall tumbling time/ hydrodynamic diameter, we employed eq. 1 to obtain 13C R2,polyplex using pfree and pbound values obtained from 1H relaxation experiment for N:P of 0.25 (Table 1) and 13C R2 values. The 13C R2,polyplex ranged from 460–840 s−1 for the helical, non-terminal residues, and 140–670 s−1 for terminal, loop, and bulge residues for C2/C6/C8. For C1′ carbons, the R2,polyplex values ranged from 380–680 s−1 for helical, non-terminal residues, and 180–450 s−1 for terminal, bulge, and loop residues. Overall tumbling times obtained using these 13C R2,polyplex values (see materials and methods) ranged bewteen ~80–280 ns. Assuming spherical shape and using Stokes-Einstein’s relation31 the diameter obtained was in the range ~10–15 nm. These values are close to the range obtained from 1H relaxation (~7.5–14 nm).
Effect on Dendrimer Structure upon Polyplex Formation
MD simulations have shown that the flexibility of PAMAM dendrimers play a crucial role in the binding interactions with nucleic acids.23 However, few atomic level experiments24,31 have been used to provide insights into how the dendrimer structure is affected upon polyplex formation.24,31 To this end, we performed 1D 1H NMR titrations of 5 μM G5-PEG with increasing TAR concentrations (0–67 μM) resulting in decreasing N:P ratios from ∞ to 0.25 (Figure 4). These titrations were focused on the resonances belonging to G5-PEG. We observe no significant chemical shift perturbations on the proton signals (a, b, c, d, e, f) from the PAMAM framework31 of G5-PEG or the PEG proton signals (i, j, k),12,31 suggesting that the interaction with RNA doesn’t significantly alter the polymer structure and dynamic flexibility. Although we do not observe large changes in chemical shift, the proton signals from the PAMAM framework broaden with decreasing N:P ratios. The proton signal d, assignable to the methylene group alpha to the tertiary amine of the PAMAM framework shows an unusual behaviour in which it initially broadens nearly out of detection as the N:P decreases, and then reappears as a broad signal at N:P < 2. This could reflect structural reorganization in the polyplex around this N:P ratio, which is also suggested by our fluorescence experiments described below. The overall broadening of the proton signals from the PAMAM framework is consistent with a dynamic equilibrium between free RNA and polyplex-bound RNA as observed from the RNA perspective. Pavan et al. also observed such broadening of PAMAM peaks of unPEGylated G5 PAMAM dendrimer upon complexation with siRNA at low N:P ratios.24 By way of contrast, the PEG proton signals in our titration spectra remain sharp with integrated area and chemical shifts remaining essentially constant. This behavior is consistent with the PEG arms of the dendrimer remaining highly flexible in the polyplex. Absence of broadening of the PEG proton peaks also suggests that broadening of both the PAMAM and RNA peaks is due to polyplex formation and broadening associated with slower tumbling of the larger particles and not due to overall viscosity changes.
Figure 4.
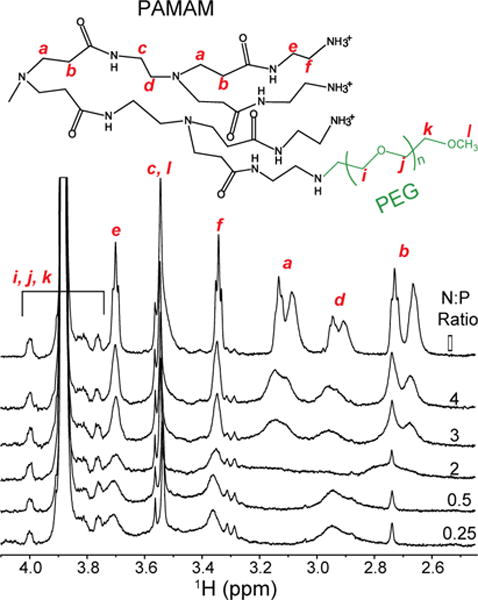
Effect of RNA interaction on the dendrimer. 1H NMR spectra of 5 μM G5-PEG titrated with increasing concentrations of TAR to decreasing N:P ratios with assignments of 1H NMR peaks of G5-PEG mapped onto its chemical structure shown above the NMR spectra. Peak assignments are based on previously published studies.12,31 Peaks representing PAMAM framework of G5-PEG are broadened while PEG peaks remain sharp suggesting that the RNA is binding to the PAMAM part while the PEG arms remain flexible.
Competitive Exchange of Polyplex-Bound RNA Measured using Fluorescence
The NMR experiments indicate that the free RNA and DNA31 exists in rapid exchange with polyplex-bound forms at N:P ≤ 1. Such a dynamic exchange behavior may help explain the mechanism by which polyplexes play a dual role of both protecting the nucleic acids from nuclease degradation as well as allowing for oligonucleotide release from the polyplexes that make it into the cells. A dynamic equilibrium between free and bound RNA does suggest that the time spent by the RNA in the free state can allow for interaction with nucleases in the cell depending on the off-rate of the polyplex equilibrium and on-rate of nuclease binding equilibrium. However, a fast exchange would allow for the RNA to spend less time as ‘free’ and ‘attackable’ by nucleases. While we have not specifically tested nuclease activity in this work, reports in literature have suggested that association with polyplexes slows down but does not completely prevent degradation by nucleases in a N:P ratio dependent manner.70,71 However, the NMR experiment is unable to probe what happens beyond N:P > 1 due to severe line broadening of the measured signals. To determine if RNA is still exchangeable at higher N:P ratios, we designed a fluorescence-based competition experiment (Figure 5). In the experiment, TAR containing a fluorescein tag on the 3′ end (TAR-FL) was mixed with increasing concentrations of G5-PEG to obtain N:P ratios ranging from 0 to 28. The fluorescence intensity was measured at a wavelength of 520 nm for each of the N:P points. We observe that the fluorescence intensity of TAR-FL decreases as the concentration of G5-PEG increases up to N:P ratio of 2, after which the intensity recovers to about 55% of initial fluorescence, and finally levels off at higher N:P ratios. A possible explanation for the initial decrease in TAR-FL fluorescence is that due to polyplex formation, RNA molecules come in close proximity leading to dye-dye quenching.
Figure 5.
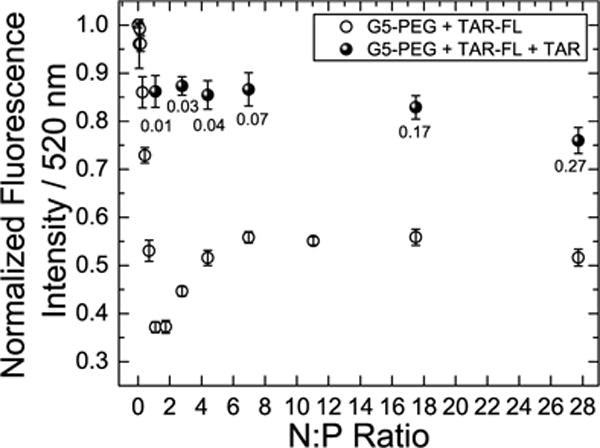
Fluorescence quenching based competitive displacement assay demonstrating release of fluorescein tagged TAR from polyplexes with G5-PEG. Open circles indicate fluorescence of polyplex mixture at different charge ratios before adding untagged TAR. Filled circles indicate fluorescence of the polyplex mixtures after adding untagged 0.3 μL of 500 μM TAR with numbers below the filled circles indicating resulting N:P ratios. The total concentration of tagged TAR in all N:P ratios was 50 nM. Buffer condition used is 50mM Tris-HCl, 50mM KCl, 0.01% Trition-X, pH 7.4.
An interesting feature of the fluorescence intensity binding curve is that the fluorescence recovers partially beyond ~N:P 2. Van Rompaey et al.72 and Zheng et al.73 have previously used similar fluorescence quenching assays to assess binding of other types of polycationic polymers with oligonucleotides. Zheng et al.73 also observed similar recovery in an assay with Tye563-labeled siRNA and hyperbranched PEI, which they attributed to molecular reorganization of the polyplexes. Interestingly in our case, beyond N:P ratio 2 we also observe in our 1H NMR titration spectra recovery of the resonance assigned to methylene group alpha to the tertiary amine of the PAMAM framework. This observation is consistent with molecular reorganization leading to conformational changes close to this region in the PAMAM framework. Increased electrostatic repulsion as more cationic polymer is added may causes an average increase in dye-dye distance. Increase in the volume of polyplex around these N:P points also may be a plausible explanation for partial reversal of fluorescence quenching. Beyond N:P of 6 the fluorescence intensity is constant. This is suggestive of no further changes in the structural organization of the polyplex particle. These results are consistent with the DLS results in which the number average size increases up to N:P 1 and remains more or less constant at N:P of 5.
Next we used this assay to test whether the RNA bound at N:P ratios greater than 1:1 remains exchangeable. Our prediction was if the RNA is exchangeable, it would be possible to rapidly recover the fluorescence intensity observed at low N:P ratios by simple addition of untagged TAR. Excess of untagged TAR was added to the preformed polyplex mixture at each N:P ratio described above and the resulting fluorescence intensity was measured. In all cases, addition of excess untagged TAR resulted in recovery of TAR-FL fluorescence intensity within < 30 min. For selected N:P ratios (1, 10, and 70) the fluorescence intensity was re-measured within 2 min of adding untagged TAR (Figure S3). The fluorescence intensity was also recovered in this case. Adding excess RNA introduces competition of binding; however, this condition also provides a model for the competition with other biomolecules when polyplex is introduced to cell culture or an organism. These data indicate that at N:P ratios >1:1, which are not probed by NMR, the polyplex RNA remains exchangeable and that this process is rapid on the fluorescence experiment time scale (< 2 min).
Comparison with Prior DNA Study
Overall, the exchange behaviour observed for polyplexes of TAR/G5-PEG is similar to that observed previously for 20-bp DNA/G5-PEG polyplexes,31 i.e. the exchange is rapid on the NMR timescale. Similar R2,polyplex values were obtained in both studies indicating presence of similar sized particles (~8–15 nm). However, we did observe differences in the 1H NMR spectra. In studies of DNA, the nucleic acid resonances broaden with increasing N:P ratio and are nearly undetectable at N:P ratio of 1. However, they reappear at N:P ratio of 5 either due to release of the DNA or increased local dynamics at the pico-nanosecond timescale. However, for the RNA polyplexes, peaks did not reappear beyond N:P of 1. Although it is difficult to hypothesize the origin of differences between RNA and DNA at these N:P ratios, there are some literature evidences suggesting local structural differences may exist between RNA and DNA polyplexes. For example, as the closest comparison, CD spectra of polyplexes of PEGylated PAMAM G4 and a double stranded RNA showed little changes at N:P of 5 and above whereas polyplexes with a 20-nucleotide antisense DNA showed significant spectral changes at same N:P ratios, suggesting conformational changes in the antisense DNA.26
As for the DLS measurements on the DNA polyplexes, a bimodal distribution was observed only for N:P ratio of 0.25 based on intensity where the second peak was observed ~8–20 nm. A unimodal distribution was observed for higher N:P ratios with the intensity average diameter ~60 nm. For the RNA, particles in the size range ~12–40 nm were observed for all N:P charge ratios in addition to ~140–200 nm particles. In addition, number average analysis indicated the smaller size particles represented the majority population. Overall, both studies suggest the presence of small sized polyplexes as well as the larger particles in the more commonly reported size range in exchange with free RNA.
Proposed Model of Exchange in the Polyplex System of RNA and Dendrimer
Combining results from NMR, DLS, and fluorescence experiments, we can propose a model for dynamic exchange illustrated in Scheme 1. The scheme shows smaller polyplexes in rapid exchange with free RNA as indicated by NMR line broadening without chemical shift perturbations. We hypothesize that this intrinsic rapid exchange in the polyplex could provide a mechanism for RNA release and protection from nuclease degradation. It is also conceivable that these dynamics affect interaction with other biomolecules in the cell such as plasma membrane components, and intracellular RNA, lipids, and proteins. For example, our recent study has shown that in polyplex treated cells the induced plasma membrane current is the same value as obtained for POCP-treated cell alone and persists even after the cells were allowed to recover by rapidly exchanging with POCP-free ECS solution. These data indicate that the POCPs are released into the cell plasma membrane and remain intercalated in the form of a stabilized pore or carpet structure.74 The size range for these smaller polyplexes were comparable between our NMR (~7.5–15 nm) and DLS (~12–40 nm) diameter measurements. Polyplex particles in this size range have been hypothesized to be the active form for effective gene delivery.75–77 The lower size limit for first pass elimination by kidneys has been estimated to be 10 nm diameter based on sieving coefficients simulated for proteins, Ficoll, and Dextran particles using pore theory of glomerular permselectivity.78 The upper size limit for particles to successfully reach target locations has been suggested75 to be ~70 nm diameter based on a study79 which showed that gylcolipid/liposome particles > 70 nm diameters were not recognized by the asialogycoprotein receptor on hepatocytes. The NMR data suggests that in the small sized polyplex particle, the RNA maintains its structural and dynamic properties without any differences localized in the flexible bulge residues and are consistent with effects observed with divalent cations.64 This is consistent with previous circular dichroism (CD) spectroscopy studies on RNA polyplexes with PEGylated G4 PAMAM26 and a variety of unPEGylated dendrimers27 where the overall CD spectral pattern of polyplexes remained similar to that of A-form RNA, minimally indicating that the RNA remains in its A-form helical secondary structure. Similar results were also inferred for transfer RNA (t-RNA) polyplexes from both CD spectroscopy and infrared (IR) spectroscopy.28 In addition, we also have evidence for presence of larger polyplex particles in the size range ~140–200 nm as measured by DLS. It is possible that these larger particles are in exchange with either free RNA or the smaller polyplex particles, as suggested by the decrease in integrated area of NMR resonances. We note that our data is consistent with the number of particles in the ~12–40 nm size range being dominant but a greater volume of material being present in the large ~140–200 nm particles. This leads us to put forward a picture of the polyplex system as an ‘ion-cloud’ like environment where the RNA and dendrimer molecules are held together by transient non-specific interactions preserving the RNA local structure and dynamics and are in dynamic exchange between free and bound forms.
Scheme 1.
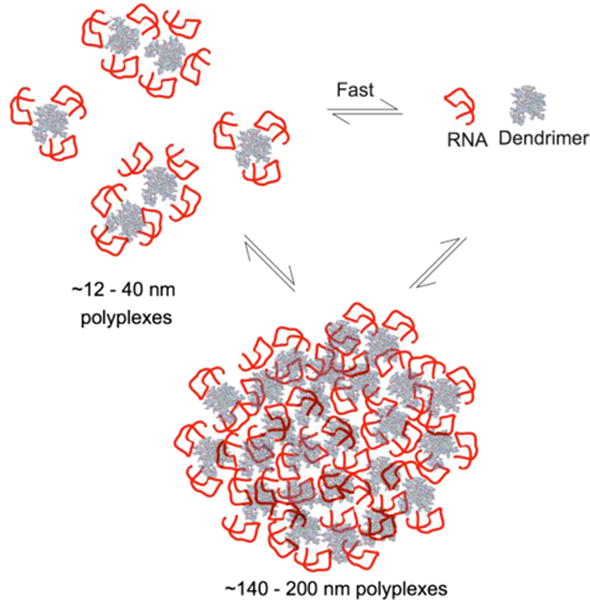
Proposed exchange model between polyplex bound forms and free RNA. As suggested by DLS, the ~12–40 nm particles represent the majority of polyplex population.
CONCLUSION
Using a combination of experimental approaches we describe a coherent picture for the structure and dynamics of nanoscopic RNA-dendrimer complexes. DLS and NMR experiments support the view that RNA-dendrimer polyplexes form two distinct populations with sizes of ~12–40 nm and ~140–200 nm. The smaller polyplexes are expected to be the more functional form as this size range falls within the proposed size limits for delivery vehicles. Interestingly, we find no structural differences for the two forms of RNA (free and bound), though the existence of these species is clear from the data. In addition to the structural properties, we study the dynamical aspects of these polyplexes. Using a combination of NMR and fluorescence spectroscopy we find strong evidence that the RNA is rapidly exchanging between free and polyplex-bound forms. This dynamic property is crucial for biological function, as the nucleic acid cargo needs to be both protected and released. The prospect of having functionally active RNA bound to a dendrimer delivery vehicle is extremely promising for future design. Lastly, we hypothesize that the small, active population of polyplexes are in free exchange with the larger aggregates observed in DLS. By considering all of these data, we propose a picture of how polyplexes may exist in solution that includes rapidly exchanging RNA molecules on smaller RNA-dendrimer polyplexes, as well as on the larger polyplexes aggregating and exchanging. The data presented here should prove useful for future engineering of polyplexes for the purpose of therapeutic nucleic acid delivery.
Supplementary Material
Acknowledgments
This project has been funded in part with Federal funds from the National Institutes of Health, National Institute of Biomedical Imaging and Bioengineering under Award RO1EB005028 given to M.M.B.H and by the NIH grant R01 AI087463-01 and Agilent Thought Leader Award given to H.M.A.
Footnotes
Supporting information
Results of intensity average based analysis of DLS data, monoexponential fits of integrated areas of 1D spectra obtained using CPMG based 1H relaxation experiment, fluorescence quenching based competition assay measured at faster time interval (Figures S1–S3) can be downloaded online. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no conflict of interest.
References
- 1.Davis ME. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keefe AD, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogris M, Wagner E. Hum Gene Ther. 2011;22:799–807. doi: 10.1089/hum.2011.065. [DOI] [PubMed] [Google Scholar]
- 5.Seyhan AA. Hum Genet. 2011;130:583–605. doi: 10.1007/s00439-011-0995-8. [DOI] [PubMed] [Google Scholar]
- 6.Burnett JC, Rossi JJ. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X, Huang L. Acc Chem Res. 2012;45:971–979. doi: 10.1021/ar200151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanasty R, Dorkin JR, Vegas A, Anderson D. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 9.Davis ME, Brewster ME. Nat Rev Drug Discov. 2004;3:1023–1035. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- 10.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 11.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Nat Rev Gene. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Klutz AM, Jacobson KA. Bioconjug Chem. 2008;19:1660–1672. doi: 10.1021/bc700483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Putnam D. Nat Mater. 2006;5:439–451. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 14.Mao S, Neu M, Germershaus O, Merkel O, Sitterberg J, Bakowsky U, Kissel T. Bioconjugate Chem. 2006;17:1209–1218. doi: 10.1021/bc060129j. [DOI] [PubMed] [Google Scholar]
- 15.Waite CL, Sparks SM, Uhrich KE, Roth CM. BMC Biotech. 2009;9 doi: 10.1186/1472-6750-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchida H, Itaka K, Nomoto T, Ishii T, Suma T, Ikegami M, Miyata K, Oba M, Nishiyama N, Kataoka K. J Am Chem Soc. 2014;136:12396–12405. doi: 10.1021/ja506194z. [DOI] [PubMed] [Google Scholar]
- 17.Aliabadi HM, Landry B, Sun C, Tang T, Uludag H. Biomaterials. 2012;33:2546–2569. doi: 10.1016/j.biomaterials.2011.11.079. [DOI] [PubMed] [Google Scholar]
- 18.Ainalem ML, Nylander T. Soft Matter. 2011;7:4577–4594. [Google Scholar]
- 19.Wan L, Manickam DS, Oupicky D, Mao G. Langmuir. 2008;24:12474–12482. doi: 10.1021/la802088y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez AP, Romero EL, Morilla MJ. Int J Pharm. 2009;380:189–200. doi: 10.1016/j.ijpharm.2009.06.035. [DOI] [PubMed] [Google Scholar]
- 21.Mills M, Orr BG, Holl MMB, Andricioaei I. J Phys Chem B. 2013;117:973–981. doi: 10.1021/jp309616t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mills M, Orr B, Holl MMB, Andricioaei I. Biophys J. 2010;98:834–842. doi: 10.1016/j.bpj.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavan GM, Albertazzi L, Danani A. J Phys Chem B. 2010;114:2667–2675. doi: 10.1021/jp100271w. [DOI] [PubMed] [Google Scholar]
- 24.Pavan GM, Posocco P, Tagliabue A, Maly M, Malek A, Danani A, Ragg E, Catapano CV, Pricl S. Chem Eur J. 2010;16:7781–7795. doi: 10.1002/chem.200903258. [DOI] [PubMed] [Google Scholar]
- 25.Prevette LE, Kodger TE, Reineke TM, Lynch ML. Langmuir. 2007;23:9773–9784. doi: 10.1021/la7009995. [DOI] [PubMed] [Google Scholar]
- 26.Reyes-Reveles J, Sedaghat-Herati R, Gilley DR, Schaeffer AM, Ghosh KC, Greene TD, Gann HE, Dowler WA, Kramer S, Dean JM, Delong RK. Biomacromolecules. 2013;14:4108–4115. doi: 10.1021/bm4012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ionov M, Lazniewska J, Dzmitruk V, Halets I, Loznikova S, Novopashina D, Apartsin E, Krasheninina O, Venyaminova A, Milowska K, Nowacka O, Gomez-Ramirez R, de la Mata FJ, Majoral JP, Shcharbin D, Bryszewska M. Int J Pharm. 2015;485:261–269. doi: 10.1016/j.ijpharm.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 28.Froehlich E, Mandeville JS, Kreplak L, Tajmir-Riahi HA. Biomacromolecules. 2011;12:2780–2787. doi: 10.1021/bm200547e. [DOI] [PubMed] [Google Scholar]
- 29.Su CJ, Chen HL, Wei MC, Peng SF, Sung HW, Ivanov VA. Biomacromolecules. 2009;10:773–783. doi: 10.1021/bm801246e. [DOI] [PubMed] [Google Scholar]
- 30.Jensen LB, Mortensen K, Pavan GM, Kasimova MR, Jensen DK, Gadzhyeva V, Nielsen HM, Foged C. Biomacromolecules. 2010;11:3571–3577. doi: 10.1021/bm101033g. [DOI] [PubMed] [Google Scholar]
- 31.Prevette LE, Nikolova EN, Al-Hashimi HM, Holl MMB. Mol Pharm. 2012;9:2743–2749. doi: 10.1021/mp3002864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Y, Li Y-B, Wang B, Lin R-Y, van Dongen M, Zurcher DM, Gu X-Y, Holl MMB, Liu G, Qi R. Mol Pharm. 2012;9:1812–1821. doi: 10.1021/mp3001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinnenthal J, Buck J, Ferner J, Wacker A, Furtig B, Schwalbe H. Acc Chem Res. 2011;44:1292–1301. doi: 10.1021/ar200137d. [DOI] [PubMed] [Google Scholar]
- 34.Mustoe AM, Brooks CL, Al-Hashimi HM. Ann Rev Biochem. 2014;83:441–466. doi: 10.1146/annurev-biochem-060713-035524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puglisi JD, Tan R, Calnan BJ, Frankel AD, Williamson JR. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, Li JR, Xi F, Jiang L. FEBS Lett. 2004;563:241–245. doi: 10.1016/S0014-5793(04)00284-4. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Guo ZP, Chen Y, Liu T, Jiang L. Chem Biol Drug Des. 2006;68:314–318. doi: 10.1111/j.1747-0285.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Science. 2006;311:653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 39.Hansen AL, Al-Hashimi HM. J Am Chem Soc. 2007;129:16072–16082. doi: 10.1021/ja0757982. [DOI] [PubMed] [Google Scholar]
- 40.Dethoff EA, Hansen AL, Musselman C, Watt ED, Andricioaei I, Al-Hashimi HM. Biophys J. 2008;95:3906–3915. doi: 10.1529/biophysj.108.140285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, Dethoff EA, Al-Hashimi HM. Proc Natl Acad Sci USA. 2014;111:9485–9490. doi: 10.1073/pnas.1407969111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meltzer AD, Tirrell DA, Jones AA, Inglefield PT, Hedstrand DM, Tomalia DA. Macromolecules. 1992;25:4541–4548. [Google Scholar]
- 43.Meltzer AD, Tirrell DA, Jones AA, Inglefield PT. Macromolecules. 1992;25:4549–4552. [Google Scholar]
- 44.Hu J, Fang M, Cheng Y, Zhang J, Wu Q, Xu T. J Phys Chem B. 2010;114:7148–7157. doi: 10.1021/jp1007889. [DOI] [PubMed] [Google Scholar]
- 45.Mullen DG, Fang M, Desai A, Baker JR, Orr BG, Banaszak Holl MM. ACS Nano. 2010;4:657–670. doi: 10.1021/nn900999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullen DG, Desai A, van Dongen MA, Barash M, Baker JR, Jr, Holl MMB. Macromolecules. 2012;45:5316–5320. doi: 10.1021/ma300485p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Dongen MA, Desai A, Orr BG, Baker JR, Holl MMB. Polymer. 2013;54:4126–4133. doi: 10.1016/j.polymer.2013.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kunath K, von Harpe A, Petersen H, Fischer D, Voigt K, Kissel T, Bickel U. Pharm Res. 2002;19:810–817. doi: 10.1023/a:1016152831963. [DOI] [PubMed] [Google Scholar]
- 49.Qi R, Gao Y, Tang Y, He R-R, Liu T-L, He Y, Sun S, Li B-Y, Li Y-B, Liu G. AAPS J. 2009;11:395–405. doi: 10.1208/s12248-009-9116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fant K, Eshjoerner EK, Jenkins A, Grossel MC, Lincoln P, Norden B. Mol Pharm. 2010;7:1734–1746. doi: 10.1021/mp1001312. [DOI] [PubMed] [Google Scholar]
- 51.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 52.Carr HY, Purcell EM. Phys Rev. 1954;94:630–638. [Google Scholar]
- 53.Meiboom S, Gill D. Rev Sci Instrum. 1958;29:688–691. [Google Scholar]
- 54.Jarymowycz VA, Stone MJ. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 55.Boisbouvier J, Wu Z, Ono A, Kainosho M, Bax A. J Biomol NMR. 2003;27:133–142. doi: 10.1023/a:1024931619957. [DOI] [PubMed] [Google Scholar]
- 56.Aboul-ela F, Karn J, Varani G. J Mol Biol. 1995;253:313–332. doi: 10.1006/jmbi.1995.0555. [DOI] [PubMed] [Google Scholar]
- 57.Hansen AL, Al-Hashimi HM. J Magn Reson. 2006;179:299–307. doi: 10.1016/j.jmr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Hassan PA, Rana S, Verma G. Langmuir. 2015;31:3–12. doi: 10.1021/la501789z. [DOI] [PubMed] [Google Scholar]
- 59.Liu X, Liu C, Laurini E, Posocco P, Pricl S, Qu F, Rocchi P, Peng L. Mol Pharm. 2012;9:470–481. doi: 10.1021/mp2006104. [DOI] [PubMed] [Google Scholar]
- 60.Jensen LB, Pavan GM, Kasimova MR, Rutherford S, Danani A, Nielsen HM, Foged C. Int J Pharm. 2011;416:410–418. doi: 10.1016/j.ijpharm.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 61.Al-Hashimi HM, Pitt SW, Majumdar A, Xu W, Patel DJ. J Mol Biol. 2003;329:867–873. doi: 10.1016/s0022-2836(03)00517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Musselman C, Al-Hashimi HM, Andricioaei I. Biophys J. 2007;93:411–422. doi: 10.1529/biophysj.107.104620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Q, Throolin R, Pitt SW, Serganov A, Al-Hashimi HM. J Am Chem Soc. 2003;125:10530–10531. doi: 10.1021/ja0363056. [DOI] [PubMed] [Google Scholar]
- 64.Casiano-Negroni A, Sun XY, Al-Hashimi HM. Biochemistry. 2007;46:6525–6535. doi: 10.1021/bi700335n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee J, Vogt CE, McBrairty M, Al-Hashimi HM. Anal Chem. 2013;85:9692–9698. doi: 10.1021/ac402038t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Nat Chem Biol. 2011;7:553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Callaghan PT. Clarendon Press. Oxford; 1991. [Google Scholar]
- 68.Dethoff EA, Petzold K, Chugh J, Casiano-Negroni A, Al-Hashimi HM. Nature. 2012;491:724–728. doi: 10.1038/nature11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stelzer AC, Kratz JD, Zhang Q, Al-Hashimi HM. Angew Chem Int Edit. 2010;49:5731–5733. doi: 10.1002/anie.201000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ainalem ML, Bartles A, Muck J, Dias RS, Carnerup AM, Zink D, Nylander T. PLOS One. 2014;9:e92692. doi: 10.1371/journal.pone.0092692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bielinska AU, Kukowska-Latallo JF, Baker JR., Jr Biochim Biophys Acta. 1997;1353:180–190. doi: 10.1016/s0167-4781(97)00069-9. [DOI] [PubMed] [Google Scholar]
- 72.Van Rompaey E, Chen Y, Muller JD, Gratton E, Van Craenenbroeck E, Engelborghs Y, De Smedt S, Demeester J. Biol Chem. 2001;382:379–386. doi: 10.1515/BC.2001.046. [DOI] [PubMed] [Google Scholar]
- 73.Zheng M, Pavan GM, Neeb M, Schaper AK, Danani A, Klebe G, Merkel OM, Kissel T. ACS Nano. 2012;6:9447–9454. doi: 10.1021/nn301966r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vaidyanathan S, Anderson KB, Merzel RL, Jacobovitz B, Kaushik MP, Kelly CN, van Dongen MA, Dougherty CA, Orr BG, Banaszak Holl MM. ACS Nano. 2015;9:6097–6109. doi: 10.1021/acsnano.5b01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bartlett DW, Davis ME. Bioconjugate Chem. 2007;18:456–468. doi: 10.1021/bc0603539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meyer M, Philipp A, Oskuee R, Schmidt C, Wagner E. J Am Chem Soc. 2008;130:3272–3275. doi: 10.1021/ja710344v. [DOI] [PubMed] [Google Scholar]
- 77.Ulasov AV, Khramtsov YV, Trusov GA, Rosenkranz AA, Sverdlov ED, Sobolev AS. Mol Ther. 2011;19:103–112. doi: 10.1038/mt.2010.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Venturoli D, Rippe B. Am J Physiol Renal Physiol. 2005;288:F605–F613. doi: 10.1152/ajprenal.00171.2004. [DOI] [PubMed] [Google Scholar]
- 79.Rensen PCN, Sliedregt LAJM, Ferns A, Kieviet E, van Rossenberg SMW, van Leeuwen SH, van Berkel TJC, Biessen EAL. J Biol Chem. 2001;276:37577–37584. doi: 10.1074/jbc.M101786200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.