

Abstract
Synthesis of phospholipids can occur de novo or via remodeling of the existing phospholipids. Synthesis of triglycerides, a form of energy storage in cells, is an end product of these pathways. Several 1-acylglycerol-3-phosphate-O-acyltransferases (AGPATs) acylate lysophosphatidic acid (LPA) at the sn-2 (carbon 2) position to produce phosphatidic acid (PA). These enzymes are involved in phospholipids and triglyceride synthesis through an evolutionary conserved process involving serial acylations of glycerol-3-phosphate. We cloned a cDNA predicted to be an AGPAT isoform (AGPAT10). This cDNA has been recently identified as glycerol-3-phosphate-O-acyltransferase isoform 3 (GPAT3). When this AGPAT10/GPAT3 cDNA was expressed in Chinese Hamster ovary cells, the protein product localizes to the endoplasmic reticulum. In vitro enzymatic activity using lysates of human embryonic kidney-293 cells infected with recombinant AGPAT10/GPAT3 adenovirus show that the protein has a robust AGPAT activity with an apparent Vmax of 2 nmol/min per mg protein, but lacks GPAT enzymatic activity. This AGPAT has similar substrate specificities for LPA and acyl-CoA as shown for another known isoform, AGPAT2. We further show that when overexpressed in human Huh-7 cells depleted of endogenous AGPAT activity by sh-RNA-AGPAT2-lentivirus, the protein again demonstrates AGPAT activity. These observations strongly suggest that the cDNA previously identified as GPAT3 has AGPAT activity and thus we prefer to identify this clone as AGPAT10 as well.
Introduction
The 1-acylglycerol-3-phosphate-O-acyltransferase (AGPAT) enzymes catalyze the conversion of lysophosphatidic acid (LPA) to phosphatidic acid (PA; Eberhardt et al. 1997, West et al. 1997, Leung 2001, Coleman & Lee 2004, McMaster & Jackson 2004, Ye et al. 2005, Agarwal et al. 2006, 2007). PA serves as a substrate for the synthesis of various other polar phospholipids (PL) such as phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidylethanolamine (PE), which are essential components of the cellular membranes. Intracellular synthesis of these PLs occurs via the Kennedy or the cytidine diphosphate-diacylglycerol pathways. In addition, PA also generates signaling molecules such as phosphatidylinositol (PI) which are involved in important cellular function(s) including insulin signaling. PA carries two fatty acids in the sn-1 and sn-2 position of the glycerol backbone. While the specificity of fatty acids at sn-1 (carbon 1) position is determined by glycerol-3-phosphate acyltransferases (GPATs), variability at sn-2 (carbon 2) position occurs due to esterification by AGPATs (Agarwal & Garg 2003, Coleman & Lee 2004, McMaster & Jackson 2004). Thus, these two enzymes are capable of producing numerous PA species with varying fatty acids at the two carbon positions.
In addition to their role in the synthesis of various glycerophospholipids, the AGPATs are also involved in producing triacylglycerol (TAG) by providing the substrate for subsequent reaction for phosphatidic acid phosphatases (PAPs), which includes both lipid phosphate phosphatases (LPPs) and lipins, and diacylglycerol acyltransferases (DGATs). In brief, glycerol-3-phosphate is esterified at the sn-1 position with mostly saturated fatty acid by GPATs, followed by usually an unsaturated fatty acid at the sn-2 position by AGPATs (Coleman & Lee 2004, McMaster & Jackson 2004). The PA thus formed is dephosphorylated by PAPs. There are two classes of PAPs, type 1 which represents lipins 1, 2, and 3 and type 2 which include LPPs 1, 2, and 3 (Brindley 2004, Donkor et al. 2007, Carman & Han 2008). While lipins can dephosphorylate PA, they have no significant phosphatase activity towards LPA, LPPs show broader substrate preference and can dephosphorylate both LPA and PA. The last step involves esterification at the sn-3 position by DGATs to produce TAG.
We recently reported mutations in the AGPAT2 gene in patients affected with congenital generalized lipodystrophy (CGL; Agarwal et al. 2002). CGL is a syndrome with complete or nearly complete loss of adipose tissue found since birth. The affected children are predisposed to develop diabetes, hepatic steatosis, and hyperlipidemia in their adult life (Agarwal & Garg 2006). This observation suggests additional roles of AGPATs in adipocyte differentiation and maturation (Gale et al. 2006), beyond its role in TAG synthesis. The objective of our search for additional AGPATs has been twofold: first to find new isoform(s) which might carry genetic lesions in patients with lipodystrophies but do not harbor any mutation in the known lipodystrophy loci, and second, to understand how these isoforms might influence the synthesis of various glycerophospholipids.
As a first step toward this goal, we isolated the 10th human cDNA encoding an AGPAT and describe its enzymatic activity. This cDNA was previously identified as LPAAT-θ by Tang et al. (2006), but they did not report any enzymatic activity. Subsequently, Cao et al. (2006) reported GPAT activity for the same protein sequence, instead of the expected AGPAT activity and labeled it as GPAT3. We determined the AGPAT and GPAT enzymatic activities using a recombinant adenovirus expression system and show that the same isoform has only AGPAT activity in our assay system. Following the Human Genome Organization’s nomenclature, we have also named it as AGPAT10. To avoid confusion, we will refer to this cDNA as AGPAT10/GPAT3.
Methods
Generation of V5 epitope-tagged wild-type AGPAT10/GPAT3 recombinant adenovirus
The construction strategy for the V5 epitope-tagged AGPAT10/GPAT3 recombinant adenovirus was as follows. The recombinant adenovirus was created using the AdEasy adenoviral system (Stratagene, La Jolla, CA, USA) as suggested by the manufacturer. Generation of human AGPAT10/GPAT3 (pDrive-AGPAT10/GPAT3) is described in the Supplementary material, see supplementary data in the online version of Journal of Molecular Endocrinology at http://jme.endo-crinology-journals.org/content/vol42/issue6/. The full length AGPAT10 cDNA was tagged with V5 epitope at the amino-terminus by amplifying the cDNA from plasmid pDrive-AGPAT10/GPAT3 with primers 5′-GCGTCGACATGGGTAAGCCTATCCCTAACCCTC-TCCTCGGTCTCGATTCTACGGAGGGCGCAGAGCT-G-3′; the sequence encoding for the V5 epitope is underlined, and the primer 6 (Supplementary material). The amplified product was cloned in TA-cloning vector, sequenced, restricted with XhoI, and cloned in the pShuttle-CMV vector at the same site. The subsequent restriction digest and sequencing confirmed that the clone was in the correct orientation. To generate recombinant adenovirus, the pShuttle-CMV-AGPAT10/GPAT3 plasmid was digested with PmeI and the pAdEasy-1 with PacI and co-transfected in HEK-293 cells using Lipofectamine-2000. Several viral pools were further propagated in the same HEK-293 cells. The viral pool that showed most enzymatic activity was selected for further amplification and purification using the Virabind adenovirus purification kit (Cell Biolabs Inc., San Diego, CA, USA). The recombinant adenovirus-β-galactosidase was generated as above.
AGPAT enzymatic activity of AGPAT10/GPAT3 in the cell lysate
For all enzymatic assays, the adenoviral recombinant protein was used. The HEK-293 cells were infected with AGPAT10/GPAT3 recombinant adenovirus at a multiplicity of infection (MOI) of 150 or more. After 48 h, the infected cells were collected and washed once with PBS. The cellular viral pellet was resuspended in the lysis buffer (100 mM Tris pH 7·4, 10 mM NaCl containing protease inhibitor cocktail (Roche). The cells were lysed with three freeze–thaw cycles and centrifuged at 3000 g for 10 min at 4 °C to remove the cell debris. Protein concentration was determined by a commercially available colorimetric assay (Bio-Rad Laboratories).
The enzyme activity was determined by measuring the conversion of [3H]-LPA to [3H]-PA as described previously (Haque et al. 2005, Agarwal et al. 2006). Briefly, the enzymatic reaction was assembled in 200 μl of 100 mM Tris–HCl buffer pH 7·4, containing the following: 10 μmol/l LPA (oleoyl-sn1-glycerol-3-phosphate, Avanti Polar Lipids, Alabaster, AL, USA), 50 μmol/l oleoyl CoA (Sigma), 1 μl of [3H]-oleoyl-LPA (specific activity 30–60 Ci/mmol; PerkinElmer Life and Analytic Sciences, Boston, MA, USA), and 1 mg/ml fatty acid free BSA. The reaction was initiated by adding 30 μg total protein (cell lysate), followed by incubation for 10 min at 37 °C. The reaction was terminated by adding 0·5 ml of 1-butanol containing 1 M HCl to extract phospholipids. The butanol extract was dried under vacuum, and the LPA and PA were resolved using the solvent system chloroform–methanol–acetic acid–water (25:10:3:1). Radioactive spots were identified by co-migration with unlabeled LPA and PA standard and visualized in iodine vapors. The [3H]-LPA and [3H]-PA spots were scraped and counted for radioactivity (Tri-Carb Liquid Scintillation Counter 3100TR; PerkinElmer Life Sciences).
In order to determine whether the protein could accept any additional lysophospholipids as substrates, we used LPC, LPE, LPS, LPG, and LPI, all containing the C18:1 fatty acid at the sn-1 position. The enzymatic activity was determined as above except that LPA was replaced by the above-mentioned lysophospholipids.
Acyl-CoA specificity
The specificity of esterification of sn-2 position of [3H]-LPA (sn-1-oleoyl-2-hydroxy-sn-glycerol-3-phosphate) was determined by substituting the fatty acids with the following acyl-CoAs. Decanoyl (C10:0)-, lauroyl (C12:0)-, myristoyl (C14:0)-, palmitoyl (C16:0)-, stearoyl (C18:0)-, oleoyl (C18:1)-, linolenoyl (C18:3; n-3)-, γ-linolenoyl (C18:3; n-6)-, arachidoyl (C20:0)-, arachidonoyl (C20:4; n-6)-, behenoyl (C22:0)-, docosahexaenoyl (C22:6; n-3)-, tricosanoyl (C23:0)-, ligneceroyl (C24:0)-, nervonoyl (C24:1; n-9), and hexacosanoyl (C26:0)-CoA. The assay conditions remained the same as described for the AGPAT activity.
sn-1-acyl-lysophosphaditic acid specificity
To determine the specificity of various LPAs for the enzymatic activity, the following LPAs with various fatty acids at sn-1 position were used for the assay: myristoyl (C14:0)-, palmitoyl (C16:0)-, arachidonoyl (C20:4)-, arachidoyl (C20:0)-, linoleoyl (C18:2)-, and linolenoyl (C18:3). The enzymatic activity of these LPA species was compared with that of sn-1-oleoyl (C18:1)-2-hydroxy-sn-glycerol-3-phosphate. The enzymatic assay was assembled as before except that unlabeled LPAs and [14C]-oleoyl-CoA were used as substrates, and the activity was determined by the formation of [14C]-PA.
Glycerol-3-phosphate acyltransferase assay
The recombinant adenovirus-GPAT1 (gift from Daniel Linden, AstraZeneca) was amplified and purified as for AGPAT10/GPAT3 adenovirus. GPAT activity was determined as described earlier (Linden et al. 2004, 2006). HEK-293 cells were infected with recombinant GPAT1-adenovirus at a MOI of 150, and the cell lysate was prepared as described above. The enzyme reaction was assembled by adding 60 μM acyl-CoA, 150–80 μM G-3-P, spiked with either [14C]-glycerol-3-phosphate (American Radiolabels, St Louis, MO, USA) or [14C]-oleoyl CoA (PerkinElmer) in a buffer mix consisting of 75 mM Tris pH 7·4, 2 mg/ml BSA, 4 mM MgCl2, 2 mM dithiothreitol, in total volume of 200 μl. The enzymatic reaction was started by adding 100 μg of cell lysate and further incubated for 20 min at room temperature. The lipid extraction procedure was the same as for the acyltransferase (AGPAT) assay. The product formed (LPA) was resolved on thin layer chromatography (TLC) using chloroform:methanol:acetic acid:H2O (50:25:8:4) as mobile phase. The LPA spot was identified by exposure to iodine vapors, while G-3-P remained at the origin; these spots were scraped and quantified. We were not able to detect any AGPAT activity in these assays.
Quantitative real-time PCR in human tissue panel
Quantitative PCR was performed using the TaqMan primer and probes, designed using primer express software, ABI PRISM 7700 sequence detection system and PRIMER EXPRESS analysis software (Livak 1997) using the human cDNA panel from Clontech. To amplify AGPAT10/GPAT3, 100 pg cDNA was added to the forward (5′ -TGACAACGGATGGATGTTATGC-3′) and reverse (5′ -GACAAGCCTTGACCATAGCTCTCT-3′) primers along with fam-labeled probe (TTGGCCAGGTTCATG) and universal mix containing AmpliTaq and appropriate buffers. The PCR was followed for 40 cycles of 94 °C for 15 s and 60 °C for 30 s. The human AGPAT6/GPAT4 was amplified as above but with the following primers: forward (5′-TGACAGCCATCATCACCTACCA-3′) reverse (5′-GAGGTATGATTGGCCACACAGA-3′) and fam-labeled probe (CTCCATACTAACCGGTGTGTCT). The cDNA was amplified in duplicate along with G3PDH as an internal control. The ΔCt value for each tissue was calculated as ΔCt=(Ct (tissue) − Ct (G3PDH)).
Western blot analysis
Western blot analysis was essentially carried out as described by Schagger (2006). Total cell lysate (40 μg) from the V5-AGPAT10/GPAT3 and beta-galactosidase were resolved on 10% Tricine gel (NuSep Ltd, Frenchs Forest, NSW, Australia) followed by transfer onto nylon membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 5% non-fat milk containing 0·2% Tween-20 and then incubated with V5 antibody conjugated to HRP (dil 1:5000, Invitrogen) for 2 h at room temperature. The membrane was washed and then incubated with ECL reagent (GE Healthcare, Piscataway, NJ, USA), and exposed to X-ray film for 1 min. The same blot was stripped and reprobed with GAPDH for equal loading.
Generation of EGFP-tagged wild-type AGPAT10/ GPAT3 expression vector
The construction of the wild-type AGPAT10/GPAT3 fusion protein with GFP was carried out using the pDrive-AGPAT10/GPAT3 wild-type plasmid as the template and primer pair 7 and 8 (Supplementary material). The amplified product and the plasmid pEGFP-N3 (Clontech) were restricted with XhoI and BamHI and cloned in the similar site. The expression plasmid was sequenced to ascertain the orientation and the correctness for the junction sequences.
Immunofluorescence microscopy
Stably expressing AGPAT10/GPAT3-EGFP CHO cells were grown on glass cover slips 1 day before the experiment. Cells were fixed and permeabilized by incubation with methanol (−20 °C) for 20 min and 0·1% Triton X-100 for 25 min at room temperature respectively. Cells were washed 3×5 min with PBS and then incubated with primary antibody (Sec61-β; Upstate, Lake Placid, NY, USA diluted 1:200) for 60 min at 37 °C in a humidified chamber. Cells were then washed 3×5 min with PBS and incubated with AlexaFluor 598 coupled fluorescent secondary antibody (Invitrogen) for 60 min at 37 °C. After incubation, cells were washed 3×5 min with PBS, counterstained with 4′-6-diamidino-2-phenylindole (DAPI) during the washes and mounted on a glass slide with Aqua Poly/Mount (Polysciences, Inc., Warrington, PA, USA). Cells were observed with DeltaVision RT Deconvolution Microscope (Applied Precision, LLC, Issaquah, WA, USA). The obtained images were deconvoluted to remove the reflective fluorescence using softWoRx software.
Generation of stably knockdown AGPAT2 Huh7 cells
The human Huh7 cells were essentially generated by infecting the cells with lentivirus expressing shRNA for AGPAT2 and selecting with puromycin. Five sh-RNA plasmid constructs, pKO.1-AGPAT2 (E3–E7), where the expression of various sh-RNAs was directed by U6 promoter, were procured from Open Biosystems, Huntsville, AL, USA. Additional viral packaging plasmids, psPax2 (packaging plasmid), and pMD2.G (envelope plasmid), were from Addgene, USA. The lentiviruses were produced by transfecting each of the five pKO.1-AGPAT2 constructs along with the packaging plasmids in 293T cells according to manufacturer’s suggestion. The culture media containing the AGPAT2-shRNA-lentivirus were collected and centrifuged at 1000 g for 10 min to remove cellular debris. The lentivirus titer was determined by Lenti-X lentivirus qRT-PCR titration kit (Clonetech). The viral titers were usually in the range of 106−108/ml. The Huh-7 cells were seeded in 6-well plate and infected with each of the above AGPAT2-shRNA-lentivirus in presence of polybrene (5 μg/ml). After 24 h of infection, the cells were selected by adding puromycin (1 μg/ml). Prior to infection of Huh-7 with lentivirus particles, susceptibility of Huh-7 cells to puromycin was determined. It was found that 1 μg/ml was the minimum concentration of puromycin required to kill Huh-7 cells completely. Selection of all the Huh-7 cells infected with various lentiviruses construct was carried out at the predetermined concentration of puromycin. All five stable cell lines generated were analyzed for expression of AGPAT2 transcript by quantitative real-time PCR as well as semiquantitative PCR. Among these, the cells infected with AGPAT2-shRNA-E5-lentivirus construct (CCGGCGAGGGTACTCGCAACGACAACTCGAGTTGTCGTTGCGAGTACCCTCGTTTTTG; the bold sequences are sense, italics the loop, and those underlined are antisense) produced the most significant knockdown of AGPAT2 mRNA. The stable Huh-7 cells expressing scramble sh-RNA was generated as above.
Characterization of the stable Huh7-AGPAT2-shRNA-E5 cells
The stable cells, Huh7-AGPAT2-shRNA-E5, Huh7-scramble-shRNA, and Huh-7 (uninfected cells) were used to determine the total AGPAT enzymatic activity. The knockdown of mRNA was analyzed by real-time PCR followed by enzymatic activity. The expression of AGPAT1 and AGPAT2 in Huh7-AGPAT2-shRNA-knockdown cells was determined by TaqMan assays on demand real-time PCR, assay number Hs00197401_m1 and Hs00197410_m1, from ABI, Foster City, CA, USA. Knockdown of AGPAT2 transcript was also determined by semi-quantitative PCR using reduced number of PCR cycles (25) and primer pair, sense 5′-TCATGTACCTCAGGGGCGTCTTC-3′ and antisense 5′-GGGGATGGCTTCCGGCACCTGCACT-3′. The PCR products were analyzed on 1% agarose gel, and spot density were quantitated using ChemiImager 5500, version 2.03 from Alpha Innotech Corporation, San Leandro, CA, USA.
Expression and determination of the enzymatic activity of recombinant adenovirus expressing human AGPAT10/GPAT3 in Huh7-AGPAT2-shRNA-E5 cells
To determine the AGPAT enzymatic activities of the human AGPAT10/GPAT3 in these knockdown cells, the cells were infected with the recombinant adeno-virus at MOI of 150. The expression of protein was allowed for 48 h. The cells were scraped in 2 ml PBS and washed once with PBS. The washed cells were resuspended in assay buffer as described above. Cells were subjected to three freeze–thaw cycles and centrifuged at 875 g for 10 min to remove the cellular debris. The supernatant was used for the enzyme assay and western blotting.
Results
Tissue distribution of human AGPAT10/GPAT3
Northern blot analysis of human tissues showed that the AGPAT10/GPAT3 mRNA was close to 3·0 kb in size in the heart, skeletal muscle, and kidney (Supplementary material). Quantitative real-time PCR revealed that AGPAT10/GPAT3 is highly expressed in the adipose tissue followed by a slightly lower expression in the testis and kidney (Fig. 1). The heart, lung, spleen, ovary, and leukocytes had about several fold less expression compared with the adipose tissue. However, tissue survey by Cao et al. (2006) shows the highest expression in kidney, followed by heart, testis and muscle with significantly decreased expression in adipose tissue. It is interesting to observe that human AGPAT6/GPAT4, which is highly homologous to AGPAT10/GPAT3, is also expressed in all the tissues examined but was either similar in expression level in the adipose tissue, kidney, and heart or about 2–6 fold less than the AGPAT10/ GPAT3 in other tissues. The expression of AGPAT6/ GPAT4 is more in pancreas than that of adipose tissue. While there is no human mRNA expression data to compare, in mouse, the highest expression is observed in brown and visceral white adipose tissue. This expression level was similar in liver tissue as well (Vergnes et al. 2006).
Figure 1.
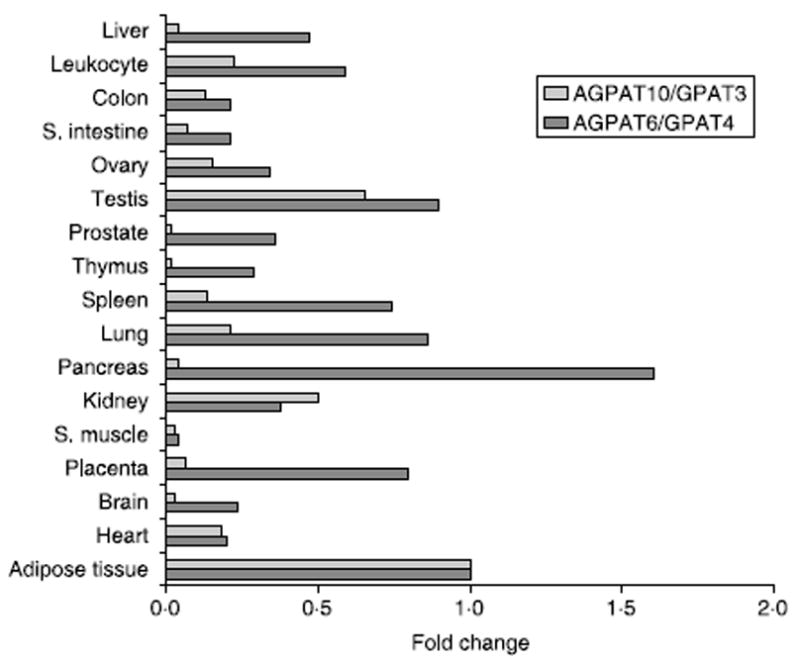
Expression of AGPAT6/GPAT4 and AGPAT10/GPAT3 in human tissue panel. Shown are the expression values relative to those observed in the adipose tissue. All values were normalized to GAPDH. S. intestine, small intestine; S. muscle, skeletal muscle. Adipose tissue is from omental fat.
Subcellular localization of AGPAT10/GPAT3-GFP
In CHO cells stably expressing the AGPAT10/GPAT3-GFP fusion protein, fluorescence microscopy showed an endoplasmic reticulum (ER) like expression pattern. This subcellular localization was confirmed by co-localizing AGPAT10/GPAT3-GEP with the ER specific protein Sec61-β (Fig. 2).
Figure 2.
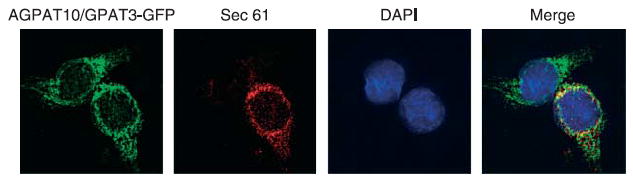
Localization of AGPAT10/GPAT3-GFP to endoplasmic reticulum in stably expressing CHO cells: Overexpressing AGPAT10/GPAT3-GFP CHO cell were fixed in methanol and incubated with antibody, Sec61, specific for endoplasmic reticulum and imaged for green and red fluorescence using fluorescence microscope. Shown is a representative cell for Sec61-β (red fluorescence), AGPAT10/GAPTβ (green fluorescence) DAPI (blue fluorescence), and the merge.
Acyltransferase activity of recombinant AGPAT10/GPAT3
As illustrated in Fig. 3A, western blot analysis showed robust expression of the human V5 epitope-tagged AGPAT10/GPAT3 protein. AGPAT and GPAT activity were carried out on the same lot of protein as subjected to western blot analysis. Figure 3B shows the AGPAT activity of AGPAT10/GPAT3 in the presence of oleoyl-CoA as the acyl donor and 1-oleoyl glycerol-3-phosphate as the acceptor. Cell lysate from similarly expressed adenovirus LacZ gene was used as negative control. AGPAT10/GPAT3 catalyzed the conversion of LPA to PA at a rate of ~2 nmol/min per mg protein. In similar assay conditions, the AGPAT2 showed twice as much AGPAT activity to that of AGPAT10/GPAT3 (data not shown). However, there was no GPAT activity detectable for AGPAT10/GPAT3 in these cell lysate. Lysates of cells infected with GPAT1 adenovirus, in contrast, showed robust conversion of G-3-P to LPA, ~4 nmol/min per mg protein (used as a positive control, Fig. 3C). In some assays, we used C12:0-acyl-CoA and C16:0-acyl-CoA to determine the GPAT activity (Supplementary material). It is evident from our results that AGPAT10/GPAT3 lacks GPAT activity in our assay system. This experiment showed that the gene AGPAT10/GPAT3 encodes an AGPAT enzyme. Therefore, experiments were carried out to further characterize this protein as an AGPAT10.
Figure 3.
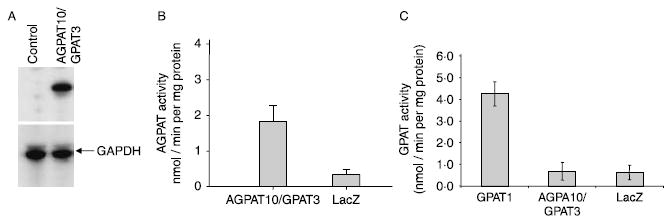
AGPAT enzymatic activity of wild-type human AGPAT10/GPAT3 in HEK-293 cells. (A) Shown is the western blot for the cell pool, used for determining the acyltransferase activity, for the V5 epitope-tagged AGPAT10/GPAT3 as probed with V5-antibody. GAPDH is included to demonstrate equal loading. (B) The enzymatic activity determined as conversion of 3H-LPA to 3H-PA in the presence of oleoyl-CoA, and expressed as product (3H-PA) formed per minute per mg protein. The LPA to PA conversion by recombinant adenovirus β-galactosidase (LacZ) was used as a control. Not shown is the conversion of substrate in the absence of enzyme. (C) Glycerol-3-phosphate acyltransferase enzymatic activity of AGPAT10/GPAT3 in the HEK-293 cell lysate. The GPAT activity was determined by incubating [14C]-glycerol-3-phosphate with an acyl-CoA. Recombinant adenovirus expressing human GPAT1 was included as a positive control. The recombinant adenovirus β-galactosidase (LacZ) was used as a control. Not shown is the conversion of substrate in the absence of enzyme.
To determine whether other lysophospholipids might also be substrate for this isoform, we determined the acyltransferase activity with LPC, LPS, LPE, LPG, and LPI. None of these PLs served as substrate for this enzyme (Fig. 4).
Figure 4.
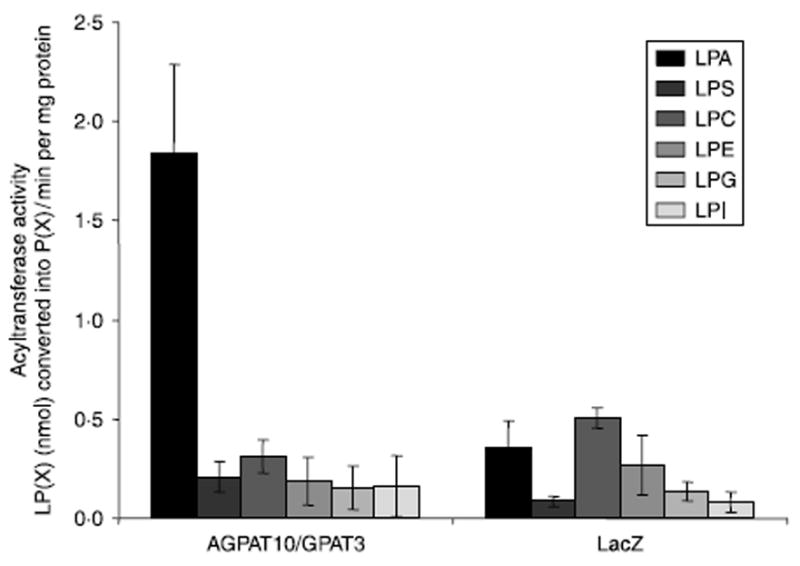
Acyltransferase activity of AGPAT10/GPAT3 for various lysophospholipids, LPA, LPS, LPC, LPE, LPG, and LPI. The enzymatic activity was determined as conversion of various lysophospholipids to their corresponding 3H-phospholipids in the presence of 3H-oleoyl-CoA, and expressed as product 3H-phospholipids formed per minute per mg protein. The recombinant adenovirus β-galactosidase (LacZ) was used as a control. Not shown is the conversion of substrate in the absence of enzyme. Shown are the mean values from three independent assays carried out in triplicate.
Substrate specificity of AGPAT10/GPAT3
To assess the substrate specificity of AGPAT10/GPAT3, LPAs with various fatty acids at the sn-1 position were incubated with adenoviral recombinant protein and [14C]-oleoyl-CoA. Wild-type AGPAT10/GPAT3 accommodated myristoyl, linoleoyl, and palmitoyl fatty acids at the sn-1 position of the LPA, synthesizing PAs at about the same rate as the preferred substrate, sn-1-oleoyl-LPA (Fig. 5A). Other LPAs, such as sn-1-linolenoyl and sn-1-arachidonoyl, were esterified at 50–70% the rate of sn-1-oleoyl-LPA. However, the sn-1-arachidoyl form of LPA was not esterified.
Figure 5.
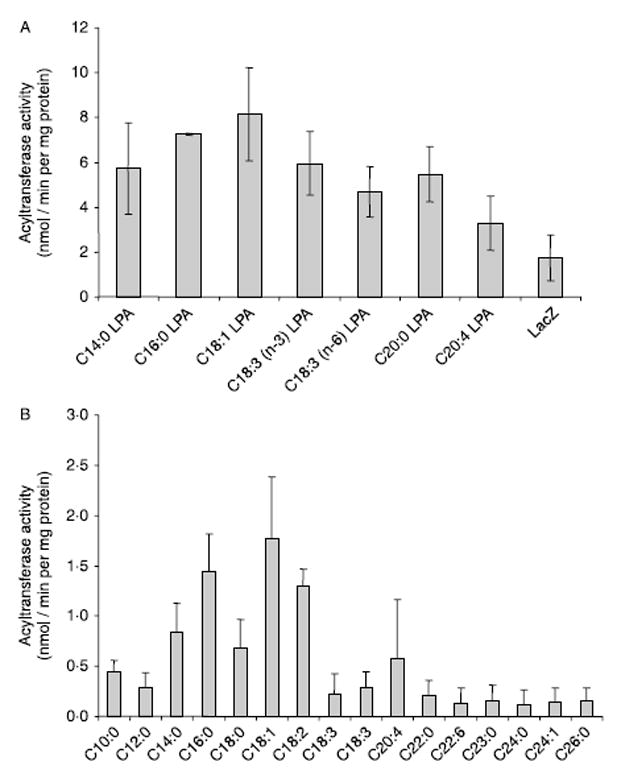
(A) LPA specificity of the recombinant human AGPAT10/GPAT3 expressed in HEK-293 cells. Specificity of human recombinant AGPAT10/GPAT3 for various species of sn-1-lysophosphatidic acid acceptors was determined using oleoyl-CoA as donor. Various species of lysophosphatidic acid with medium and long chain fatty acids at sn-1 position were used as acceptors. Activity was expressed as product (3H-PA) formed per minute per mg protein. All enzymatic activities were determined in two independent experiments in triplicate. (B) Acyl-CoA specificity of recombinant human AGPAT10/GPAT3 expressed in HEK-293 cells. Specificity of human recombinant AGPAT10/GPAT3 for acyl-CoA donors was determined using sn-1-oleoyl-lysophosphatidic acid as an acceptor and various medium and long chain fatty acyl-CoA as donors. All enzymatic activities were determined in two independent experiments in triplicate. The n-3, n-6, n-9 indicate the position of the first double bond from the CH3 end of the molecule.
To assess the specificity of AGPAT10/GPAT3 for the other substrate, the fatty acids which esterify the sn-2 position, cell lysates were incubated with [3H]-LPA with various unlabeled acyl-CoAs. Wild-type AGPAT10/ GPAT3 used palmitoyl-CoA, myristoyl-CoA and stearoyl-CoA at the rate of about 25% of the preferred substrate oleoyl-CoA (Fig. 5B). All other acyl-CoAs were poor substrates compared with oleoyl-CoA.
Knowing the preferred LPA and acyl-CoA for the enzyme AGPAT10/GPAT3, we then systematically determined the optimum temperature, pH, time of incubation, and protein concentration (Supplementary material) to further undertake the kinetic studies. The apparent Km and Vmax for LPA was determined using 1-oleoyl-lysophosphatidic acid and 1-oleoyl-CoA as shown in Fig. 6 and in Table 1.
Figure 6.
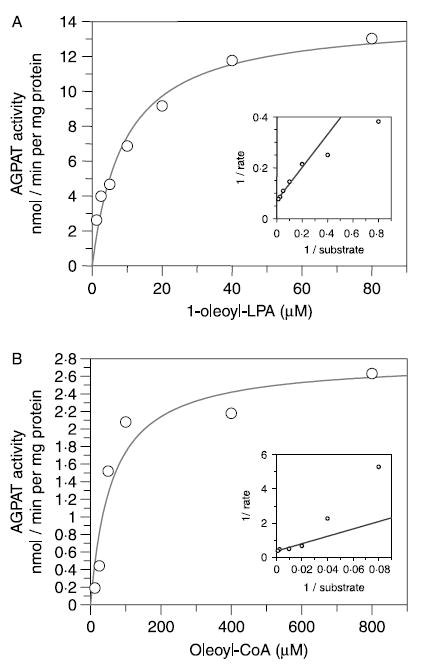
Kinetic studies of the recombinant human AGPAT10 expressed in HEK-293 cells. (A) The saturation curve of AGPAT10/GPAT3 enzymatic activity as determined by incubating increasing concentrations of LPA with fixed amount of [14C]-oleoyl-CoA. The data were transformed to fit the Lineweaver–Burk plot to determine the Km and Vmax (shown as an insert). The enzymatic activities were determined in triplicate with each experiment carried out in triplicate. (B) The saturation curve of AGPAT10/GPAT3 enzymatic activity was determined by varying the concentration of oleoyl-LPA spiked with known amount of [14C]-oleoyl-CoA. The enzymatic data were transformed as above to obtain the Km and Vmax (shown as an insert). The values were determined using GraFit software (Erithacus Software Ltd, Surrey, UK).
Table 1.
Shown are the apparent affinity (Km) and maximum velocity (Vmax) of 1-acylglycerol-3-phosphate-O-acyltransferase isoform 10/glycerol-3-phosphate acyltransferase isoform 3 (AGPAT10/GPAT3) (this study) and those of AGPAT2 (data from Hollenback et al. 2006)
|
Km (μM)
|
Vmax (nmol/min per mg protein)
|
Vmax/Km
|
|
|---|---|---|---|
| Substrate | |||
| AGPAT10/GPAT3 | |||
| Acyl-CoA | 2·7 | 60 | 22 |
| LPA | 9·2 | 14·19 | 1·5 |
| AGPAT2 | |||
| Acyl-CoA | 0·4 | 210 | 525 |
| LPA | 2 | 200 | 100 |
|
|
|||
Human AGPAT10/GPAT3 was expressed in HEK-293 cells using recombinant adenovirus, whereas human AGPAT2 was expressed in Sf9 cells using recombinant baculovirus. Kinetics for both the proteins was determined using unpurified recombinant proteins.
Acyltransferase activities in Huh-7-AGPAT2 knockdown cells
To further demonstrate the AGPAT activity of the AGPAT10/GPAT3, we created human liver AGPAT2 knockdown cells. Huh7-shRNA-AGPAT2-E5 cell pool showed about 50 and 70% reduced expression by either method (Supplementary material). In order to determine if the knockdown of AGPAT2 has resulted in any change in mRNA levels of AGPAT1, quantification of mRNA expression was carried out along with that of AGPAT2. We did observe a 40% increase in the mRNA expression of AGPAT1 isoform (data not shown). Further characterization of the Huh7-shRNA-AGPAT2-E5 was carried out by determining the acyltransferase enzymatic activity. AGPAT assay showed that there was almost 80% decrease in acyltransferase activity compared with Huh-7 cells (Supplementary material). These Huh7-shRNA-AGPAT2-E5 acyltransferase knockdown cells were used to determine the AGPAT activity.
Acyltransferase over expression of AGPAT10/GPAT3 in Huh7-shRNA-AGPAT2-E5 cell line
Major contributor towards the AGPAT activity in liver cells is the expression of AGPAT2 (unpublished data). Hence, in this study, we created a model cell line of Huh-7 with knock down of AGPAT2 transcript. Construction of the Huh-7 liver cell with 80% reduced AGPAT enzymatic activity enabled us to investigate activity of overexpressed AGPAT10 in this liver cell line. Adenovirus containing the expression construct for AGPAT10/GPAT3 was infected in Huh7-shRNA-AGPAT2-E5 cell line along with adenovirus LacZ or adenovirus expressing the GPAT1. Figure 7A shows the activity profile for AGPAT activity. In this assay, a fourfold increase in the AGPAT activity was evident with AGPAT10/GPAT3 construct. With the same lysate, experiments were carried out to determine if there was any GPAT activity, but we were unable to detect any GPAT activity in these cells (Fig. 7B). However, lysate from cells infected with the recombinant adenovirus GPAT1 showed a tenfold increase in the GPAT activity.
Figure 7.
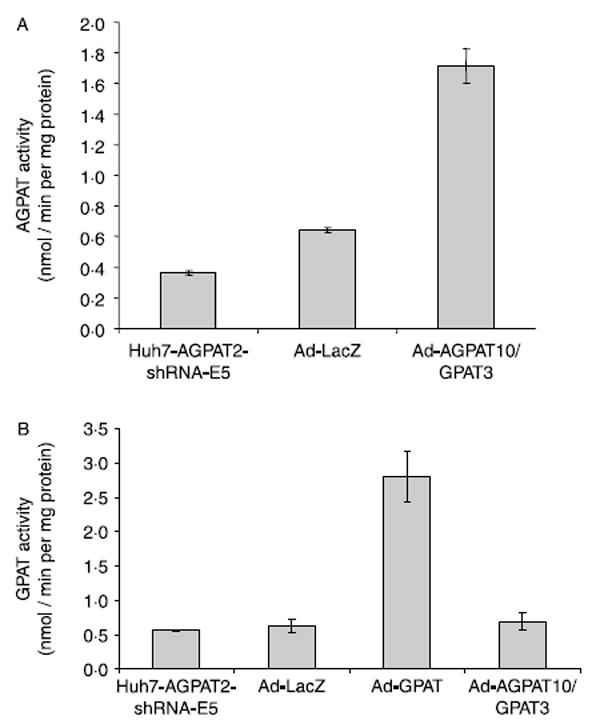
Acyltransferase activities in Huh 7-AGPAT2-shRNA knockdown E5 cells. (A) Shown is the mean AGPAT activity in cell lysates with overexpression of AGPAT10/GPAT3, as compared with the activity of cell lysates from adenovirus expressing LacZ and human Huh-7 cells where endogenous AGPAT2 was inhibited using shRNA as controls. (B) Cell lysate from the same experiment was used to determine the GPAT activity for AGPAT10/GPAT3 as compared with the activity of adenovirus expressing LacZ and human Huh-7 cells where endogenous AGPAT2 was inhibited using shRNA. Shown also is the enzymatic activity for GPAT1 which was expressed separately but in the same cell line.
Discussion
The sequencing of the human genome and the availability of bioinformatics tools helped us in cloning AGPAT10/GPAT3. The human gene for AGPAT10/GPAT3 is located on chromosome 4q21.23, spans over 69 kb, and consists of 13 exons (Supplementary material). When aligned with other known sn-2 acyltransferases, it appears that AGPAT10/GPAT3 is very closely aligned with human AGPAT6/GPAT4 (Supplementary material). The human AGPAT10/GPAT3 is highly conserved with mouse, zebrafish, Caenorhabditis elegans, and Arabidopsis (Supplementary material). Real-time quantitative PCR on a tissue panel for both the AGPAT6/GPAT4 and AGPAT10/GPAT3 revealed that both isoforms are expressed similarly in the various tissues we examined. The highest expression was observed in the omental adipose tissue with a 1·5-fold less expression in the testis. A twofold decreased expression of AGPAT10/GPAT3 was observed in the kidney. In all other tissues, the expression of AGPAT10/GPAT3 was less than fivefold or more. In our tissue panel, AGPAT6/GPAT4 is also highly expressed in liver, only about a fold less than adipose tissue, whereas the expression of AGPAT10/GPAT3 is several fold less than AGPAT6/GPAT4. The human AGPAT10/GPAT3 protein localizes to ER when expressed in CHO cells as observed by others (Cao et al. 2006).
The recombinant AGPAT10/GPAT3 protein shows a robust sn-2 acyltransferase activity in vitro, using LPA as an acyl acceptor and acyl-CoA as an acyl donor. Among various LPAs used as an acyl acceptor with oleoyl-CoA as the acyl donor, 1-oleoyl-LPA was the preferred substrate. LPAs with palmitoyl (C16:0), linolenoyl (C18:3, n-3) and arachidonoyl (C20:4) had 50% reduced activity. Similarly, with 1-oleoyl-LPA as acyl acceptor, and various acyl donors, oleic acid (C18:1) emerged as the most efficient acyl donor; whereas palmitoyl (C16:0) and linolenoyl (C18:2) were 57% and myristoyl (C14:0) and stearoyl (C18:0) were only 50% of the maximal activity. These data show that the substrate preference for AGPAT10/GPAT3 in vitro remains 1-oleoyl-LPA and oleic acid similar to that observed for AGPAT2 (Hollenback et al. 2006). AGPAT10/GPAT3 did not recognize various other lysophospholipids such as LPS, LPC, LPE, LPG, and LPI. Since all the lysophospholipids carry oleic acid at the sn-1 position, it remains unclear if the AGPAT10/GPAT3 might use these lysophospholipids if various other fatty acids are present at sn-1 position of the phospholipids.
The enzyme kinetic studies show that AGPAT10/GPAT3 has an apparent Km of 9·2 μM at half maximal velocity with an apparent Vmax of 14·19 nmol/min per mg protein for LPA. The apparent Km and Vmax was several fold lower for acyl-CoA, being 60 μM and 2·7 nmol/min per mg protein, respectively (Table 1). Although the preferred substrates for AGPAT10/GPAT3 remain similar to those for AGPAT2, the kinetics of the two enzymes is quite different (Hollenback et al. 2006). Using human recombinant AGPAT2 obtained from insect Sf9 cells, the apparent Km and Vmax for 1-oleoyl-CoA was 0·4 μM and 210 nmol/min per mg protein respectively. The enzyme kinetics of AGPAT2 for the other substrate LPA was 2·0 μM (Km) with the Vmax of 200 nmol/min per mg protein (Hollenback et al. 2006). This indicates that the AGPAT2 has about sevenfold higher apparent affinity for the same substrate (acyl-CoA) than AGPAT10/GPAT3 and nearly fourfold higher turnover rate. Although the vectors employed to obtain recombinant proteins are different, our studies indicate that AGPAT2 will be the preferred enzyme over AGPAT10/GPAT3 for acylation in tissues where it is expressed more than the AGPAT2 isoform, particularly in adipocytes where AGPAT2 is expressed in very high level.
While our study was in progress, the same cDNA, protein sequence, was reported to encode a protein with acyl-CoA: GPAT activity and was named GPAT3 (Cao et al. 2006). Inhibition of GPAT3 by siRNA also reduced the accumulation of triglyceride in 3T3-L1 cells. However, whether enzymatic activity was decreased as well, is unclear (Cao et al. 2006). We attempted to demonstrate the GPAT activity with the same recombinant protein expressed in HEK-293 cells, under similar assay conditions and with various fatty acids (data not shown), but were unable to substantiate GPAT activity for this cDNA. As a positive control in our experiment, we employed recombinant adenovirus expressing GPAT1 which showed similar level of GPAT activity as reported earlier (Linden et al. 2004, 2006). While there is no significant explanation why the same clone is producing two different enzymatic activities, it might be argued that our adenovirus expression system is more robust than the plasmid based vectors, which tend to produce lower amounts of protein and enzymatic activity. It is of interest to note that the two expression systems show different conversion rates (Fig. 3C).
As shown in the previous study (Cao et al. 2006), the AGPAT10/GPAT3 enzyme is involved in the biosynthesis of TAG in adipocytes. Whether the increase in the TAG synthesis is due to its AGPAT activity or also to increased expression of GPAT enzyme remains unclear. The closely related isoform, AGPAT6, which is also shown to have GPAT activity (AGPAT6/GPAT4; Chen et al. 2008, Nagle et al. 2008), when deleted in cellular model or in animals should show significantly decreased TAG biosynthesis in adipose tissue. However, AGPAT6/GPAT4 null mice are reported to have minimal loss of adipose tissue: only reduced sub-dermal adipose tissue was observed (Beigneux et al. 2006, Vergnes et al. 2006). This observation suggests that there is redundancy for activity of this acyltransferase. The alignment of two proteins, AGPAT6/GPAT4 and AGPAT10/GPAT3, did not reveal any significant differences in the proteins’ catalytic sites and putative substrate binding sites which might indicate the differences in their enzymatic activities (Supplementary material). At this time, we cannot ascertain why AGPAT6/GPAT4 demonstrates GPAT activity; however, attempts to assay AGPAT activity has been reported. We have employed a robust expression system (recombinant adenovirus) to generate recombinant protein for our assays. We have not studied AGPAT6/GPAT4 protein using our expression system and thus cannot comment. To the best of our knowledge, such a robust expression system for AGPAT6/GPAT4 is lacking. Future studies employing robust expression systems might resolve this issue.
As shown in Table 1, the significantly higher catalytic activity of another isoform, AGPAT2, will most efficiently acylate LPA to PA for phospholipids and TAG synthesis. In this context, it is interesting to note that the AGPAT2 deficiency in humans leads to complete loss of adipose tissue (Agarwal & Garg 2006), the phenotype of Agpat2 null mice is awaited. In adipose tissue, AGPAT2 is highly expressed, about 13-fold more than AGPAT10/GPAT3 (unpublished), and would appear to be the main TAG and PL synthesizer. Our interpretation of the AGPAT10/GPAT3 is that, although the enzyme is located in the ER, it may not be in the same ‘micro domain’ of the ER where other enzymes involved in the biosynthesis of TAG are localized. Future studies including development of AGPAT10/GPAT3 knockdown or overexpression mouse models may ascertain the precise role of AGPAT10/GPAT3 in phospholipids and TAG biosynthesis.
Supplementary Material
Acknowledgments
We thank Ruth Giselle Huet and Katie Tunison for technical assistance, Daniel Linden, AstraZeneca, Sweden, for recombinant adenovirus-GPAT1, Richard Auchus, MD, PhD for critical review of the manuscript.
Funding
This work was supported by the National Institutes of Health grants R01-DK54387 and by the Southwestern Medical Foundation.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Agarwal AK, Garg A. Congenital generalized lipodystrophy: significance of triglyceride biosynthetic pathways. Trends in Endocrinology and Metabolism. 2003;14:214–221. doi: 10.1016/s1043-2760(03)00078-x. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Garg A. Genetic disorders of adipose tissue development, differentiation, and death. Annual Review of Genomics and Human Genetics. 2006;7:175–199. doi: 10.1146/annurev.genom.7.080505.115715. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Arioglu E, de Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nature Genetics. 2002;31:21–23. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Barnes RI, Garg A. Functional characterization of human 1-acylglycerol-3-phosphate acyltransferase isoform 8: cloning, tissue distribution, gene structure and enzymatic activity. Archives of Biochemistry and Biophysics. 2006;449:64–76. doi: 10.1016/j.abb.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Sukumaran S, Bartz R, Barnes RI, Garg A. Functional characterization of human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 9: cloning, tissue distribution, gene structure, and enzymatic activity. Journal of Endocrinology. 2007;193:445–457. doi: 10.1677/JOE-07-0027. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Vergnes L, Qiao X, Quatela S, Davis R, Watkins SM, Coleman RA, Walzem RL, Philips M, Reue K, et al. Agpat6–a novel lipid biosynthetic gene required for triacylglycerol production in mammary epithelium. Journal of Lipid Research. 2006;47:734–744. doi: 10.1194/jlr.M500556-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley DN. Lipid phosphate phosphatases and related proteins: signaling functions in development, cell division, and cancer. Journal of Cellular Biochemistry. 2004;92:900–912. doi: 10.1002/jcb.20126. [DOI] [PubMed] [Google Scholar]
- Cao J, Li JL, Li D, Tobin JF, Gimeno RE. Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. PNAS. 2006;103:19695–19700. doi: 10.1073/pnas.0609140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman GM, Han GS. Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. Journal of Biological Chemistry. 2009;284:2593–2597. doi: 10.1074/jbc.R800059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Kuo MS, Li S, Bui HH, Peake DA, Sanders PE, Thibodeaux SJ, Chu S, Qian YW, Zhao Y, et al. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. Journal of Biological Chemistry. 2008;283:10048–10057. doi: 10.1074/jbc.M708151200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Progress in Lipid Research. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. Journal of Biological Chemistry. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- Eberhardt C, Gray PW, Tjoelker LW. Human lysophosphatidic acid acyltransferase. cDNA cloning, expression, and localization to chromosome 9q34.3. Journal of Biological Chemistry. 1997;272:20299–20305. doi: 10.1074/jbc.272.32.20299. [DOI] [PubMed] [Google Scholar]
- Gale SE, Frolov A, Han X, Bickel PE, Cao L, Bowcock A, Schaffer JE, Ory DS. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. Journal of Biological Chemistry. 2006;281:11082–11089. doi: 10.1074/jbc.M509612200. [DOI] [PubMed] [Google Scholar]
- Haque W, Garg A, Agarwal AK. Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochemical and Biophysical Research Communications. 2005;327:446–453. doi: 10.1016/j.bbrc.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Hollenback D, Bonham L, Law L, Rossnagle E, Romero L, Carew H, Tompkins CK, Leung DW, Singer JW, White T. Substrate specificity of lysophosphatidic acid acyltransferase beta – evidence from membrane and whole cell assays. Journal of Lipid Research. 2006;47:593–604. doi: 10.1194/jlr.M500435-JLR200. [DOI] [PubMed] [Google Scholar]
- Leung DW. The structure and functions of human lysophosphatidic acid acyltransferases. Frontiers in Bioscience. 2001;6:d944–d953. doi: 10.2741/leung. [DOI] [PubMed] [Google Scholar]
- Linden D, William-Olsson L, Rhedin M, Asztely AK, Clapham JC, Schreyer J. Overexpression of mitochondrial GPAT in rat hepatocytes leads to decreased fatty acid oxidation and increased glycerolipid biosynthesis. Journal of Lipid Research. 2004;45:1279–1288. doi: 10.1194/jlr.M400010-JLR200. [DOI] [PubMed] [Google Scholar]
- Linden D, William-Olsson L, Ahnmark A, Ekroos K, Hallberg C, Sjogren HP, Becker B, Svensson L, Clapham JC, Oscarsson J, et al. Liver-directed overexpression of mitochondrial glycerol-3-phosphate acyltransferase results in hepatic steatosis, increased triacylglycerol secretion and reduced fatty acid oxidation. FASEB Journal. 2006;20:434–443. doi: 10.1096/fj.05-4568com. [DOI] [PubMed] [Google Scholar]
- Livak KJ. ABI Prism 7700 Sequencer detection system. User Bulletin 2. PE Applied Biosystems. 1997:1–36. [Google Scholar]
- McMaster CR, Jackson TR. Phospholipid synthesis in mammalian cells. In: Daum G, editor. Lipid Metabolism and Membrane Biogenesis. Berlin, Heidelberg: Springer-Verlag; 2004. pp. 5–30. [Google Scholar]
- Nagle CA, Vergnes L, Dejong H, Wang S, Lewin TM, Reue K, Coleman RA. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6– / – mice. Journal of Lipid Research. 2008;49:823–831. doi: 10.1194/jlr.M700592-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H. Tricine-SDS-PAGE. Nature Protocols. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- Tang W, Yuan J, Chen X, Gu X, Luo K, Li J, Wan B, Wang Y, Yu L. Identification of a novel human lysophosphatidic acid acyltransferase, LPAAT-theta, which activates mTOR pathway. Journal of Biochemistry and Molecular Biology. 2006;39:626–635. doi: 10.5483/bmbrep.2006.39.5.626. [DOI] [PubMed] [Google Scholar]
- Vergnes L, Beigneux AP, Davis R, Watkins SM, Young SG, Reue K. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. Journal of Lipid Research. 2006;47:745–754. doi: 10.1194/jlr.M500553-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J, Tompkins CK, Balantac N, Nudelman E, Meengs B, White T, Bursten S, Coleman J, Kumar A, Singer JW, et al. Cloning and expression of two human lysophosphatidic acid acyltransferase cDNAs that enhance cytokine-induced signaling responses in cells. DNA and Cell Biology. 1997;16:691–701. doi: 10.1089/dna.1997.16.691. [DOI] [PubMed] [Google Scholar]
- Ye GM, Chen C, Huang S, Han DD, Guo JH, Wan B, Yu L. Cloning and characterization a novel human 1-acyl-sn-glycerol-3-phosphate acyltransferase gene AGPAT7. DNA Sequence. 2005;16:386–390. doi: 10.1080/10425170500213712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.