

Abstract
Within hours after stroke, potentially cytotoxic pro-inflammatory mediators are elevated within the brain; thus, one potential therapeutic strategy is to reduce them and skew the brain toward an anti-inflammatory state. Because interleukin-4 (IL-4) treatment induces an anti-inflammatory, “alternative-activation” state in microglia and macrophages in vitro, we tested the hypothesis that early supplementation of the brain with IL-4 can shift it toward an anti-inflammatory state and reduce damage after transient focal ischemia. Adult male rat striata were injected with endothelin-1, with or without co-injection of IL-4. Inflammation, glial responses and damage to neurons and white matter were quantified from 1 to 7 days later. At 1 day, IL-4 treatment increased striatal expression of several anti-inflammatory markers (ARG1, CCL22, CD163, PPARγ), increased phagocytic (Iba1-positive, CD68-positive) microglia/macrophages, and increased VEGF-A-positive infiltrating neutrophils in the infarcts. At 7 days, there was evidence of sustained, propagating responses. IL-4 increased CD206, CD200R1, IL-4Rα, STAT6, PPARγ, CD11b, and TLR2 expression and increased microglia/macrophages in the infarct and astrogliosis outside the infarct. Neurodegeneration and myelin damage were not reduced, however. The sustained immune and glial responses when resolution and repair processes have begun warrant further studies of IL-4 treatment regimens and long-term outcomes.
Keywords: Alternative activation, Gliosis, Interleukin 4, Neuroinflammation, Neutrophil entry, Phagocytic microglia, Transient brain ischemia.
INTRODUCTION
Stroke is the third most prevalent cause of death worldwide and a leading contributor to long term disability (1). The first several days are a critical period, with numerous detrimental outcomes for which modifying treatments are being sought. In animal models of transient ischemia, neurons in the infarct degenerate within a week (2, 3). During this time the brain mounts an inflammatory response that begins with microglia and astrocytes and is followed by entry of circulating innate immune cells (4, 5). The inflammatory response includes an initial release of cytokines, chemokines, and reactive oxygen and nitrogen species that can exacerbate damage, alter the blood-brain barrier, and recruit additional immune cells. Some toxic pro-inflammatory cytokines increase within hours in the infarct (6). This suggests that therapies targeting inflammation should be administered quickly after a stroke (7). However, broad-spectrum anti-inflammatory treatments have not translated well in early clinical trials (7). In pursuing new therapeutic approaches and focusing on more selective targets; a major concern is that not enough is known about inflammatory responses after cerebral ischemia.
A more recent experimental approach is to manipulate “activation” of innate immune cells to modify brain inflammation. Microglia and infiltrating macrophages and neutrophils rapidly respond to environmental cues but the outcomes are complex. Macrophages (8, 9) and microglia (10, 11) exhibit a range of responses from pro-inflammatory (often called M1 or classical activation) to multiple anti-inflammatory or resolving (often called M2) states. Neutrophils contribute to the early pro-inflammatory response, entering the brain by 1–3 days after ischemia in rodent models, after which they are phagocytosed by microglia and macrophages (12, 13). Astrocytes form a glial scar to help contain the damage but also contribute to the inflammatory response (14). Because all of these cell types potentially contribute to neuroinflammation, identifying and testing therapeutic targets requires a better understanding of cellular responses in vivo.
Much of our understanding of macrophage and microglial activation comes from in vitro studies on isolated cells. Classical activation (M1) is commonly evoked by bacterial lipopolysaccharide (LPS), with or without interferon-γ. This state is regarded as pro-inflammatory and cytotoxic, and is often monitored by upregulation of tumor necrosis factor α (TNF), inducible nitric oxide synthase (iNOS/NOS2), interleukin (IL)-1β, and IL-6. Alternative activation (also called M2a), is thought to help in tissue repair and homeostasis, and can be experimentally induced by IL-4. It is usually monitored by upregulation of 1 or more of arginase 1 (ARG1), Ym1 (chitinase 3-like 3), “found in inflammatory zone 1” (FIZZ1), CD206 (mannose receptor), chemokine (C-C motif) ligand 22 (CCL22), and CD163 (haptoglobin-hemoglobin scavenger receptor (10, 15, 16). In the healthy brain, IL-4 levels are very low (17, 18), and microglia can respond to in vivo IL-4 injection (19). IL-4 receptors have also been detected on neutrophils (20), astrocytes (18), T lymphocytes (21), endothelial cells (22), oligodendrocytes (23), and some neurons (24, 25); all of these potentially could respond to injected IL-4.
Many reports show that harmful pro-inflammatory mediators are elevated in the brain within minutes to hours after ischemia (26, 27). Therefore, we tested the hypothesis that injecting IL-4 into the ipsilateral striatum at the onset of ischemia would skew the brain toward an anti-inflammatory state, and reduce damage. To assess the outcome, we monitored molecular and cellular responses, and damage to neurons and white matter during the first week, the time during which a pronounced inflammatory reaction develops.
MATERIALS AND METHODS
Transient Focal Ischemia and IL-4 Treatment
All experimental procedures were performed in accordance with the University Health Network animal care committee (Animal Use Protocol no. 1132), under guidelines established by the Canadian Council on Animal Care. We used a popular rat model of transient focal ischemia in which the vasoconstrictor, endothelin-1 (ET-1), is injected into the striatum. Adult male Sprague-Dawley rats at 3‒4 months old (250‒300 g) (Charles River; St Constant, QC, Canada) were anesthetized using isoflurane (5% induction, 1.5%–2% maintenance), and placed in a stereotaxic instrument (Stoelting, Wood Dale, IL). Body temperature was controlled with a thermostatically regulated heating pad; heart rate, respiratory rate and arterial oxygen saturation were monitored during the surgery and recovery from the anesthetic using the MouseOx system (Starr Life Sciences, St. Laurent, QC, Canada). A 2-cm-long incision was made to expose the skull. A 1-mm-diameter burr hole was drilled (0.2 mm anterior and 3.5 mm lateral to bregma) and a 27-gauge needle was lowered into the center of the right striatum (6 mm ventral). ET-1 (Calbiochem, EMD Biosciences, Gibbstown, NJ) (400 pmol in 2 μl sterile saline), was injected at a rate of 500 nl/minute using an UltraMicroPump II (World Precision Instruments, Sarasota, FL). At the end of each injection, the needle was left in place for 5 minutes to minimize backflow. Because ET-1 potency can vary, we first tested several concentrations before conducting the experimental surgeries. We found that 400 pmol created a reproducible ischemic lesion that was restricted to the striatum, as monitored by staining with 2% TTC (2,3,5-triphenyl tetrazolium chloride). This is consistent with our previous work using this model (28–31).
The ET-1 model produces several hallmark features of injury seen in humans after ischemic stroke, including accumulation of activated innate immune cells (microglia, neutrophils, macrophages), neuron death, axon damage, demyelination, and formation of a glial scar (29, 30, 32, 33). We favor the ET-1 model because, in our hands, there is very little mortality, the reproducible lesion can be confined to the striatum, and we have previously characterized some aspects of white-matter damage and microglia/macrophage and astrocyte responses during the first 7 days (28, 30).
In an attempt to skew the ipsilateral striatum toward an anti-inflammatory state at the onset of ischemia, we co-injected ET-1 and 500 ng rat recombinant IL-4 (R&D Systems, Burlington, ON, Canada). The IL-4 dose was selected based on a pilot study (50 ng, 500 ng, 1 µg), which showed that 500 ng was the lowest dose that consistently upregulated expression of anti-inflammatory genes in the striatum. When IL-4 was used, it was mixed with sterile saline containing ET-1 and injected at the same final volume (2 μl). The ischemia control was 2 μl of ET-1 diluted in saline. Sham control animals (no ischemia) were injected with 2 μl saline alone. None of the saline-injected animals died after surgery; 3/29 and 1/31 died following ET-1 and ET-1 + IL-4 surgeries, respectively.
RNA Analysis
Rats were transcardially perfused with 120 ml phosphate buffered saline (PBS). The brains were removed from rats injected with saline (control), ET-1 (ischemia), or ET-1 + IL-4 (treatment group). The ipsilateral striatum was isolated, immediately frozen on dry ice, and stored at ‒40 °C. Three to 6 animals were used at each time point (1, 3, 7 days post-ischemia) for each treatment group. Striata were individually homogenized in TRIzol (Invitrogen, Life Technologies, Burlington, ON, Canada) and total RNA was extracted and purified using the RNeasy Mini Kit (Qiagen, Mississauga, ON, Canada). RNA samples were stored at –80°C until used.
nCounter Gene Expression Assay
For multiplexed analysis, RNA samples (200 ng/striatum) were analyzed using the Nanostring nCounter System at the Princess Margaret Genomics Centre (Toronto, ON, Canada; http://www.pmgenomics.ca/pmgenomics), last accessed 23 August 2016. This assay is quantitative, relatively high-throughput, and has high sensitivity, similar to real-time RT-PCR (34). NanoString nCounter Technologies created a code set containing capture and reporter probes that recognize complementary sequences of 35‒50 base pairs in mRNA for the specified genes (Table). Results were subjected to technical and biological normalizations as follows: Using nSolver software (version 1.1; www.nanostring.com, last accessed 23 August 2016), raw counts were normalized to positive control probes, and then to 3 housekeeping genes that are considered to be useful for stroke studies (35): Hprt1 (hypoxanthine-guanine phosphoribosyltransferase), Sdha (succinate dehydrogenase complex subunit A), and Ywhaz (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide). Expression of each gene was then compared between treatment groups (saline, ET-1, ET-1 + IL-4) and across time (1, 3, 7 days) within each treatment group.
Quantitative Real Time RT-PCR
For neutrophil chemoattractant molecules, mRNA expression was determined by quantitative real time RT-PCR (qRT-PCR) because we added these molecules after the NanoString analysis was completed. cDNA was reverse transcribed from 0.8 μg of RNA using 200 U of SuperScriptII RNase and 0.5 mM dNTPs and 0.5 μM oligodT according to the manufacturer’s instructions (Invitrogen). “Primer3Output” (http://bioinfo.ut.ee/primer3-0.4.0, last accessed 23 August 2016) was used to design the primers as follows. Cxcl1 (chemokine (C-X-C motif) ligand 1) [GenBank:NM_030845.1] forward: 5′-TCGATGGTCGTTCAATTCC-3′ and reverse: 5′-TCATCTCTCCGCCCTTCTT-3′. Cxcl2 [GenBank:NM_053647.1] forward: 5′-CCAGACAGAAGTCATAGCCACTC-3′ and reverse: 5′-CTTTCCAGGTCAGTTAGCCTTG-3′. Ccl3 [GenBank: NM_013025.2] forward: 5′-TGCCCTTGCTGTTCTTCTCT-3′ and reverse: 5′-GGCTGCTGGTCTCAAAATAGTC-3′. Hprt1 [GenBank: NM_012583.2] 5′-TCTCCATCACGTTCCTTTCC-3′ and reverse: 5′-CAAGGGCATATCCAACAACA-3′. Amplification was carried out using an ABI PRISM 7700 Sequence Detection System (PE Biosystems, Foster City, CA) with the following parameters: 95°C for 2 minutes, 40 cycles at 95°C for 5 seconds, 60°C for 30 seconds; followed by dissociation at 95°C for 15 seconds, 60°C for 15 seconds, 95°C for 15 seconds. “No template” and “no amplification” controls were included for each gene. A single peak on each melt curve confirmed amplification specificity. The threshold cycle value for each gene was normalized to that of the housekeeping gene, Hprt1.
Immunohistochemistry
Rats were transcardially perfused with 120 ml PBS, followed by 120 ml 4% paraformaldehyde. Three to 5 animals were used at each time point for each treatment group. The whole brain was harvested, placed in 4% paraformaldehyde solution for 24 hours and cryoprotected by incubating first in 10% sucrose for 6 hours, and then in 30% sucrose until the brain sank. Fixation and cryoprotection steps were carried out at 4 °C. Brains were embedded in freezing medium (Dako-Canada, Mississauga, ON, Canada), flash frozen in a slurry of dry ice and 95% ethanol, and stored at ‒40 °C until coronal sections (16 μm thick) were cut using a cryostat (Model CM350S, Leica, Richmond Hill, ON, Canada). Sections were placed on gelatin-coated slides (1% gelatin, 0.5% chromium potassium sulfate), and stored at ‒40 °C.
For immunohistochemical analysis, slides were allowed to warm to room temperature and then were rehydrated in phosphate buffered Triton X-100 solution (PBT) containing 0.1 mol/l PBS, 0.1% bovine serum albumin, and 0.2% Triton X-100, pH 7.5. The 16-μm-thick sections were outlined with a Pap pen (Ted Pella, Inc. Redding, CA). Following a 2-hour incubation in 10% donkey serum to block non-specific binding, each primary antibody was added as follows. Microglia and macrophages were detected using either a rabbit polyclonal antibody against “ionized calcium-binding adapter-1” ([Iba1] 1:1000; Wako, Osaka, Japan) or a mouse monoclonal antibody against CD11b (OX-42; 1:200; Abcam, Cambridge, MA). Neutrophils were labeled with a rabbit polyclonal antibody against polymorphonuclear leukocyte antigen ([PMN] 1:10,000; Cedarlane, Burlington, ON, Canada). A mouse monoclonal antibody against the lysosome-associated glycoprotein, CD68 (1:200; Serotec, Raleigh, NC) was used as an indicator of phagocytic cells. Astrocytes were identified using a rabbit polyclonal antibody against glial fibrillary acidic protein ([GFAP] 1:500; Sigma-Aldrich, Oakville, ON, Canada). Healthy myelin was labeled with a mouse monoclonal antibody that recognizes myelin basic protein ([MBP] 1:100; Sigma-Aldrich), as before (29, 30, 33). Damaged myelin was labeled with a rabbit polyclonal antibody against “degraded myelin basic protein” ([dMBP] 1:250; Chemicon, Temecula, CA), as before (29, 30, 33). A mouse monoclonal antibody was used to detect vascular endothelial growth factor A ([VEGF-A] 1:100; Santa Cruz Biotechnology, Santa Cruz, CA). After overnight incubation with each primary antibody, followed by washing in PBT (3 × 15 minutes each), slides were incubated for 2 hours with Dylight-conjugated secondary antibodies: donkey anti-mouse-488, donkey anti-rabbit-594 (1:400; Jackson Immunoresearch, West Grove, PA). All sections were then incubated for 10 minutes with the nuclear stain 4′-6-diamidino-2-phenylindole ([DAPI] 1:5000 Sigma-Aldrich), then washed in PBS (3 × 15 minutes each). Slides were cover-slipped using mounting medium (Dako-Canada) and stored at ‒4 °C until imaged.
Fluoro-Jade B Histochemistry
To assess degenerating neurons, we stained tissue sections with a polyanionic fluorescein derivative, Fluoro-Jade B (FJB) (Histochem, Inc., Jefferson, AR), which is commonly used after CNS injury (36, 37). Frozen sections from 3 animals from each group (ET-1, ET-1 + IL-4) were stained at each time point (1, 3, 7 days). Slides were immersed for 3 minutes in 100% ethanol, 1 minute in 70% ethanol, and rinsed for 1 minute in distilled water. They were then immersed in a 0.06% solution of potassium permanganate, shaken gently for 15 minutes, rinsed for 1 minute in distilled water, and then placed in 0.001% FJB for 30 minutes with gentle shaking, before rinsing in distilled water (3 × 1 minute each). The slides were left to dry in a fume hood for 30 minutes, cleared in 100% xylene (3 × 2 minutes each), cover-slipped with DPX mounting medium (Sigma-Aldrich) and stored at ‒4 °C until imaged.
Sampling Procedures, Quantification and Statistical Analysis
Labeled sections (immunohistochemistry, FJB) were examined with a confocal microscope (LSM 700 META, Zeiss, Oberkochen, Germany). Results were quantified using ImageJ software (Version 1.33K, NIH). To reduce variability, labeling of all tissue sections was done in a single batch, and all imaging for a given stain was completed on the same day. Pinhole and exposure times were determined at the start of each imaging session and held constant throughout. All cell counts were made manually by an observer blinded to the treatment. For each animal, cell staining was averaged from 4 sampling boxes of 320 µm2 each (CD11b; VEGF-A, PMN), 5 sampling boxes of 300 µm2 (MBP, dMBP), or 6 sampling boxes of 200 µm2 (Iba1, CD68; PMN, CD68; FJB; GFAP) taken from the caudate putamen. Results were then averaged for 3–5 animals. To count total numbers of microglia/macrophages (Iba1-positive or CD11b-positive) or neutrophils (PMN-positive), only cells with visible DAPI-stained nuclei were included. To identify cells that were likely phagocytic, sections were double labeled with Iba1 and CD68 (microglia/macrophages) or with PMN and CD68 (neutrophils). Following immunostaining, tissue sections were stained with DAPI to label nuclei to aid in cell counting. Similarly, we assessed the number of VEGF-A expressing neutrophils as PMN-positive, VEGF-A-positive. For CD68, MPB, dMBP, and GFAP staining, digital image analysis was conducted much as previously described (38). That is, area fractions were quantified in ImageJ (http://rsbweb.nih.gov/ij, last accessed 23 August 2016) after subtracting the background fluorescence and converting the images to gray scale. The threshold was adjusted to capture pixels representing cells or staining areas of interest. Then the area fraction was measured. For MBP and dMBP staining, area fractions were not adjusted to a threshold; instead, values represent the proportion of labeled pixels in the entire image (% of total).
Statistical Analyses
All statistical analyses were carried out using GraphPad Prism software (version 6.0, San Diego, CA). All graphical data are reported as the mean ± SE. Statistical differences between saline (sham controls), ET-1 (ischemic controls), and ET-1 + IL-4 (treated animals) were assessed using a 2-way ANOVA, with time (1, 3, 7 days) and treatment as independent variables. When comparing only ischemic (ET-1) versus treated animals (ET-1 + IL-4), we used a 1-way ANOVA (when also comparing with saline) or Student t-test (for ET-1 vs ET-1 + IL-4 only). Post-hoc tests were Bonferroni’s (for 2-way ANOVA) or Tukey’s (for 1-way ANOVA).
RESULTS
Early IL-4 Treatment Skewed the Striatum Toward an anti-Inflammatory Phenotype After Ischemia
First, we monitored effects of ischemia by comparing the striatum of saline-injected control rats with the ipsilateral striatum of ET-1-injected rats at 1, 3, and 7 days. Then, in a separate, treated cohort, IL-4 was injected into the striatum at the same time and location as ET-1 to ensure that it was rapidly delivered to the site of damage. Because IL-4 levels are normally very low (17), the injected IL-4 is expected to predominate. To assess whether early IL-4 treatment skewed the brain response, we compared transcript expression of a panel of genes (Table 1), which were chosen as explained in the “Introduction” and “Discussion” sections.
In saline-injected controls, both pro-inflammatory (Fig. 1A) and anti-inflammatory genes (Fig. 1B) were expressed at low levels (arbitrary cutoff <100 counts/200 ng mRNA sample) at all times tested. The IL-4 receptor (Ilr4) and signaling molecule (Stat6; signal transducer and activator of transcription 6) were moderately expressed. (1) Ischemia alone (ET-1) evoked an early pro-inflammatory response, with increases in transcript levels at 1 day in Tnf (TNF-α 4-fold), Nos2 (6.4-fold), and Il6 (11.5-fold). Among the anti-inflammatory genes, only Chi3l3 (Ym1) increased (5.1-fold at 7 days); however, because the entire ipsilateral striatum was sampled, specific cellular responses will be underestimated. At 3 and 7 days, several genes were trending toward increased expression: Arg1, Ppargγ (peroxisome proliferator-activated receptor-γ), IL4r, Stat6, and Mrc1 (CD206), which might reflect later immune cell responses. (2) IL-4 treatment (ET-1 + IL-4) had a striking effect on the transcript levels of genes associated with an anti-inflammatory state (Fig. 1B). Relative to ischemic animals at 1 day, expression was increased for Arg1 (5.8-fold), Ccl22 (16.5-fold), Cd163 (5.3-fold), and Pparγ (3.4-fold). Arg1 remained elevated at 3 days (3.1-fold). Several genes were elevated at 7 days: Pparγ (1.8-fold), IL4r (2.2-fold), Stat6 (1.8-fold), and Mrc1 (2.2-fold). These results indicate that early IL-4 injection successfully skewed the ischemic striatum toward an anti-inflammatory state, and that several effects on gene expression lasted for 7 days, the longest time examined. Interestingly, transcript expression of the pro-inflammatory mediators were relatively low and IL-4 treatment did not significantly change them (Fig. 1A).
FIGURE 1.

IL-4 skews the infarcted striatum toward an anti-inflammatory phenotype. Rats were injected with saline (sham control, black bars), ET-1 (ischemic control; white bars) or ET-1 + 500 ng of rat recombinant IL-4 (ET-1 + IL-4; gray bars). Transcript levels of pro-inflammatory (A), anti-inflammatory (B) and other immunomodulatory molecules (C) were quantified in the ipsilateral striatum at 1, 3, and 7 days. The symbols represent: * differences from saline-injected shams; † time-dependent changes within a treatment group; # IL-4 effects. One symbol indicates p < 0.05; 2 symbols, p < 0.01; 3 symbols, p < 0.001.
Next, we examined expression of several immunomodulatory molecules and receptors: Itgam (CD11b), CD68, toll-like receptor 2 (Tlr2), Tlr4, Cx3cr1 (fractalkine receptor), Cd200r1, transforming growth factor beta 1 (Tgfβ1), and heme oxygenase-1 (Hmox1 (HO-1)). In saline-injected animals, their transcript levels were very low (Fig. 1C). Ischemia evoked a delayed increase in most of these genes, and by 7 days, all (except Tlr4 and Hmox1) were significantly elevated: Itgam by 4-fold, Cd68 (6.4-fold), Cx3cr1 (3.1-fold), Cd200r1 (5.2-fold), Tgfβ1 (4.2-fold), and Tlr2 (3.8-fold). IL-4 treatment further increased transcript levels of several genes at 7 days compared with ischemia alone: Itgam (by a further 1.6-fold), Cd200r1 (1.8-fold), Tlr2 (1.7-fold), Tlr4 (1.7-fold), and Hmox1 (3.6-fold). These gene responses provide evidence that a single IL-4 injection at the time of ischemia induction can produce long-lasting changes. The next question was whether immune cell responses are sustained.
Effects of IL-4 Treatment on Microglia/Macrophages in the Infarct
We studied the striatum because it a common site of lacunar infarcts; and its unique architecture facilitates analysis of damage to neurons and myelinated axon bundles in the same location. Figure 2A shows the naïve striatum, illustrating the appearance of MBP-labeled myelinated white-matter tracts and Iba-1-labeled “resting” microglia. Microglial tiling, which reflects their territorial organization, shows most cell bodies in the neuropil region, with processes often extended into white-matter bundles. The infarct shows rapid damage to neurons and myelinated axon bundles, maximal accumulation of inflammatory cells (microglia, macrophages, neutrophils). Because this was the location where IL-4 was injected, that is where it likely exerted its greatest effects. Using the same ischemia model, we previously demonstrated that the infarct corresponds with numerous cellular responses, including decreased NeuN and GFAP staining at 1 day, accumulation of Iba1- and CD68-labeled microglia/macrophages within the boundaries of the GFAP-labeled glial scar (beginning at 1 day; massive by 3 and 7 days), accumulation of SC1/hevin and amyloid precursor protein (APP) in damaged axon bundles, as well as pronounced vimentin accumulation at the edge of the ischemic infarct (28, 30). In addition, we have shown that there is progressive loss of normal MBP and increase in degraded MBP in the infarct (29, 30).
FIGURE 2.

IL-4 treatment increases phagocytic microglia/macrophages in the infarct. (A) Images of the naïve rat striatum show microglia labeled with Iba1 (red), and white-matter bundles labeled with MBP (green). Scale bar = 50 μm and applies to all main images in panels A, B, D. (B) Representative images of the ischemic infarct at 1 and 7 days after ET-1–induced transient focal ischemia, with and without IL-4 treatment, labeled for CD11b (microglia/macrophages; green). Nuclei are labeled with DAPI (blue). In the 7 day ischemia image, the higher-magnification inset illustrates how cells were recognized for quantification (scale bar = 10 μm). (C) Summary of the number of CD11b-positive microglia/macrophages at 1 and 7 days. (D) IL-4 treatment increases CD68-labeled microglia/macrophages at 1 day after ischemia. Representative images from the infarct with and without IL-4 treatment at 1, 3, and 7 days. Microglia/macrophages were labeled for Iba1 (red) and the glycoprotein, CD68 (green). The images at 1 day show examples of CD68-positive Iba1-positive cells (open arrows) and CD68-positive Iba1-negative cells (closed arrows). Scale bar = 100 μm and applies to all images. (E) Summary of CD68 labeling expressed as area fraction at 1, 3, and 7 days. F: The 1 day tissue was further analyzed by counting the number of CD68-positive cells and double-labeled microglia/macrophages (Iba1-positive, CD68-positive), as well as the percentage of total Iba1-positive microglia/macrophages that were CD68-positive. For all graphs, values are shown as mean ± SE. The symbols represent: * differences from saline-injected shams; † time-dependent changes within a treatment group; # IL-4 effects. One symbol indicates p < 0.05; 3 symbols, p < 0.001.
Here, we asked whether IL-4 treatment affects the accumulation of microglia/macrophages in the infarct. At 1 day after ischemia, CD11b-positive microglia/macrophages were evident, and by 7 days they had massively infiltrated the infarct (Fig. 2B). Because CD11b staining was mainly on the cell surface (see green ring in Fig. 2B, inset), it was relatively easy to count CD11b-positive cells. At 1 day after ischemia, there were similar numbers of CD11b-positive cells with and without IL-4 treatment, and these numbers were similar to saline-injected animals (Fig. 2C). At 7 days, ischemia evoked a 12-fold increase in CD11b-positive cells; and IL-4 treatment doubled their numbers. Therefore, IL-4 increased the number of microglia/macrophages that infiltrate the infarct 1 week after ischemia.
Next, we asked whether IL-4 treatment affects the numbers of microglia/macrophages expressing CD68, a lysosomal glycoprotein that indicates increased phagocytic activity (39). Microglia do not express CD68 in the healthy brain but activated microglia and macrophages become CD68-positive after acute CNS damage, including focal ischemia and intracerebral hemorrhage (ICH) (30, 31, 40). Similar to CD11b staining (Fig. 2B), Iba1-positive microglia/macrophages were prevalent in the infarct (Fig. 2D), and their densities increased with time. By 7 days, Iba1-positive microglia/macrophages completely filled the infarct, both inside and outside white-matter bundles, as previously shown (30). CD68 staining also progressively increased in the infarct and CD68-positive cells filled the white-matter bundles (Fig. 2D). IL-4 treatment did not significantly change the area fraction of CD68 staining (Fig. 2E). However, at 1 day, when the cells were easy to count using these stains, IL-4 treatment increased the numbers of CD68-positive and double-labeled cells (Iba1-positive, CD68-positive) (Fig. 2F). The 1.7-fold increase in the percentage of CD68-labeled microglia/macrophages suggests that IL-4 increased their phagocytic activity. Finally, at 1 day after ischemia we noted that IL-4 treatment increased (from 18% to 42%) a population of CD68-positive cells that did not label with Iba1, and thus, were not microglia or macrophages. Therefore, we next sought to identify these cells.
IL-4 Treatment Increased Neutrophils in the Infarct
In this model of ischemia, we and others have shown that neutrophils enter the infarct by 1 day and that their numbers decrease by 3 days (13, 30). Here, we asked whether the CD68-positive cells that did not label with Iba1were neutrophils. Using an antibody directed against PMN leukocytes, we observed many PMN-positive neutrophils in the infarct at 1 day (Fig. 3A). IL-4 treatment increased their numbers by 2.6-fold, and similarly increased (by 2.4-fold) the number of double-labeled (PMN-positive CD68-positive) neutrophils (Fig. 3B). IL-4 did not change the percentage of neutrophils that were CD68-positive.
FIGURE 3.
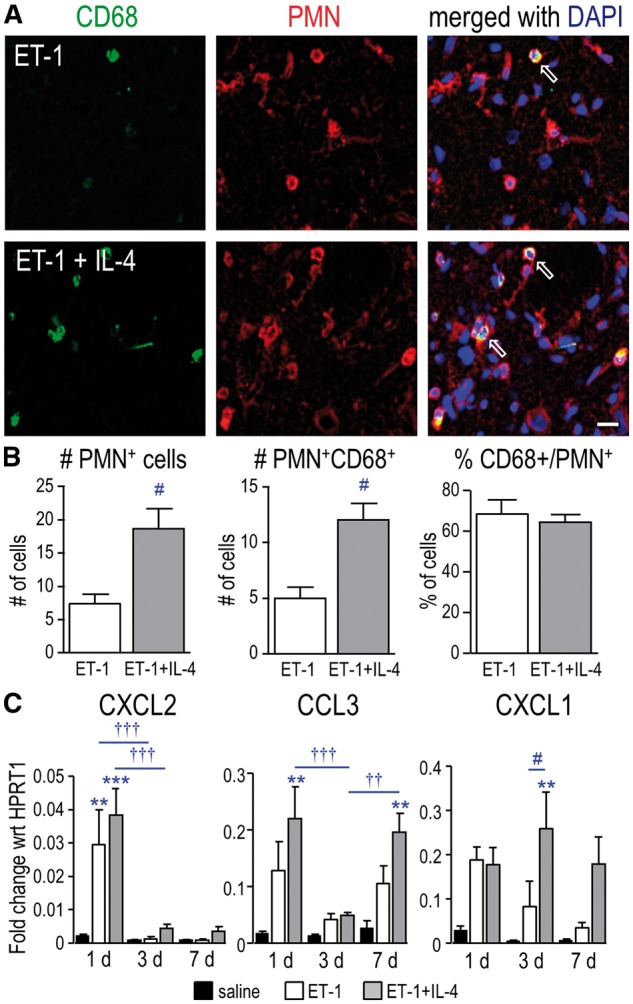
IL-4 treatment increases neutrophils in the infarct at 1 day after ischemia. (A) Representative images from the infarct at 1 day. Cells were labeled with antibodies against CD68 (green) and PMN leukocytes (neutrophils; red). Nuclei were labeled with DAPI (blue). Examples show double-labeled PMN-positive, CD68-positive cells (open arrows). Scale bar = 20 μm and applies to all panels. (B) Summary of the numbers of PMN-positive cells and double-labeled cells (PMN-positive, CD68-positive) per 200 µm2, as well as the percentage of total PMN-positive cells that were CD68-positive at 1 day. (C) Transcript levels of neutrophil chemoattractant molecules were determined by qRT-PCR at 1, 3, and 7 days. The symbols represent: * differences from saline-injected shams; † time-dependent changes within a treatment group; and # IL-4 effects. One symbol indicates p < 0.05; 2 symbols, p < 0.01; 3 symbols, p < 0.001.
Because IL-4 treatment apparently increased neutrophil entry at 1 day without altering their phagocytic phenotype, we next asked whether it evoked an early increase in expression of molecules known to influence neutrophil extravasation and/or vascular permeability. CXCL2, CCL3, and CXCL1 are well-known neutrophil chemoattractants (41). At 1 day after ischemia, transcript levels of all 3 molecules increased but IL-4 treatment did not further affect them (Fig. 3C). At later times, only Cxcl1 expression was increased in IL-4 treated animals. VEGF-A regulates neutrophil transmigration in vitro (42) and in vivo (43). Strikingly, at 1 day after ischemia, IL-4 treatment increased transcript levels of Vegfa (Fig. 4A), increased the number of VEGF-A–labeled neutrophils (PMN-positive, VEGF-A-positive cells; Fig. 4B) by 5-fold (Fig. 4C), and doubled the percentage of neutrophils expressing VEGF-A. Thus, IL-4 treatment increased neutrophil entry.
FIGURE 4.
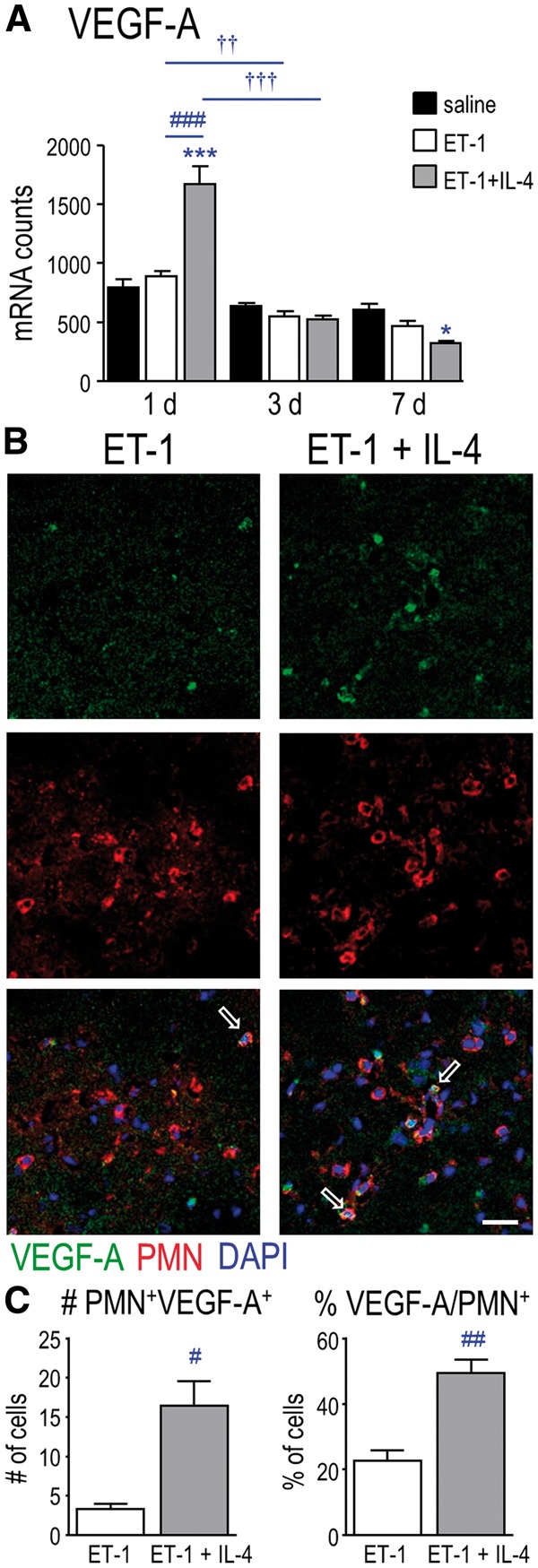
IL-4 treatment increased VEGF-A expression in neutrophils at 1 day after ischemia. (A) Transcript levels of Vegfa were monitored at 1, 3, and 7 days. (B) Representative images from the infarct 1 day after ischemia. Cells were labeled for VEGF-A (green) and PMN (neutrophils; red). Nuclei were labeled with DAPI (blue). Examples show double-labeled PMN-positive, VEGF-A-positive cells (open arrows). Scale bar = 25 μm and applies to all panels. (C) Summary of double-labeled cells (PMN-positive, VEGF-A-positive) per 320 µm2, and the percentage of PMN-positive cells that were VEGF-A-positive at 1 day. For all graphs, 1 symbol indicates p < 0.05; 2 symbols; p < 0.01; 3 symbols, p < 0.001.
IL-4 Treatment Did Not Prevent Early Neurodegeneration or Myelin Damage
If early IL-4 treatment was beneficial, we had expected that it would greatly decrease damage to neurons and white matter after ischemia. FJB labels degenerating neurons (36, 37) and, in the undamaged contralateral striatum, there were no FJB-labeled neurons at 1 day (Fig. 5A), 3 or 7 days (not shown). In contrast, in the infarct, there were many FJB-positive cells, and their numbers peaked at 1 day (Fig. 5A, B). Rather than reducing neurodegeneration, IL-4 treatment increased the number of FJB-positive cells at 1 day (by 29%), and this coincided with the increased neutrophil entry shown in Figure 3. As neurons were lost over time (3 and 7 days); the number of FJB-positive cells decreased with and without treatment.
FIGURE 5.
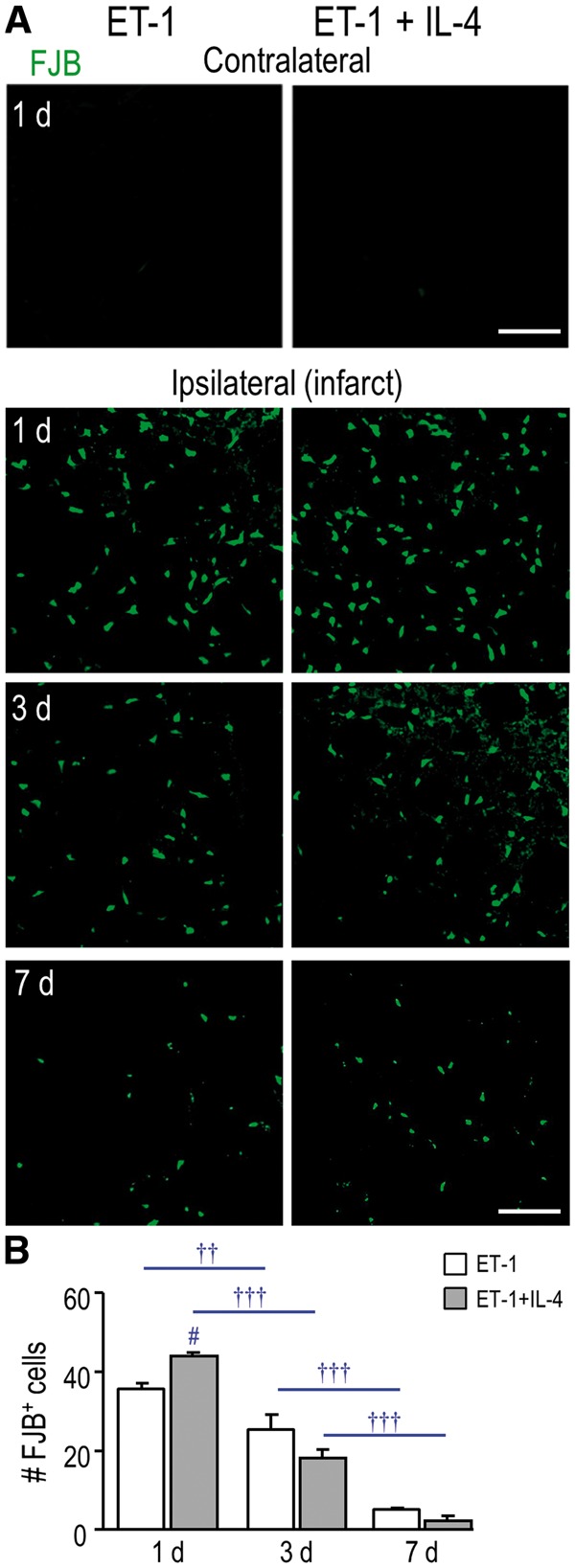
IL-4 treatment did not rescue neurodegeneration in the infarct. (A) Representative images show FJB (green), which was used to identify degenerating neurons. The contralateral and ipsilateral striatum (infarct region) from the same rat are shown (either untreated or IL-4 treated) for 1 day. The ipsilateral infarct is also shown for 3 and 7 days. Scale bar = 100 μm and applies to all images. (B) Summary of the number of FJB-labeled cells per 200 µm2 in the infarct at 1, 3, and 7 days. The symbols represent: † time-dependent changes within a treatment group; # IL-4 effects. One symbol indicates p < 0.05; 2 symbols, p < 0.01; 3 symbols, p < 0.001.
Using this ischemia model, we previously showed that white-matter damage progresses over the first week, as indicated by decreased staining for MBP and increased dMBP (30, 33). At 1 day after ischemia, normal MBP staining was seen in the white-matter bundles and neuropil (Fig. 6A). By 3 days, bundles in the infarct contained both MBP and dMBP, and by 7 days, only dMBP was seen. IL-4 treatment did not alter these temporal changes in MBP and dMPB staining (Fig. 6B and C). Thus, the progressive myelin damage seen during the first week in this model was not rescued by IL-4 treatment.
FIGURE 6.
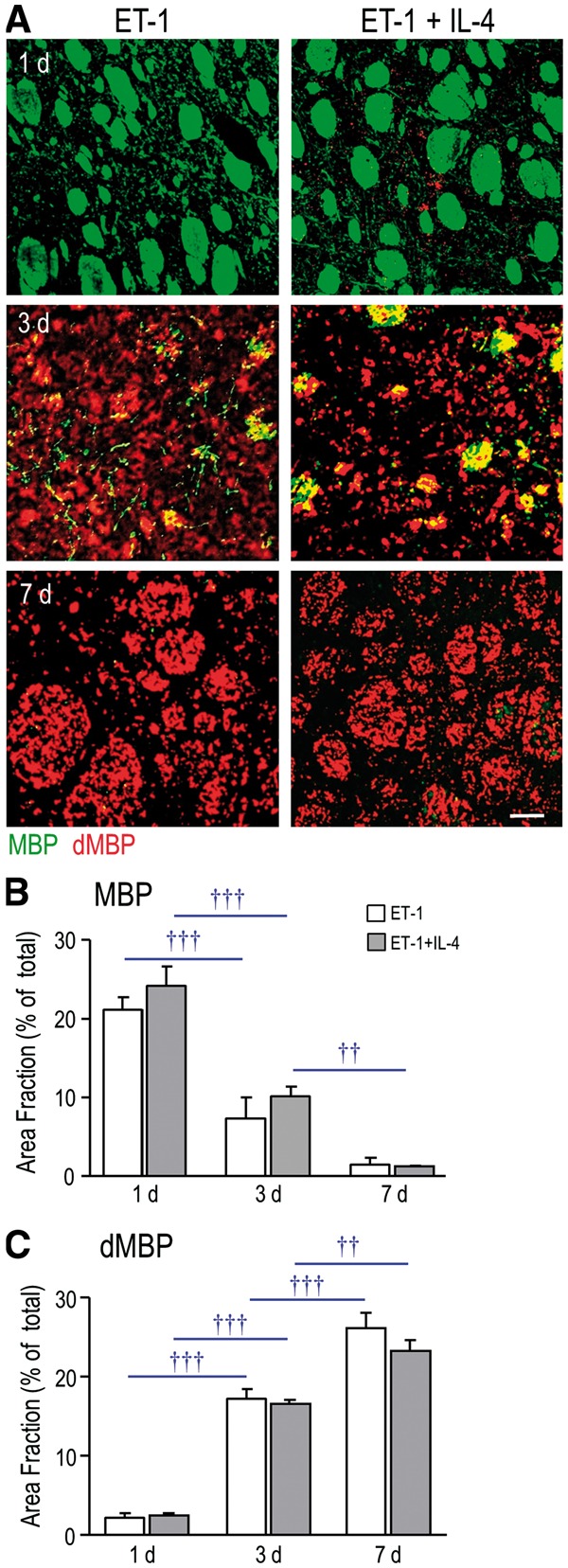
IL-4 treatment did not rescue the progressive myelin damage after ischemia. (A) Representative images of MBP (green) and dMBP (red) staining after ischemia, with and without IL-4 treatment. Scale bar = 50 μm and applies to all images. (B, C) Summary of area fraction of staining (stained area/total area) per 300 µm2 for MBP and dMBP. Time-dependent changes within each treatment group are indicated as †† p < 0.01 and ††† p < 0.001.
IL-4 Treatment Increased Astrogliosis Outside the Infarct
In this ischemia model, reactive astrocytes form a glial scar that begins by 3 days, is well formed by 7 days, and clearly delineates the infarct region in which neurons and astrocytes die (28, 30). In the present study, ischemia alone increased GFAP transcript expression at 3 days, and IL-4 treatment increased it at 7 days (Fig. 7A). Subsequently, we compared GFAP staining in several locations. As expected, in the contralateral striatum, staining was typical of non-activated astrocytes, with GFAP in the cell body and along thin primary processes (Fig. 7B). In contrast, astrocytes within glial scars lose their individual territorial domains and acquire long processes that overlap and are oriented toward the lesion (44). At 7 days after ischemia there was a pronounced glial scar at the edge of the infarct (Fig. 7B), which was accompanied by a 2-fold increase in the area fraction of GFAP staining. GFAP staining in the glial scar was not affected by IL-4 treatment (Fig. 7C). However, in the surrounding striatum beyond the glial scar, reactive astrocytes possessed hypertrophied processes, and IL-4 treatment had striking effects. GFAP staining was much more prominent (Fig. 7B) and the area fraction of staining increased 1.7-fold (Fig. 7C), indicating that IL-4 treatment increased the astroglial response in brain tissue outside the infarct.
FIGURE 7.
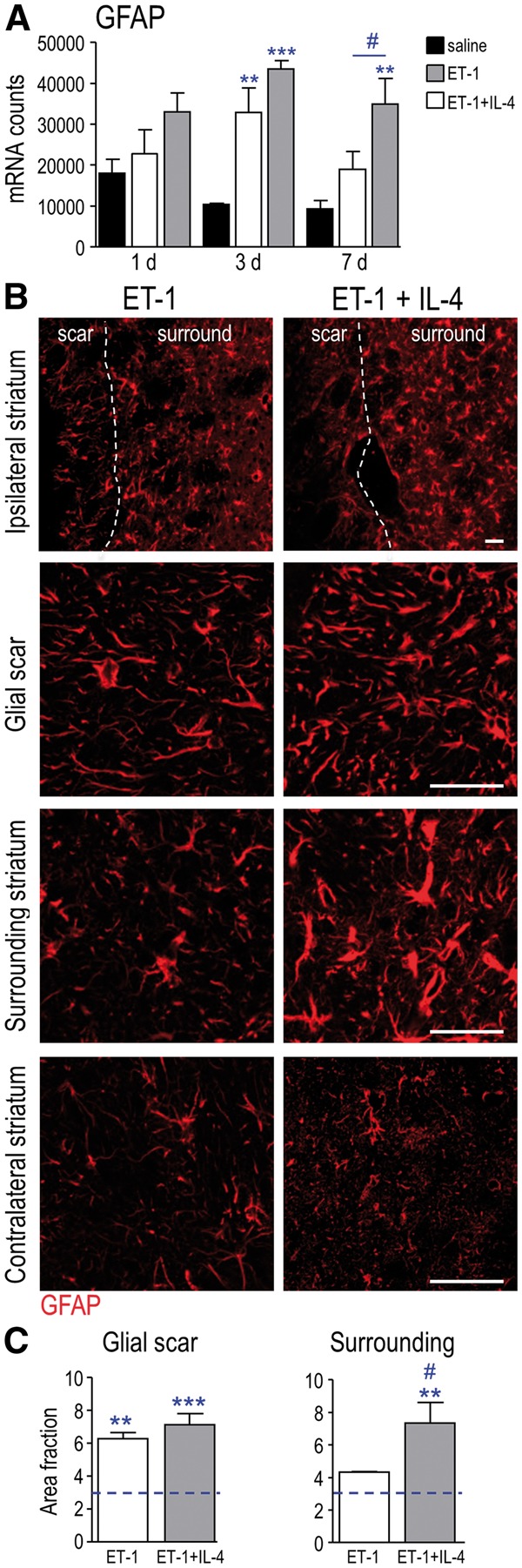
Effects of IL-4 treatment on glial scar formation and astrogliosis. (A) Transcript expression of GFAP from saline-injected sham controls; and at 1, 3, and 7 days after ischemia, with or without IL-4 treatment. (B) Representative images of immunostaining for GFAP at 7 days after ischemia, showing the approximate boundary of the glial scar (dashed line). Images displaying each region (glial scar, surrounding striatum, contralateral striatum) were obtained from the same untreated (or IL-4–treated) rat. Scale bar = 50 μm and applies to all images. (C) Summary of area fraction of GFAP staining per 200 µm2 at 7 days after ischemia. The dashed line indicates GFAP staining in saline-injected sham control animals. The symbols represent: * differences from saline-injected shams; # IL-4 effects. One symbol indicates p < 0.05; 2 symbols, p < 0.01; 3 symbols, p < 0.001.
DISCUSSION
There were several salient findings of this study: (1) At 1 day after ET-1–induced ischemia, the ipsilateral striatum was skewed toward a pro-inflammatory state, with elevated TNF, NOS2/iNOS and IL-6. A single injection of recombinant IL-4 (with ET-1) increased several anti-inflammatory mediators for up to 7 days, but the pro-inflammatory mediators were not affected. Overall, IL-4 injection evoked a mixed inflammatory milieu at 1 day and a more anti-inflammatory profile at later times. (2) By 1 day after ischemia, IL-4 treatment nearly doubled (to 70%) microglia/macrophages that expressed the phagocytic marker, CD68. By 7 days, IL-4 increased the total number of microglia/macrophages in the infarct. (3) IL-4 treatment resulted in a 2-fold increase in neutrophil infiltration that was accompanied by their increased expression of VEGF-A. (4) IL-4 treatment did not reduce degeneration of striatal neurons or the profound progression of myelin damage over the 7-day period after ischemia. (5) By 7 days, when a glial scar was well formed, IL-4 treatment increased the astrocytic response beyond the scar, further into the striatum.
Gene Profiling
We first examined expression of selected inflammation-related genes to assess time-dependent responses to ischemia and asked whether IL-4 injection skewed the brain toward an anti-inflammatory state. Without treatment, at the earliest time examined (1 day), the ischemic striatum was skewed toward a pro-inflammatory state, with elevated levels of TNF, NOS2/iNOS and IL-6. Then, several anti-inflammatory genes increased by 7 days. This temporal profile is consistent with reports that pro-inflammatory mediators are rapidly elevated after ischemia (26, 27), and that anti-inflammatory mediators are elevated by 1–7 days, depending on the molecule (26, 45).
IL-4 injection increased the anti-inflammatory profile at 1 day (elevated CD163, ARG1, CCL22, PPARγ), and, importantly, some effects were long lasting. At 7 days after IL-4 injection, there was increased expression of several immunomodulatory molecules and receptors (IL-4Rα, STAT6, CD206, PPARγ, CD11b, CD200R1, TLR2, TLR4, HO-1). We think it most likely that these changes result from sustained effects on inflammatory cells and from propagating cellular responses, not from residual IL-4. One possibility is that IL-4 signaling perpetuates alternative activation of microglia and macrophages. For instance, IL-4 increases expression of its receptor, IL-4Rα, in rat microglia (46, 47); and the downstream signaling molecule, STAT6, binds to the promoter of many PPARγ-regulated genes and increases their expression (48). Long-term maintenance of alternative activation can result from an IL-4-mediated increase in PPARγ (49); hence, PPARγ agonists are being assessed for their anti-inflammatory and neuroprotective effects in ischemia models (50). Here, we found that IL-4 treatment increased IL-4Rα, STAT6 and PPARγ, i.e. molecules that are expected to prolong alternative activation. IL-4 treatment greatly increased CCL22 at 1 day. CCL22 increased as early as 6 hours after ICH in the rat striatum (29), and by 5 days in the murine middle cerebral artery occlusion (MCAo) ischemia model (45). Finally, some IL-4–induced changes in inflammatory gene expression might result from increased numbers of neutrophils at early times; and from increased microglia/macrophages and astrogliosis at later times.
Very few studies have assessed effects of IL-4 treatment on acute inflammatory responses in the brain, but some anti-inflammatory effects have been seen, as in this study. After transient ischemia (MCA/common carotid artery occlusion), subcutaneous IL-4 injection increased expression of IL-4Rα, CD163 and PPARγ in the mouse cortex at 1 and 3 days (47). IL-4 injection into the brain increased M2 markers (Arg1, IL-13) 1 day after ICH in rats (51). However, in a viral encephalitis model, wild-type and IL-4 knockout mice had the same protein levels of TNF (52).
IL-4 is considered a negative regulator of pro-inflammatory responses (18) but in vivo effects might depend on the stroke model. At 1 day after ICH, IL-4 injection reduced the M1 markers, iNOS, IL-1β, IL-6, and TNF (51); whereas, after transient ischemia, we did not detect a significant decrease in these markers. One possibility is that differing cell types and proportions respond to IL-4. In a study of naïve mice, microglia were the primary responders but only ∼18% of striatal microglia responded after IL-4 was injected into the third ventricle (19). Among endogenous CNS cells, microglia and astrocytes are considered most likely to respond to IL-4 (18), but other cells might also respond and contribute to the molecular profile. Oligodendrocytes and some neurons express IL-4 receptors (23–25), and one must also consider responses of blood vessels and infiltrating cells after ischemia. For example, IL-4 receptors are found on human endothelial cells (22) and neutrophils (20). For these reasons, we assessed specific cellular responses to IL-4 treatment.
Microglia/Macrophages
A single IL-4 injection increased the number of microglia/macrophages in the infarcts at 7 days. Without being able to distinguish activated microglia from infiltrating monocytes, this increase might be due to increased macrophage infiltration, microglia migration or proliferation or both. IL-4 increases proliferation of murine microglia (53) and migration of rat microglia (54) in vitro. After transient ischemia in mice, entry of blood-borne monocytes began by 1 day, was most prominent at about 4 days, and was considered maximal by 7 days (55). Interestingly, we found that IL-4-treated rats had markedly elevated CXCL1 expression at 3 days. While CXCL1 is mainly known as a neutrophil chemoattractant (56), it contributes to monocyte transendothelial migration in vitro, and its expression is elevated in monocytes from patients with Alzheimer disease (AD) and in the APP/PS1 mouse model of AD (57).
After CNS damage, microglia phagocytose damaged tissue and infiltrating cells, including neutrophils (7, 13, 58). Our results suggest that IL-4 treatment increased the ability of microglia/macrophages to phagocytose. At 1 day after ischemia, when degenerating neurons and infiltrating neutrophils had increased, IL-4 treated rats had nearly 4 times as many Iba1-positive cells expressing CD68. Direct effects of IL-4 on microglia at this early time and at the site of IL-4 injection are likely, but further interactions between cells and damaged tissue are inevitable complications of in vivo studies. There is little directly relevant, stroke-related literature. However, Iba1-positive, CD68-positive microglia/macrophages preferentially accumulated in damaged white-matter tracts in this ischemia model (30), and murine microglia isolated after transient MCAo showed increased phagocytosis of latex beads at 1 and 3 days (59). Cultured microglia express many phagocytosis-related receptors, with complex changes after IL-4 treatment, i.e. decreased CD11b, SR-A, FcγRIa, TREM2 and, surprisingly, CD68, and increased P2Y6 and the inhibitory receptor, FcγRIIb; and decreased inhibitory receptor, signal regulatory protein-α (SIRPα) (60). In vitro results might depend on the target being phagocytosed. IL-4 increased the ability of rat microglia to phagocytose apoptotic neurons (47), but not myelin debris (60).
Neutrophils
Neutrophils respond rapidly to tissue damage and are short-lived (61). We previously found that they were prevalent in the infarct at 1 day and decreased by 3 days (30). Here, IL-4 treatment substantially increased their numbers at 1 day, and most of them were CD68-positive, (i.e. phagocytic). While phagocytosis can be helpful after cerebral infarction, neutrophils can harm bystander tissue. Their numbers in the brain positively correlate with the infarct volume after ischemia (13), and they can exacerbate damage through release of proteolytic enzymes, cytokines, and reactive oxygen species (61). Therefore, reducing the impact of neutrophils is one avenue being investigated in clinical stroke trials (12).
There is indirect evidence that IL-4 facilitates neutrophil extravasation. In the periphery, IL-4 injection increased recruitment of murine neutrophils and monocytes to a dorsal subcutaneous air pouch (62). In the murine CNS, IL-4 deletion reduced neutrophil entry and meningoencephalitis after viral infection (52). Neutrophil recruitment is initiated by elevated levels of chemoattractants, including CXCL1 and CXCL2, which are synthesized by LPS-stimulated M1 microglia (63) and macrophages (56). Consistent with a role in early neutrophil entry, we found increased CXCL1 and CXCL2 expression at 1 day after ischemia. IL-4 treatment did not alter their levels and, instead, it increased both VEGF-A mRNA expression and the proportion of neutrophils containing VEGF-A protein. VEGF-A regulates angiogenesis, vascular permeability (64), neutrophil transmigration (42, 43), and increases blood vessel leakiness after ischemia (65). VEGF-A-responsive neutrophils might have a pro-angiogenic profile (CD49d-positive, VEGFR1high, CXCR4high) that differs from those responding to pro-inflammatory chemokines (43). After ischemia alone, we did not see elevated VEGF-A transcript expression, possibly because the blood-brain barrier is not breached in the ET-1 model (32).
Neutrophils are removed from brain tissue by phagocytosis. In a mouse permanent MCAo stroke model, their removal by microglia/macrophages decreased the infarct size, and clearance was increased by the PPARγ agonist, rosiglitazone (66). IL-4 upregulates PPARγ (67), and we observed a substantial PPARγ induction at 1 and 7 days after IL-4 treatment. One possibility is that the increased neutrophil accumulation at 1 day in IL-4-treated animals provokes a later increase in microglia/macrophages to promote phagocytosis. Thus, the observed increase in CD68-positive microglia/macrophages 1 day after IL-4 treatment might also be a response to increased neutrophil-derived myeloperoxidase (13).
Damage to Striatal Neurons and White Matter Tracts
After transient ischemia in the rat striatum, we previously observed progressive and extensive myelin damage in the first week (30). This time course mirrored the increase in phagocytic (CD68-positive) microglia/macrophages (this study), and their increase inside damaged white-matter bundles (30). IL-4 treatment did not reduce this progressive myelin damage. As in the present study, neurodegeneration peaked at 1 day in the transient MCAo model (2) and in the forelimb motor cortex of ET-1–treated animals (3). Rather than rescuing them, IL-4 modestly increased the number of degenerating striatal neurons at 1 day, and this was coincident with the observed increase in phagocytic neutrophils. It is hard to compare these results with the limited literature concerning IL-4 treatment after ischemia, particularly in rats.
After transient MCAo in mice, infarct volumes and neurological deficits at 1 day were greater in IL-4 knockout mice; and IL-4 injection 30 minutes before ischemia reduced deficits in knockout, but not wild-type, mice (68). At early times (<3 days), similar sensorimotor deficits were seen in wild type and IL-4 knockout mice but later, the knockout mice had greater sensorimotor, learning and memory deficits (69). In that study, IL-4-mediated protective mechanisms were delayed. Multiple subcutaneous IL-4 injections (at 2, 24, 36, 72 hours) improved neurological scores at 2 weeks but not at 1 day (47); and 7 days of ventricular IL-4 infusion produced functional benefits weeks later (69). Thus, benefits of IL-4 supplementation might only be seen at later times, during the repair phase. One surprising inconsistency is that, in the same model, IL-4 was secreted early after ischemia by stressed neurons in the penumbra but not by dying neurons in the infarct, and it decreased by 24 hours (47). In the future, it will be important to examine the sensitivity of inhibitory versus excitatory neurons. The transient MCAo model causes extensive damage to the cortex, which mainly contains excitatory neurons. In our studies of transient ischemia in the striatum, 90%–95% of the cell bodies belong to medium spiny inhibitory neurons (70).
TABLE.
Target Sequences Used to Design Custom CodeSet for nCounter Gene Expression Assay
| Gene | GenBank Accession no. | Target sequence |
|---|---|---|
| Arg1 | NM_053336.2 | ACGGGAAGGTAATCATAAGCCAGAGACTGACTACCTTAAACCACCGAAATAAATGTGAATACATCGCATAAAAGTCATCTGGGGCATCACAGCAAACCGA |
| Ccl22 | NM_057203.1 | TACATCCGTCACCCTCTGCCACCACGTTTCGTGAAGGAGTTCTACTGGACCTCAAAGTCCTGCCGCAAGCCTGGCGTCGTTTTGATAACCATCAAGAACC |
| Cd68 | NM_001031638.1 | CTCTCATTCCCTTACGGACAGCTTACCTTTGGATTCAAACAGGACCGACATCAGAGCCACAGTACAGTCTACCTTAACTACATGGCAGTGGAATACAATG |
| Cd163 | NM_001107887.1 | AGTTTCCTCAAGAGGAGAGGTCTTGATACATCAAGTTCAGTACCAAGAGATGGATTCGAAGACGGATGATCTGGACTTGCTGAAATCCTCGGGTTGGCAT |
| Cd200r1 | NM_023953.1 | CTGCTTTTGGAGAACTTCTCACGTAGCAGTACTCTTGATCTGGGGGGTCTTCGCGGCTGAGTCAAGTTGTCCTGATAAGAATCAAACAATGCAGAACAAT |
| Chi3l3 (Ym1) | NM_001191712.1 | AAGCGTTTGAGAAAGAATCTACTGAGCAAGAAATCCCAAGGCTGCTTCTCACTGCCACAGTAGCTGGAGTCATTGACACAATCCAGTCTGGTTACAAGAT |
| Cx3cr1 | NM_133534.1 | ATGTGCAAGCTCACGACTGCTTTCTTCTTCATTGGCTTCTTTGGGGGCATATTCTTCATCACCGTCATCAGCATCGACCGGTACCTCGCCATCGTCCTGG |
| Gfap | NM_017009.1 | TACAGACTTTCTCCAACCTCCAGATCCGAGAAACCAGCCTGGACACCAAATCTGTGTCAGAAGGCCACCTCAAGAGGAACATCGTGGTAAAGACGGTGGA |
| Hmox1 (HO-1) | NM_012580.2 | CTAGTTCATCCCAGACACCGCTCCTGCGATGGGTCCTCACACTCAGTTTCCTGTTGGCGACCGTGGCAGTGGGAATTTATGCCATGTAAATGCAGTGTTG |
| Hprt1 | NM_012583.2 | AGCTTCCTCCTCAGACCGCTTTTCCCGCGAGCCGACCGGTTCTGTCATGTCGACCCTCAGTCCCAGCGTCGTGATTAGTGATGATGAACCAGGTTATGAC |
| Il6 | NM_012589.1 | GGAACAGCTATGAAGTTTCTCTCCGCAAGAGACTTCCAGCCAGTTGCCTTCTTGGGACTGATGTTGTTGACAGCCACTGCCTTCCCTACTTCACAAGTCC |
| Il4r | NM_133380.2 | GGGTGTCAGCATCTCCTGCATCTGCATCCTATTGTTTTGCCTGACCTGTTACTTCAGCATTATCAAGATTAAGAAGATATGGTGGGACCAGATTCCCACT |
| Itgam (Cd11b) | NM_012711.1 | CATCCCTTCCTTCAACAGTAAAGAAATATTCAACGTCACCCTCCAGGGCAATCTGCTATTTGACTGGTACATCGAGACTTCTCATGACCACCTCCTGCTT |
| Mrc1 (Cd206) | NM_001106123.1 | CTTTGGAATCAAGGGCACAGAGCTATATTTTAACTATGGCAACAGGCAAGAAAAGAATATCAAGCTTTACAAAGGTTCCGGTTTGTGGAGCAGATGGAAG |
| Nos2 | NM_012611.2 | ACGGGACACAGTGTCGCTGGTTTGAAACTTCTCAGCCACCTTGGTGAGGGGACTGGACTTTTAGAGACGCTTCTGAGGTTCCTCAGGCTTGGGTCTTGTT |
| Pparg | NM_013124.1 | TTTATAGCTGTCATTATTCTCAGTGGAGACCGCCCAGGCTTGCTGAACGTGAAGCCCATCGAGGACATCCAAGACAACCTGCTGCAGGCCCTGGAACTCC |
| Sdha | NM_130428.1 | CCTCCGATTAAGGCAAATGCTGGAGAAGAGTCGGTTATGAATCTTGACAAGTTGAGATTTGCTGATGGAAGTGTAAGAACATCAGAGCTGCGCCTCAGCA |
| Stat6 | NM_001044250.1 | GTGGTTTGATGGTGTCCTGGACCTCACTAAACGCTGTCTTCGGAGCTACTGGTCAGATCGGCTGATCATCGGCTTTATCAGTAAGCAATATGTCACTAGC |
| Tgfb1 | NM_021578.2 | CGCCTGCAGAGATTCAAGTCAACTGTGGAGCAACACGTAGAACTCTACCAGAAATATAGCAACAATTCCTGGCGTTACCTTGGTAACCGGCTGCTGACCC |
| Tlr2 | NM_198769.2 | TTTACAAACCCTTAGGGTAGGAAATGTTGACACTTTCAGTGAGATAAGGAGAATAGATTTTGCTGGGCTGACCTCTCTCAACGAACTTGAAATTCAGGTA |
| Tlr4 | NM_019178.1 | GTCAGTGTGCTTGTGGTAGCCACTGTAGCATTTCTGATATACCACTTCTATTTTCACCTGATACTTATTGCTGGCTGTAAAAAGTACAGCAGAGGAGAAA |
| Tnf | NM_012675.2 | GGTGATCGGTCCCAACAAGGAGGAGAAGTTCCCAAATGGGCTCCCTCTCATCAGTTCCATGGCCCAGACCCTCACACTCAGATCATCTTCTCAAAACTCG |
| Vegfa | NM_001110334.1 | GTGACCAAGCACGGTGGTCCCTCGTGGAACTGGATTCGCCATTTTCTTATATTTGCTGCTAAATCGCCAAGCCCGGAAGATTAGGGAGTTTTGTTTCTGG |
| Ywhaz | NM_013011.3 | TCCTGAACTCCCCAGAGAAAGCCTGCTCTCTTGCAAAAACAGCTTTTGATGAAGCCATTGCTGAACTTGATACATTAAGTGAAGAGTCGTACAAAGACAG |
Astrogliosis
Astrogliosis, which is a prominent response after focal ischemia, includes increased GFAP expression, process hypertrophy, and formation of a glial scar that is thought to confine tissue damage (14, 71). We confirmed our previous finding that a glial scar was well formed by 7 days in this ischemia model (28, 30), and found that IL-4 treatment increased the astrocytic response in the striatum beyond the scar. The timing (7 days) coincided with an IL-4–mediated increase in accumulation of microglia/macrophages in the infarct. Astrogliosis can have beneficial effects (neuroprotection, restricting damage and CNS inflammation, promoting blood-brain barrier repair) or harmful effects (exacerbating inflammation), depending on the signaling cascades initiated over time after the precipitating insult (14). There is some evidence that IL-4 can have longer term effects on astrogliosis and functional outcomes. When an adeno-associated virus-IL-4 vector was intracranially injected to boost IL-4 levels in the brain there was increased astrogliosis in the hippocampus of APP/PS1 transgenic mice (72).
SUMMARY AND CONCLUSIONS
Injecting IL-4 into the rat striatum at the onset of transient ischemia had both expected and unexpected effects and, in interpreting these outcomes, it is helpful to delineate the time frame. At early times, a single IL-4 injection skewed the ischemic striatum from an early pro-inflammatory state to a mixed inflammatory state on day 1. Although other cells express IL-4 receptors, effects of IL-4 injection at this early time likely involve microglia (most macrophages enter later). Thus, IL-4 apparently increased the numbers of phagocytic microglia and neutrophils in the infarct. Surprisingly, IL-4 treatment modestly increased early degeneration of striatal neurons, and we think this is likely due to the increase in neutrophils.
At later times, some IL-4–mediated anti-inflammatory changes were sustained for the entire 7-day period. Long-lasting outcomes are expected to have complex origins involving cellular and chemical crosstalk, including other cell types that express IL-4 receptors. At 7 days, IL-4 increased the number of microglia/macrophages in the infarct. The progressive myelin damage was not rescued by 7 days, but we do not know if behavioral outcomes might be improved with a longer treatment and study duration designed to affect resolution and repair processes. In principle, the IL-4-mediated increase in astrogliosis might reflect one or more mechanisms, including a direct interaction of IL-4 with astrocytic IL-4 receptors or response to changes in gene expression or increased neutrophils, neurodegeneration, or phagocytic microglia/macrophages. Based on the late timing and location outside the infarct and site of IL-4 injection, we favor an indirect mechanism.
Our results of attempting to force the brain into an anti-inflammatory state soon after ischemia (including sustained effects observed at 7 days after a single IL-4 injection) warrant further study of other IL-4 treatment regimes, and of long-term cellular and behavioral outcomes over weeks to months.
ACKNOWLEDGEMENTS
We are very grateful to Xiaoping Zhu for expert technical assistance. Part of this work was presented at the Canadian Association for Neuroscience Meeting (Montreal, May 2014), and the Society for Neuroscience Meeting (Chicago, October 2015.
REFERENCES
- 1.Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015;385:117–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu F, Schafer DP, McCullough LD. TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Meth 2009;179:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguemeni C, Gomez-Smith M, Jeffers MS, et al. Time course of neuronal death following endothelin-1 induced focal ischemia in rats. J Neurosci Meth 2015;242:72–6 [DOI] [PubMed] [Google Scholar]
- 4.Amantea D, Nappi G, Bernardi G, et al. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. Febs J 2009;276:13–26 [DOI] [PubMed] [Google Scholar]
- 5.Kriz J, Lalancette-Hebert M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol 2009;117:497–509 [DOI] [PubMed] [Google Scholar]
- 6.Offner H, Subramanian S, Parker SM, et al. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab 2006;26:654–65 [DOI] [PubMed] [Google Scholar]
- 7.Fu Y, Liu Q, Anrather J, et al. Immune interventions in stroke. Nature Rev 2015;11:524–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003;3:23–35 [DOI] [PubMed] [Google Scholar]
- 9.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The J Clin Invest 2012;122:787–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colton C, Wilcock DM. Assessing activation states in microglia. CNS Neurol Dis Drug Targ 2010;9:174–91 [DOI] [PubMed] [Google Scholar]
- 11.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Molec Neurobiol 2016;53:1181–94 [DOI] [PubMed] [Google Scholar]
- 12.Jickling GC, Liu D, Ander BP, et al. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 2015;35:888–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weston RM, Jones NM, Jarrott B, et al. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab 2007;27:100–14 [DOI] [PubMed] [Google Scholar]
- 14.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 2015;16:249–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010;32:593–604 [DOI] [PubMed] [Google Scholar]
- 16.Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Ann Rev Immunol 2013;31:317–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diab A, Zhu J, Lindquist L, et al. Haemophilus influenzae and Streptococcus pneumoniae induce different intracerebral mRNA cytokine patterns during the course of experimental bacterial meningitis. Clin Exp Immunol 1997;109:233–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gadani SP, Cronk JC, Norris GT, et al. IL-4 in the brain: a cytokine to remember. J Immunol 2012;189:4213–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepe G, Calderazzi G, De Maglie M, et al. Heterogeneous induction of microglia M2a phenotype by central administration of interleukin-4. J Neuroinflam 2014;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Girard D, Paquin R, Beaulieu AD. Responsiveness of human neutrophils to interleukin-4: induction of cytoskeletal rearrangements, de novo protein synthesis and delay of apoptosis. Biochem J 1997;325:147–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zola H, Flego L, Weedon H. Expression of IL-4 receptor on human T and B lymphocytes. Cell Immunol 1993;150:149–58 [DOI] [PubMed] [Google Scholar]
- 22.Palmer-Crocker RL, Hughes CC, Pober JS. IL-4 and IL-13 activate the JAK2 tyrosine kinase and Stat6 in cultured human vascular endothelial cells through a common pathway that does not involve the gamma c chain. J Clin Invest 1996;98:604–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cannella B, Raine CS. Multiple sclerosis: cytokine receptors on oligodendrocytes predict innate regulation. Ann Neurol 2004;55:46–57 [DOI] [PubMed] [Google Scholar]
- 24.Nolan Y, Maher FO, Martin DS, et al. Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J Biol Chem 2005;280:9354–62 [DOI] [PubMed] [Google Scholar]
- 25.Walsh JT, Hendrix S, Boato F, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest 2015;125:699–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin R, Liu L, Zhang S, et al. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res 2013;6:834–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 2009;7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lively S, Moxon-Emre I, Schlichter LC. SC1/hevin and reactive gliosis after transient ischemic stroke in young and aged rats. J Neuropathol Exp Neurol 2011;70:913–29 [DOI] [PubMed] [Google Scholar]
- 29.Lively S, Schlichter LC. SC1/hevin identifies early white matter injury after ischemia and intracerebral hemorrhage in young and aged rats. J Neuropathol Exp Neurol 2012;71:480–93 [DOI] [PubMed] [Google Scholar]
- 30.Moxon-Emre I, Schlichter LC. Evolution of inflammation and white matter injury in a model of transient focal ischemia. J Neuropathol Exp Neurol 2010;69:1–15 [DOI] [PubMed] [Google Scholar]
- 31.Moxon-Emre I, Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol 2011;70:218–35 [DOI] [PubMed] [Google Scholar]
- 32.Hughes PM, Anthony DC, Ruddin M, et al. Focal lesions in the rat central nervous system induced by endothelin-1. J Neuropathol Exp Neurol 2003;62:1276–86 [DOI] [PubMed] [Google Scholar]
- 33.Schlichter LC, Hutchings S, Lively S. Inflammation and white matter injury in animal models of ischemic stroke In: Baltan S, et al. eds. White Matter Injury in Stroke and CNS Disease, Vol. 4 New York: Springer New York, 2014:461–504 [Google Scholar]
- 34.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26:317–25 [DOI] [PubMed] [Google Scholar]
- 35.Gubern C, Hurtado O, Rodriguez R, et al. Validation of housekeeping genes for quantitative real-time PCR in in-vivo and in-vitro models of cerebral ischaemia. BMC Molec Biol 2009;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmuck G, Kahl R. The use of Fluoro-Jade in primary neuronal cell cultures. ArchToxicol 2009;83:397–403 [DOI] [PubMed] [Google Scholar]
- 37.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 2000;874:123–30 [DOI] [PubMed] [Google Scholar]
- 38.Donnelly DJ, Gensel JC, Ankeny DP, et al. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Meth 2009;181:36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Damoiseaux JG, Dopp EA, Calame W, et al. Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 1994;83:140–7 [PMC free article] [PubMed] [Google Scholar]
- 40.Wasserman JK, Yang H, Schlichter LC. Glial responses, neuron death and lesion resolution after intracerebral hemorrhage in young vs. aged rats. Eur J Neurosci 2008;28:1316–28 [DOI] [PubMed] [Google Scholar]
- 41.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol 2011;32:452–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TH, Avraham H, Lee SH, et al. Vascular endothelial growth factor modulates neutrophil transendothelial migration via up-regulation of interleukin-8 in human brain microvascular endothelial cells. J Biol Chem 2002;277:10445–51 [DOI] [PubMed] [Google Scholar]
- 43.Massena S, Christoffersson G, Vagesjo E, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015;126:2016–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 2010;119:7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063–70 [DOI] [PubMed] [Google Scholar]
- 46.Liu BS, Ferreira R, Lively S, et al. Microglial SK3 and SK4 currents and activation state are modulated by the neuroprotective drug, riluzole. J Neuroimmune Pharmacol 2013;8:227–37 [DOI] [PubMed] [Google Scholar]
- 47.Zhao X, Wang H, Sun G, et al. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci 2015;35:11281–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szanto A, Balint BL, Nagy ZS, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity 2010;33:699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chawla A. Control of macrophage activation and function by PPARs. Circ Res 2010;106:1559–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci 2008;13:1813–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Ding S, Huang W, et al. Interleukin-4 ameliorates the functional recovery of intracerebral hemorrhage through the alternative activation of microglia/macrophage. Front Neurosci 2016;10:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vilela MC, Campos RD, Mansur DS, et al. Role of IL-4 in an experimental model of encephalitis induced by intracranial inoculation of herpes simplex virus-1 (HSV-1). Arq Neuro-Psiquiatria 2011;69:237–41 [DOI] [PubMed] [Google Scholar]
- 53.Suzumura A, Sawada M, Itoh Y, et al. Interleukin-4 induces proliferation and activation of microglia but suppresses their induction of class II major histocompatibility complex antigen expression. J Neuroimmunol 1994;53:209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lively S, Schlichter LC. The microglial activation state regulates migration and roles of matrix-dissolving enzymes for invasion. J Neuroinflammation 2013;10:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schilling M, Besselmann M, Leonhard C, et al. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003;183:25–33 [DOI] [PubMed] [Google Scholar]
- 56.De Filippo K, Dudeck A, Hasenberg M, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013;121:4930–7 [DOI] [PubMed] [Google Scholar]
- 57.Zhang K, Tian L, Liu L, et al. CXCL1 contributes to beta-amyloid-induced transendothelial migration of monocytes in Alzheimer’s disease. PLoS One 2013;8:e72744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neumann J, Sauerzweig S, Ronicke R, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci 2008;28:5965–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritzel RM, Patel AR, Grenier JM, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 2015;12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siddiqui TA, Lively S, Schlichter LC. Complex molecular and functional outcomes of single versus sequential cytokine stimulation of rat microglia. J Neuroinflammation 2016;13:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wright HL, Moots RJ, Bucknall RC, et al. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010;49:1618–31 [DOI] [PubMed] [Google Scholar]
- 62.Ratthe C, Ennaciri J, Garces Goncalves DM, et al. Interleukin (IL)-4 induces leukocyte infiltration in vivo by an indirect mechanism. Mediators Inflamm 2009;2009:193970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwarz JM, Bilbo SD. LPS elicits a much larger and broader inflammatory response than Escherichia coli infection within the hippocampus of neonatal rats. Neurosci Lett 2011;497:110–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bates DO, Hillman NJ, Williams B, et al. Regulation of microvascular permeability by vascular endothelial growth factors. J Anat 2002;200:581–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest 2000;106:829–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cuartero MI, Ballesteros I, Moraga A, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 2013;44:3498–508 [DOI] [PubMed] [Google Scholar]
- 67.Nelms K, Keegan AD, Zamorano J, et al. The IL-4 receptor: signaling mechanisms and biologic functions. Ann Rev Immunol 1999;17:701–38 [DOI] [PubMed] [Google Scholar]
- 68.Xiong X, Barreto GE, Xu L, et al. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 2011;42:2026–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, Liu J, Zhao S, et al. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke 2016;47:498–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerfen CR. Synaptic organization of the striatum. J Electron Microsc Tech 1988;10:265–81 [DOI] [PubMed] [Google Scholar]
- 71.Ding S. Dynamic reactive astrocytes after focal ischemia. Neural Regen Res 2014;9:2048–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Latta CH, Sudduth TL, Weekman EM, et al. Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-beta using BV2 microglial cells and APP/PS1 transgenic mice. J Neuroinflammation 2015;12:41. [DOI] [PMC free article] [PubMed] [Google Scholar]