

The authors used gene array in combination with functional in vitro assays to study mesenchymal stromal cells (MSCs) from donors with type 1 diabetes (T1D) and healthy controls. Gene expression (cytokine secretion, immunomodulatory activity, and wound healing potential) differed between the groups. Cell count, growth kinetics, and colony-forming unit-fibroblast capacity were comparable, and MSCs from the two groups were similar in immunosuppressive activity, migratory capacity, and hemocompatibility.
Keywords: Mesenchymal stem cells, Diabetes, Cellular therapy, Immunosuppression, Adult human bone marrow
Abstract
Bone marrow mesenchymal stromal cells (BM-MSCs) have been characterized and used in many clinical studies based on their immunomodulatory and regenerative properties. We have recently reported the benefit of autologous MSC systemic therapy in the treatment of type 1 diabetes mellitus (T1D). Compared with allogeneic cells, use of autologous products reduces the risk of eliciting undesired complications in the recipient, including rejection, immunization, and transmission of viruses and prions; however, comparable potency of autologous cells is required for this treatment approach to remain feasible. To date, no analysis has been reported that phenotypically and functionally characterizes MSCs derived from newly diagnosed and late-stage T1D donors in vitro with respect to their suitability for systemic immunotherapy. In this study, we used gene array in combination with functional in vitro assays to address these questions. MSCs from T1D donors and healthy controls were expanded from BM aspirates. BM mononuclear cell counts and growth kinetics were comparable between the groups, with equivalent colony-forming unit-fibroblast capacity. Gene microarrays demonstrated differential gene expression between healthy and late-stage T1D donors in relation to cytokine secretion, immunomodulatory activity, and wound healing potential. Despite transcriptional differences, T1D MSCs did not demonstrate a significant difference from healthy controls in immunosuppressive activity, migratory capacity, or hemocompatibility. We conclude that despite differential gene expression, expanded MSCs from T1D donors are phenotypically and functionally similar to healthy control MSCs with regard to their immunomodulatory and migratory potential, indicating their suitability for use in autologous systemic therapy.
Significance
The potential for mesenchymal stromal cells (MSCs) as a cell-based therapy in the treatment of immunologic disorders has been well established. Recent studies reported the clinical potential for autologous MSCs as a systemic therapy in the treatment of type I diabetes mellitus (T1D). The current study compared the genotypic and phenotypic profiles of bone marrow-derived MSCs from T1D and healthy donors as autologous (compared with allogeneic) therapy provides distinct advantages, such as reduced risk of immune reaction and transmission of infectious agents. The findings of the current study demonstrate that despite moderate differences in T1D MSCs at the gene level, these cells can be expanded in culture to an extent corresponding to that of MSCs derived from healthy donors. No functional difference in terms of immunosuppressive activity, blood compatibility, or migratory capacity was evident between the groups. The study findings also show that autologous MSC therapy holds promise as a T1D treatment and should be evaluated further in clinical trials.
Introduction
Type 1 diabetes mellitus (T1D) is a multifactorial chronic immune-mediated disease leading to the progressive destruction of insulin-producing β-cells in the pancreas [1–3]. Addressing this inflammatory response may provide an opportunity for T1D therapy, with the aim of controlling or arresting the progression of β-cell destruction and restoring glycemic control and immune hemostasis [4].
Systemic infusions with multipotent mesenchymal stromal cells (MSCs) have been studied alone and in combination with different therapeutic strategies for β-cell replacement [5], with several preclinical studies suggesting that MSC infusion protects mice from the development of experimentally induced diabetes [6–9]. Lee et al. demonstrated that infused human MSCs support murine insulin production, lower blood glucose levels, and improve kidney disease in the non-obese diabetic (NOD) mouse model [6]. A lack of human insulin in this model suggests that MSCs do not differentiate into β-cells, but may instead promote the function and repair of remaining endogenous islets [6].
MSCs are rapidly cleared from the blood circulation after infusion, suggesting that these cells are able to trigger the instant blood-mediated inflammatory reaction (IBMIR), which is characterized by rapid lysis of the infused cells, involving platelet consumption and complement and coagulation activation [10]. Despite the potential loss of MSCs, a complex and sophisticated immunosuppressive environment including the release of paracrine factors and vesicles may be initiated following IBMIR.
Notably, microvesicles released from MSCs have been shown to inhibit a proinflammatory response to an islet antigenic stimulus in T1D in vitro [11], as well as improving graft versus host disease symptoms in a patient study [12]. The strategy to use MSC therapy in diabetes is further supported by the knowledge that when coculturing islets with MSCs, islet survival and insulin secretion are enhanced in vitro [13], and in vivo, islet survival, function, and angiogenesis after transplantation are also improved [14].
Inflammatory mediators in T1D patients, as well as the uremic microenvironment in subjects with end-stage diabetic nephropathy, can potentially alter bone marrow MSC (BM-MSC) phenotype and therapeutic properties [2–4, 15, 16]. We have recently presented a phase I/IIa trial on the use of autologous MSCs in patients with newly diagnosed T1D [17]. In our clinical study, we observed no side effects with MSC adoptive transfer; and C-peptide responses to a mixed-meal tolerance test during the first year after treatment were preserved or even increased. This study, in addition to other clinical trials using autologous MSC transplantation, demonstrates the safety of such cellular therapies [17–20].
Where therapeutically efficacious, autologous cellular therapies offer distinct advantages over an allogeneic approach, with a reduced risk of human leukocyte antigen (HLA) immunity, rejection, and transmission of donor-derived infection or disease [17]. To date, however, no comparative evaluation of MSCs derived from T1D and healthy donors with respect to their therapeutic potential has been reported. In this study, we investigated whether BM-MSCs from T1D donors offer a therapeutic cell source, for immunotherapy via systemic intravenous delivery, equivalent to BM-MSCs derived from healthy donors. BM-MSCs were isolated from T1D and healthy donors and phenotypically and functionally characterized using a variety of in vitro assays in combination with gene array expression profiling.
Materials and Methods
MSC Donors, Isolation, Expansion, and Characterization of Therapeutic MSCs
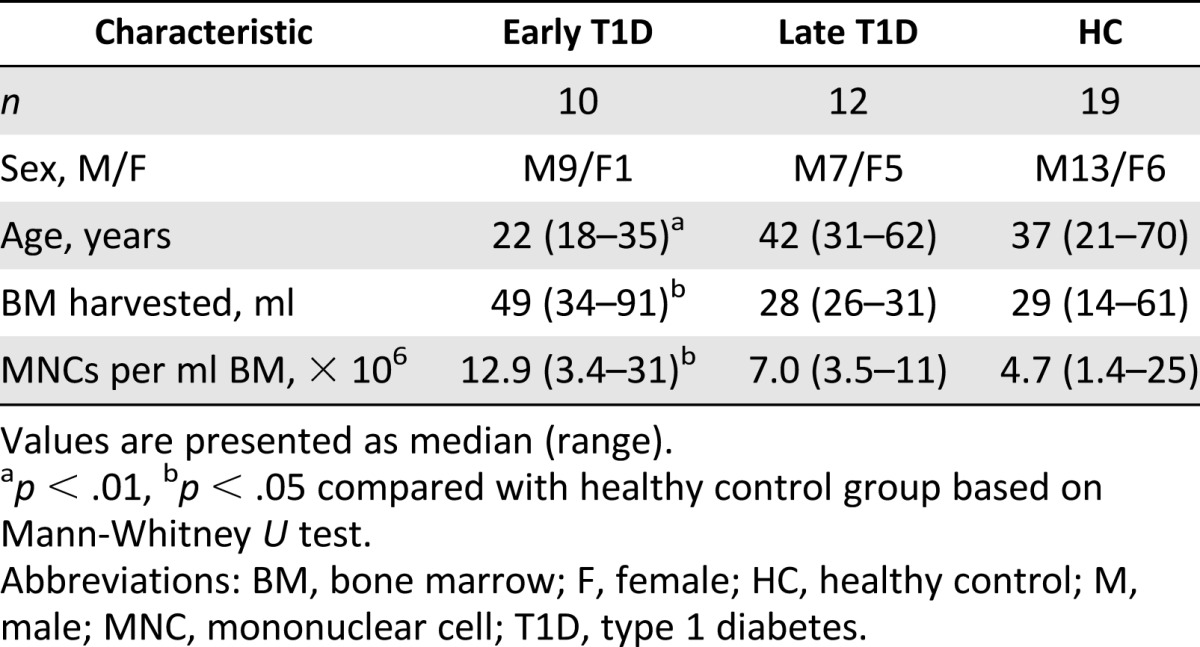
Expansion and characterization of MSCs was performed according to guidelines of the European Blood and Marrow Transplantation Group and approved by the Swedish National Board of Health and Welfare, as described previously [21–23]. MSCs were harvested from healthy volunteer donors or diabetic patients undergoing autologous MSC treatment in the context of clinical trials. Simultaneous consent for in vitro investigations of the cell characteristics was obtained in accordance with the Helsinki convention. Iliac crest BM aspirates were collected from 19 healthy control donors (HCs), 10 patients diagnosed with T1D less than 6 weeks earlier (early-stage T1D [ET1D]), and 12 patients with late-stage T1D (LT1D) and severe renal failure (Table 1). The clinical status of patients was previously described [17].
Table 1.
Bone marrow donor characteristics of HC and T1D donors
To isolate MSCs, BM-MNCs were separated over a gradient of Percoll (GE Healthcare, Uppsala, Sweden, http://www.gelifesciences.com), washed, and resuspended in Dulbecco’s modified Eagle’s medium low-glucose medium (Thermo Fisher Scientific Life Sciences, Waltham, MA, http://www.thermofisher.com) supplemented with 5% pooled human platelet-rich plasma and 100 IU/ml penicillin, 0.1 mg/ml streptomycin, and 0.25 μg/ml Fungizone (culture medium) and plated at a density of 1.6 × 105 cells per cm2. When the cultures reached confluence (>80%), the cells were detached by 0.05% Trypsin-EDTA (Thermo Fisher Scientific Life Sciences), replated at a density of 4.0 × 103 cells per cm2, and cultured for up to five passages.
All isolated MSCs from T1D donors and HCs were confirmed by flow cytometry to express the MSC marker profile according to the International Society for Cellular Therapy guidelines (positive for CD73, CD90, CD105, and HLA-I and negative for CD14, CD34, CD45, and HLA-II) (supplemental online Fig. 1A). The following antibodies from BD Biosciences (Stockholm, Sweden, http://www.bd.com/se) were used: CD14/45 (fluorescein isothiocyanate [FITC]/phycoerythrin [PE]) #342408, CD73 (PE) #550257, CD90 (FITC) #555595, IgG1/2a (FITC/PE) #342409, and CD34 (PE) #345802. CD105 (PE) #326040 was purchased from Ancell Corp. (Stillwater, MN, http://www.ancell.com). HLA-I (PE) #R7000 and HLA-II (FITC) #F0817 were purchased from Dako Sweden (Solna, Sweden, http://www.dako.com). Retained capacity to differentiate into both osteogenic and adipogenic lineages was assessed as previously described [22] and demonstrated by Alizarin red and Oil Red O staining (supplemental online Fig. 1B, 1C).
CFU-F Capacity and Growth Kinetics
The number of colony-forming units-fibroblast (CFU-F) relative to the number of seeded MSCs after plastic adherence and expansion for one passage was recorded (n = 4 per group; passage 1 [P1]). Briefly, 100 MSCs at P0 were seeded into one well of a six-well plate, and colonies of more than 50 cells were counted after 14 days after fixation with 70% ethanol and staining with 0.1% (wt/vol) crystal violet.
Growth kinetics were monitored for five sequential passages by calculating cell population doublings (PDs) at each passage using the formula log n/log 2, where n is the number of cells at harvest divided by the number of cells seeded. The cumulative PD was calculated to express the proliferative capacity of each donor. PD rate at clinically relevant P1 was calculated by dividing the number of days in culture with the number of PDs and is expressed as PDs per week.
Gene Expression Analysis With Microarray and Real-Time Quantitative Polymerase Chain Reaction
RNA was extracted from MSCs (P1–P3) obtained from HCs, ET1D, and LT1D donors (n = 3 per group) using an miRNA Mini Kit (Qiagen, Hilden, Germany, http://www.qiagen.com). The RNA was labeled and amplified (one-cycle amplification) according to Affymetrix GeneChip Expression Analysis Technical Manual. Chips were scanned using the GeneChip Scanner 3000. Human Genome U133 plus 2.0 Chips were normalized by robust multiarray average using invariant set normalization [24].
Probe level expression values were calculated using the PM-MM model provided by the DNA-Chip Analyzer (dCHIP) software (http://www.dchip.org). Differentially expressed genes were defined with a cutoff of 1.5-fold change, with a lower bound of 90% confidence interval (CI). Mapping of KEGG pathways for biochemical pathway analysis of microarray data was performed with the Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics resources v6.7 (http://david.abcc.ncifcrf.gov/tools.jsp) [25].
For gene expression analysis with quantitative real-time polymerase chain reaction (qRT-PCR), total RNA was extracted from MSCs of HC and LT1D (n = 5 per group) using the RNeasy Mini Kit (Qiagen). cDNA was synthesized using the High Capacity cDNA Reverse Transcription kit (4368814; Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com), and qRT-PCR was performed with Fast SYBR Green Master Mix (Applied Biosystems). Amplification was performed on a CFX384 C1000 Touch Real-time system (Bio-Rad, Hercules, CA, http://www.bio-rad.com). Expression levels were normalized to β-actin levels and are presented as normalized expression or as fold change over unscratched controls. The primers used were actin: forward AGCTACGAGCTGCCTGAC, reverse AAGGTAGTTTCGTGGATGC; tissue factor pathway inhibitor (TFPI): forward GGAAGAAGATCCTGGAATATGTCGG, reverse CTTGGTTGATTGCGGGAGTCAGGGAG; plasminogen activator, urokinase (PLAU): forward CACGCAAGGGGAGATGAA, reverse ACAGCATTTTGGTGGTGACTT; cyclooxygenase 2 (COX2): forward CTTCACGCATCAGTTTTTCAAG, reverse TCACCGTAAATATGATTTAAGTCCAC; leukemia inhibitory factor (LIF): forward TGCCAATGCCCTCTTTATTC, reverse GTCCAGGTTGTTGGGGAAC; FBJ murine osteosarcoma viral oncogene homolog (FOS): forward ACTACCACTCACCCGCAGAC, reverse CCAGGTCCGTGCAGAAGT; bone morphogenetic protein-2 (BMP-2): forward CGGACTGCGGTCTCCTAA, reverse GGAAGCAGCAACGCTAGAAG; nuclear factor (NF)-κB: forward CTGGCAGCTCTTCTCAAAGC, reverse TCCAGGTCATAGAGAGGCTCA; NF-κB inhibitor (IKB): forward CATCCGATGGCACAATCA, reverse CTGGATCTCCAGGCACCA; and transforming growth factor (TGF)-β: forward ACTACTACGCCAAGGAGGTCAC, reverse TGCTTGAACTTGTCATAGATTTCG.
Functional Analyses
MSCs isolated from HC, ET1D, and LT1D donors were assessed using a number of in vitro functional assays to indicate whether these cell sources demonstrated equivalent efficacy in respects relevant for successful immunotherapy.
Secretion of Immunomodulatory Cytokines and Chemokines
The secretion of trophic mediators in response to licensing by proinflammatory stimuli was assessed in HC and T1D MSCs (n = 4 donors per group). MSCs were seeded at a concentration of 1 × 105 cells per 2 ml culture medium and allowed to adhere overnight. Culture medium was removed and replaced with fresh medium ± 100 U/ml interferon (IFN)-γ and 10 ng/ml tumor necrosis factor (TNF)-α. Cells were incubated for 3 days, before the media was removed and spun for 5 minutes at 500g to remove cellular debris. Levels of secreted interleukin (IL)-6, prostaglandin E2 (PGE2), hepatocyte growth factor (HGF), chemokine (C-X-C motif) ligand 1 (CXCL1), and CXCL6 were quantified by enzyme-linked immunosorbent assay (ELISA) according to the supplier’s instructions (R&D Systems, Minneapolis, MN, http://www.rndsystems.com). Indoleamine 2,3-dioxygenase (IDO) activity was measured by quantification of the tryptophan metabolite l-kynurenine, as previously described [26].
Suppression of T-Cell Activation and Effector Function by T1D MSCs
Peripheral blood mononuclear cells were prepared by centrifugation of heparinized blood on Ficoll-Isopaque (AlereTechnologies AS, Oslo, Norway, http://www.axis-shield-density-gradient-media.com), and untouched CD3+ T cells were subsequently isolated by magnetic-activated cell sorting (Human Pan T Cell Isolation Kit; Miltenyi Biotec Norden, Lund, Sweden, http://www.miltenyibiotec.com). T cells were cultured in RPMI 1640 medium supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), l-glutamine (2 mM; Thermo Fisher Scientific Life Sciences), and 10% heat-inactivated pooled human blood type AB serum and activated using anti-CD2/CD3/CD28 microbeads (Miltenyi Biotec) at a 1:2 bead-to-cell ratio. MSCs (P2; n = 4 donors per group) were added at a 1:10 ratio to T cells either in direct contact or in 0.4 μm PET Transwell membrane inserts and cultured for 3 days. CD25 was used as a marker of MSC-mediated suppression of T-cell activation as assessed by flow cytometry (CD3 [V450] #560365 and CD25 [PE] #555432 purchased from BD Biosciences). Fifty thousand gated events were recorded per sample and analyzed using FlowJo software (v7.6; FlowJo, Ashland, OR, http://www.flowjo.com).
Suppression of T-cell effector function was assessed by measurement of the proinflammatory cytokines TNF-α, IL-2, and IFN-γ in the coculture supernatants by ELISA (R&D Systems).
In Vitro Scratch Wound Healing Assay
Migration in response to wounding was assessed in vitro using the scratch assay as previously described [27]. Briefly, HC and LT1D MSCs (where significant gene expression differences in the gene ontology term [GO term] stress and wounding was detected by array; n = 4 donors per group) were seeded in duplicates into a 24-well plate at a density of 1.7 × 105 cells per well in 1 ml culture medium (density previously optimized for 100% confluence upon adherence and spreading). Cells were allowed to adhere overnight before removal of the medium and introduction of a full-depth scratch using a pipette tip (average wound width 0.9 mm). Wounded cells were washed twice with phosphate-buffered saline (PBS) to remove cellular debris before replacing with 1 ml of fresh culture medium. Wound closure was visualized by acquiring transmitted light images (10-millisecond exposure) in three viewpoints per well at ×5 optical magnification at 37°C using a Leica DMI6000 wide-field microscope with an EM-CCD 16-bit camera (Evolve; Andor Technology, Windsor, CT, http://www.andor.com) at 30-minute intervals for 24 hours. Degree of wound closure was assessed by measuring wound width, using Image J 1.46r software (NIH, Bethesda, MD, http://imagej.nih.gov/ij), every 4 hours after scratching. Data are expressed as percentage wound closure compared to initial wound width. At the end of the 24-hour imaging, cell lysates were harvested with RLT buffer (Qiagen) for gene expression analysis with qRT-PCR. A parallel plate without scratch wounds, incubated in corresponding conditions, was used as a control.
Assessment of Cell Surface Expression of Complement Inhibitors by T1D MSCs
Expression levels of the complement inhibitors CD46, CD55, and CD59 were assessed on the cell surface of HC and T1D MSCs (P2; n = 4 donors per group) by flow cytometry. Briefly, cells were expanded to P2 as described above, trypsinized, and stained using CD46 (FITC) #315304, CD55 (PE-cyanine 7) #311314, and CD59 (PE) #304707 (all purchased from BioLegend, San Diego, CA, http://www.biolegend.com). Viability was assessed using LIVE/DEAD Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific Life Sciences). Thirty thousand events were recorded in the live cell gate and analyzed using FlowJo software.
In Vitro Assessment of MSC Blood Compatibility
Aliquots of freshly thawed clinical grade MSCs from HCs or LT1D donors (n = 5 per group) were exposed to human blood using the Chandler whole blood loop system, consisting of tubing with a heparinized inner surface (Corline Systems, Uppsala, Sweden, http://www.corline.se), as described previously [10, 28–31]. Fresh non-anticoagulated ABO-compatible human blood was obtained from healthy volunteers, who had given informed consent in accordance with the Helsinki protocol and had received no medication for at least 10 days. Pieces of tubing containing 7 ml human blood were prepared [31] and supplemented with 100 µl PBS containing 5% AB plasma with or without freshly thawed MSCs. To determine the time course of the reaction between the blood and the cells, 1 ml samples from each blood tube were collected before and at 5, 15, and 30 minutes after cell addition. Reactions were stopped by addition of 10 mM EDTA (pH 7.4). Platelet counts were obtained for each sample using a cell counter (Beckman-Coulter, Nyon, Switzerland, http://www.beckmancoulter.com). The remaining sample volume was centrifuged at 3,000g for 20 minutes at 4°C. Plasma samples were collected and stored at −80°C, and formation of thrombin-antithrombin complex, complement C3 activation fragment a (C3a), and soluble C5b-9 complex (sC5b-9) was measured by ELISA [10].
Statistical Analysis
Comparisons between HCs and T1D MSCs were statistically analyzed using Student’s t test or Mann-Whitney U test where data did not fulfill requirements for parametric testing (normal distribution and equal variances). Time-related changes in the groups were analyzed using repeated-measures analysis of variance or Friedman test, whereas pairwise comparisons in the groups were analyzed using paired t test or Wilcoxon signed rank test. p < .05 was considered statistically significant (Prism 5.0; GraphPad, San Diego, CA, http://www.graphpad.com; SPSS 22; IBM, Armonk, NY, http://www.ibm.com).
Results
No Differences in Growth Kinetics Between HC and T1D MSCs In Vitro
Isolated MSCs from BM aspirates of 10 ET1D donors, 12 LT1D donors, and 19 HCs (Table 1) were initially compared with regard to number of MNCs, colony-forming capacity, and growth kinetics (Fig. 1). Linear regression analysis demonstrated a significant age-related decline in the number of BM-MNCs per milliliter of BM in both diabetes and HC donors (p < .01) (Fig. 1A). No significant difference in colony-forming capacity of T1D donors at P1 was evident compared with HCs (Fig. 1B).
Figure 1.

Growth characteristics of MSCs from HC and T1D donors. (A): Age-dependent decline in BM-MNCs per milliliter of BM aspirated from HCs (n = 19), ET1D donors (n = 10), and LT1D donors (n = 12). (B): CFU-F per 100 MSCs at P1 for HC (n = 4), ET1D (n = 4), and T1D donors (n = 4). (C): Growth kinetics (mean ± SD) for MSCs isolated from healthy and T1D donors P1–5 (n = 4 each). (D): Population doubling rate per week for cells from HC (n = 4), ET1D (n = 4), and LT1D (n = 4) donors at P1. Box plot whiskers indicate minimum to maximum. ∗∗, p < .01. Abbreviations: BM-MNC, bone marrow mononuclear cell; CFU-F, colony-forming unit fibroblast; ET1D, early-stage T1D; HC, healthy control; LT1D, late-stage T1D; MSC, mesenchymal stromal cell; ns, not significant; P, passage; T1D, type 1 diabetes.
A comparative analysis of growth kinetics was performed to investigate whether expansion rates and a comparative number of cells for therapeutic usage (in our experience this is generally P2–P3 [32]) could be generated in an equivalent time frame. No significant difference in growth potential or kinetic rate (at P1) between HCs and ET1D or LT1D MSCs was apparent (Fig. 1C, 1D). These data suggest that expansion capacity is comparable between the T1D and HC MSCs, and therefore a sufficient therapeutic number of cells can be expanded in a time frame similar to that of HCs.
Identification of Differentially Expressed Genes
Whole-genome microarray analysis revealed that only 30 transcripts were more than 1.5-fold differentially expressed in MSCs from ET1D donors, and 211 of 47,000 were differentially expressed in LT1D donors compared with HCs (Fig. 2; supplemental online Tables 1, 2). Among these, 177 genes were upregulated and 31 genes were downregulated. GO term analysis of differentially expressed genes in LT1D suggested expression changes in genes enriched in pathways related to growth factor activity, multicellular organismal development and process, response to external stimulus, stress and wounding, adenylate kinase, the TGF-β-signaling pathway, and chromosome 20p12 genes (supplemental online Table 3). Enrichment of the pathway chromosome Y genes was identified from the differentially expressed genes in ET1D compared with HC because of a skewed distribution of the sex of the donors. No other pathway was enriched in ET1D.
Figure 2.

Microarray analysis on global gene expression pattern of MSCs. Heatmap of gene expression in MSCs from ET1D donors (n = 3), LT1D donors (n = 3), and HCs (n = 3). Clustering shows 1.5-fold up- and down-regulated genes (using lower bound of 90% CI) of MSCs from T1D donors compared with HC MSCs. Red represents upregulated and blue downregulated expression. See supplemental online Tables 1 and 2 for complete lists of differentially expressed genes in MSCs from ET1D and LT1D donors, respectively. Abbreviations: CI, confidence interval; ET1D, early-stage T1D; HC, healthy control; LT1D, late-stage T1D; MSC, mesenchymal stromal cell; T1D, type 1 diabetes.
Migration Response to In Vitro Wounding Is Not Affected in T1D MSCs
Results from the whole-genome microarray GO term analysis (supplemental online Table 3) suggested differential expression of genes associated with response to wounding. To assess whether the LT1D MSCs had a differential migratory response to wounding, as suggested by the gene array, an in vitro scratch wound assay was performed. Despite the observed differences in gene expression, no difference in time-dependent wound closure between HC and LT1D MSCs (Fig. 3A, 3B) was observed.
Figure 3.

In vitro scratch wound healing. (A): Confluent cultures of MSCs from HC or LT1D (n = 4 per group) donors were subjected to scratch wounding, and wound closure was monitored by 24-hour live cell imaging. (B): Migration in response to scratch wounding was assessed by quantifying reduction in wound width every 4 hours after initial scratching (mean ± SD). Time-dependent differences in wound closure between MSCs from HC and LT1D donors were analyzed by repeated-measures analysis of variance. (C): Basic expression levels (mean ± SD) of genes related to wounding were studied with qRT-PCR analysis in unscratched control cultures of MSCs from HC and LT1D donors. (D): Fold change in expression levels of wound response genes in scratched MSC cultures compared with unscratched control cultures of the same donors at the 24-hour end point. Box plot whiskers indicate minimum to maximum; asterisks indicate significant difference in gene expression compared with unscratched cultures. ∗, p < .05; ∗∗, p < .01. Abbreviations: HC, healthy control; LT1D, late-stage T1D; MSC, mesenchymal stromal cell; ns, not significant; qRT-PCR, quantitative real-time polymerase chain reaction; T1D, type 1 diabetes.
qRT-PCR confirmed the array findings, demonstrating a threefold higher expression of FOS (p < .05) and sixfold higher expression of BMP-2 (p < .05) in LT1D compared with HC MSCs at resting unscratched state (Fig. 3C). Twenty-four hours after wounding, no differences between HC and T1D MSCs in expression levels of any of the nine genes analyzed were detected. Compared with expression levels in unscratched cultures, there was a significant decrease in FOS levels in both HC and LT1D MSCs 24 hours after initial wounding (p < .05 for both), and in LIF (p < .05) and BMP-2 (p < .01) in LT1D MSCs, indicating normalization of expression levels of these genes in response to scratch wounding (Fig. 3D).
Preserved Immunomodulatory Phenotype of T1D MSCs
HC, ET1D, and LT1D MSCs constitutively secreted comparable levels of IL-6, CXCL1, CXCL6, and PGE2 and demonstrated similar IDO activity (Fig. 4A–4D, 4F). Baseline levels of HGF were lower in ET1D MSCs compared with LT1D (p < .05) (Fig. 4E).
Figure 4.
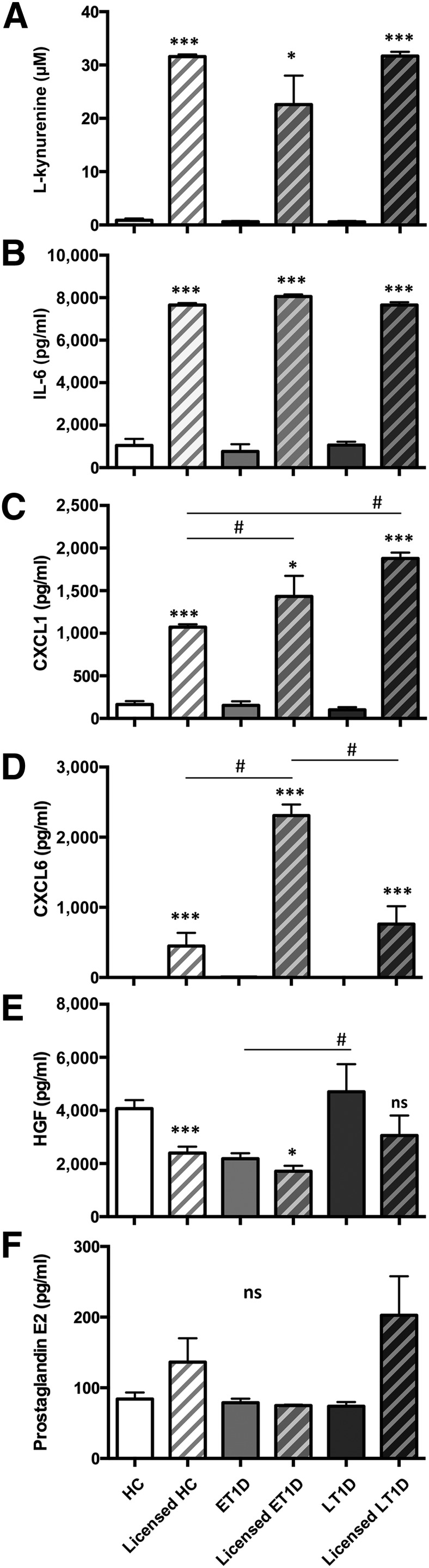
Trophic properties of HC and T1D MSCs in response to licensing. P2 MSCs from HC, ET1D, and T1D donors (n = 4 per group) were evaluated for trophic properties at baseline and after exposure to proinflammatory cytokines IFN-γ and TNF-α (licensed MSCs). (A) IDO activity was assessed by measurement of l-kynurenine (µM). Detection of representative secreted trophic and immunomodulatory factors (pg/ml), IL-6 (B), CXCL1 (C), CXCL6 (D), HGF (E), and PGE2 (F) (mean ± SEM). The effect of licensing in each donor group was analyzed using paired t-test (B, D–F) or Wilcoxon signed rank test (A, C); ∗, p < .05; ∗∗∗, p < .001. Differences between donor groups were analyzed with one-way analysis of variance with Tukey’s post hoc test; #, p < .05. Abbreviations: CXCL, chemokine (C-X-C motif) ligand; ET1D, early-stage T1D; HC, healthy control; HGF, hepatocyte growth factor; IL, interleukin; LT1D, late-stage T1D; MSC, mesenchymal stromal cell; ns, not significant; P, passage; T1D, type 1 diabetes.
Licensing of the MSCs to an anti-inflammatory phenotype by exposure to IFN-γ and TNF-α induced a significant upregulation of IDO activity and IL-6 secretion to comparable levels in HC, ET1D, and LT1D MSCs (p < .001, except IDO ET1D vs licensed ET1D: p < .05) (Fig. 4A, 4B). In both T1D groups, CXCL1 upregulation in response to licensing was higher than in HC MSCs (p < .05) (Fig. 4C), and CXCL6 was significantly higher in the licensed ET1D MSCs compared with both HCs and LT1D MSCs (p < .05) (Fig. 4D). HGF was significantly downregulated in HC and ET1D MSCs compared with baseline levels (p < .001 and p < .05, respectively) (Fig. 4E), but no response was seen in the LT1D group (Fig. 4E). PGE2 secretion was not altered in licensed cells from either HC or T1D groups compared with their respective baseline levels (Fig. 4F).
Despite differences in the responsiveness of the MSC secretome to proinflammatory cytokine exposure, all HCs and T1D donors were able to actively suppress the activation of T cells in vitro (p < .05) (Fig. 5A). Notably, ET1D MSCs were significantly more efficient in suppressing T-cell activation via their secretome than HCs (p < .05) (Fig. 5A) despite their overall lower secretion levels of some soluble factors, as highlighted in the gene array and ELISAs. MSCs from the LT1D donors were more efficacious in suppressing T-cell activation in both contact and Transwell settings compared with corresponding HCs (p < .05) (Fig. 5A).
Figure 5.

Suppression of T-cell activation and effector function with exposure to HC and T1D MSCs. MSCs from HC, ET1D, or LT1D donors (n = 4 per group) were cocultured with antibody-activated T cells in direct contact and separated by Transwell membrane inserts. (A): MSCs suppress T-cell activation as assessed by decreased CD25 expression in contact and Transwell cultures (MFI). Secretion of T-cell effector function molecules IL-2 (B), IFN-γ (C), and TNF-α (D) was suppressed in MSC coculture; most evidently by MSC produced soluble factors (mean ± SEM). All MSCs were compared with T cells only using one-way analysis of variance with Dunnett’s post hoc test; comparisons between MSC donor groups were analyzed using one-way analysis of variance with Tukey’s post hoc test; ∗, p < .05; ∗∗, p < .01; ∗∗∗, p < .001. Abbreviations: BM-MNC, bone marrow mononuclear cell; ET1D, early-stage T1D; HC, healthy control; IFN, interferon; IL, interleukin; LT1D, late-stage T1D; MFI, median fluorescence intensity; MSC, mesenchymal stromal cell; P, passage; T1D, type 1 diabetes; TNF, tumor necrosis factor.
Comparative analysis of T-cell effector function demonstrated that MSCs from all donors exerted comparable suppression on T-cell secretion of IL-2 (p < .001) (Fig. 5B) and IFN-γ, with IFN-γ being suppressed only when the cells were not in direct contact (p < .001) (Fig. 5C). TNF-α levels were suppressed in Transwell settings compared with contact conditions (HC and ET1D, p < .01; LT1D p < .05) (Fig. 5D). Only HC and LT1D MSCs in Transwell suppressed TNF-α levels compared with the T cells only control (HC p < .05; LT1D p < .01) (Fig. 5D).
In Vitro Hemocompatibility of T1D MSCs
To test triggering of IBMIR and complement activation of HCs and T1D MSCs, we investigated the expression of the complement inhibitors CD46, CD55, and CD59 on the cell surface of MSCs, as well as the clotting formation upon whole blood exposure in the Chandler loop model.
Flow cytometry confirmed that MSCs from HCs and T1D donors expressed similar relative levels of CD46 on their cell surface (Fig. 6A). LT1D MSCs expressed significantly higher levels of CD55 compared with both HCs and ET1D donors (p < .05) (Fig. 6B). CD59 levels were, however, significantly lower in both ET1D and LT1D donors compared with HCs (p < .05) (Fig. 6C), but comparable between the T1D subsets.
Figure 6.

Hemocompatibility profiling of MSCs from HC and T1D donors. Cell surface expression of complement inhibitors CD46 (A), CD55 (B), and CD59 (C) was assessed by flow cytometry on HC, ET1D, and LT1D MSCs (n = 4 donors per group) at P2. Data are expressed as median fluorescence intensity (MFI) ± SEM and analyzed using Mann-Whitney U test; ∗, p < .05. Early passage MSCs (P2–3) from HC or T1D donors were tested for triggering of the instant blood mediated inflammatory reaction (IBMIR) after exposure to nonanticoagulated whole blood in the Chandler blood loop model (n = 10 per group). (D): Representative photographs of clot formation from 4 donors from each group after a 60-min incubation of blood with HC or T1D MSCs (15,000 cells per milliliter) or PBS buffer as negative control. (E): Detection of coagulation and complement activation markers after treatment of blood with MSCs (mean ± SD): free platelets (% relative to PBS, 30 minutes time point), and ELISA quantification of TAT, complement C3 activation fragment a (C3a), and soluble C5b-9 complex (sC5b-9). Significance was analyzed with paired t test (same blood donor exposed to different MSCs); ∗, p < .05. Abbreviations: BM-MNC, bone marrow mononuclear cell; C3a, complement C3 activation fragment a; CFU-F, colony-forming unit fibroblast; Cy7, cyanine 7; ET1D, early-stage T1D; FITC, fluorescein isothiocyanate; HC, healthy control; LT1D, late-stage T1D; MFI, median fluorescence intensity; MSC, mesenchymal stromal cell; P, passage; PBS, phosphate-buffered saline; PE, phycoerythrin; T1D, type 1 diabetes; TAT, thrombin-antithrombin complex.
Despite differential cell surface expression of the complement inhibitors between the HC and T1D MSC groups, non-anticoagulated whole blood exposure revealed similar clot formation for both HC and LT1D donor groups (Fig. 6D), with LT1D MSCs demonstrating lower platelet consumption (p < .05) (Fig. 6E) and thrombin formation (p < .05) (Fig. 6F), indicating an overall improved hemocompatibility compared with HC MSCs. No significant difference in complement activation products C3a and sC5b-9 was demonstrated (Fig. 6G and 6H).
Discussion
In this study, we demonstrate that BM-MSCs derived from HC and T1D donors have comparable phenotypic and in vitro functional profiles. Their overall similar in vitro properties, long-term growth kinetics, and immunomodulatory capacity indicate that BM-MSCs from T1D donors are suitable for autologous cell therapy. These findings agree with our recent clinical experience that autologous MSC treatment is safe in T1D patients and may even help to maintain C-peptide secretion for at least 1 year [17]. The option of an autologous cellular approach is preferential, where therapeutically efficacious, reducing the risk of HLA immunity, rejection, and potential transmission of donor-derived infection or disease.
MSCs derived for this study demonstrated typical counts and an age-dependent decline of BM-MNCs for HC and T1D donors at isolation. No difference in CFU-F efficacy at P1 and comparable in vitro growth kinetics confirmed that sufficient numbers of cells for therapeutic efficacy could be yielded from a volume of BM and in a time frame comparable to those of HCs.
Global gene expression analysis indicated moderate differences between ET1D MSCs and HCs. MSCs derived from LT1D donors demonstrated differential expression (>1.5-fold) of 211 genes in categories such as response to external stimulus, stress, wounding, and growth factor activity, suggesting a state of disease memory in these cells. To ascertain whether these genotypic differences correspond to a change in functionality, HC MSCs and T1D MSCs were further investigated using a number of specific in vitro assays.
Upregulation of several genes involved in wound healing and stress responses in LT1D MSCs was confirmed with qRT-PCR analysis of resting MSCs. However, the in vitro scratch wound assay showed no difference in the rate or extent of wound closure between HC and LT1D MSCs. In contrast to the in vivo situation, in which wound healing involves a range of cell types and signaling molecules, the in vitro assay reflects the migrational capacity of MSCs in response to wounding alone. Our results indicate that the MSCs are functionally unaltered in this respect when investigated under in vitro conditions. It is therefore possible to conceive that delayed wound healing in T1D patients may be due primarily to impaired fibroblast functionality in the diabetic tissue or potential impairment in MSC-stromal crosstalk in the orchestration of the wound healing response under diabetic conditions in vivo [33]. This hypothesis is supported by gene expression data demonstrating differences in levels of FOS, which has been implicated in initiation of re-epithelialization [34], and BMP-2, which has been linked to recruitment and migration of MSCs [35] and the stromal response to injury [36].
In establishing a suitable cell source for systemic immunotherapy, understanding of the trophic and immunomodulatory properties of the MSC source is central. Long-term exposure to the diabetic environment has been suggested to induce disease memory in BM-MSCs [37], characterized by an increased expression of proinflammatory markers such as IL-6. Although our mRNA expression data supports a fourfold increase of IL-6 in LT1D MSCs compared with HCs, baseline secretion of IL-6 protein, along with PGE2, CXCL1, CXCL6, and IDO activity, were not significantly changed in the diabetes MSCs. We do report significantly lower secretion of HGF by ET1D MSCs compared with the LT1D donors, but no significant change from the HCs was evident.
Exposure of MSCs to proinflammatory cytokines such as IFN-γ and TNF-α has been reported to induce an anti-inflammatory MSC phenotype [22, 38]. In this study, we investigated whether responsiveness to such licensing was affected in T1D MSCs compared with HCs. IDO and IL-6 have been reported as two of the primary soluble immunomodulatory factors implicated in MSC-mediated immunosuppression [39–41]. Here we report significant upregulation of both factors in response to licensing, but no difference in the level of secretion between MSC donor groups.
Moderate downregulation of HGF was evident in HC and ET1D MSCs in response to inflammation. In contrast, LT1D MSCs demonstrated no significant decrease in secretion compared with baseline controls with proinflammatory cytokine exposure. Differences were also evident in CXCL1 secretion, with T1D MSCs secreting higher levels compared with HCs, and in CXCL6 secretion, with ET1D MSCs also secreting higher levels in response to licensing compared with both HCs and LT1D MSCs. These differences in chemokine expression suggest no loss in responsiveness to inflammatory stimuli and a comparable or increased ability to recruit innate immune cells such as monocytes, neutrophils, and eosinophils.
Experience from animal models suggests that MSC infusions slow T1D progression by supporting damaged tissues and modulating immune responses [6–9]. In our study, both HCs and T1D MSCs demonstrated a similar suppression of T-cell activation, with some indication of a potentially enhanced immunosuppressive effect with LT1D MSCs. In addition, similar suppression of proinflammatory cytokine production is reported between MSC donor groups, particularly in Transwell culture, in which soluble factors and microvesicles released by the MSCs play a pivotal role. These data suggest that the in vitro immunomodulatory activity of the MSCs is not compromised after exposure to the diabetic microenvironment in vivo; further supporting the above similarities in trophic factor levels between HCs and T1D MSCs.
Soria and coworkers recently reported on the altered hemocompatibility of diabetic donor adipose tissue-derived MSCs [42] and suggested the need to include appropriate safety testing of autologous MSC products before clinical use. We found that LT1D MSCs had similar to favorable hemocompatibility after exposure to human whole blood. In addition to their potentially enhanced immunosuppressive activity, LT1D MSCs demonstrated significantly lower platelet consumption and thrombin formation in comparison with HC MSCs.
Conclusion
In determining an appropriate cell source for systemic cell-based immunotherapy, knowledge of immunomodulatory potential and hemocompatibility is essential. In this study, we demonstrate that despite transcriptional differences between HC and T1D MSCs, no functional differences in terms of migratory capacity, immunomodulation, or hemocompatibility were evident. Notably, we have recently published a clinical trial using the same donor MSCs as investigated here for the autologous treatment of newly diagnosed T1D patients [17]. This study demonstrated no side effects after MSC infusion and, during the first year, preserved or even increased C-peptide response to a mixed-meal tolerance test. We conclude, therefore, that autologous BM is a suitable source for clinical grade production of therapeutic MSCs in the systemic treatment of T1D and should be evaluated in larger clinical trials.
Supplementary Material
Acknowledgments
This study was supported by grants from Swedish Cancer Society, Children’s Cancer Foundation, Swedish Medical Research Council, VINNOVA, Stockholm County Council (ALF), Cancer Society in Stockholm, Swedish Society of Medicine, Tobias Foundation, Karolinska Institutet, Juvenile Diabetes Foundation International, AFA Insurance, Swedish Diabetes Association, Swedish Juvenile Diabetes Foundation, Novo Nordisk Foundation, and Diabetes Wellness Sverige.
Author Contributions
L.C.D., J.J.A., N.H., and G.M.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; C.G., I.B., and M.S.: collection and assembly of data; H.Q.: collection and assembly of data, data analysis and interpretation; B.N.: conception and design, final approval of manuscript; L.E.K. and K.T.S.: provision of study material; P.-O.C.: conception and design, provision of study material; O.K.: conception and design, provision of study material, final manuscript approval; K.L.B: conception and design, financial support, provision of study material, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
O.K. and K.L.B. are cofounders and owners of iCell Science AB. The other authors indicated no potential conflicts of interest.
References
- 1.Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:2. doi: 10.1101/cshperspect.a007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 3.Mangialardi G, Oikawa A, Reni C, et al. Bone marrow microenvironment: A newly recognized target for diabetes-induced cellular damage. Endocr Metab Immune Disord Drug Targets. 2012;12:159–167. doi: 10.2174/187153012800493530. [DOI] [PubMed] [Google Scholar]
- 4.Chhabra P, Brayman KL. Stem cell therapy to cure type 1 diabetes: From hype to hope. Stem Cells Translational Medicine. 2013;2:328–336. doi: 10.5966/sctm.2012-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hematti P, Kim J, Stein AP, et al. Potential role of mesenchymal stromal cells in pancreatic islet transplantation. Transplant Rev (Orlando) 2013;27:21–29. doi: 10.1016/j.trre.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Lee RH, Seo MJ, Reger RL, et al. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci USA. 2006;103:17438–17443. doi: 10.1073/pnas.0608249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urbán VS, Kiss J, Kovács J, et al. Mesenchymal stem cells cooperate with bone marrow cells in therapy of diabetes. Stem Cells. 2008;26:244–253. doi: 10.1634/stemcells.2007-0267. [DOI] [PubMed] [Google Scholar]
- 8.Gabr MM, Zakaria MM, Refaie AF, et al. Insulin-producing cells from adult human bone marrow mesenchymal stem cells control streptozotocin-induced diabetes in nude mice. Cell Transplant. 2013;22:133–145. doi: 10.3727/096368912X647162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho JH, Tseng TC, Ma WH, et al. Multiple intravenous transplantations of mesenchymal stem cells effectively restore long-term blood glucose homeostasis by hepatic engraftment and β-cell differentiation in streptozocin-induced diabetic mice. Cell Transplant. 2012;21:997–1009. doi: 10.3727/096368911X603611. [DOI] [PubMed] [Google Scholar]
- 10.Moll G, Rasmusson-Duprez I, von Bahr L, et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells. 2012;30:1565–1574. doi: 10.1002/stem.1111. [DOI] [PubMed] [Google Scholar]
- 11.Favaro E, Carpanetto A, Lamorte S, et al. Human mesenchymal stem cell-derived microvesicles modulate T cell response to islet antigen glutamic acid decarboxylase in patients with type 1 diabetes. Diabetologia. 2014;57:1664–1673. doi: 10.1007/s00125-014-3262-4. [DOI] [PubMed] [Google Scholar]
- 12.Kordelas L, Rebmann V, Ludwig AK, et al. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970–973. doi: 10.1038/leu.2014.41. [DOI] [PubMed] [Google Scholar]
- 13.Jung EJ, Kim SC, Wee YM, et al. Bone marrow-derived mesenchymal stromal cells support rat pancreatic islet survival and insulin secretory function in vitro. Cytotherapy. 2011;13:19–29. doi: 10.3109/14653249.2010.518608. [DOI] [PubMed] [Google Scholar]
- 14.Park KS, Kim YS, Kim JH, et al. Trophic molecules derived from human mesenchymal stem cells enhance survival, function, and angiogenesis of isolated islets after transplantation. Transplantation. 2010;89:509–517. doi: 10.1097/TP.0b013e3181c7dc99. [DOI] [PubMed] [Google Scholar]
- 15.Jeker LT, Bour-Jordan H, Bluestone JA. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb Perspect Med. 2012;2:a007807. doi: 10.1101/cshperspect.a007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Jia S, Geoffrey R, et al. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180:1929–1937. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson PO, Schwarcz E, Korsgren O, et al. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes. 2015;64:587–592. doi: 10.2337/db14-0656. [DOI] [PubMed] [Google Scholar]
- 18.Cai J, Wu Z, Xu X, et al. Umbilical cord mesenchymal stromal cell with autologous bone marrow cell transplantation in established type 1 diabetes: A pilot randomized controlled open-label clinical study to assess safety and impact on insulin secretion. Diabetes Care. 2016;39:149–157. doi: 10.2337/dc15-0171. [DOI] [PubMed] [Google Scholar]
- 19.Oh KW, Moon C, Kim HY, et al. Phase I trial of repeated intrathecal autologous bone marrow-derived mesenchymal stromal cells in amyotrophic lateral sclerosis. Stem Cells Translational Medicine. 2015;4:590–597. doi: 10.5966/sctm.2014-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mudrabettu C, Kumar V, Rakha A, et al. Safety and efficacy of autologous mesenchymal stromal cells transplantation in patients undergoing living donor kidney transplantation: A pilot study. Nephrology (Carlton) 2015;20:25–33. doi: 10.1111/nep.12338. [DOI] [PubMed] [Google Scholar]
- 21.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 22.Le Blanc K, Tammik L, Sundberg B, et al. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol. 2003;57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x. [DOI] [PubMed] [Google Scholar]
- 23.Le Blanc K, Samuelsson H, Gustafsson B, et al. Transplantation of mesenchymal stem cells to enhance engraftment of hematopoietic stem cells. Leukemia. 2007;21:1733–1738. doi: 10.1038/sj.leu.2404777. [DOI] [PubMed] [Google Scholar]
- 24.Qian H, Badaloni A, Chiara F, et al. Molecular characterization of prospectively isolated multipotent mesenchymal progenitors provides new insight into the cellular identity of mesenchymal stem cells in mouse bone marrow. Mol Cell Biol. 2013;33:661–677. doi: 10.1128/MCB.01287-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 26.Davies LC, Lönnies H, Locke M, et al. Oral mucosal progenitor cells are potently immunosuppressive in a dose-independent manner. Stem Cells Dev. 2012;21:1478–1487. doi: 10.1089/scd.2011.0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enoch S, Peake MA, Wall I, et al. ‘Young’ oral fibroblasts are geno/phenotypically distinct. J Dent Res. 2010;89:1407–1413. doi: 10.1177/0022034510377796. [DOI] [PubMed] [Google Scholar]
- 28.Nilsson B, Larsson R, Hong J, et al. Compstatin inhibits complement and cellular activation in whole blood in two models of extracorporeal circulation. Blood. 1998;92:1661–1667. [PubMed] [Google Scholar]
- 29.Bennet W, Sundberg B, Groth CG, et al. Incompatibility between human blood and isolated islets of Langerhans: A finding with implications for clinical intraportal islet transplantation? Diabetes. 1999;48:1907–1914. doi: 10.2337/diabetes.48.10.1907. [DOI] [PubMed] [Google Scholar]
- 30.Gustafson EK, Elgue G, Hughes RD, et al. The instant blood-mediated inflammatory reaction characterized in hepatocyte transplantation. Transplantation. 2011;91:632–638. doi: 10.1097/TP.0b013e31820ae459. [DOI] [PubMed] [Google Scholar]
- 31.Ekdahl KN, Hong J, Hamad OA, et al. Evaluation of the blood compatibility of materials, cells, and tissues: Basic concepts, test models, and practical guidelines. Adv Exp Med Biol. 2013;735:257–270. doi: 10.1007/978-1-4614-4118-2_18. [DOI] [PubMed] [Google Scholar]
- 32.von Bahr L, Sundberg B, Lönnies L, et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol Blood Marrow Transplant. 2012;18:557–564. doi: 10.1016/j.bbmt.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 33.Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med. 2006;23:594–608. doi: 10.1111/j.1464-5491.2006.01773.x. [DOI] [PubMed] [Google Scholar]
- 34.Gangnuss S, Cowin AJ, Daehn IS, et al. Regulation of MAPK activation, AP-1 transcription factor expression and keratinocyte differentiation in wounded fetal skin. J Invest Dermatol. 2004;122:791–804. doi: 10.1111/j.0022-202X.2004.22319.x. [DOI] [PubMed] [Google Scholar]
- 35.Fiedler J, Röderer G, Günther KP, et al. BMP-2, BMP-4, and PDGF-bb stimulate chemotactic migration of primary human mesenchymal progenitor cells. J Cell Biochem. 2002;87:305–312. doi: 10.1002/jcb.10309. [DOI] [PubMed] [Google Scholar]
- 36.Peake MA, Caley M, Giles PJ, et al. Identification of a transcriptional signature for the wound healing continuum. Wound Rep Regen. 2014;22:399–405. doi: 10.1111/wrr.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madhira SL, Challa SS, Chalasani M, et al. Promise(s) of mesenchymal stem cells as an in vitro model system to depict pre-diabetic/diabetic milieu in WNIN/GR-Ob mutant rats. PLoS One. 2012;7:e48061. doi: 10.1371/journal.pone.0048061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernardo ME, Fibbe WE. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Deng Y, Yi S, Wang G, et al. Umbilical cord-derived mesenchymal stem cells instruct dendritic cells to acquire tolerogenic phenotypes through the IL-6-mediated upregulation of SOCS1. Stem Cells Dev. 2014;23:2080–2092. doi: 10.1089/scd.2013.0559. [DOI] [PubMed] [Google Scholar]
- 40.Meisel R, Zibert A, Laryea M, et al. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 41.Melief SM, Geutskens SB, Fibbe WE, et al. Multipotent stromal cells skew monocytes towards an anti-inflammatory interleukin-10-producing phenotype by production of interleukin-6. Haematologica. 2013;98:888–895. doi: 10.3324/haematol.2012.078055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Acosta L, Hmadcha A, Escacena N, et al. Adipose mesenchymal stromal cells isolated from type 2 diabetic patients display reduced fibrinolytic activity. Diabetes. 2013;62:4266–4269. doi: 10.2337/db13-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.