

Abstract
Objective:
To determine the genetic cause of slowly progressive cerebellar ataxia, sensorineural deafness, and hypergonadotropic hypogonadism in 5 patients from 3 different families.
Methods:
The patients comprised 2 sib pairs and 1 sporadic patient. Clinical assessment included history, physical examination, and brain MRI. Linkage analysis was performed separately on the 2 sets of sib pairs using single nucleotide polymorphism microarrays, followed by analysis of the intersection of the regions. Exome sequencing was performed on 1 affected patient with variant filtering and prioritization undertaken using these intersected regions.
Results:
Using a combination of sequencing technologies, we identified compound heterozygous mutations in HSD17B4 in all 5 affected patients. In all 3 families, peroxisomal D-bifunctional protein (DBP) deficiency was caused by compound heterozygosity for 1 nonsense/deletion mutation and 1 missense mutation.
Conclusions:
We describe 5 patients with juvenile DBP deficiency from 3 different families, bringing the total number of reported patients to 14, from 8 families. This report broadens and consolidates the phenotype associated with juvenile DBP deficiency.
Peroxisomal D-bifunctional protein (DBP) deficiency is an autosomal recessive disorder that compromises peroxisomal fatty acid oxidation. DBP is crucial to the β-oxidation of fatty acids in the peroxisome, catalyzing the second and third steps of this process which are performed, respectively, by a 2-enoyl-coenzyme A (CoA) hydratase unit and a 3-hydroxyacyl-CoA dehydrogenase unit.1 DBP deficiency is typically associated with a severe clinical presentation comprising neonatal hypotonia, seizures, and severely impaired psychomotor development.1 Dysmorphism, loss of hearing and vision, and CNS abnormalities also commonly occur.
Infant onset DBP deficiency has been classified into 3 subgroups based on the relative impairment of the 2 DBP functions.2 Type I deficiency represents a total deficiency of both the hydratase and hydroxyacyl-CoA dehydrogenase activities. Type II is an isolated deficiency of the hydratase activity, and type III is an isolated deficiency of the hydroxyacyl-CoA dehydrogenase activity. The 3 subgroups share a similar spectrum of clinical features but differ in severity: nearly all infants with type I deficiency die in the first year of life, whereas those with types II and III often survive beyond 10 years.1
Recently, a less severe form of DBP has been described in 5 families3–7 and designated juvenile DBP deficiency,5 or alternatively DBP deficiency type IV6 or multifunctional protein ataxia syndrome.8 Here, we describe 5 affected individuals from 3 new families affected by juvenile DBP deficiency and expand the published phenotypic spectrum for this disorder.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Royal Children's Hospital Human Research Ethics Committee (approval number 28097). Participants were recruited following review of clinical details obtained from medical records and family interviews. Each participant signed an informed consent document before enrollment.
Genetic analysis.
Genetic studies were performed essentially as described previously.9 Genomic DNA isolated from the peripheral blood and single nucleotide polymorphism (SNP) genotype data were generated. Parametric multipoint linkage analysis was performed specifying a rare recessive disease model. Genomic DNA was analyzed by whole-exome capture (Illumina TruSeq capture kit, San Diego, CA), and massively parallel sequencing (MPS) was performed on a HiSeq2000 (Axeq Technologies, Macrogen, Seoul, Korea). Reads were aligned to the reference genome with Novoalign, and variants were identified with SAMtools and annotated by ANNOVAR. Variants were filtered in a step-wise manner against exclusion criteria including linkage region, minor allele frequency (≤0.01, 1000 Genomes10), and functional impact. Inclusion criteria included predicted damaging effects on the protein using PolyPhen-2 and SIFT. Sanger sequence analysis of HSD17B4 was performed by PCR amplification (primer sequences available on request). Primary fibroblast cells were analyzed by Western blot as previously described11 using anti-HSD17B4 (1:1000 HPA021479; Sigma-Aldrich, St. Louis, MO) and anti–β-actin antibody (1:10000 A5441; Sigma-Aldrich). The reference sequences used for HSD17B4 were NG_008182.1, NM_000414.3, and NP_000405.1.
RESULTS
Clinical descriptions.
The clinical details of 5 affected individuals from 3 pedigrees are presented in table 1 and the family pedigrees in figure 1A.
Table 1.
Summary of clinical details of affected individuals in this study and previously reported in the literature

Figure 1. Compound heterozygous mutations in HSD17B4 cause juvenile peroxisomal D-bifunctional protein deficiency.

(A) Pedigree structure of families with cerebellar ataxia with hypergonadotropic hypogonadism. Affected individuals are represented by black symbols. DNA from individuals that was used in this study is indicated (*). Single nucleotide polymorphism arrays and linkage were performed on samples I-2, II-1, II-2 (family 1), and II-1 and II-2 (family 2); WES was performed on sample II-2 (family 2). (B) A schematic representation of HSD17B4 indicating the dehydrogenase, hydratase, and SCP2 domains. The position and type of all compound heterozygous mutations identified in the 8 families reported with juvenile peroxisomal D-bifunctional protein deficiency to date (table 2) are indicated. A = pedigree 1, B = pedigree 2, C = pedigree 3, and D–H correspond to families 4–8, respectively. (C) Immunoblot analysis with an HSD17B4-specific antibody (1:1000, HPA021479) identified a ∼80 kDa full length and ∼35 kDA processed dehydrogenase domain protein in control fibroblasts (C1 and C2) that was significantly reduced in extracts derived from affected individuals II-1 and II-2 (families 1 and 2). An antibody directed against β-actin confirmed equivalent protein loading. FB = fibroblast. FB = fibroblast.
Pedigree 1, comprising 2 affected sisters of Italian descent (II-1 and II-2), was published previously under the designation “Cerebellar ataxia with hypergonadotropic hypogonadism” (OMIM #605672),12 prior to the identification of HSD17B4 mutations. Briefly, the sisters, aged 57 and 63 years at the most recent follow-up, both presented with adult-onset progressive cerebellar ataxia and secondary amenorrhea due to hypergonadotropic hypogonadism. Intellect was normal, peripheral sensory neuropathy was observed, and there was sensorineural deafness with vestibular hypofunction. Neurologic symptoms were observed in the third decade, and secondary amenorrhea was described at 16 and 32 years of age, respectively. Brain MRI on both sisters showed marked atrophy of the cerebellum, involving both hemispheres and vermis. Additional metabolic studies were undertaken in the younger sister (II-2). Plasma levels of very long-chain fatty acids (VLCFAs) were normal: C24:0/C22:0 ratio was 0.762 (reference 0.550–1.115) and C26:0/C22:0 ratio was 0.020 (reference <0.035). Plasma phytanic acid level was 0.1 mg/100 mL (reference <0.7). Urine bile acid profile was normal.
Pedigree 2 includes 2 affected siblings, a male and a female, along with 3 unaffected female siblings born to nonconsanguineous parents of Lebanese descent. The male sib (II-2) presented at age 29 years with a 5-year history of progressive ataxia. His workmates had noticed impaired balance and slurred speech, of which he was unaware. Increased falls and deteriorating handwriting had been noted for 3 years. He had not noted any visual or hearing impairment or sensory symptoms, and there was no erectile dysfunction.
Examination revealed a very broad-based ataxic gait with mild cerebellar dysarthria. There was bilateral horizontal gaze-evoked nystagmus, but horizontal saccades appeared normal. Upward saccades were hypometric, and downward saccades hypermetric. Tone and power were normal in the limbs, and deep tendon reflexes were reduced but present without enhancement. There was bilateral appendicular ataxia, with notably slow and hypermetric ballistic tracking (“finger chase”) movements. Vibration and pinprick perception were intact distally, but 2-point discrimination was clearly impaired at the great toes (2.5 cm). Brain MRI at age 25 years showed mild cerebellar atrophy with vermal emphasis and normal brainstem and cerebrum. Audiology showed moderate high-frequency hearing loss. At follow-up assessment 1 year later, his testosterone, luteinizing hormone (LH), follicle-stimulating hormone (FSH), and sperm count were normal. He had naturally conceived 2 sons and a daughter. Five years later, he required a wheelchair for mobility outside the house. His vibration perception threshold was well below the fifth percentile for his age, but pinprick sensation remained intact.
The sister of the family proband (II-1) presented at age 31 years with a 4-year history of dysarthria and impaired balance, although she reported always sensing some imbalance. She had been referred to an ENT specialist, who had discovered moderate high-frequency sensorineural hearing loss (figure 2A). Ovarian failure had been diagnosed on investigation for primary amenorrhea. Examination revealed a mildly broad-based gait and mild cerebellar dysarthria. Visual smooth pursuit was normal, and impersistent horizontal gaze-evoked nystagmus was evident on right gaze (only). Saccades to target were hypermetric to the right and into down gaze. Tone, power, and reflexes were normal in the limbs. There was mild appendicular ataxia. Vibration and pinprick perception and 2-point discrimination were normal at the great toes. Brain MRI performed at age 31 years showed clear pancerebellar atrophy, with normal brainstem and cerebrum (figure 2B). At age 37 years, she reported tinnitus, mild dysphagia, and worsening dysarthria and ataxia. There was evidence of sensory neuropathy with reduced vibration perception and 2-point discrimination at the great toes, although pinprick perception was preserved and ankle jerks were present.
Figure 2. Loss of HSD17B4 results in sensorineural deafness and slowly progressive cerebellar ataxia.

(A) Audiogram from patient II-2, pedigree 2 showing high-frequency sensorineural hearing loss. (B) Brain MRI from the same patient showing marked cerebellar atrophy. PTA = pure-tone audiometry.
Pedigree 3 includes 1 affected individual (II-1), a 33-year-old man born to nonconsanguineous parents of Italian ancestry. His younger brother is unaffected. He presented at age 6 years with bilateral sensorineural hearing loss, for which he has used hearing aids from age 7 years. Beginning at age 14 years, he developed a very slowly progressive cerebellar ataxia. A decline in gait was first noted in the early teenage years, with incoordination and occasional falls becoming gradually more evident. There have not been any problems with vision, or with motor weakness or sensory disturbance. He has never had a tremor or involuntary movements, and there is no history of myoclonus or seizures. His cognitive development is entirely normal. Clinical examination revealed gait ataxia, dysarthria, and limb ataxia, consistent with cerebellar disease. Although his ocular movements have a full range, visual pursuit was saccadic, with saccades being hypermetric in the horizontal plane, and there was marked gaze-evoked nystagmus, particularly on left gaze. Tongue movements were slightly slow. There was no evidence of motor weakness or sensory loss. Serial brain MRI demonstrated progressive pancerebellar atrophy with normal brainstem appearance. Nerve conduction studies did not show evidence of peripheral neuropathy. He was infertile due to hypergonadotropic hypogonadism with elevated FSH (33.8 IU/L N 1.5–12.4) and normal LH (7.6 IU/L N 1.7–8.6). Serum testosterone was at the lower end of the normal range (9.6 nmol/L N 8.3–30.2). A semen analysis showed azoospermia, and a testicular biopsy revealed no sperm and germ cell arrest. Serum phytanic acid was normal.
Mutation identification and characterization.
DNA from the 2 siblings from pedigree 1 and their mother were hybridized to Affymetrix Genome-wide 6.0 SNP chips. From the genotyping data, we identified regions where the affected siblings were sharing both maternal and paternal haplotypes (IBD2 regions), thereby eliminating candidate genes over 3 quarters of the genome. In addition, no copy number variants of interest were detected in this family.
DNA from the 2 affected siblings from pedigree 2 was hybridized to Illumina Human 610-quad beadchips. From the genotyping data, we identified IBD2 regions between the siblings which covered approximately 30% of the autosome, with 40 separate intervals. The male sib then underwent MPS, and variant filtering was restricted to these linkage regions. As the suspected disease model was compound heterozygous, the filtering process only included genes that had 2 potential causative mutations. This filtering resulted in 20 candidate genes containing 40 potential causative mutations.
Linkage regions from pedigrees 1 and 2 were then intersected, and subsequent variant filtering and prioritization was undertaken using these regions. This analysis identified HSD17B4 as a promising candidate, with 2 mutations identified in the male sib. The first variant (NM_000414(HSD17B4_v003):c.661C>T, p.(Leu221Phe) is a missense mutation, absent in population databases, that is predicted to be possibly damaging/deleterious by PolyPhen-2 and SIFT, respectively. The second variant (c.1132 G>T, p.Gly378*) is a nonsense mutation, most likely resulting in nonsense-mediated decay or a truncated protein product. The 2 variants were confirmed by Sanger sequencing in both siblings, and each parent was found to carry one of the variants, consistent with a compound heterozygous disease model.
Sanger sequencing was then undertaken in the 2 sisters from pedigree 1 and their unaffected mother. The same 2 variants were identified in both affected sisters. The first variant (c.58+1 G>A) is predicted to cause loss of the exon 1 donor splice site. The second variant (c.293A>AG, p.Asn98Ser) is a missense mutation predicted to be damaging/deleterious. The mother of the sisters was sequenced and found to have the c.293A>AG, p.Asn98Ser variant but not the c.58+1 G>GA variant. The father was not available for the study.
Sanger sequencing of HSD17B4 was then performed in pedigree 3. Two variants were identified in the affected male. The first variant in exon 4 (c.186_187del:pIle62Metfs*12) is a 2-base pair deletion (AA) causing a frameshift and a premature stop codon. The second variant is a missense mutation (c.680A>G;p.His227Arg) that is predicted to be probably damaging/deleterious. Each variant was present in one unaffected parent, consistent with a compound heterozygous disease model. Only one of the 2 mutations (c.680A>G;p.His227Arg) was present in his unaffected brother.
To determine the functional effect of mutations in HSD17B4, Western blot analysis of primary fibroblasts derived from all affected members in families 1 and 2 was performed. The 736 amino acid, 79.6 kDa protein encoded by HSD17B4 includes conserved dehydrogenase, hydratase, and SCP2 domains (figure 1B). In control fibroblast cells, an intense band of the expected size (approximately 80 kDa) was detected using an anti-HSD17B4 antibody. In addition, a band consistent with the ∼35 kDA processed dehydrogenase domain13 was also observed. The relative abundance of both of these proteins was significantly reduced in patient fibroblasts from families 1 and 2 (figure 1B).
DISCUSSION
The reported phenotype of juvenile DBP deficiency is characterized by childhood or early adult-onset sensorineural deafness, ataxia, and hypergonadotropic hypogonadism and overlaps with that of Perrault syndrome (sensorineural deafness and, in females, ovarian dysgenesis).3–7 Here, we describe 5 patients with juvenile (type IV) DBP deficiency from 3 different families, bringing the total number of reported patients to 14, from 8 families. We previously classified the family reported here as pedigree 1 as being affected by “Cerebellar ataxia with hypergonadotropic hypogonadism” (OMIM #605672)12; however, it is now clear that this is the same disorder as juvenile DBP deficiency.
This report broadens and consolidates the phenotype associated with juvenile DBP deficiency. The 3 cardinal manifestations of juvenile DBP deficiency are ataxia associated with cerebellar atrophy on MRI, sensorineural deafness, and hypergonadotropic hypogonadism. The disorder is slowly progressive with the onset of hearing loss and ataxia typically in the first decade of life; however, symptoms may escape recognition until the second or even the third decade. Peripheral neuropathy, affecting large diameter sensory fibers clinically, typically develops some years after the onset of deafness and ataxia. Ambulation is progressively impaired, with some patients requiring the use of a wheelchair between the third and sixth decades. Notably, our oldest patient was ambulant with a walker in her 60s.
Gonadal dysfunction in juvenile DBP deficiency shows greater variability. Of the 8 females identified with this disorder, 5 have had primary ovarian insufficiency, whereas 3 have undergone spontaneous puberty, followed some years later by premature menopause; none has had children. Of the 6 males with juvenile DBP deficiency, information about gonadal function is available for only 4. From previous reports, 2 males have been described as having low testosterone levels,4,5 of whom 1 had a semen analysis showing azoospermia.4 We report similar findings in one of our affected males (pedigree 3, II:1) who had a testosterone level at the lower end of the normal range associated with an elevated FSH level, and a testicular biopsy that showed germ cell arrest. In contrast, our other affected male had normal testicular function and fathered 3 children. Our data suggest that infertility is not a certain outcome for male individuals with DBP deficiency.
Juvenile DBP deficiency shares some features with the severe neonatal forms of DBP deficiency, notably the presence of hearing loss, cerebellar atrophy, and peripheral neuropathy; however, a number of features of neonatal onset DBP deficiency are absent from juvenile DBP deficiency, including hypotonia, seizures, visual loss, liver disease, and cortical malformations.1 Notably, all 5 of our patients had normal intelligence, although learning difficulties and intellectual disability have been described in other patients. The 3 siblings previously reported5 differ somewhat from the other juvenile DBP patients with respect to the presence of intellectual disability (ranging from mild to severe), spasticity, and white matter changes on MRI; these findings may be specific to the combination of mutations in these siblings.
It is important that the routine peroxisomal screening tests (VLCFA and phytanic acid levels), which are helpful in diagnosing the infantile forms of DBP deficiency, have, when tested, been consistently normal in juvenile DBP deficiency,3,5,7 including both of our patients who were tested. In the absence of a biochemical screening test, diagnosis of juvenile DBP deficiency relies on the recognition of the characteristic clinical presentation. It is therefore likely that juvenile DBP deficiency is an underdiagnosed entity and that further cases will come to light with the increased use of exome sequencing.
In common with other disorders of peroxisomal function, in DBP deficiency, our understanding of the links between cellular mechanisms and disease pathogenesis is incomplete. For some reason, the cerebellum is exquisitely sensitive to perturbations in peroxisomal function and cerebellar pathologies, including hypoplasia, atrophy, and demyelination, are common features of peroxisomal disorders.8 It is possible that neurodegeneration results from a combination of both the accumulation of toxic peroxisomal metabolic substrates (e.g., VLCFAs and phytanic acid) and deficiency of peroxisomal synthetic products; however, studies in knockout mice have failed to correlate neurodegeneration with the level of any candidate metabolite.8 It has been hypothesized that peroxisome dysfunction leads to widespread molecular and metabolic changes within the cell, including altered cell signaling, increased oxidative stress, and impaired mitochondrial function, and that these in turn lead to altered neuronal function and neuronal cell death.14
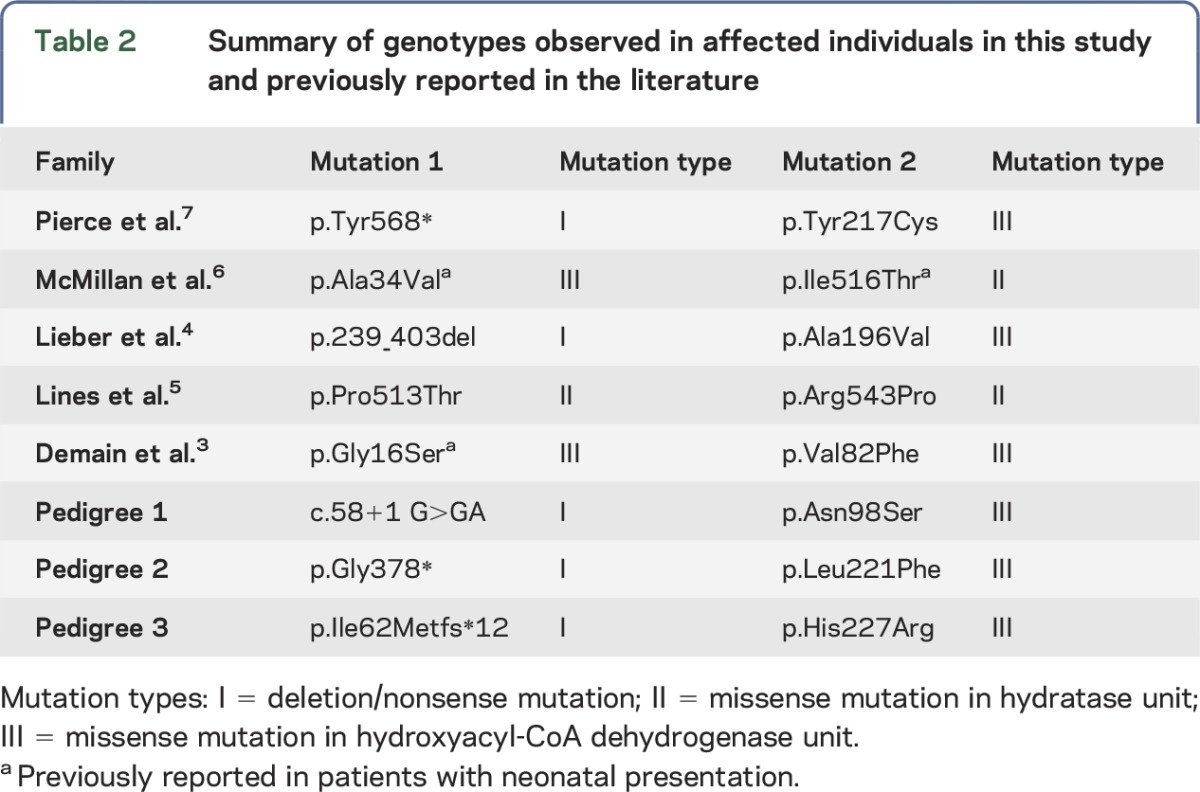
There is a correlation between DBP subgroups and genotype, with type I deficiency associated with nonsense and deletion mutations, type II deficiency associated with missense mutations in the hydratase unit, and type III deficiency associated with missense mutations in the hydroxyacyl-CoA dehydrogenase unit.13 Analysis of the genotypes of the 8 families with juvenile DBP deficiency (table 2) demonstrates that all affected individuals are compound heterozygotes, either for 2 missense mutations or for 1 missense mutation and 1 nonsense/deletion mutation. Of interest, 5 of the 8 families, including the 3 families reported here, are compound heterozygous for 1 nonsense/deletion mutation (the latter typically associated with type I DBP deficiency) and 1 missense mutation in the hydroxyacyl-CoA dehydrogenase unit, making this the most common genotypic category for juvenile DBP deficiency. Of the other families, one is compound heterozygote for 2 missense mutations in the hydroxyacyl-CoA dehydrogenase unit and another is compound heterozygous for 2 missense mutations in the hydratase unit. Although it was previously suggested that juvenile DBP deficiency might be specifically associated with the presence of 1 mutation in each of the hydratase and hydroxyacyl-CoA dehydrogenase units,6 only 1 family has been described with this combination of mutation types. Notably, 3 of the mutations reported in juvenile DBP deficiency have previously been reported in patients with neonatal DBP deficiency, indicating genotypic overlap between the subtypes of DBP deficiency.
Table 2.
Summary of genotypes observed in affected individuals in this study and previously reported in the literature
Juvenile DBP deficiency shares phenotypic features with Perrault syndrome and has been described by some authors as a type of Perrault syndrome.3,7 Perrault syndrome is an autosomal recessive syndrome, the cardinal features of which are sensorineural hearing loss in both sexes and primary ovarian insufficiency in females. Additional neurologic features such as ataxia, neuropathy, and intellectual disability are present in some patients, but are not required for diagnosis. Apart from HSD17B4, the other 4 genes so far associated with Perrault syndrome all encode mitochondrial proteins.3 Although there is obvious clinical overlap between juvenile DBP deficiency and Perrault syndrome, we believe that the conditions are sufficiently clinically and molecularly distinct for juvenile DBP deficiency not to be classified as a subtype of Perrault syndrome.
ACKNOWLEDGMENT
The authors thank the families involved in this research and acknowledge the generous support of the Lefroy and Handbury families.
GLOSSARY
- CoA
coenzyme A
- DBP
D-bifunctional protein
- FSH
follicle-stimulating hormone
- LH
luteinizing hormone
- MPS
massively parallel sequencing
- SNP
single nucleotide polymorphism
- VLCFA
very long-chain fatty acid
AUTHOR CONTRIBUTIONS
D.J.A. formulated the study, performed patient recruitment, provided and interpreted the clinical data, and cowrote the manuscript. A.P.L.M. performed molecular analysis and revised the manuscript. E.S. contributed to the design of the study, provided and interpreted the clinical data, and revised the manuscript. R.T. performed bioinformatic analysis, interpreted the data, and revised the manuscript. G.G. performed molecular analysis and revised the manuscript. M.B.D. contributed to the design of the study, provided and interpreted the clinical data, and revised the manuscript. K.P. performed patient recruitment, collected clinical data, and revised the manuscript. C.B. performed bioinformatic analysis, interpreted the data, and revised the manuscript. R.J.L. contributed to the design of the study, provided and interpreted the clinical data, and revised the manuscript. M.B. contributed to the design of the study, performed bioinformatic analysis, interpreted the data, and revised the manuscript. P.J.L. formulated the study, interpreted the data, and cowrote the manuscript.
STUDY FUNDING
This work was funded in part by National Health and Medical Research Council Australia Program Grant 490037 to D.J.A. and M.B. A.P.L.M. and R.T. are supported by APA scholarships funded by the Australian Government. M.B. is supported by an ARC Future Fellowship (FT100100764), and P.J.L. is supported by an NHMRC Career Development Fellowship (APP1032364). R.J.L. is supported by a Melbourne Children's Clinician-Scientist Fellowship. This work was made possible through the Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
DISCLOSURE
Dr. Amor has received research support from Cancer Council Australia, NHMRC, and Cancer Australia & Prostate Cancer Foundation of Australia. Ms. Marsh reports no disclosures. Dr. Storey has served on the scientific advisory board of the Bethlehem-Griffiths Foundation (received payment in kind); has served on the editorial board of Cerebellum and Ataxias; and has received research support from the National Health and Medical Research Council of Australia and NIH. Mr. Tankard has received research support from the Australian Postgraduate Award scholarship. Ms. Gillies has received research support from NHMRC. Dr. Delatycki has served on the editorial boards of BMC Neurology and the Journal of Pediatric Genetics and has received research support from NHMRC, FARA USA, and FARA Australia. Ms. Pope has received research support from NHMRC. Ms. Bromhead reports no disclosures. Dr. Leventer has received research support from NHMRC and the Campbell Edwards Trust. Dr. Bahlo has served on the scientific advisory boards of the Macular Telangiectasia (MacTel) Consortium and the Department of Health (Australian Government); has received travel funding/speaker honoraria for the 7th International Conference on Genomics & Bio-IT APAC 2012, the International Mathematics Institute conference (Pacific Region), the Australian Statistical Society Conference, the International Stroke Genetics Meeting, the Human Genome Meeting, AGTA, and the MacTel Consortium Meeting; has served on the editorial board of Statistical Applications in Genetics and Molecular Biology; holds a patent for Method of determining response to treatment with immunomodulatory composition Details genetic markers that predict response to Hepatitis C treatment; has been a consultant for the MacTel Consortium, the Human Genetics Society of Australia, the Australian Statistical Society-Institute of Mathematical Statistics, the International Conference on Systems Biology, the Sixth Barossa Meeting, and the 8th International Conference on Genomics & Bio-IT APAC; and has received research support from NHMRC, the Australian Research Council (ARC), the Victorian Life Sciences Computing Initiative (VLSCI), the Royal Melbourne Hospital Home Lottery Research Awards, the Leukemia Foundation National Research Program, the ARC Future Fellowship, ARC Linkage Grant, DEST International Science Linkages grant, the Human Genetics Society of Australia, the Epilepsy Society of Australia, the Royal Statistical Society, the Institute of Mathematical Statistics, the Biometrics Society of Australasia, the Australian Statistics Society, the American Society of Human Genetics, and the International Genetic Epidemiology Society. Dr. Lockhart has served on the editorial boards of Genetics Research International and Open Access (OA) Genetics and has received research support from NHMRC. Go to Neurology.org/ng for full disclosures.
REFERENCES
- 1.Ferdinandusse S, Denis S, Mooyer PA, et al. Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann Neurol 2006;59:92–104. [DOI] [PubMed] [Google Scholar]
- 2.Wanders RJA, Barth PG, Heymans HSA. Single peroxisomal enzyme deficiencies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Molecular and Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 2001:3219–3256. [Google Scholar]
- 3.Demain LA, Urquhart JE, O'Sullivan J, et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet Epub 2016 Mar 11. doi: 10.1111/cge.12776. [DOI] [PubMed]
- 4.Lieber DS, Hershman SG, Slate NG, et al. Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4-deficiency. BMC Med Genet 2014;15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lines MA, Jobling R, Brady L, et al. Peroxisomal D-bifunctional protein deficiency: three adults diagnosed by whole-exome sequencing. Neurology 2014;82:963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMillan HJ, Worthylake T, Schwartzentruber J, et al. Specific combination of compound heterozygous mutations in 17beta-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency. Orphanet J Rare Dis 2012;7:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierce SB, Walsh T, Chisholm KM, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet 2010;87:282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Munter S, Verheijden S, Regal L, Baes M. Peroxisomal disorders: a review on cerebellar pathologies. Brain Pathol 2015;25:663–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson GR, Sim JC, McLean C, et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology. Am J Hum Genet 2014;95:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson GR, Sunley J, Smith KR, et al. Mutations in SH3PXD2B cause Borrone dermato-cardio-skeletal syndrome. Eur J Hum Genet 2014;22:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amor DJ, Delatycki MB, Gardner RJM, Storey E. New variant of familial cerebellar ataxia with hypergonadotropic hypogonadism and sensorineural deafness. Am J Med Genet 2001;99:29–33. [DOI] [PubMed] [Google Scholar]
- 13.Ferdinandusse S, Ylianttila MS, Gloerich J, et al. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am J Hum Genet 2006;78:112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crane DI. Revisiting the neuropathogenesis of Zellweger syndrome. Neurochem Int 2014;69:1–8. [DOI] [PubMed] [Google Scholar]