

Abstract
Historically, glutamate uptake in the CNS was mainly attributed to glial cells for three reasons: 1) none of the glutamate transporters were found to be located in presynaptic terminals of excitatory synapses; 2) the putative glial transporters, GLT-1 and GLAST are expressed at high levels in astrocytes; 3) studies of the constitutive GLT-1 knockout as well as pharmacological studies demonstrated that >90% of glutamate uptake into forebrain synaptosomes is mediated by the operation of GLT-1. Here we summarize the history leading up to the recognition of GLT-1a as a presynaptic glutamate transporter. A major issue now is understanding the physiological and pathophysiologial significance of the expression of GLT-1 in presynaptic terminals. To elucidate the cell-type specific functions of GLT-1, a conditional knockout was generated with which to inactivate the GLT-1 gene in different cell types using Cre/lox technology. Astrocytic knockout led to an 80% reduction of GLT-1 expression, resulting in intractable seizures and early mortality as seen also in the constitutive knockout. Neuronal knockout was associated with no obvious phenotype. Surprisingly, synaptosomal uptake capacity (Vmax) was found to be significantly reduced, by 40%, in the neuronal knockout, indicating that the contribution of neuronal GLT-1 to synaptosomal uptake is disproportionate to its protein expression (5–10%). Conversely, the contribution of astrocytic GLT-1 to synaptosomal uptake was much lower than expected. In contrast, the loss of uptake into liposomes prepared from brain protein from astrocyte and neuronal knockouts was proportionate with the loss of GLT-1 protein, suggesting that a large portion of GLT-1 in astrocytic membranes in synaptosomal preparations is not functional, possibly because of a failure to reseal. These results suggest the need to reinterpret many previous studies using synaptosomal uptake to investigate glutamate transport itself as well as changes in glutamate homeostasis associated with normal functions, neurodegeneration, and response to drugs.
Keywords: neuronal glutamate transport, glutamate homeostasis, GLT-1, conditional knockout
Introduction
In the mammalian brain, small molecules called neurotransmitters function to transmit signals across chemical synapses. There are 4 lines of evidence that need to be assembled to establish a substance as a neurotransmitter—synthesis in the presynaptic terminal, release from the presynaptic terminal by action potentials, the presence of receptors on the postsynaptic membrane that produce a response that can be measured postsynaptically, and the presence of a clearance mechanism for the transmitter in the extracellular space. Transmitter clearance is required for several reasons: 1) to prevent the accumulation of the transmitter in the extracellular space; 2) to prevent the metabolic waste of leaving the transmitter in the extracellular space unrecovered for cellular utilization; 3) to restore transmitter to presynaptic terminals that might become depleted of transmitter.
Glutamate is now widely accepted as the excitatory neurotransmitter in the mammalian brain, but its role as a neurotransmitter was slow to become established because of its central role in intermediary metabolism, its ubiquitous distribution, and presence in high concentration in the brain. One approach to testing the hypothesis that glutamate is a neurotransmitter that was taken in the 1970’s was to focus on the requirement that a transmitter must have a specific clearance mechanism in the terminals from which it is released. This approach was pioneered by Julie Axelrod and colleagues, who demonstrated monoamine uptake systems in synaptosome preparations, and also that the uptake that was observed was specifically into presynaptic terminals. With this as a background, Wofsey, Kuhar, and Snyder reasoned: “Transport systems into nerve terminals have been described for several putative neurotransmitters, including norepinephrine, serotonin, GABA. If certain amino acids are neurotransmitters, then perhaps analogous uptake systems might transport them into nerve terminals. If such is the case, the exogenous amino acid which would label selectively a “neurotransmitter pool” should be more highly localized in synaptosomal fractions than would be the endogenous amino acids, which would include metabolic as well as transmitter pools” (Wofsey et al., 1971). These authors showed that 3H-glutamate and aspartate accumulate in a unique low density synaptosomal fraction containing pinched off nerve terminals, a similar result to one obtained using brain slices (Kuhar and Snyder, 1970). It was subsequently shown that the uptake system for glutamate into brain synaptosomes is a high affinity sodium dependent carrier (Bennett et al., 1972; Logan and Snyder, 1971). The conclusions of these studies were reinforced by EM autoradiographic studies demonstrating uptake of 3H-glutamate into axon terminals of pigeon and rat (Beart, 1976a, b; Divac et al., 1977; Iversen and Storm-Mathisen, 1976; Storm-Mathisen, 1977; Storm-Mathisen and Iversen, 1979). These studies, in aggregate, provided biochemical evidence for a neurotransmitter pool of glutamate that was at least in part sustained by a specific high affinity transport system, and were important in providing part of the foundation of evidence to support the identification of glutamate as the excitatory neurotransmitter (Fonnum et al., 1981).
In 1992, the 3 glutamate transporters of the forebrain were cloned, and, none of them proved to be localized in presynaptic terminals of excitatory synapses. However, astrocyte membranes adjacent to synapses expressed high levels of the transporters GLT-1 and GLAST, and eventually the notion that there is a glutamate transporter in presynaptic terminals was abandoned as being unsupported by molecular evidence and biologically unnecessary. A detailed history of these developments has been provided in a comprehensive review by Niels Danbolt (Danbolt, 2001) concerning glutamate transport that appeared in 2001 in which the problem was alluded to as the “elusive presynaptic glutamate transporter”. The present review represents a brief overview of the project started by Snyder and colleagues over 40 years ago to characterize a presynaptic glutamate transport system in the brain and a more detailed update to cover the time period after 2001. For a more detailed coverage of the period before 2001, the reader is referred to the review by Niels Danbolt.
1.1 Glutamate transporter family
Excitatory neuronal activity is accompanied by release of glutamate into the synaptic cleft (Seifert et al., 2006) (Fig. 1). The amino acid L-glutamate is not only the major excitatory neurotransmitter in the central nervous system (CNS) but also a potent neurotoxin (Billups et al., 1998; Choi and Rothman, 1990; Lipton and Rosenberg, 1994; Meldrum and Garthwaite, 1990; Olney, 1990). To maintain low, non-toxic extracellular glutamate concentrations and ensure the fidelity of synaptic transmission, in addition to diffusion, rapid glutamate removal from the synaptic cleft is essential. In the case of excitatory synapses in the mammalian central nervous system clearance is provided by glutamate transporters. By controlling the concentration profile of glutamate in and around the synaptic cleft after release (Huang and Bergles, 2004), glutamate transporters play an important role in preserving the signaling functions of synapses (Katagiri et al., 2001; Stoffel et al., 2004), regulating the activation of nearby metabotropic receptors (Huang et al., 2004; Otis et al., 2004; Scanziani et al., 1997), controlling crosstalk between excitatory synapses (Arnth-Jensen et al., 2002; Asztely et al., 1997; Rusakov and Kullmann, 1998), and in some regions may shape the kinetics of excitatory postsynaptic currents (EPSCs) (Barbour et al., 1994; Takahashi et al., 1995; Tong and Jahr, 1994; Tzingounis and Wadiche, 2007).
Figure 1. Schematic diagram of the glutamatergic synapse showing locations of the glutamate transporter subtypes.

GLT-1 is the major glutamate transporter and is primarily expressed in astrocytes (Lehre et al., 1995; Rothstein et al., 1994) but also in neurons, in axon terminals (Chen et al., 2004). In the hippocampus, the expression of GLT-1 protein in axon terminals is approximately 5–10% of total GLT-1 protein expression (Furness et al., 2008). GLAST is thought to be exclusively a glial transporter (Rothstein et al., 1994; Storck et al., 1992). EAAC1 is a neuronal glutamate transporter with a somatodendritic localization (Holmseth et al., 2012; Rothstein et al., 1994; Stoffel et al., 2004).
The stoichiometry of glutamate transport is determined by the transmembrane concentration gradients for sodium (Na+), potassium (K+), and protons (H+), and the membrane potential. These electrochemical gradients are maintained by the Na+/K+-ATPase and provide the driving force for glutamate uptake. It is generally accepted that GLT-1 co-transports one Glu− with three Na+ and one H+, and countertransports one K+ per cycle (Levy et al., 1998; Zerangue and Kavanaugh, 1996) making it an electrogenic transport process. Under physiological conditions, the inwardly directed Na+ gradient and outwardly directed K+ gradient, as well as the interior negative membrane potential favor the accumulation of glutamate into the cell against its concentration gradient (Kanner, 2006; Zerangue and Kavanaugh, 1996).
Two large families of high affinity, sodium dependent transporters resident in the plasma membrane have been identified. One family includes chloride dependent transporters such as those specific for catecholamines and for glycine (Amara, 1992). A separate family of potassium dependent glutamate transporters includes five members: GLT-1, isolated from rat (Pines and Kanner, 1990), whose human counterpart is excitatory amino acid transporter 2 (EAAT2) (Arriza et al., 1994); GLAST, isolated from rat (Stoffel et al., 1992), whose human counterpart is EAAT1 (Arriza et al., 1994); EAAC1, first isolated from rabbit (Kanai and Hediger, 1992), whose human counterpart is EAAT3 (Arriza et al., 1994); EAAT4, expressed predominantly in the cerebellum (Fairman et al., 1995), and EAAT5, expressed predominantly in the retina (Arriza et al., 1997). These glutamate transporters transport only the stereoisomer L-glutamate but both L- and D-aspartate.
1.2. Glutamate transporters in axon terminals implicated in the pathophysiology of ischemia
At the same time that the mammalian glutamate transporters were being cloned, evidence emerged that excessive glutamate is released from nerve terminals during ischemia (Madl, 1995; Madl and Burgesser, 1993). Nerve terminals contain higher glutamate concentrations than astroglia (Lipton, 1999). Previous experiments showed dependence of excitotoxicity in the striatum and hippocampus upon intact glutamatergic projections (Benveniste et al., 1989; McBean and Roberts, 1984) suggesting that release of glutamate from these terminals plays an important role in the pathogenesis of excitotoxicity, although alternative explanations focusing on postsynaptic effects of deafferentation were also considered (Buchan and Pulsinelli, 1990b; Kaur et al., 2006). Abundant evidence suggested that glutamate uptake in the striatum occurs predominantly in terminals of the corticostriatal pathway (Cross et al., 1986; Divac et al., 1977; Fonnum et al., 1981). In animals, destruction of afferent excitatory pathways markedly potentiated the toxicity of exogenously applied glutamate (Kohler and Schwarcz, 1981; McBean and Roberts, 1985). In addition, intact axon terminals appeared to be required for global ischemic injury to CA1 (Benveniste et al., 1989; Buchan and Pulsinelli, 1990a), and glutamate was shown to be released from axon terminals with ischemia (Benveniste et al., 1989). These data suggested that glutamate transporters in axon terminals might play an essential role in excitotoxicity in the striatum and hippocampus. However, by lesioning the cerebral cortex Levy (1995) and colleagues found both a reduced synaptosomal uptake, as well as decreased expression levels of GLT-1 and GLAST in the striatum. These findings suggested that astroglial expression of GLT-1 and GLAST is controlled, at least in part, by neurons. This hypothesis was soon confirmed by in vitro studies (Gegelashvili et al., 1997; Robinson et al., 1998; Swanson et al., 1997).
1.3. GLT-1
GLT-1 is the major transporter in the brain and represents 1% of total brain protein (Lehre and Danbolt, 1998). Deletion of the GLT-1 gene eliminates 95% of the glutamate uptake activity in forebrain synaptosomes and leads to premature death due to intractable seizures (Tanaka et al., 1997).
Multiple studies have demonstrated that GLT-1 mRNA is expressed in neurons in addition to astrocytes (Berger et al., 2005; Berger and Hediger, 1998; Schmitt et al., 1996; Torp et al., 1994; Trotti et al., 1995). In the mature brain, GLT-1 protein is highly expressed in glial membranes (Danbolt et al., 1992; Lehre et al., 1995) and was thought to be exclusively associated with astrocytes (Lehre et al., 1995; Rothstein et al., 1994). Chaudhry and colleagues (1995) found weak GLT-1 immunogold labeling over neuronal membranes at excitatory synapses in the hippocampus but were undecided about whether this labeling was above background, and if so, was not due to spatial spread of the heavy labeling of adjacent astrocyte membranes. In reviewing these data, the conclusion reached was that the apparent neuronal labeling was not significant and therefore axon terminals do not express GLT-1 (Danbolt, 2001).
This notion was in apparent contradiction to a long series of pharmacological studies of synaptosomal glutamate or aspartate uptake, beginning with work by Ferkany and Coyle (Ferkany and Coyle, 1986) and extended by Robinson (Robinson et al., 1991; Robinson et al., 1993) and others (Bridges et al., 1999; Koch et al., 1999a; Koch et al., 1999b) showing that glutamate uptake in synaptosomes prepared from the forebrain is almost completely blocked by the specific GLT-1 inhibitor dihydrokainate (DHK). Genetic evidence for the importance of GLT-1 in synaptosomal uptake came from studies using the pan GLT-1 knockout, as already mentioned (Tanaka et al., 1997). To reconcile these data with the apparent absence of GLT-1 protein in neuronal membranes, the consensus developed that synaptosomal uptake represents uptake across glial membranes. The conclusion that neurons don’t have a glutamate clearance mechanism was supported by electrophysiological studies that showed that no transporter currents could be measured in pyramidal cell bodies. The view that emerged was that the job of glutamate clearance at synapses was the exclusive province of astrocytes (Bergles et al., 1999; Bergles and Jahr, 1997; Huang and Bergles, 2004). However, expression of GLT-1 protein in neurons in vivo had been found in studies of the retina (Euler and Wassle, 1995; Rauen and Kanner, 1994; Rauen et al., 1996) and under pathological conditions, such as after hypoxia (Martin et al., 1997).
2. GLT-1 expression in neurons
2.1. GLT-1 in neurons in culture
The toxicity of glutamate to cerebral neurons in culture is predominantly mediated by NMDA receptors (Choi et al., 1988). However, a problem that was not fully recognized or appreciated was that the potency of glutamate as an excitoxic agonist in the mixed cultures of neurons and astrocytes that were typically used for these studies was about 100 fold less than the affinity of glutamate for the NMDA receptor (Rosenberg and Aizenman, 1989; Rosenberg et al., 1992). The anomalous low potency in mixed cultures was due to a well-characterized problem of pharmacological properties being distorted by the presence of an active uptake system which, in fact, had already been demonstrated in a biochemical study of NMDA receptor mediated activation of cGMP synthesis in the cerebellum (Garthwaite, 1985). Since the anomalously low excitotoxic potency of glutamate was shown to be due to the operation of a glutamate transport system in mixed cultures of astrocytes and neurons that was not operating in the neuronal cultures, it was easy to ascribe the transport system responsible to astrocytes. However, an electron microscopic examination of the two types of cultures revealed that in the mixed cultures synapses were sequestered deep in the culture by a network of astrocyte processes at the surface of the culture, whereas in the neuronal cultures, synapses were exposed directly to the extracellular medium (Harris and Rosenberg, 1993). Therefore, it was conceivable that in mixed cultures, synapses were protected by their sequestration by astrocyte processes, and within the protected space provided by astrocytes, either neuronal or astrocyte glutamate transporters (or both) might be operating to prevent extracellular glutamate from reaching the high density of glutamate receptors at or around synapses. This perspective led to an effort to characterize glutamate transport in neurons using the relatively pure neuronal cultures that had been used for the excitotoxicity studies, as well as in pure astrocyte cultures and mixed cultures of neurons and astrocytes.
Pharmacological studies showed that bulk uptake of radioactive substrate for glutamate transporters (either L-glutamate or D-aspartate) into neuronal cultures was inhibited by DHK (Wang et al., 1998). Since neuronal cultures invariably contain a small proportion of astrocytes, the question remained whether glutamate uptake assayed in these cultures was actually due to uptake into neurons. Neuronal uptake was clearly observed using radioactive [3H]-D-aspartate autoradiography (unpublished data, Fig. 2, A-F). Panel A shows a low power view (125×) of cultures. Silver grains outlining many neuronal processes are evident, as well as a few cell bodies (two are present near the center of the field). In addition, many grains are present diffusely but not uniformly in the background and without revealing any recognizable cellular detail. Parts of several large astrocytes are also revealed by the grains, for example in the upper right and lower left. Panel B shows another low power view, clearly showing a neuron in the center of the field that has labeling over its cell body as well as several processes that radiate out from it. Again, a part of an astrocyte is shown in the lower right corner. Panels C and D show at higher magnification (250×) similarly treated cultures. Panel C is centered on one or possibly two very large astrocytes with many thick processes darkly labeled by silver grains. The heavy background labeling surrounding the astrocyte is also seen here, as it is in panel D. In addition, panel D shows several labeled neuronal cell bodies as well as processes. The apparent holes in the heavily labeled background constitute neuronal cell bodies that are not labeled. Panels E and F show cultures that have been exposed to [3H]-D-aspartate in the presence of 500 μM DHK. In this case, the astrocytes are still heavily labeled as can be seen in panel E. However the background labeling is now very light, for example in the culture surrounding the central astrocytes. Panel F shows that this background labeling has not been eliminated altogether, and many neuronal cell bodies are in fact lightly labeled with silver grains. In control experiments, it was found that 1 mM PDC eliminated virtually all labeling with [3H]-D-aspartate so that the density of silver grains was not significantly different than in cultures that went through the whole autoradiographic procedure but were never exposed to tracer. Taken together, these observations established that there is a DHK sensitive glutamate transporter expressed in forebrain neurons in culture.
Figure 2. [3H]-D-aspartate uptake autoradiography in rat neuronal cultures.
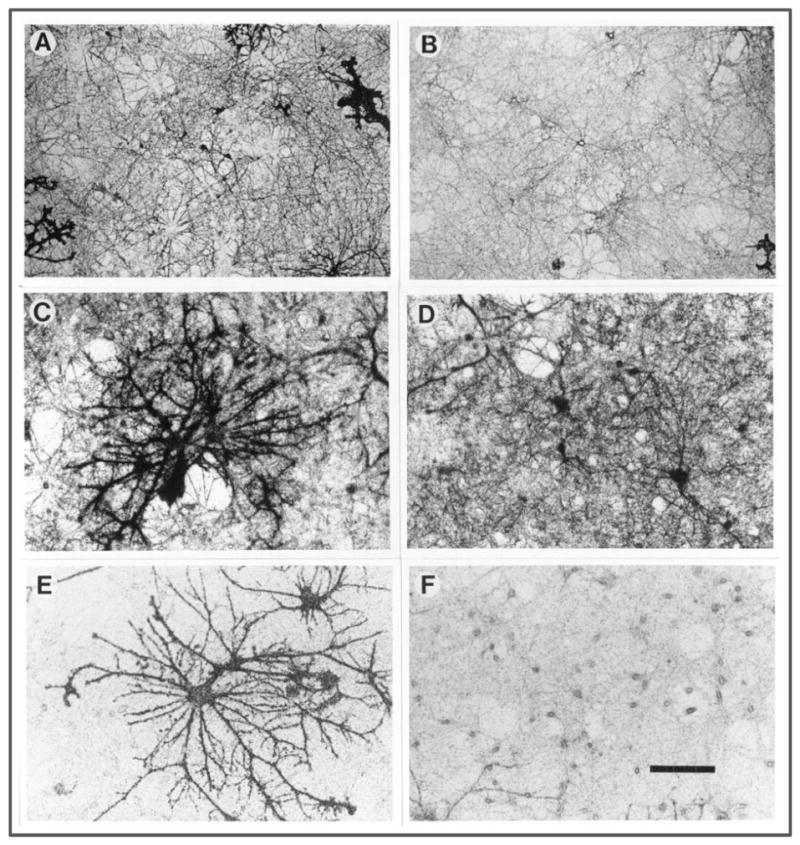
Neuronal cultures derived from embryonic rat forebrain were exposed to [3H]-D-aspartate for 5 minutes, after which they were fixed for 24 hours, dehydrated in ethanol, dried, dipped in emulsion, and exposed for 4 weeks (Kriegstein and Dichter, 1984). All photomicrography was done using brightfield illumination. Scale bar represents 240 microns in A and B, and 120 microns in C – F.
A, B) Low magnification view (125×) of cultures exposed to [3H]-D-aspartate
C, D) Higher power view (250×) of cultures exposed to [3H]-D-aspartate
E, F) Higher power view of cultures exposed to [3H]-D-aspartate in the presence of 500 μM DHK
Since DHK sensitivity of GLT-1 was shown to be due to a specific DNA cassette in the GLT-1 gene (Zhang and Kanner, 1999), it was conceivable that there was a novel glutamate transporter expressed in neurons containing this DHK sensitivity conferring cassette, or a GLT-1 variant in neurons different from the form described by Pines (1992) which might not be recognized by available antibodies. To identify whether a novel glutamate transporter or transporter variant was expressed in neurons, a cDNA library was prepared from forebrain neuronal cultures and screened using a homology strategy (Chen et al., 2002) similar to that used by Amara and colleagues to discover EAAT4 which is highly expressed in the cerebellum and produces the different pharmacology observed in cerebellar synaptosomes compared with forebrain synaptosomes (Fairman et al., 1995). By this approach, a variant form of GLT-1 was cloned from the neuronal cDNA library. This variant form, produced by alternate polyadenylation, contains a longer 3-UTR different from the originally published GLT-1 and extends to a stop codon between exons 9 and 10. The original GLT-1 form as was designated as “GLT-1a” (GLT-1a; GenBank accession number AY069978) and the variant GLT-1 form as “GLT-1b” (GLT-1b; GenBank accession number AF451299). Northern blot analysis of forebrain RNA confirmed the expression of GLT-1b in vivo and showed high levels of expression in the cortex, hippocampus, striatum, thalamus, and midbrain. Two other groups reported on the identification of the GLT-1b variant form, using different approaches (Reye et al., 2002a; Reye et al., 2002b; Schmitt et al., 2002). An isoform of GLT-1 with a similar C-terminus to GLT-1 b had previously been cloned from mouse liver (Utsunomiya-Tate et al., 1997). Danbolt and colleagues (Holmseth et al., 2009) determined the relative levels of expression of GLT-1a, GLT-1b, and another variant form, GLT-1c in the rat forebrain. GLT-1b and GLT-1c were approximately 6% and 1% of GLT-1a expression. No labeling of GLT-1b was found in spines or nerve terminals.
Other in vitro studies also obtained evidence for the expression of GLT-1 in neurons. In mixed rat hippocampal microcultures of the forebrain Mennerick and colleagues (Mennerick et al., 1998) performed studies using a combination of antibodies against the N- and C-terminus of GLT-1. They found a significant expression GLT-1 exclusively in excitatory neurons. Examining the functionality of the transporter, they found DHK sensitive D-aspartate currents. Notably, in this study GLT-1 was found to be exclusively localized in neurons in postsynaptic processes. Suchak and colleagues (Suchak et al., 2003) detected GLT-1 mRNAs in primary cultures of mouse cortical or striatal neurons using reverse-transcriptase PCR. Western blotting of synaptosomes from adult mouse and rat demonstrated GLT-1 protein (Suchak et al., 2003). In her dissertation, Y.H. Huang found that photolysis of MNI-D-aspartate elicited glutamate transporter currents in hippocampal CA3 but not CA1 pyramidal neurons. This photolysis-evoked transporter current was absent in GLT-1 knockout mice and in the presence of WAY 213613, a selective antagonist of GLT-1, indicating that the current was mediated by GLT-1. The current could be evoked at the soma as well as the apical and basal dendrites of CA3 pyramidal neurons (Huang, 2006).
2.2. In situ hybridization and electron microscopic localization of GLT-1a in neurons
Several groups (Berger and Hediger, 1998; Schmitt et al., 1996) described the occurrence of GLT-1 mRNA expression in neurons in the mature brain, prominently in the CA3 region of the hippocampus, but without associated expression of the protein (Danbolt, 2001). To determine which variant form of GLT-1 occurs in neurons, Chen et al. (Chen et al., 2004) performed GLT-1 variant specific in situ hybridization [Fig. 3, I from (Chen et al., 2004)]. Directly comparing the expression of GLT-1a and GLT-1b mRNA in the hippocampus showed, somewhat surprisingly, that GLT-1a mRNA is the predominant form in neurons in this region and throughout the brain (Berger et al., 2005). These results encouraged a re-examination of the expression of GLT-1a in neurons using pre-embedding EM-ICC (Aoki et al., 2000) and avoiding glutaraldehyde fixation that might destroy immunoreactive epitopes on the GLT-1 protein. Semi-quantitative analysis of the subcellular distribution of GLT-1 demonstrated that GLT-1a was present in 14–29% of axon terminals in the hippocampus, depending on the region [Fig. 2, 3, 4 from (Chen et al., 2004)].
Figure 3. Demonstration that GLT-1a isoform is expressed in axon terminals in the hippocampus.

I) in situ hybridization with probes directed at the different 3 UTRs of GLT-1a and GLT-1b in the hippocampus [see (Chen et al., 2004)]. GLT-1mRNA was labeled in hippocampus using probes covering the full-length GLT-1 coding sequence (pan-GLT-1 probe) (A, D, E, J,M, N ) or specific stretches of the 3-UTR sequence of GLT-1a (B, F, G, K, O, P) and GLT-1b (C, H, I, L, Q, R). The top two rows (A–I) illustrate single in situ hybridization labeling obtained with AP detection. The bottom two rows (J–R) show double hybridized sections in which the fluorescent labeling of astrocytic mRNA is blocked or attenuated, because of quenching by the AP reaction product associated with a GLAST probe. The second (D–I) and fourth (M–R) rows show magnified views of the CA1 (left panel; D, F, H, M, O, Q) and CA3 (right panel; E, G, I, N, P, R) subfields of the hippocampi shown in the row above, as outlined by the boxes shown in A. A, D, E, The pan-GLT-1 probe produces prominent neuronal labeling for GLT-1 in pyramidal cell neurons in the CA3 (E) subfield but not in CA1 ( D). Astrocytes throughout the hippocampus and dentate gyrus are also labeled. B, F, G, GLT-1a labeling shows a similar pattern of neuronal labeling, although at reduced intensity. Astrocytic labeling is present but reduced compared with that in A. C, H, I, GLT-1b labeling is prominent in astrocytes throughout the hippocampus. CA3 neurons are not clearly labeled (I). J,M, N, Fluorescent labeling using the pan-GLT-1 probe highlights the strong labeling of CA3 neurons (J, N). Astrocytic labeling in the CA1 region (M) is blocked by the AP deposit produced by the cohybridized GLAST probe, which only labels astrocytes. K, O, P, Strong GLT-1a fluorescent labeling in CA3 neurons (K, P) but not in CA1 neurons (O). L, Q, R, The differential double-in situ technique reveals weak, but distinct, GLT-1b fluorescent labeling in the CA3 region (R and arrows in L) but not in CA1 (Q). Note that the photographic exposure time was longer for L, Q, and R compared with J, K, and M-P because of the much weaker signal obtained with the GLT-1b probe. The lower signal/noise ratio with the GLT-1b probe is evident based on the more apparent background labeling in L, Q, and R. The labeling by the GLT-1b probe is locally very restricted, because of the low abundance of the message and the amplification technique that gains sensitivity at the expense of resolution. The area of apparent labeling under the arrow shafts in L is attributable to a bubble under the section. These findings demonstrate that the GLT-1a isoform is expressed in CA3 neurons. The GLT-1b transcript is weakly expressed in the CA3 region. The transcript for GLT-1a appears to be predominant in neurons. Bars: L, 500μm; R, 100μm.
II) Electron microscopic immunocytochemistry using an anti-GLT-1a antibody [see (Chen et al., 2004)]. Electron microscopy reveals GLT-1a immunoreactivity specifically in axon terminals, astrocytes, and dendrites. Immunoreactivity is visualized by the flocculent, electron-dense, peroxidase reaction product along membranes. A and B show examples of immunoreactive terminals (T) forming asymmetric axo-spinous junctions. Here and in other panels, the large open arrows point to PSDs, whereas small arrows point to portions of neurons exhibiting high concentrations of immunoreactivity. C shows the specificity of labeling of axon terminals and astrocytic processes within a single section. Immunocytochemical conditions that produced astrocytic labeling, as reported previously, also resulted in detection of GLT-1a within some, but not all, axo-spinous excitatory junctions, and only rarely within dendrites. The field shown in C contains four axo-spinous synapses, two of which exhibit immunoreactivity within the axon terminal. The other two synapses show no detectable levels of immunoreactivity within the terminals (UT, unlabeled terminal). In the same field, astrocytic processes surround the dendritic shaft portions of the spine receiving synaptic input from the labeled terminal. These astrocytic processes exhibit immunoreactivity along the intracellular surface (arrowheads). D shows an example of a dendritic shaft exhibiting immunoreactivity along the smooth endoplasmic reticulum near a PSD. E shows complete absence of immunoreactivity in terminals, astrocytes (*), and spines after preadsorption of the antibody with the synthetic peptide against which the antibody was generated. Scale bars: A, C–E, 500 nm; B, 625 nm.
Figure 4. Generation of a conditional knockout of GLT-1 and characterization of effects of inactivation of GLT-1 in neurons and in astrocytes.

A) a conditional knockout of GLT-1 was generated inserting loxP sites flanking exon 4: B) strategies for producing mouse lines in which GLT-1 is inactivated in neurons and in astrocytes; C&D) body weight and survival in the pan GLT-1 KO (Tanaka et al., 1997); E&F) body weight and survival for the conditional astrocytic GLT-1 knockout (Petr et al., 2015); G&H) body weight and survival for the conditional neuronal GLT-1 KO [(Petr et al., 2015); I&J) Saturation analysis of 3H-L-glutamate uptake into forebrain synaptosomes prepared from the astrocytic GLT-1 KO (I) and the neuronal GLT-1 KO (J) (Petr et al., 2015).
2.3. Quantitative studies of GLT-1a expression and function in the hippocampus
To validate these findings and further investigate the distribution of GLT-1 protein and uptake activity Furness et al. (2008) used a combined approach of direct demonstration of GLT-1a using anti-GLT-1a immunogold as well as anti-D-aspartate immunocytochemistry following exposure of hippocampal slices to D-aspartate. In this study about ¾ of axon terminals in the hippocampus were found to accumulate D-aspartate, and this accumulation was shown to be by a DHK-sensitive transporter that was absent in GLT-1 knockout mice. It was further found that about 80% GLT-1 is localized in astrocyte membranes; only about 6% was detected in the plasma membrane of synaptic terminals. Paradoxically, given the low levels of GLT-1 protein expression in axon terminals, but anticipating results that were later obtained using a conditional GLT-1 knockout mouse (Petr et al., 2015) (see below), more than half of the D-aspartate transported into hippocampal slices was found in axon terminals.
In addition to the studies in the hippocampus, more recent studies have demonstrated GLT-1 in presynaptic terminals the rat somatic sensory cortex (Melone et al., 2009), in the human cortex (Melone et al., 2011) and in the rat striatum (Petr et al., 2013). In a developmental study DeSilva et al. (2012) determined the cellular and temporal expression of GLT-1 in the developing human cerebral cortex.
3. Conditional knockout of GLT-1
These findings led to the more general question of what is the function of GLT-1 in axon terminals. Since GLT-1 in neurons represents only about 5% of its total expression, is its function or functions, if any, related to glutamate clearance or does the transporter expressed in this special location participate in some other aspect of glutamate signaling, metabolism, or development?
To pursue these questions and dissect the contributions of astrocytic and neuronal GLT-1 to glutamate homeostasis in the intact brain, a conditional knockout was generated [Fig. 4, A, B, from (Petr et al., 2015)]. The conditional knockout was based on the pan knockout Tanaka et al. (1997) (Fig. 4, B, box), in which the mouse gene encoding GLT-1 was disrupted by homologous recombination resulting in replacement of exon 4 by the neomycin resistance gene. In the conditional knockout, loxP sites flanking exon 4 were inserted, allowing the use Cre-recombinase to delete exon 4 in cells or regions of interest using specific Cre drivers.
GLT-1 was inactivated in neurons through Cre expression driven by the synapsin I promoter (JAX Stock no. 003966), and in astrocytes through the tamoxifen-inducible human GFAP Cre/ERT driver (Casper et al., 2007). To restrict the expression of Cre to astrocytes, which is necessary since some neuronal progenitor cells in embryonic animals express GFAP, tamoxifen was applied postnatally (P5–P9), as described by (Ganat et al., 2006). Consistent with the view that most GLT-1 is expressed in astrocytes, elimination of GLT-1 from astrocytes resulted in loss of about 80% of GLT-1 protein. In contrast, when GLT-1 protein expression was assayed in the neuronal GLT-1 knockout, no detectable change was found, consistent with the view that neurons express only a small fraction of GLT-1 protein in the brain. To confirm that the cell-type specific knockouts were effective and specific, hippocampal slices were assayed using LM and EM-ICC. In the astrocyte knockout, there was an 85% reduction in the number of GLT-1 labeled astrocyte processes but no change in the density of axon terminals labeled with GLT-1. In the neuronal KO there was a decrease by about 90% of axon terminals labeled with GLT-1 immunoreactivity, but no change in density of astrocyte processes that were labeled (Petr et al., 2015).
3.1. Phenotype of conditional GLT-1 knockouts
The pan GLT-1 knockout is lethal, producing intractable seizures leading to death with a 50% mortality at six weeks of age on the 129XC57Bl/6 background [(Tanaka et al., 1997) (Fig. 4, C, D from (Tanaka et al., 1997)]. The phenotype of epilepsy was also found in the conditional astrocyte knockout [Fig. 4, E, F from (Petr et al., 2015)]. These mice also had reduced weight gain and reduced life span, with 50% survival at 25 weeks of age [Fig. 4, G, H from (Petr et al., 2015)]. The neuronal GLT-1 knockout did not have epilepsy or compromised life span, at least up to 1 year. Interestingly, on a general assessment of behavior called the SHIRPA battery (Brooks and Dunnett, 2009), no abnormalities either in the astrocyte knockout or the neuronal knockout were found.
3.2. Synaptosomal uptake
To assess the function of the conditional knockout of GLT-1 in astrocytes and in neurons radioactive glutamate uptake into a crude synaptosomal preparation made from forebrain tissue was measured [Fig. 4, I, J from (Petr et al., 2015)]. Surprisingly, only a marginal and not significant reduction (about 15%) of glutamate uptake was found in the astrocytic knockout. In contrast, when GLT-1 was deleted in neurons, synaptosomal glutamate uptake capacity (Vmax) was reduced significantly by about 40%. These results indicate that the bulk of the GLT-1 protein in a crude synaptosome preparation, namely that fraction that is associated with astrocytes, does not significantly contribute to glutamate uptake into synaptosomes, whereas the GLT-1 protein associated with neuronal membranes, which is a fraction too small to detect by comparing immunoblots of neuronal GLT-1 knockout in the brain with that of wildtype littermates, seems to account for 40% of synaptosomal glutamate uptake.
3.2.1. Numerology
It is a useful exercise to compare the fraction of synaptosomal uptake of glutamate associated with neuronal and astrocytic GLT-1, as determined by conditional knockout of GLT-1 in neurons and in astrocytes as reported in Petr et al. (2015). There are several ways to make this comparison: a) GLT-1 mediates 95% of synaptosomal uptake, but the neuronal component (40%) plus the astrocyte component (15%), each measured by effect of the neuronal and astrocytic knockout, respectively do not add up to 95%; b) the remainder uptake in the astrocyte knockout experiments, which logically is the neuronal component, is 85%, but the neuronal component determined by neuronal knockout is 40%; c) the remainder uptake in the neuronal knockout experiments, which logically is the astrocyte component, is 60%, but, the astrocyte component determined by astrocyte knockout is 15%. The simplest explanation for these differences in estimation of the astrocyte contribution to glutamate uptake is partial excision of the GLT-1 gene by GFAP-CreERT. However, when reduction in glutamate uptake in synaptosomes in the astrocyte knockouts was plotted as a function of reduction in GLT-1 protein in immunoblots on the same synaptosomal membranes, no correlation was found despite a variation in remainder protein from nearly undetectable to 40% (Sun and Rosenberg, unpublished data)..
It is suspected that the difference in the apparent astrocyte contribution obtained from the neuronal GLT-1 knockout (60%) and the astrocyte knockout (15%) may be due to compensatory changes, that is, an increase in GLT-1 mediated uptake in neurons in compensation for deletion of GLT-1 from astrocytes and vice versa. Interestingly, immunolabeling for GLT-1 revealed the suggestion of an increased expression within axon terminals in GFAP/Cre+ brains compared to the controls (12.5 ± 3% for the GFAP/Cre- and 20 ± 3% for the GFAP/Cre+ brains) that, however, did not achieve significance.
3.2.2. The contributions of neuronal and astrocytic GLT-1 to synaptosomal uptake are not proportionate to their levels of protein expression
The major question, however, is why is the glutamate uptake activity by neuronal GLT-1 so high and by astrocyte GLT-1 so low, in proportion to protein expression in neurons and astrocytes, respectively, in the synaptosome preparations? Several possible explanations were considered. The synaptosomal preparation used for the experiments cited was a crude preparation in which the P2 pellet was resuspended, having discarded that material in the P1 pellet. A trivial explanation would be that in the preparation of synaptosomes, a large portion of the astrocyte membranes containing (astrocytic) GLT-1 was discarded in the P1 pellet, or in the supernatant from the P2 pellet. However, no difference in the effect of the neuronal GLT-1 knockout was observed comparing the unspun homogenate, the P2 pellet, or the resuspended P1 pellet. Very little uptake activity was detected in the supernatant above the P2 pellet.
Another possibility was that glutamate taken up into presynaptic terminals and into astrocytic vesicles might have different metabolic fates. If glutamate were metabolized inside nerve terminals but not astrocytic vesicles, then the accumulation of glutamate within astrocyte vesicles might retard transport. However, no difference was found in the effect of neuronal GLT-1 knockout using L-3H-glutamate or the non-metabolizable D-3H-aspartate as the tracers.
The study of transport using radioactive substrates is subject to the problem that the uptake of radioactive material reflects two processes, heteroexchange of radioactive substrate with cold substrate on the other side of the membrane bounding the two compartments. Therefore, neuronal uptake of radioactive glutamate might have a greater component of heteroexchange than astrocytic uptake. The two processes, heteroexchange and net uptake, can be differentiated by using an ion carrier, such as nigericin, that equilibrates cations across the membrane. The nigericin sensitive component of uptake thus represents net uptake, and it was found that the fraction of uptake sensitive to nigericin was similar in the neuronal GLT-1 knockout (representing astrocytic uptake) and in the astrocytic GLT-1 knockout (representing neuronal GLT-1 uptake). Therefore a predominance of heteroexchange mediated by neuronal GLT-1 does not seem to be the explanation for the disproportionately large fraction of uptake mediated by neuronal GLT-1. This conclusion is supported by the findings of Zhou et al (Zhou et al., 2014) that the relative rates of net glutamate uptake and heteroexchange mediated by GLT-1 are similar.
Finally, 2 possibilities remained. It was conceivable that neuronal GLT-1 was in some way modified as to be more active than astrocytic GLT-1. Another possibility was that for some reason astrocytic membranes might be less likely to reseal to form transport competent vesicles. For vesicles to demonstrate uptake, they must be able to reseal to present a permeability barrier that can sustain gradients of ions and glutamate across it. To address these issues, brain protein was reconstituted into liposomes, and uptake assayed into these liposomes. In the liposome preparation, uptake of glutamate was proportionate to protein expression. An approximate 80% loss of protein was observed in the liposomes from astrocytic GLT-1 knockout mice compared to controls, and similarly, an approximate 80% loss of uptake activity was observed. Neuronal GLT-1 knockout did not produce a detectable change in GLT-1 protein, and, did not produce a detectable effect on glutamate uptake. These results argue against the possibility of a post-translational modification of GLT-1 in neurons that increases activity and is stable to the reconstitution procedure. They argue for the possibility that astrocytic GLT-1 is underestimated in synaptosomal preparations because a large fraction of astrocytic GLT-1 is expressed in membranes that do not reseal to form transport competent structures or is not able to mediate transport in its native environment for reasons yet to be determined.
4. Conclusion
GLT-1 is expressed in many populations of neurons at the mRNA level, and it seems likely, but certainly not established, that where there is GLT-1 mRNA there will also be protein expression. The eminent population geneticist J.B.S. Haldane (1963) wrote in a review of a publication, towards the end of his life: “I suppose the process of acceptance will pass through the usual four stages: 1) this is worthless nonsense; 2) this is an interesting, but perverse, point of view; 3) this is true, but quite unimportant; 4) I always said so.” Regarding the particular issue at hand, that is, whether presynaptic glutamatergic terminals have a glutamate uptake system, and if so, its identity, we have seen the proposition that GLT-1 is the presynaptic glutamate transporter pass through stage 1, and is probably lodged somewhere between stages 2 and 3. The critical question is the one implicit in Haldane’s stage 3--what are the functions (if any) of GLT-1 expressed in neurons and, specifically, in axon terminals. A related question is why do some neurons express GLT-1 presynaptically, while others do not? The use of conditional GLT-1 knockout mice should help address these questions through a combination of behavioral, biochemical, electrophysiological, genomic and proteomic studies to fully characterize the neuronal GLT-1 knockout. This work is in progress. Glutamate homeostasis is now thought to be very important in the regulation of a number of neurobiological processes, and in each of these, it would be reasonable to ask what are the relative roles of astrocytic and neuronal GLT-1.
Highlights.
Review of 40 year history of effort to characterize presynaptic glutamate transport
GLT-1a is the glutamate transporter in excitatory terminals
A conditional GLT-1 knockout confirms its presence and function in axon terminals
Mismatch between neuronal and astrocytic GLT-1 uptake and protein expression
Acknowledgments
The authors wish to acknowledge support by the William Randolph Hearst Foundation, Hereditary Disease Foundation, Children’s Hospital Intellectual and Developmental Disabilities Research Center (IDDRC) core grant HD 18655, NINDS RO1NS066019, and NIMH R21MH104318.
Abbreviations
- DHK
dihydrokainate
- EAAT1 (GLAST)
Excitatory amino acid transporter 1
- EAAT2 (GLT-1
slc1a2), Excitatory amino acid transporter 2
- EAAT3 (EAAC1)
Excitatory amino acid transporter 3
- EAAT4
Excitatory amino acid transporter 4
- EAAT5
Excitatory amino acid transporter 5
- EM-ICC
electron microscopy immunocytochemistry
- H+
proton
- K+
potassium
- LM-ICC
light microscopy immunocytochemistry
- LTD
long-term depression
- LTP
long-term-potentiation
- mGluR
metabotropic glutamate receptor
- nGLT-1−/− (synGLT-1 KO)
conditional neuronal GLT-1 knockout
- Na+
sodium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amara SG. Neurotransmitter transporters. A tale of two families. Nature. 1992;360:420–421. doi: 10.1038/360420d0. [DOI] [PubMed] [Google Scholar]
- Aoki C, Rodrigues S, Kurose H. Use of electron microscopy in the detection of adrenergic receptors. Meth Mol Biol. 2000;126:535–563. doi: 10.1385/1-59259-684-3:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nature Neuroscience. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Beart PM. The autoradiographic localization of L-(3H) glutamate in synaptosomal preparations. Brain Research. 1976a;103:350–355. doi: 10.1016/0006-8993(76)90804-0. [DOI] [PubMed] [Google Scholar]
- Beart PM. An evaluation of L-glutamate as the transmitter released from optic nerve terminals of the pigeon. Brain Research. 1976b;110:99–114. doi: 10.1016/0006-8993(76)90211-0. [DOI] [PubMed] [Google Scholar]
- Bennett JP, Jr, Logan WJ, Snyder SH. Amino acid neurotransmitter candidates: sodium-dependent high-affinity uptake by unique synaptosomal fractions. Science. 1972;178:997–999. doi: 10.1126/science.178.4064.997. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Jorgensen MB, Sandberg M, Christensen T, Hagberg H, Diemer NH. Ischemic damage in hippocampal CA1 is dependent on glutamate release and intact innervation from CA3. Journal of Cerebral Blood Flow and Metabolism. 1989;9:629–639. doi: 10.1038/jcbfm.1989.90. [DOI] [PubMed] [Google Scholar]
- Berger UV, DeSilva TM, Chen W, Rosenberg PA. Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. Journal of Comparative Neurology. 2005;492:78–89. doi: 10.1002/cne.20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol (Berl) 1998;198:13–30. doi: 10.1007/s004290050161. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE. Clearance of glutamate inside the synapse and beyond. Current Opinion in Neurobiology. 1999;9:293–298. doi: 10.1016/s0959-4388(99)80043-9. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Billups B, Rossi D, Oshima T, Warr O, Takahashi M, Sarantis M, Szatkowski M, Attwell D. Physiological and pathological operation of glutamate transporters. Prog Brain Res. 1998;116:45–57. doi: 10.1016/s0079-6123(08)60429-x. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Kavanaugh MP, Chamberlin AR. A pharmacological review of competitive inhibitors and substrates of high-affinity, sodium-dependent glutamate transport in the central nervous system. Current Pharmaceutical Design. 1999;5:363–379. [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nat Rev Neurosci. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- Buchan AM, Pulsinelli WA. Septo-hippocampal deafferentation protects CA1 neurons against ischemic injury. Brain Research. 1990a;512:7–14. doi: 10.1016/0006-8993(90)91163-b. [DOI] [PubMed] [Google Scholar]
- Buchan AM, Pulsinelli WA. Septo-hippocampal deafferentation protects CA1 neurons against ischemic injury. Brain Research. 1990b;512:7–14. doi: 10.1016/0006-8993(90)91163-b. [DOI] [PubMed] [Google Scholar]
- Casper KB, Jones K, McCarthy KD. Characterization of astrocyte-specific conditional knockouts. Genesis. 2007;45:292–299. doi: 10.1002/dvg.20287. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, Campagne MV, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. Journal of Neuroscience. 2002;22:2142–2152. doi: 10.1523/JNEUROSCI.22-06-02142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. Journal of Neuroscience. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Koh J, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. Journal of Neuroscience. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annual Review of Neuroscience. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Slater P, Reynolds GP. Reduced high-affinity glutamate uptake sites in the brains of patients with Huntington’s disease. Neuroscience Letters. 1986;67:198–202. doi: 10.1016/0304-3940(86)90397-6. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Progress in Neurobiology. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-t. [DOI] [PubMed] [Google Scholar]
- DeSilva TM, Borenstein NS, Volpe JJ, Kinney HC, Rosenberg PA. Expression of EAAT2 in neurons and protoplasmic astrocytes during human cortical development. Journal of Comparative Neurology. 2012;520:3912–3932. doi: 10.1002/cne.23130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divac I, Fonnum F, Storm-Mathisen J. High affinity uptake of glutamate in terminals of corticostriatal axons. Nature. 1977;266:377–378. doi: 10.1038/266377a0. [DOI] [PubMed] [Google Scholar]
- Euler T, Wassle H. Immunocytochemical identification of cone bipolar cells in the rat retina. Journal of Comparative Neurology. 1995;361:461–478. doi: 10.1002/cne.903610310. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- Ferkany J, Coyle JT. Heterogeneity of sodium-dependent excitatory amino acid uptake mechanisms in rat brain. Journal of Neuroscience Research. 1986;16:491–503. doi: 10.1002/jnr.490160305. [DOI] [PubMed] [Google Scholar]
- Fonnum F, Storm-Mathisen J, Divac I. Biochemical evidence for glutamate as neurotransmitter in corticostriatal and corticothalamic fibres in rat brain. Neuroscience. 1981;6:863–873. doi: 10.1016/0306-4522(81)90168-8. [DOI] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Attwell D, Danbolt NC. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2) Neuroscience. 2008;157:80–94. doi: 10.1016/j.neuroscience.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, Silbereis J, Cave C, Ngu H, Anderson GM, Ohkubo Y, Ment LR, Vaccarino FM. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. Journal of Neuroscience. 2006;26:8609–8621. doi: 10.1523/JNEUROSCI.2532-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. Cellular uptake disguises action of L-glutamate on N-methyl-D-aspartate receptors. Br J Pharmac. 1985;85:297–307. doi: 10.1111/j.1476-5381.1985.tb08860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. Journal of Neurochemistry. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. The truth about death Journal of Genetics. 1963;58:464. [Google Scholar]
- Harris KM, Rosenberg PA. Localization of synapses in rat cerebral cortex in dissociated cell culture. Neuroscience. 1993;53:495–508. doi: 10.1016/0306-4522(93)90214-z. [DOI] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, Lehre KP, Danbolt NC. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci. 2012;32:6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, Danbolt NC. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience. 2009;162:1055–1071. doi: 10.1016/j.neuroscience.2009.03.048. [DOI] [PubMed] [Google Scholar]
- Huang Y. PhD. Johns Hopkins University; Baltimore, Maryland: 2006. Glutamate transporter function at excitatory synapses. [Google Scholar]
- Huang YH, Bergles DE. Glutamate transporters bring competition to the synapse. Current Opinion in Neurobiology. 2004;14:346–352. doi: 10.1016/j.conb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Huang YH, Sinha SR, Tanaka K, Rothstein JD, Bergles DE. Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons. J Neurosci. 2004;24:4551–4559. doi: 10.1523/JNEUROSCI.5217-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen LL, Storm-Mathisen J. Uptake of (3H)glutamate in excitatory nerve endings in the hippocampal formation of the rat. Act Physiol Scand. 1976;96:22A–23A. doi: 10.1016/0306-4522(79)90154-4. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- Kanner BI. Structure and function of sodium-coupled GABA and glutamate transporters. Journal of Membrane Biology. 2006;213:89–100. doi: 10.1007/s00232-006-0877-5. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. European Journal of Neuroscience. 2001;14:547–553. doi: 10.1046/j.0953-816x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- Kaur J, Zhao Z, Geransar RM, Papadakis M, Buchan AM. Prior deafferentation confers long term protection to CA1 against transient forebrain ischemia and sustains GluR2 expression. Brain Research. 2006;1075:201–212. doi: 10.1016/j.brainres.2005.12.123. [DOI] [PubMed] [Google Scholar]
- Koch HP, Chamberlin AR, Bridges RJ. Nontransportable inhibitors attenuate reversal of glutamate uptake in synaptosomes following a metabolic insult. Molecular Pharmacology. 1999a;55:1044–1048. doi: 10.1124/mol.55.6.1044. [DOI] [PubMed] [Google Scholar]
- Koch HP, Kavanaugh MP, Esslinger CS, Zerangue N, Humphrey JM, Amara SG, Chamberlin AR, Bridges RJ. Differentiation of substrate and nonsubstrate inhibitors of the high-affinity, sodium-dependent glutamate transporters. Molecular Pharmacology. 1999b;56:1095–1104. doi: 10.1124/mol.56.6.1095. [DOI] [PubMed] [Google Scholar]
- Kohler C, Schwarcz R. Monosodium glutamate: increased neurotoxicity after removal of neuronal re-uptake sites. Brain Research. 1981;211:485–491. doi: 10.1016/0006-8993(81)90978-1. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Dichter MA. Neuron generation in dissociated cell cultures from fetal rat cerebral cortex. Brain Research. 1984;295:184–189. doi: 10.1016/0006-8993(84)90829-1. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Snyder SH. The subcellular distribution of free H3-glutamic acid in rat cerebral cortical slices. Journal of Pharmacology and Experimental Therapeutics. 1970;171:141–152. [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. Journal of Neuroscience. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Attwell D, Hoover F, Ash JF, Bjoras M, Danbolt NC. Inducible expression of the GLT-1 glutamate transporter in a CHO cell line selected for low endogenous glutamate uptake. FEBS Letters. 1998;422:339–342. doi: 10.1016/s0014-5793(98)00036-2. [DOI] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Walaas SI, Storm-Mathisen J, Danbolt NC. Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur J Neurosci. 1995;7:2036–2041. doi: 10.1111/j.1460-9568.1995.tb00626.x. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiological Reviews. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Logan WJ, Snyder SH. Unique high affinity system for glycine, glutamaic and aspartic acids in central nervous tissue in the rat. Nature. 1971;234:297–299. doi: 10.1038/234297b0. [DOI] [PubMed] [Google Scholar]
- Madl JE. Increased glutamate uptake by glial cells during ATP depletion in hippocampal slices. Society for Neuroscience Abstracts 21(#803.2) 1995:2044. [Google Scholar]
- Madl JE, Burgesser K. Adenosine triphosphate depletion reverses sodium-dependent, neuronal uptake of glutamate in rat hippocampal slices. Journal of Neuroscience. 1993;13:4429–4444. doi: 10.1523/JNEUROSCI.13-10-04429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, Traystman RJ. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997;42:335–348. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- McBean GJ, Roberts PJ. Chronic infusion of L-glutamate causes neurotoxicity in rat striatum. Brain Research. 1984;290:372–375. doi: 10.1016/0006-8993(84)90959-4. [DOI] [PubMed] [Google Scholar]
- McBean GJ, Roberts PJ. Neurotoxicity of L-glutamate and DL-threo-3-hydroxyaspartate in the rat striatum. J Neurochem. 1985;44:247–254. doi: 10.1111/j.1471-4159.1985.tb07137.x. [DOI] [PubMed] [Google Scholar]
- Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Conti F. Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia. 2009;57:108–117. doi: 10.1002/glia.20744. [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Ducati A, Iacoangeli M, Conti F. Cellular and Synaptic Localization of EAAT2a in Human Cerebral Cortex. Front Neuroanat. 2011;4:151. doi: 10.3389/fnana.2010.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF. Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J Neurosci. 1998;18:4490–4499. doi: 10.1523/JNEUROSCI.18-12-04490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW. Excitotoxicity: an overview. Can Dis Wkly Rep. 1990;16(Suppl 1E):47–57. discussion 57–48. [PubMed] [Google Scholar]
- Otis TS, Brasnjo G, Dzubay JA, Pratap M. Interactions between glutamate transporters and metabotropic glutamate receptors at excitatory synapses in the cerebellar cortex. Neurochemistry International. 2004;45:537–544. doi: 10.1016/j.neuint.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Petr GT, Schultheis LA, Hussey KC, Sun Y, Dubinsky JM, Aoki C, Rosenberg PA. Decreased expression of GLT-1 in the R6/2 model of Huntington’s disease does not worsen disease progression. European Journal of Neuroscience. 2013;38:2477–2490. doi: 10.1111/ejn.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. Journal of Neuroscience. 2015;35:5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Pines G, Kanner BI. Counterflow of L-glutamate in plasma membrane vesicles and reconstituted preparations from rat brain. Biochemistry. 1990;29:11209–11214. doi: 10.1021/bi00503a008. [DOI] [PubMed] [Google Scholar]
- Rauen T, Kanner BI. Localization of the glutamate transporter GLT-1 in rat and macaque monkey retinae. Neurosci Lett. 1994;169:137–140. doi: 10.1016/0304-3940(94)90375-1. [DOI] [PubMed] [Google Scholar]
- Rauen T, Rothstein JD, Wassle H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996;286:325–336. doi: 10.1007/s004410050702. [DOI] [PubMed] [Google Scholar]
- Reye P, Sullivan R, Pow DV. Distribution of two splice variants of the glutamate transporter GLT-1 in the developing rat retina. Journal of Comparative Neurology. 2002a;447:323–330. doi: 10.1002/cne.10218. [DOI] [PubMed] [Google Scholar]
- Reye P, Sullivan R, Scott H, Pow DV. Distribution of two splice variants of the glutamate transporter GLT-1 in rat brain and pituitary. Glia. 2002b;38:246–255. doi: 10.1002/glia.10059. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Hunter-Ensor M, Sinor J. Pharmacologically distinct sodium-dependent l-[3H]glutamate transport processes in rat brain. Brain Research. 1991;544:196–202. doi: 10.1016/0006-8993(91)90054-y. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Sinor JD, Dowd LA, Kerwin JF., Jr Subtypes of sodium-dependent high-affinity L[3H]glutamate transport activity: Pharmacologic specificity and regulation by sodium and potassium. Journal of Neurochemistry. 1993;60:167–179. doi: 10.1111/j.1471-4159.1993.tb05835.x. [DOI] [PubMed] [Google Scholar]
- Robinson S, Tani M, Strieter RM, Ransohoff RN, Miller RH. The chemokine growth-regulated oncogene-α promotes spinal cord oligodendrocyte precursor proliferation. Journal of Neuroscience. 1998;18:10457–10463. doi: 10.1523/JNEUROSCI.18-24-10457.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neuroscience Letters. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Amin S, Leitner M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. Journal of Neuroscience. 1992;12:56–61. doi: 10.1523/JNEUROSCI.12-01-00056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Lesch KP, Kugler P. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience. 2002;109:45–61. doi: 10.1016/s0306-4522(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Puschel B, Jons T, Kugler P. Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience. 1996;71:989–1004. doi: 10.1016/0306-4522(95)00477-7. [DOI] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Stoffel W, Korner R, Wachtmann D, Keller BU. Functional analysis of glutamate transporters in excitatory synaptic transmission of GLAST1 and GLAST1/EAAC1 deficient mice. Brain Res Mol Brain Res. 2004;128:170–181. doi: 10.1016/j.molbrainres.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Stoffel W, Muller R, Binczek E, Hofmann K. Mouse apolipoprotein AI. cDNA-derived primary structure, gene organisation and complete nucleotide sequence. Biol Chem Hoppe Seyler. 1992;373:187–193. doi: 10.1515/bchm3.1992.373.1.187. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm-Mathisen J. Glutamic acid and excitatory nerve endings: reduction of glutamic acid uptake after axotomy. Brain Research. 1977;120:379–386. doi: 10.1016/0006-8993(77)90918-0. [DOI] [PubMed] [Google Scholar]
- Storm-Mathisen J, Iversen LL. Uptake of [3H]Glutamic acid in excitatory nerve endings: light and electronmicroscopic observations in the hippocampal formation of the rat. Neuroscience. 1979;4:1237–1253. doi: 10.1016/0306-4522(79)90154-4. [DOI] [PubMed] [Google Scholar]
- Suchak SK, Baloyianni NV, Perkinton MS, Williams RJ, Meldrum BS, Rattray M. The ‘glial’ glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings. J Neurochem. 2003;84:522–532. doi: 10.1046/j.1471-4159.2003.01553.x. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. Journal of Neuroscience. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kovalchuk Y, Attwell D. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J Neurosci. 1995;15:5693–5702. doi: 10.1523/JNEUROSCI.15-08-05693.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994;13:1195–1203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Torp R, Danbolt NC, Babaie E, Bjoras M, Storm-Mathisen J, Ottersen OP. Differential expression of two glial glutamate transporters in the rat brain: in situ hybridization study. European Journal of Neuroscience. 1994;6:936–942. doi: 10.1111/j.1460-9568.1994.tb00587.x. [DOI] [PubMed] [Google Scholar]
- Trotti D, Volterra A, Lehre KP, Rossi D, Gjesdal O, Racagni G, Danbolt NC. Arachidonic acid inhibits a purified and reconstituted glutamate transporter directly from the water phase and not via the phospholipid membrane. Journal of Biological Chemistry. 1995;270:9890–9895. doi: 10.1074/jbc.270.17.9890. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y. Tissue specific variants of glutamate transporter GLT-1. FEBS Letters. 1997;416:312–316. doi: 10.1016/s0014-5793(97)01232-5. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chung HJ, Schnuer J, Pratt K, Zable AC, Kavanaugh MP, Rosenberg PA. High affinity glutamate transport in rat cortical neurons in culture. Molecular Pharmacology. 1998;53:88–96. doi: 10.1124/mol.53.1.88. [DOI] [PubMed] [Google Scholar]
- Wofsey AR, Kuhar MJ, Snyder SH. A unique synaptosomal fraction, which accumulates glutamic and aspartic acids, in brain tissue. Proceedings of the National Academy of Sciences of the United States of America. 1971;68:1102–1106. doi: 10.1073/pnas.68.6.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kanner BI. Two serine residues of the glutamate transporter GLT-1 are crucial for coupling the fluxes of sodium and the neurotransmitter. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1710–1715. doi: 10.1073/pnas.96.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang X, Tzingounis AV, Danbolt NC, Larsson HP. EAAT2 (GLT-1; slc1a2) glutamate transporters reconstituted in liposomes argues against heteroexchange being substantially faster than net uptake. Journal of Neuroscience. 2014;34:13472–13485. doi: 10.1523/JNEUROSCI.2282-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]