

Abstract
Acute myeloid leukemia (AML) is a biologically complex and molecularly and clinically heterogeneous disease, and its incidence increases with age. Cytogenetics and mutation testing remain important prognostic tools for treatment after induction therapy. The post-induction treatment is dependent on risk stratification. Despite rapid advances in determination of gene mutations involved in the pathophysiology and biology of AML, and the rapid development of new drugs, treatment improvements changed slowly over the past 30 years, with the majority of patients eventually experiencing relapse and dying of their disease. Allogenic hematopoietic stem cell transplantation remains the best chance of cure for patients with intermediate- or high-risk disease. This review gives an overview about advances in prognostic markers and novel treatment options for AML, focusing on new prognostic and probably therapeutic mutations, and novel drug therapies such as tyrosine kinase inhibitors.
Keywords: Acute myeloid leukemia, mutations, targeted therapies, tyrosine kinase inhibitors, review
Acute myeloid leukaemia (AML) is a heterogeneous disorder characterized by clonal expansion of myeloid progenitors (blasts) in the bone marrow and peripheral blood. Formerly, AML had a very poor prognosis. Due to improvements in therapeutic regimens and supportive care (e.g. anti-infective agents, and transfusion support), AML is now cured in approximately 35-40% of patients younger than 60 years (1). For those over 60 years old, the prognosis has improved but remains poor (2). Chromosomal abnormalities (e.g. deletions, translocations) are identified in approximately 50% of all adult patients with primary AML and have long been recognized as the genetic events that cause and promote this disease (1,3). Certain cytogenetic abnormalities, including t(8;21)(q22;q22), t(15;17)(q22;q12) and inv(16)(p13.1;q22), are associated with longer remission and survival, while alterations of chromosomes 5, 7, complex karyotype (described as >3 chromosomal abnormalities) and 11q23 are associated with poor response to therapy and shorter overall survival (1). In contrast, about 40-50% of all AML cases are cytogenetically normal (CN-AML) (3). Although, this group has an intermediate risk of relapse, a substantial heterogeneity is found in this population regarding clinical outcome. Molecular screening of this AML category is important in risk stratification and therapy decisions (4). The identification of new mutations in AML has raised prognostic and probably therapeutic implications for patients with AML.
Prognosis/Risk Stratification
Age and performance status in addition to chromosomal and molecular aberrations remain the most important tools for outcome prediction in AML (3). In 2010, the European Leukaemia Net classification scheme was created in an effort to standardize risk stratification in adult patients with AML by incorporating cytogenetic and known molecular abnormalities (4). Patients are classified into one of four risk groups: favorable, intermediate 1, intermediate 2 and adverse (Table I).
Table I. Eurpean Leukaemia Net risk group [adapted from (4)].
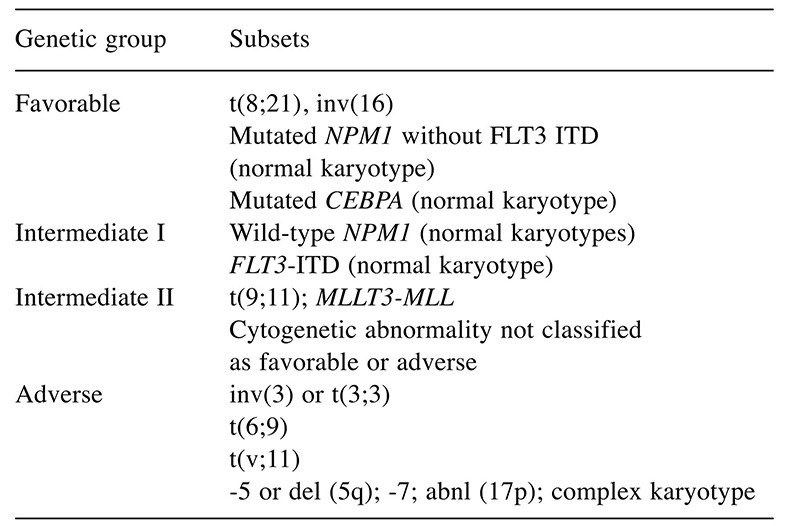
abnl, Abnormalities; CEBPA, CCAAT/enhancer-binding protein alpha; del, deletion; FLT3 ITD, Fms-related tyrosine kinase 3 internal tandem duplications; MLLT3-MLL, mixed lineage leukemia; NPM1, nucleophosmin 1
Standard Therapy for AML
Intensive induction chemotherapy has remained unchanged for more than four decades and is the backbone of therapy. For young adults (age <60 years) and fit elderly patients the intensive anthracycline and cytarabine regimen ‘7+3’ induction therapy is the standard of care (5,6). The aim of induction chemotherapy is to achieve morphological complete remission (CR) (5). Thus, patients with de novo AML achieve CR in 65-73% of cases using standard induction with '7+3', while only 38-62% of patients over 60 years of age with AML achieve CR (5,6). Consolidation or post-induction therapy is given to prevent relapse and eradicate minimal residual leukemia in the bone marrow after induction as a bridge to transplant or to achieve cure. Assessment of minimal residual disease using real-time polymerase chain reaction or next-generation sequencing is increasingly being used to help track treatment response and has been shown to be superior than morphology alone in predicting impending relapse (7,8).
In general, there are two main strategies for consolidation; chemotherapy (including targeted agents) and hematopoietic stem cell transplantation (HSCT) (9,10). Both strategies can be used alone or most commonly in combination depending on the type of leukemia, the fitness of the patient and the availability of a stem cell donor. One of the most important treatment decisions in AML is to estimate the benefit and risk associated with allogeneic HSCT in first remission for a given patient.
Transplantation offers the best means of preventing AML recurrence, but remains associated with higher treatment-related morbidity and mortality, especially in older patients (11). In patients with favorable-risk AML, the relapse risk may be low enough and the salvage rate high enough to postpone HSCT to second remission. This strategy has been validated in several donor versus no-donor studies (12,13). In these studies, favorable patients (i.e. those with core binding factor AML (CBF-AML)) from the no-donor group performed, as well as those from the donor group, whereas all other patients appeared to benefit from undergoing HSCT. A study by the European Society for Blood and Marrow Transplantation examined the role of reduced intensity (RIC) conditioning versus myeloablative regimens (MAC) to younger patients aged 40-60 years in first CR1 (14). Among 2,974 patients, 1,638 had MAC and 1,336 RIC transplants. Overall survival was higher in patients with RIC with low-risk cytogenetics but not in the intermediate- or poor-risk AML. Relapse incidence was lower with MAC in poor- and intermediate-risk AML. Non-relapse mortality was higher in MAC in all cytogenetic risk groups. They confirmed lower relapse but higher non-relapse mortality risks with MAC. MAC is not superior in patients with higher risk cytogenetics, but is inferior to RIC in the small cohort of patients with AML with low-risk cytogenetics (14).
New Mutations with Prognostic and Therapeutic Implications in AML
During the past decade, several studies have shown that the presence or absence of specific gene mutations or changes in gene expression can further classify AML cases and have an effect on patient prognosis (3,4,15-17). This is particularly relevant for patients with CN-AML. With the technique of next-generation sequencing, the genetic landscape of CN-AML has been more defined with each case having an average of 13 mutations, eight of which are random ‘passenger’ mutations and five of which are recurrent ‘driver’ mutations (15,16). Key molecular abnormalities have been identified and are now used to predict outcome and help guide treatment for patients with AML. In a very recent article by Papaemmanuil et al. the role of mutations and their correlation with pathophysiology was examined in a large cohort of 1,540 patients with AML (17). They identified 5,234 driver mutations across 76 genes or genomic regions, with two or more drivers being identified in 86% of the patients. Patterns of co-mutation compartmentalized the cohort into 11 classes, each with distinct diagnostic features and clinical outcomes. In addition to currently defined AML sub-groups, three heterogeneous genomic categories emerged: AML with mutations in genes encoding chromatin, RNA-splicing regulators, or both (in 18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (in 13%); and, provisionally, AML with isocitrate dehydrogenase (IDH2) R172 mutations (in 1%). Patients with chromatin–spliceosome and TP53–aneuploidy AML had poor outcomes, with the various class-defining mutations contributing independently and additively to the outcome. They found gene–gene interactions that were especially pronounced for nucleophosmin 1 (NPM1)-mutated AML, in which patterns of co-mutation identified groups with a favorable or adverse prognosis (17). In our review, we focus on the most relevant AML mutations (Table II).
Table II. New relevant mutations in acute myeloid leukaemia.
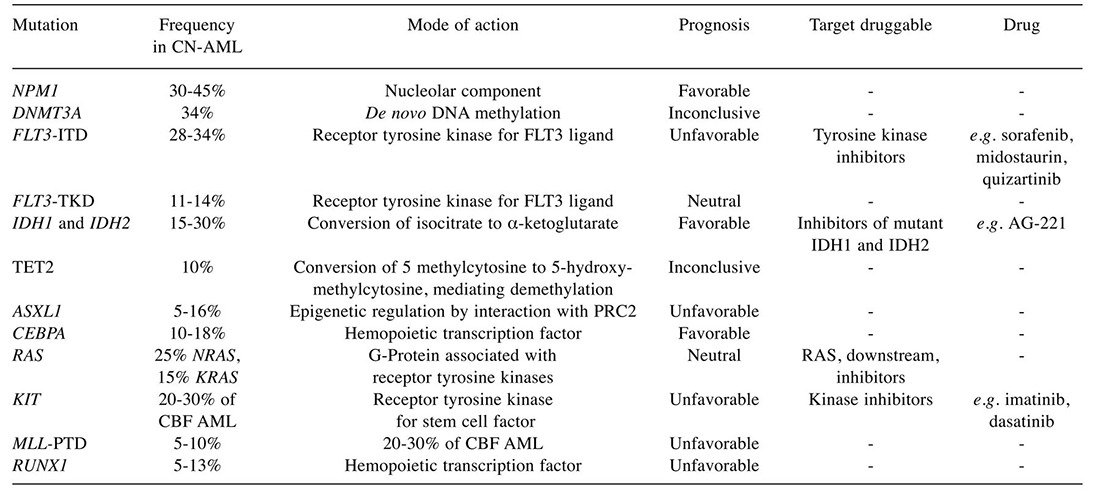
ASXL1, Additional sex comb-like 1; CBF, core-binding factor; CEBPA, CCAAT/enhancer-binding protein alpha; CN-AML; cytogenetically normal acute myeloid leukaemia; DNMT3A, DNA methyl-transferase 3A; FLT3 ITD, Fms-related tyrosine kinase 3-internal tandem duplications; FLT3 TKD, Fms-related tyrosine kinase 3-tyrosine kinase domain; IDH1 and IDH2, Isocitrate dehydrogenase 1,2; KIT, CD117; MLL PTD, mixed lineage leukaemia partial tandem duplication; NPM1, nucleophosmin 1; PRC2, polycomb repressive complex 2; RAS, RAS viral (v-ras) oncogene homologue; RUNX1, Runt-related transcription factor 1; TET2, ten-11 translocation 2
Fms-like Tyrosine Kinase 3 (FLT3) Mutations
FLT3 is a class III family receptor tyrosine kinase acting as a cytokine receptor for FLT3 ligand. FLT3 was found to be strongly expressed in hematopoietic stem cells with important roles in cell survival and proliferation (18,19). FLT3 mutations are among the most frequent mutations observed in AML and two types are distinguished. Internal tandem duplications (ITD) of FLT3 can be identified in about 20% of patients with AML, and in 28-34% of those with CN-AML; in the latter, they predict poor outcome (1,3,4,19,20). These mutations are mostly located in the juxtamembrane domain. In 28% of cases, they are found in the tyrosine kinase domain (TKD), and predict a particularly poor prognosis. The ITDs in FLT3 constitutively activate the tyrosine kinase by interfering with the auto-inhibitory function of the juxtamembrane domain and lead to enhanced rat sarcoma (RAS), mitogen-activated protein kinases (MAPK), and signal transducer and activator of transcription 5 (STAT5) signaling (19-22). They predict increased frequency of relapse, and shorter overall survival. Both types of mutations constitutively activate FLT3 signaling, promoting blast proliferation (19-21). This effect on prognosis is modulated by the ratio of mutated to wild-type alleles, with inferior outcome in the presence of a higher load of internal tandem duplications in FLT3. Evidence is emerging that patients with AML with these mutations benefit from allogeneic HSCT in first CR, that is recommended for this group (4). Furthermore, FLT3 ITD mutations have been associated with increased risk of relapse, while the prognostic relevance of FLT3 TKD mutations is controversial (22). The degree to which FLT3 ITD is a biomarker associated with poor outcome is determined by the binding site and FLT3 ITD allelic burden (20,22,23). Studies have shown that non-juxtamembrane (JM) ITD are worse than JM domain ITD mutation and higher mutant to wild-type allelic ratios were significantly associated with lower CR rates (22,23). Currently, tyrosine kinase inhibitors (TKI) are being tested in patients with FLT3-mutated AML. Unfortunately, when used alone, TKIs showed only a transient reduction of blasts, and even if initially effective, subsequent acquisition of secondary mutations induces resistance over time (24). Several small- molecule inhibitors of FLT3 have been developed with mixed results. First-generation drugs include multi-kinase inhibitors such as midostaurin, lestaurtinib, tandutinib sunitinib and sorafenib. When used as single agents they have limited anti-leukemia activity, mostly leading only to transient reduction of blood and bone marrow blasts, and increased toxicity (25). In a randomized trial of 224 patients with FLT3-mutated AML, in first relapse, lestaurtinib did not increase the response rate or prolong survival (26). Single-agent use with midostantrum, tandutinib and KW2449 in phase I/II trials were also not clinically effective (27-29). Combination therapy using FLT3 inhibitors with chemotherapy have also been conducted. Serve et al. reported a randomized trial of 201 newly diagnosed older patients with AML, using the addition of sorafenib to induction and consolidation therapy. Unfortunately, sorafenib did not improve outcomes and patients did worse in the sorafenib arm due to higher treatment-related mortality and lower CR rates (30). A recent phase II study of sorafenib in combination with 5-azacitadine in relapsed/refractory FLT3 ITD-mutant AML demonstrated a response rate of 46%, mostly consisting of CR or CR with incomplete count recovery (31). Sunitinib added to induction and consolidation chemotherapy in older patients with AML and FLT3-activating mutations showed some effectiveness, with CR rates of 53% (8/15) and 71% (5/7) for patients with FLT3 ITD and FLT3 TKD mutations, respectively. The 13 patients who achieved CR went on to be consolidated with high-dose cytarabine and 7/13 received sunitinib maintenance. The median overall survival in this study was 18.8 months (32). In a recent randomized, double-blind, placebo-controlled, phase II study by Röllig et al., the efficacy and tolerability of sorafenib versus placebo in addition to standard chemotherapy in patients with AML aged 60 years or younger was examined (33). A total of 267 patients were included in the primary analysis (placebo, n=133; sorafenib, n=134). With a median follow-up of 36 months, median event-free survival was 9 months in the placebo group versus 21 months in the sorafenib group, corresponding to a 3-year event-free survival of 22% versus 40% respectively.
Second-generation agents, promising to have better potency and fewer side-effects, include quizartinib and crenolanib, which are still undergoing clinical investigation. Drug resistance has become the major challenge in treating patients with a single FLT3 inhibitor. The point mutations identified which lead to resistance include N676, F691, and D835 within the kinase domain of FLT3 ITD (34). The novel FLT3 inhibitors, G-749 and ASP2215 (gilteritinib; active against both FLT3 ITD and D835 mutations), have recently been shown to provide sustained inhibition of FLT3 phosphorylation and increased ability to overcome drug resistance in pre-clinical trials but further studies are needed to determine if it will have clinical efficacy (35,36). Some drugs are discussed in detail below.
Midostaurin. Midostaurin (PKC-412) is a moderately potent inhibitor of FLT3–ITD and FLT3 TKD mutations and inhibits other kinases such as stem cell growth factor receptor (c-KIT), platelet-derived growth factor receptor B (PDGFRB), vascular endothelial growth factor receptor 2 (VEGFR2), and protein kinase C. A phase II study of midostaurin monotherapy for patients with relapsed and refractory FLT3-positive AML showed minimal clinical activity; only one patient achieved a partial remission (37). A follow-up phase IB study combined midostaurin with induction chemotherapy (7+3) without regard to FLT3 status and demonstrated that the combination was safe and well tolerated; complete remissions were seen in 92% of patients with FLT3 ITD compared with 74% of patients with wild-type FLT3 (38). On the basis of these data, the RATIFY study, an international randomized phase III study of midostaurin or placebo in combination with induction and consolidation chemotherapy, was designed. The outcomes, reported at the 2015 American Society of Hematology (ASH) Annual Meeting, showed improved 5-year overall survival in the midostaurin arm (51.4% versus 44.2%), regardless of whether patients were censored at the time of stem cell transplant, despite no difference in the rates of complete remission at 60 days (39). The superiority of midostaurin/chemotherapy over placebo/chemotherapy was consistent regardless of allelic burden (high vs. low), FLT3 ITD, and FLT3 TKD. Patients receiving midostaurin had an increased frequency of grade 3-4 desquamating rash. The overall survival benefit in combination with the favorable toxicity profile makes midostaurin in combination with induction and consolidation chemotherapy the new standard of care for patients with FLT3-mutated AML.
Quizartinib. More potent inhibitors of FLT3 ITD, and FLT3 TKD are in development. Quizartinib is a selective inhibitor of FLT3 ITD, but lacks activity against FLT3 TKD. The composite CR rate in phase II studies of quizartinib as a single agent in relapsed and refractory FLT3-mutated AML ranged between 44% and 54% (40-43). The median duration of response was between 11.3 and 12.7 weeks. Many patients who received quizartinib acquire mutations in the TKD of the FLT3 gene (D835 and F691) and resistance to ongoing treatment. Because of this, quizartinib may be best used as monotherapy ‘bridge’ to a potentially curative allogeneic bone marrow transplant. A randomized open-label phase III study of quizartinib versus chemotherapy (NCT02039726) with a primary endpoint of overall survival is currently accruing patients, and a randomized double-blind study of quizartinib or placebo in combination with induction and consolidation chemotherapy is in development (NCT02668653).
Gilteritinib. Gilteritinib (ASP-2215), also a potent inhibitor of FLT3, differs from quizartinib in its ability to inhibit both FLT3 ITD and FLT3 TKD mutations. Results of a phase I/II study among 165 patients receiving 80 mg or greater of gilteritinib were reported at the 2015 ASH Annual Meeting (36). Among patients with FLT3 ITD, the composite CR rate was 46%, and was 9% for those with FLT3 TKD. Responses were similar regardless of whether patients had received prior FLT3-directed therapy. The median duration of response was 15.9 weeks, similar to what is seen in clinical studies with quizartinib. A phase I study of ASP2215 in combination with induction and consolidation chemotherapy is ongoing (NCT02236013), and a randomized phase III study of ASP2215 versus salvage chemotherapy is accruing patients (NCT02421939).
Nucleophosmin 1 (NPM1) Mutations
The nucleolar protein NPM1 is involved in many cellular functions such as ribosome biogenesis, DNA repair, and regulation of apoptosis. Mutations result in aberrant localization of the protein to the cytosol; an N-terminal nucleolar localization signal is disrupted and an export signal created instead. Mutations in the NPM1 gene are among the most common genetic changes in AML (occurring in 25-35% of patients), especially CN-AML (present in 45-64% of cytogenetically normal patients) (16,44,45). In the absence of FLT3 ITD mutations, NPM1 mutations are associated with improved outcome for patients with CN-AML, even in those older than 60 years. Current ELN recommendations for diagnosis and treatment of AML class NPM1-mutated, FLT3-wild-type CN-AML as a favorable risk condition and discourage allogeneic HSCT in first CR (4). NPM1 mutations result in the aberrant expression of the NPM1 protein in the cytoplasm rather than the nucleus, stimulating myeloid proliferation and leukemia development (45-47). Clinically, the mutation is associated with monocytic morphology and in absence of FLT3 or FLT3 ITD predicts favorable overall survival. The reason for improved survival remains unclear, however, it NPM1 mutations have been associated with chemosensitivity to intensive chemotherapy in both young and old patients, which may account for improved outcome (48). NPM1 mutations are associated with other recurrent genetic abnormalities such as +8, DNA methyl-transferase 3A (DNMT3A) mutations, FLT3 ITD (40% of the time), FLT3 TKD (10-15%) and IDH mutations (25% of the time) (16,49,50).
DNA Methyltansferase 3A (DNMT3A) Mutations
DNMT3A methyltransferase 3A is an epigenetic regulator mediating de novo DNA methylation of cytosine residues. Recurrent DNMT3A mutations were first identified at residue R882 by candidate gene next-generation sequencing in 2010, accounting for about 50% of DNMT3A mutations; subsequent whole-genome and whole-exome sequencing approaches revealed additional mutations throughout the complete DNMT3A coding sequence (51). DNMT3A mutations are the most frequent recurrent gene mutations in AML after NPM1 and FLT3 mutations. This trio of FLT3, NPM1, and DNMT3A mutations was commonly found occurring together in the 200 AML genomes analyzed by the Cancer Genome Atlas project (15). Mutations in DNMT3A gene occurs in 18-22% of all AML cases and in about 34% of CN-AML (51). Missense mutations affecting arginine codon 882 (R882-DNMT3A) are more common than those affecting other codons (non-R882-DNMT3A) causing a defect in normal hematopoiesis and proper methylation (19,51). Recently, DNMT3A mutations were identified as pre-leukemic mutations, arising early in AML evolution and persisting in times of remission (52). The prognostic significance of DNMT3A mutations is therefore thought to be adverse. Initial studies showed unfavorable impact on outcome in CN-AML (49). However, these effects were age-related. Younger patients with non-R882-DNMT3A mutations had shorter disease-free and overall survival, whereas older patients with R882-DNMT3A mutations had shorter disease-free and overall survival after adjustment for other clinical and molecular prognosticators (49). A larger study involving more than 1700 AML cases found no significant impact of DNMT3A mutation on survival end-points (53). Recently, it was reported that patients with DNMT3A-mutated AML have an inferior survival when treated with standard-dose anthracycline induction therapy. Sehgal et al. concluded that this group should be considered for high-dose induction therapy (54). High-dose daunorubicin, compared to standard-dose daunorubicin, improved the rate of survival among patients with DNMT3A or NPM1 mutation or mixed lineage leukaemia (MLL) translocations but not among patients with wild-type DNMT3A, NPM1, and MLL (55).
IDH Mutations
Mutations of IDH1 and IDH2 gene are gain-of-function mutations which cause loss of the physiological enzyme function and create a novel ability of the enzymes to convert α-ketoglutarate into 2-hydroxyglutarate. Specifically recurrent mutations affecting the highly conserved arginine (R) residue at codon 132 (R132) of IDH1 and at codons R140 and R172 of IDH2 have been identified in 15-20% of all AML and 25% to 30% of patients with CN-AML (16,55,56). IDH mutations are oncogenic. They are found more frequently in older patients (50). IDH, the enzyme that converts isocitrate to alpha-ketoglutarate in the mitochondria (IDH2) or the cytoplasm (IDH1) as part of the citric acid cycle, is mutated in 15% and 10% of patients with de novo AML, respectively (57). The prevalence of IDH mutations increases with age (50). IDH mutations, in particular IDH1, are associated with lower disease-free and overall survival in CN-AML cases with NPM1 mutations and wild-type FLT3 (50,56). Orally available, selective, potent inhibitors of mutated IDH are currently being tested in phase I and II studies in AML with promising results (58). Mutant IDH enzymes acquire neomorphic activity and catalyze the conversion of alpha-ketoglutarate into beta-hydroxygutarate (2-HG). Increased intracellular 2-HG causes inhibition of ten–eleven translocation (TET) enzymes and subsequent arrest in myeloblast maturation (59-61). Inhibitors of mutant IDH1 and mutant IDH2 are currently in phase I clinical trials (NCT02381886, NCT01915498, and NCT02074839). Interim results of a phase I/II study of the IDH2 inhibitor AG-221 (Agios/Celgene), presented at the 2015 ASH Annual Meeting, demonstrated an overall response rate of 37% among 159 patients with relapsed/refractory AML with a composite CR of 27%. The duration of therapy response was 6.9 months (62). Similarly, a phase I study of the IDH1 inhibitor AG-120 demonstrated an overall response rate of 35%, with a composite CR rate of 33% (63). Accrual has started on a phase I study exploring the safety of combining AG-120 and AG-221 with both induction and consolidation chemotherapy and with 5-azacitidine (NCT02632708 and NCT0267792).
TET2 Mutations
TET2 mutations are found throughout the range of myeloid malignancies, including myelodysplastic syndrome and myeloproliferative neoplasms. TET2 mutations are detected in about 10% of patients with AML, but the prognostic relevance remains controversial (64,65). TET2 catalytic activity converts 5-methylcytosine to 5-hydroxymethyl cytosine in an α-ketoglutarate-dependent reaction. 5-hydroxymethyl cytosine is an intermediate to DNA demethylation and could serve as a new epigenetic mark associated with transcriptional regulation of promoter regions (64). Accordingly, TET2 mutant AML displays increased promoter methylation. In general, TET2 mutations are loss-of-function mutations. Overall, despite several studies, their prognostic significance remains unclear. Metzeler et al. reported TET2 mutations as being an adverse factor for CR and overall survival (65). However Gaidzik et al. did not find a prognostic effect of TET2 mutations (66).
Runt-Related Transcription Factor (RUNX1) Mutations
The RUNX1 gene is part of the t(8;21) fusion gene in core-binding factor (CBF) leukemia and is also affected by recurrent gene mutations in AML (67). Reported RUNX1 mutation frequencies vary between 5% and 90% (in AML associated with trisomy 21), probably owing to differing study populations. RUNX1 mutations in AML are associated with poor outcomes, which contrasts with the favorable prognostic effect of gene fusions involving RUNX1 (68,69). The differential prognostic impact of chromosomal and mutational lesions in RUNX1 emphasizes the importance of a complete assessment of genetic factors in AML pathogenesis. RUNX1 has been shown to be essential in normal hematopoiesis (67). Also known as AML1 protein or CBF subunit α-2 (CBFA2), RUNX1 is located at chromosome 21 and is frequently translocated with the RUNX1T1 (ETO/MTG8) gene located on chromosome 8q22, creating a fusion protein RUNX1–RUNX1T1 (AML–ETO) or t(8;21)(q22;q22) AML (68). In addition to chromosome translocations, RUNX1 mutations are commonly associated with trisomy 13, trisomy 21, absence of NPM1 and CN-AML in older patients (16). In general, studies have shown RUNX1 mutations are associated with resistance to standard induction therapy, leading to inferior overall survival for both younger and older patients (69).
CCAAT Enhancer Binding Protein α (CEBPA) Mutations
CEBPA is a transcription factor involved in lineage specification; it is crucial for the development of myeloid progenitors to differentiated neutrophils. Mutations in CEBPA are specific to AML, and are not reported in other cancers; however, overexpression of wild-type CEBPA is seen in acute lymphoblastic leukemia (ALL) with translocation t(4;19). CEBPA mutations are found in 6-10% of all AML and 15-19% of CN-AML, commonly in association with del(9q) (1,70). CEBPA is a critical transcription factor that controls gene expression during hematopoiesis (71). Importantly, only bi-allelic mutation, not single, CEBPA mutations predicted a higher CR and favorable overall survival, occurring in 4-5% of AML (72). AML with a single CEBPA mutation is associated with survival similar to that of AML with wild-type CEBPA (16,73).
Additional Sex Comb-like 1 (ASXL1) Mutations
ASXL1 is a transcriptional regulator which can either repress or activate transcription. ASXL1 mutations were first identified in 2009 by copy number analysis of epigenetic regulators using comparative genome hybridisation. They have since been found throughout myeloid malignancies, mainly chronic myelomonocytic leukaemia, myelodysplastic syndrome, and myeloproliferative neoplasms (74). ASXL1 mutations are five times more common in older (≥60 years) patients (16.2%) than those younger than 60 years (74); these mutations are associated with poor outcome in all studies reported to date (74). ASXL1 mutations are loss-of-function mutations that occur in 5-11% of all AML cases (74). The function of ASXL1 protein is not fully understood but it is suggested that it may be involved in epigenetic regulation (DNA and histone modifications) (19,66). Among older patients, ASXL1 mutations are associated with t(8;21), wild-type NPM1, absence of FLT3 ITD, mutated CEBPA, and overall inferior complete remission and overall survival (75,76).
MLL Mutations
The MLL gene at chromosome 11q23 encodes for a protein which has histone methyltransferase activity that coordinates chromatin modification as part of a regulatory complex (77). Translocations affecting the MLL gene lead to aggressive acute lymphoblastic and myeloid leukemia with poor prognosis. A duplication of the region between exon 5 and 11 or between exons 5 and 12, which include the DNA binding motifs of MLL, is inserted in frame into intron 4, and the histone methyltransferase activity of MLL is preserved. Partial tandem duplication in MLL was the first mutation identified to confer an adverse prognosis for AML patients with normal cytogenetics: most patients with this genotype relapsed within a year. In addition to translocations, partial in tandem duplications of the MLL gene have been demonstrated most often in adult de novo CN-AML and in trisomy 11 AML cases (78,79). In adult CN-AML, the frequency of MLL rearrangement is 11% with the presence of the MLL partial in tandem duplications associated with a worse prognosis (i.e. shorter duration of remission) when compared to CN-AML without such duplication (80).
Tumor Protein p53 (TP53) Mutations
TP53 is known as a prototypic tumor-suppressor gene because of its crucial role in cell cycle control. TP53 mutations are the commonest genetic changes implicated in human cancer (81). In AML, TP53 changes are rare and closely related to a complex karyotype, in which they are the strongest prognostic factor. Mutations and deletions of the tumor-suppressor gene TP53 are primarily associated in AML with complex karyotype and are rare in patients without chromosomal deletions (81). TP53 mutation is found in 8-14% of AML cases. In general, TP53 mutations confer a very adverse prognosis with documented chemoresistance (81).
c-KIT Mutations
The KIT tyrosine kinase receptor is a 145 kDa transmembrane protein critical to normal hematopoiesis (82). This mutation is rare in AML (<5%) however it is present approximately 22-29% of the time in CBF mutations (i.e. AML harboring t(8;21)(q22;q22) or inv(16)(p13.1q22) or corresponding respective fusion genes (RUNX1–RUNX1T1 and CBFB–myosin heavy chain 11 (MYH11). KIT mutations have been shown to confer higher relapse risk and lower overall survival (83). The KIT mutation in the codon D816 in particular has been associated with unfavorable disease-free and overall survival, particularly in patients with t(8;21)(q22;q22) (83). Prospective studies later confirmed that patients with CBF AML harboring KIT mutations have shorter overall survival than patients with wild type KIT for those with t(8;21)(q22;q22) but not for patients with inv(16)(p13.1q22) (84). Remarkably KIT can be targeted pharmacologically by using tyrosine kinase inhibitors, such as dasatinb (82). Preliminary results were presented recently at the American Society of Hematology Annual Meeting from a phase II trial that combined the KIT inhibitor, dasatinib with standard chemotherapy for newly diagnosed patients with CBF AML. After a median follow-up of 21 months, patients with KIT mutations who received dasatinib with standard chemotherapy showed similar outcomes to those with wild-type KIT (85).
A phase II study by Boissel et al. aimed to evaluate dasatinib as maintenance therapy in patients with CBF AML in first hematologic CR, but at higher risk of relapse due to molecular disease persistence or recurrence. A total of 26 patients aged 18-60 years old previously included in the CBF-2006 trial were eligible to receive dasatinib 140 mg daily if they had a poor initial molecular response (n=18) or molecular recurrence (n=8). The tolerance of dasatinib as maintenance therapy was satisfactory. The 2-year disease-free survival in this high-risk population of patients was 25.7%. All but one patient with molecular recurrence presented subsequent hematological relapse. Patients with slow initial molecular response had a similar disease-free survival when treated with dasatinib (40.2% at 2 years) and without any maintenance (50.0% at 2 years). The disappearance of KIT gene mutations at relapse suggests that clonal devolution may in part explain the absence of efficacy observed with single-agent dasatinib in these patients (86). More studies are needed to evaluate the long-term outcomes of KIT inhibitors in CBF AML.
Novel Targets and Drugs
B-Cell lymphoma 2 (BCL2) inhibitors. BCL2 overexpression has been implicated in the maintenance and survival of AML cells in vitro and is associated with resistance to chemotherapy and poor survival among patients with AML. The BCL2 inhibitor venetoclax (ABT-199) led to a CR/CR with incomplete blood count recovery in five of 32 patients in a phase II clinical study (87). Preclinical models suggest that the combination of venetoclax and hypomethylating agents are synergistic. On the basis of preclinical data, a phase I study of the combination of venetoclax and decitabine or 5-azacitidine was initiated. The overall response rate (34 patients) was 76%, with 71% of patients having a CR or CR with incomplete blood count recovery, without differences in response between patients who received decitabine or patients who received 5-azacitidine (88). In a study by Jacque et al., cancer cells were found to require glutamine in order to adapt to increased biosynthetic activity (89). The limiting step in intracellular glutamine catabolism involves its conversion to glutamate by glutaminase. Different glutaminase isoforms are encoded by the genes GLS1 and GLS2 in humans. Glutaminolysis inhibition activated mitochondrial apoptosis and synergistically sensitized leukemic cells to priming with the BCL2 inhibitor ABT-199. These findings show that targeting glutamine addiction via GLS1 inhibition offers a potential novel therapeutic strategy for AML.
Nuclear exporter inhibitors. The anti-leukemic efficacy of reversible inhibitors of the major nuclear export receptor, chromosome region maintenance 1 (CRM1, also termed XPO1) has brought much excitement (19). CRM1 is a major nuclear exporter protein which mediates the export and inactivation of several tumor suppressors such as p53, p73, forkhead box protein O1 (FOXO1), RB1 and p21 cyclin-dependent kinase inhibitor 1A (CDKN1A) among others (90). CRM1 has been shown to be up-regulated in a range of solid tumors and hematological malignancies, including AML (91,92). Preclinical studies indicate that treatment of AML cell lines, patient samples and AML xenografts with novel CRM1 inhibitors (Selinexor) induces strong anti-leukemic effects (93,94). Based on these studies, phase I/II clinical trials are currently ongoing to assess the safety, tolerability and activity of selinexor in AML patients (NCT02212561).
Immune therapies. Novel antibody therapies are revolutionary in the treatment of leukemia and currently under development in AML. Monoclonal antibodies being explored include CD33 (gemtuzumab ozogamicin) (95) and bispecific antibodies such as AMG 330 (anti-CD33 and CD3) (96). In a study by Amadori et al., single-agent gemtuzumab ozogamicin (GO) was compared with best supportive care (BSC) including hydroxyurea as first-line therapy in older patients with AML unsuitable for intensive chemotherapy (97). A total of 237 patients were randomly assigned (118 to GO and 119 to BSC). The median OS was 4.9 months in the GO group and 3.6 months in the BSC group; the 1-year OS rate was 24.3% with GO and 9.7% with BSC. The overall survival benefit with GO was consistent across most subgroups, and was especially apparent in patients with high CD33 expression status, in those with favorable/intermediate cytogenetic risk profile, and in women. Overall, CR plus CR with incomplete recovery of peripheral blood counts occurred in 30 out of 111 (27%) GO recipients. The rates of serious adverse events (AEs) were similar in the two groups, and no excess mortality was observed with GO.
Chimeric antigen receptor (CAR)-transduced T-cells (CARTs) are T-cells engineered to express a specific antigen receptor target designed against a specific cell-surface antigen. CD123 has been found to be expressed on the majority of AML blasts but also on normal hematopoietic cells. Preclinical data show that targeting CD123 via CARTs results in rejection of human AML and myeloablation in mouse models (98-100).
In a recent study by Davids et al., the immune checkpoint blockade established by targeting cytotoxic T-lymphocyte-associated protein 4 with ipilimumab was examined in patients after allogeneic hematopoietic stem cell transplantation. Their early-phase data showed that administration of ipilimumab was feasible in patients with recurrent hematological cancers after allogeneic HSCT, although immune-mediated toxic effects and GvHD occurred. Durable responses were observed in association with several histological subtypes of these cancers, including extramedullary acute myeloid leukemia (101).
Conclusion
AML is complex disease with a diverse genetic landscape. The field is rapidly expanding with increased understanding of the pathophysiology and potential new drug targets. Despite our improvements in targeted therapy, it has become apparent that single-drug options may be less likely to succeed over multiple-drug targets. Although allogeneic stem cell transplant has traditionally been considered to be the best strategy in this setting, the available data suggest that it may not be the most effective strategy for eradicating minimal residual disease. Novel agents such as molecularly targeted drugs (FLT3 or IDH inhibitors) or monoclonal antibody-based agents, including antibody–drug conjugates and bispecific antibodies, and, potentially, checkpoint inhibitors and chimeric antigen receptor T-cells, may improve therapeutic strategies for eradicating persistent minimal residual disease after cytotoxic regimens.
Conflicts of Interest
No Author has any conflict of interest to report in regard to this study.
References
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukaemia. N Engl J Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 2.Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, Kumar R Cavenagh J, Schuh AC, Candoni A, Récher C, Sandhu I, Bernal del Castillo T, Al-Ali HK, Martinelli G, Falantes J, Noppeney R, Stone RM, Minden MD, McIntyre H, Songer S, Lucy LM, Beach CL, Döhner H. International phase 3 study of azacitidine vs. conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126:291–299. doi: 10.1182/blood-2015-01-621664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer SC, Levine RL. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol. 2014;15:e382–394. doi: 10.1016/S1470-2045(14)70008-7. [DOI] [PubMed] [Google Scholar]
- 4.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD. European LeukemiaNet. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 5.Löwenberg B, Ossenkoppele GJ, van Putten W, Schouten HC, Graux C, Ferrant A, Sonneveld P, Maertens J, Jongen-Lavrencic M, von Lilienfeld-Toal M, Biemond BJ, Vellenga E, van Marwijk Kooy M, Verdonck LF, Beck J, Döhner H, Gratwohl A, Pabst T, Verhoef G. Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON); German AML Study Group (AMLSG); Swiss Group for Clinical Cancer Research (SAKK) Collaborative Group. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med. 2009;361:1235–1248. doi: 10.1056/NEJMoa0901409. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, Racevskis J, Dewald GW, Ketterling RP, Bennett JM, Rowe JM, Lazarus HM, Tallman MS. Anthracycline dose intensification in acute myeloid leukaemia. N Engl J Med. 2009;361:1249–1259. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukaemia: Which platforms are ready for “prime time”. Blood. 2014;124:3345–3355. doi: 10.1182/blood-2014-05-577593. [DOI] [PubMed] [Google Scholar]
- 8.Kohlmann A, Nadarajah N, Alpermann T, Grossmann V, Schindela S, Dicker F, Roller A, Kern W, Haferlach C, Schnittger S, Haferlach T. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukaemia patients with resistant disease. Leukaemia. 2014;28:129–137. doi: 10.1038/leu.2013.239. [DOI] [PubMed] [Google Scholar]
- 9.Passweg JR, Baldomero H, Bader P, Bonini C, Cesaro S, Dreger P, Duarte RF, Dufour C, Kuball J, Farge-Bancel D, Gennery A, Kröger N, Lanza F, Nagler A, Sureda A, Mohty M. Hematopoietic stem cell transplantation in Europe 2014: more than 40 000 transplants annually. Bone Marrow Transplant. 2016;51:786–792. doi: 10.1038/bmt.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornelissen JJ, Versluis J, Passweg JR, van Putten WL, Manz MG, Maertens J, Beverloo HB, Valk PJ, van Marwijk Kooy M, Wijermans PW, Schaafsma MR, Biemond BJ, Vekemans MC, Breems DA, Verdonck LF, Fey MF, Jongen-Lavrencic M, Janssen JJ, Huls G, Kuball J, Pabst T, Graux C, Schouten HC, Gratwohl A, Vellenga E, Ossenkoppele G, Löwenberg B, HOVON , SAKK Leukemia Groups SAKK Leukemia Groups: Comparative therapeutic value of post-remission approaches in patients with acute myeloid leukaemia aged 40-60 years. Leukaemia. 2015;29:1041–1050. doi: 10.1038/leu.2014.332. [DOI] [PubMed] [Google Scholar]
- 11.Versluis J, Hazenberg CL, Passweg JR, van Putten WL, Maertens J, Biemond BJ, Theobald M, Graux C, Kuball J, Schouten HC, Pabst T, Löwenberg B, Ossenkoppele G, Vellenga E, Cornelissen JJ, HOVON , SAKK Leukemia Groups Post-remission treatment with allogeneic stem cell transplantation in patients aged 60 years and older with acute myeloid leukaemia: a time-dependent analysis. Lancet Haematol. 2015;2:e427–436. doi: 10.1016/S2352-3026(15)00148-9. [DOI] [PubMed] [Google Scholar]
- 12.Cornelissen JJ, van Putten WL, Verdonck LF, Theobald M, Jacky E, Daenen SM, van Marwijk Kooy M, Wijermans P, Schouten H, Huijgens PC, van der Lelie H, Fey M, Ferrant A, Maertens J, Gratwohl A, Löwenberg B. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukaemia in young and middle-aged adults: benefits for whom? Blood. 2007;109:3658–3666. doi: 10.1182/blood-2006-06-025627. [DOI] [PubMed] [Google Scholar]
- 13.Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, Wadleigh M, DeAngelo DJ, Stone RM, Sakamaki H, Appelbaum FR, Döhner H, Antin JH, Soiffer RJ, Cutler C. Allogeneic stem cell transplantation for acute myeloid leukaemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301:2349–2361. doi: 10.1001/jama.2009.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passweg JR, Labopin M, Cornelissen J, Volin L, Socié G, Huynh A, Tabrizi R, Wu D, Craddock C, Schaap N, Kuball J, Chevallier P, Cahn JY, Blaise D, Ghavamzadeh A, Bilger K, Ciceri F, Schmid C, Giebel S, Nagler A, Mohty M. Acute Leukemia Working Party of the European Blood and Marrow Transplant Group (EBMT). Conditioning intensity in middle-aged patients with AML in first CR: no advantage for myeloablative regimens irrespective of the risk group-an observational analysis by the Acute Leukemia Working Party of the EBMT. Bone Marrow Transplant. 2015;50:1063–1068. doi: 10.1038/bmt.2015.121. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukaemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–486. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 17.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, Gundem G, Van Loo P, Martincorena I, Ganly P, Mudie L, McLaren S, O’Meara S, Raine K, Jones DR, Teague JW, Butler AP, Greaves MF, Ganser A, Döhner K, Schlenk RF, Döhner H, Campbell PJ. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maroc N, Rottapel R, Rosnet O, Marchetto S, Lavezzi C, Mannoni P, Birnbaum D, Dubreuil P. Biochemical characterization and analysis of the transforming potential of the FLT3/FLK2 receptor tyrosine kinase. Oncogene. 1993;8:909–918. [PubMed] [Google Scholar]
- 19.Saultz JN, Garzon R. Acute myeloid leukemia: a concise review. J Clin Med. 2016;5:33. doi: 10.3390/jcm5030033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukaemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- 21.Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, Groner S, Späth D, Krauter J. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–2392. doi: 10.1182/blood-2009-03-209999. [DOI] [PubMed] [Google Scholar]
- 22.Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, Linch DC. Medical Research Council Adult Leukaemia Working Party: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–2784. doi: 10.1182/blood-2007-08-109090. [DOI] [PubMed] [Google Scholar]
- 23.Pratcorona M, Brunet S, Nomdedéu J, Ribera JM, Tormo M, Duarte R, Escoda L, Guàrdia R, Queipo de Llano MP, Salamero O, Bargay J, Pedro C, Martí JM, Torrebadell M, Díaz-Beyá M, Camós M, Colomer D, Hoyos M, Sierra J, Esteve J; J, Grupo Cooperativo Para el Estudio y Tratamiento de las Leucemias Agudas Mieloblásticas Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood. 2013;121:2734–2738. doi: 10.1182/blood-2012-06-431122. [DOI] [PubMed] [Google Scholar]
- 24.Small D. Targeting FLT3 for the treatment of leukaemia. Semin Hematol. 2008;45:17–21. doi: 10.1053/j.seminhematol.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sudhindra A, Smith CC. FLT3 inhibitors in AML: Are we there yet? Curr Hematol Malig Rep. 2014;9:174–185. doi: 10.1007/s11899-014-0203-8. [DOI] [PubMed] [Google Scholar]
- 26.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, Erba H, Stuart RK, Baccarani M, Cripe LD, Tallman MS, Meloni G, Godley LA, Langston AA, Amadori S, Lewis ID, Nagler A, Stone R, Yee K, Advani A, Douer D, Wiktor-Jedrzejczak W, Juliusson G, Litzow MR, Petersdorf S, Sanz M, Kantarjian HM, Sato T, Tremmel L, Bensen-Kennedy DM, Small D, Smith BD. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3-mutant AML in first relapse. Blood. 2011;117:3294–3301. doi: 10.1182/blood-2010-08-301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 28.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 29.Pratz KW, Cortes J, Roboz GJ, Rao N, Arowojolu O, Stine A, Shiotsu Y, Shudo A, Akinaga S, Small D, Karp JE, Levis M. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood. 2009;113:3938–3946. doi: 10.1182/blood-2008-09-177030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, Brunnberg U, Schaich M, Ottmann O, Duyster J, Wandt H, Fischer T, Giagounidis A, Neubauer A, Reichle A, Aulitzky W, Noppeney R, Blau I, Kunzmann V, Stuhlmann R, Krämer A, Kreuzer KA, Brandts C, Steffen B, Thiede C, Müller-Tidow C, Ehninger G, Berdel WE. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31:3110–3118. doi: 10.1200/JCO.2012.46.4990. [DOI] [PubMed] [Google Scholar]
- 31.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, Pierce S, Daver N, Garcia-Manero G, Faderl S, Nazha A, Konopleva M, Borthakur G, Burger J, Kadia T, Dellasala S, Andreeff M, Cortes J, Kantarjian H, Levis M. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT3 internal tandem duplication mutation. Blood. 2013;121:4655–4662. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiedler W, Kayser S, Kebenko M, Janning M, Krauter J, Schittenhelm M, Götze K, Weber D, Göhring G, Teleanu V, Thol F, Heuser M, Döhner K, Ganser A, Döhner H, Schlenk RF. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169:694–700. doi: 10.1111/bjh.13353. [DOI] [PubMed] [Google Scholar]
- 33.Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, Baldus CD, Brandts CH, Kunzmann V, Einsele H, Krämer A, Schäfer-Eckart K, Neubauer A, Burchert A, Giagounidis A, Krause SW, Mackensen A, Aulitzky W, Herbst R, Hänel M, Kiani A, Frickhofen N, Kullmer J, Kaiser U, Link H, Geer T, Reichle A, Junghanß C, Repp R, Heits F, Dürk H, Hase J, Klut IM, Illmer T, Bornhäuser M, Schaich M, Parmentier S, Görner M, Thiede C, von Bonin M, Schetelig J, Kramer M, Berdel WE, Ehninger G; Study Alliance Leukaemia. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–1699. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 34.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HK, Kim HW, Lee IY, Lee J, Lee J, Jung DS, Lee SY, Park SH, Hwang H, Choi JS, Kim JH, Kim SW, Kim JK, Cools J, Koh JS, Song HJ. G-749, a novel FLT3 kinase inhibitor, can overcome drug resistance for the treatment of acute myeloid leukaemia. Blood. 2014;123:2209–2219. doi: 10.1182/blood-2013-04-493916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altman JK, Perl AE, Cortes JE, Levis MJ, Smith CC, Litzow MR, Baer MR, Claxton DF, Erba HP, Gill SC, Goldberg SL, Jurcic JG, Larson RA, Lui C, Ritchie EK, Sargent B, Schiller GJ, Spira AI, Strickland SA, Tibes R, Ustun C, Wang ES, Stuart RK, Baldus CD, Rollig C, Neubauer A, Martinelli G, Bahceci E. Antileukemic activity and tolerability of ASP2215 80 mg and greater in FLT3 mutation-positive subjects with relapsed or refractory acute myeloid leukaemia: results from a phase 1/2, open-label, dose-escalation/dose-response study. Blood. 2015;126:321. [Google Scholar]
- 37.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD. Patients with acute myeloid leukaemia and an activating mutation in FLT3 respond to a small molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 38.Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, Cortes J, Kantarjian HM, DeAngelo DJ, Huntsman-Labed A, Dutreix C, del Corral A, Giles F. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukaemia. Leukaemia. 2012;26:2061–2068. doi: 10.1038/leu.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stone RM, Mandrekar S, Sanford BL, Geyer S, Bloomfield CD, Döhner K, Thiede C, Marcucci G, Lo-Coco F, Klisovic RB, Wei A, Sierra J, Sanz MA, Brandwein JM, de Witte T, Niederwieser D, Appelbaum FR, Medeiros BC, Tallman MS, Krauter J, Schlenk RF, Ganser A, Serve H, Ehninger G, Amadori S, Larson RA, Döhner H. The multi-kinase inhibitor midostaurin prolongs survival compared with placebo in combination with daunorubicin/cytarabine induction, high-dose C consolidation, and as maintenance therapy in newly diagnosed acute myeloid leukemia patients age 18-60 with FLT3 mutations: An international prospective randomized placebo-controlled double-blind trial (CALGB 10603/RATIFY [Alliance]) Blood. 2015;126:6. [Google Scholar]
- 40.Levis MJ, Perl AE, Dombret H, Döhner H, Steffen B, Rousselot P, Martinelli G, Estey EH, Burnett AK, Gammon G, Trone D, Leo E, Cortes JE. Final results of a phase 2 open label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukaemia after second-line chemotherapy or hematopoietic stem cell transplantation. Blood. 2012;120:673. [Google Scholar]
- 41.Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, Gammon G, Trone D, Armstrong RC, James J, Levis M. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukaemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31:3681–3687. doi: 10.1200/JCO.2013.48.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortes JE, Perl AE, Dombret H, Kayser S, Steffen B, Rousselot P, Martinelli G, Estey EH, Burnett AK, Gammon G, Trone D, Leo E, Levis MJ. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥60 years of age with FLT3 ITD-positive or -negative relapsed/refractory acute myeloid leukemia. Blood. 2012;120:48. [Google Scholar]
- 43.Schiller GJ, Tallman MS, Goldberg SL, Perl AE, Marie JP, Martinelli G, Larson RA, Russell N, Trone D, Gammon G, Levis MJ, Cortes JE. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukaemia. J Clin Oncol. 2014;32:5s. [Google Scholar]
- 44.Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, Haferlach T, Hiddemann W, Falini B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukaemia with a normal karyotype. Blood. 2005;106:3733–3739. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 45.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukaemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): Biologic and clinical features. Blood. 2007;109:874–885. doi: 10.1182/blood-2006-07-012252. [DOI] [PubMed] [Google Scholar]
- 46.Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A, Bigerna B, Pasqualucci L, Mannucci R, Rosati R, Gorello P, Diverio D, Roti G, Tiacci E, Cazzaniga G, Biondi A, Schnittger S, Haferlach T, Hiddemann W, Martelli MF, Gu W, Mecucci C, Nicoletti I. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc + AML. Blood. 2006;107:4514–4523. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 47.Cheng K, Sportoletti P, Ito K, Clohessy JG, Teruya-Feldstein J, Kutok JL, Pandolfi PP. The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood. 2010;115:3341–3345. doi: 10.1182/blood-2009-03-208587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, Bullinger L, Fröhling S, Döhner H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 49.Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrózek K, Radmacher MD, Kohlschmidt J, Nicolet D, Whitman SP, Wu YZ, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Moore JO, Caligiuri MA, Larson RA, Bloomfield CD. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol. 2012;30:742–750. doi: 10.1200/JCO.2011.39.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, Späth D, Kayser S, Zucknick M, Götze K, Horst HA, Germing U, Döhner H, Döhner K. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukaemia and confer adverse prognosis in cytogenetically normal acute myeloid leukaemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 51.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, Swift GW, Reed JP, Alldredge PA, Wylie T, Walker J, Kalicki J, Watson MA, Heath S, Shannon WD, Varghese N, Nagarajan R, Westervelt P, Tomasson MH, Link DC, Graubert TA, DiPersio JF, Mardis ER, Wilson RK. DNMT3A mutations in acute myeloid leukaemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, McLeod JL, Doedens M, Medeiros JJ, Marke R, Kim HJ, Lee K, McPherson JD, Hudson TJ, HALT Pan-Leukemia Gene Panel Consortium , Brown AM, Yousif F, Trinh QM, Stein LD, Minden MD, Wang JC, Dick JE. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaidzik VI, Schlenk RF, Paschka P, Stölzle A, Späth D, Kuendgen A, von Lilienfeld-Toal M, Brugger W, Derigs HG, Kremers S, Greil R, Raghavachar A, Ringhoffer M, Salih HR, Wattad M, Kirchen HG, Runde V, Heil G, Petzer AL, Girschikofsky M, Heuser M, Kayser S, Goehring G, Teleanu MV, Schlegelberger B, Ganser A, Krauter J, Bullinger L, Döhner H, Döhner K. Clinical impact of DNMT3A mutations in younger adult patient with acute myeloid leukaemia: A comprehensive analysis of the AML study Group (AMLSG) Blood. 2013;121:4769–4777. doi: 10.1182/blood-2012-10-461624. [DOI] [PubMed] [Google Scholar]
- 54.Sehgal AR, Gimotty PA, Zhao J, Hsu JM, Daber R, Morrissette JD, Luger S, Loren AW, Carroll M. DNMT3A mutational status affects the results of dose-escalated induction therapy in acute myelogenous leukaemia. Clin Cancer Res. 2015;21:1614–1620. doi: 10.1158/1078-0432.CCR-14-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O, Levine RL. Prognostic relevance of integrated genetic profiling in acute myeloid leukaemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukaemia: A cancer and leukaemia group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, Gale RE. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011;118:409–412. doi: 10.1182/blood-2010-12-322479. [DOI] [PubMed] [Google Scholar]
- 58.Fathi AT, Wander SA, Faramand R, Emadi A. Biochemical, epigenetic, and metabolic approaches to target IDH mutations in acute myeloid leukaemia. Semin Hematol. 2015;52:165–171. doi: 10.1053/j.seminhematol.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 59.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukaemia associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stein EM, DiNardo C, Altman JK, Collins R, DeAngelo DJ, Kantarjian HM, Sekeres MA, Fathi AT, Flinn IW, Frankel AE, Levine RL, Medeiros BC, Patel MR, Pollyea D, Roboz GJ, Stone RM, Swords RT, Tallman MS, Yen K, Attar EC, Xu Q, Tosolini A, Mei JM, Thakurta A, Knight RD, De Botton S. Safety and efficacy of AG-221, a potent inhibitor of mutant IDH2 that promotes differentiation of myeloid cells in patients with advanced hematologic malignancies: results of a phase 1/2 trial. Blood. 2015;126:323. [Google Scholar]
- 63.DiNardo C, de Botton S, Pollyea DA, Stein EM, Fathi AT, Roboz GJ, Collins R, Swords RT, Flinn IW, Altman JK, Tallman MS, Kantarjian HM, Derti A, Goldwasser M, Prahl M, Wu B, Yen K, Agresta S, Stone RM. Molecular profiling and relationship with clinical response in patients with IDH1 mutation-positive hematologic malignancies receiving AG-120, a first-in-class potent inhibitor of mutant IDH1, in addition to data from the completed dose escalation portion of the phase 1 study. Blood. 2015;126:1306. [Google Scholar]
- 64.Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, Lee MC, Ko BS, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, Hsu SC, Chen YC, Chang YC, Kuo YY, Kuo KT, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–3810. doi: 10.1182/blood-2011-02-339747. [DOI] [PubMed] [Google Scholar]
- 65.Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, Curfman J, Holland KB, Schwind S, Whitman SP, Wu YZ, Blum W, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD. TET2 mutations improve the new European Leukemia Net risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaidzik VI, Paschka P, Späth D, Habdank M, Köhne CH, Germing U, von Lilienfeld-Toal M, Held G, Horst HA, Haase D, Bentz M, Götze K, Döhner H, Schlenk RF, Bullinger L, Döhner K. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30:1350–1357. doi: 10.1200/JCO.2011.39.2886. [DOI] [PubMed] [Google Scholar]
- 67.Meyers S, Downing JR, Hiebert SW. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: The runt homology domain is required for DNA binding and protein–protein interactions. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang JL, Hou HA, Chem CY, Liu CY, Chou WC, Tseng MH. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukaemia: Prognostic implication and interaction with other gene alternations. Blood. 2009;114:5352–5361. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
- 69.Mendler JH, Maharry K, Radmacher MD, Mrózek K, Becker H, Metzeler KH, Schwind S, Whitman SP, Khalife J, Kohlschmidt J, Nicolet D, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microRNA expression signatures. J Clin Oncol. 2012;30:3109–3118. doi: 10.1200/JCO.2011.40.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: Are we ready for a prognostically prioritized molecular classification. Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–628. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C, Weissmann S, Dicker F, Kohlmann A, Schindela S, Kern W, Haferlach T, Schnittger S. The role of different genetic subtypes of CEBPA-mutated AML. Leukemia. 2014;28:794–803. doi: 10.1038/leu.2013.273. [DOI] [PubMed] [Google Scholar]
- 73.Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, Nicolet D, Whitman SP, Wu YZ, Schwind S, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–6929. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alpermann T, Haferlach C, Eder C, Nadarajah N, Meggendorfer M, Kern W, Haferlach T, Schnittger S. AML with gain of chromosome 8 as the sole chromosomal abnormality (+8sole) is associated with a specific molecular mutation pattern including ASXL1 mutations in 46.8% of the patients. Leuk Res. 2015;39:265–272. doi: 10.1016/j.leukres.2014.11.026. [DOI] [PubMed] [Google Scholar]
- 76.Micol JB, Duployez N, Boissel N, Petit A, Geffroy S, Nibourel O, Lacombe C, Lapillonne H, Etancelin P, Figeac M, Renneville A, Castaigne S, Leverger G, Ifrah N, Dombret H, Preudhomme C, Abdel-Wahab O, Jourdan E. Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1-RUNX1T1 chromosomal translocations. Blood. 2014;124:1445–1449. doi: 10.1182/blood-2014-04-571018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ernest P, Wang J, Korsmeyer SJ. The role of MLL in hema-topoiesis and leukaemia. Curr Opin Hematol. 2002;9:282–287. doi: 10.1097/00062752-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 78.Caligiuri MA, Schichman SA, Strout MP, Mrózek K, Baer MR, Frankel SR, Barcos M, Herzig GP, Croce CM, Bloomfield CD. Molecular rearrangement of the ALL1 gene in acute myeloid leukemia without cytogenetic evidence of 11q23 chromosomal translocations. Cancer Res. 1994;54:370–373. [PubMed] [Google Scholar]
- 79.Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, Knuutila S, Mrózek K, Oberkircher AR, Marcucci G, de la Chapelle A, Elonen E, Block AW, Rao PN, Herzig GP, Powell BL, Ruutu T, Schiffer CA, Bloomfield CD. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res. 1998;58:55–59. [PubMed] [Google Scholar]
- 80.Döhner K, Tobis K, Ulrich R, Fröhling S, Benner A, Schlenk RF, Döhner H. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol. 2002;20:3254–3261. doi: 10.1200/JCO.2002.09.088. [DOI] [PubMed] [Google Scholar]
- 81.Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22:1539–1541. doi: 10.1038/leu.2008.143. [DOI] [PubMed] [Google Scholar]
- 82.Sattler M, Salgia R. Targeting c-KIT mutations: Basic science to novel therapies. Leuk Res. 2004;28:11–20. doi: 10.1016/j.leukres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 83.Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA, Vukosavljevic T, Perrotti D, Vardiman JW, Carroll AJ, Kolitz JE, Larson RA, Bloomfield CD, Cancer and Leukemia Group B Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24:3904–3911. doi: 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
- 84.Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, Raffoux E, Leblanc T, Thomas X, Hermine O, Quesnel B, Baruchel A, Leverger G, Dombret H, Preudhomme C, Acute Leukemia French Association (ALFA); Leucémies Aiguës Myéloblastiques de l’Enfant (LAME) Cooperative Groups Incidence and prognostic impact of c-KIT, FLT3, and RAS gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia 2006. 2006;20:965–970. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 85.Marcucci G, Geyer S, Zhao W, Caroll AJ, Bucci D, Uy GL, Blum W, Pardee T, Wetzler M, Stock W, Kolitz JE, Eisfeld AK, Bloomfield CD, Stone RM, Larson RA. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukaemia (AML): Results from CLGB 10801 (Alliance) Blood. 2014;124:8. [Google Scholar]
- 86.Boissel N, Renneville A, Leguay T, Lefebvre PC, Recher C, Lecerf T, Delabesse E, Berthon C, Blanchet O, Prebet T, Pautas C, Chevallier P, Leprêtre S, Girault S, Bonmati C, Guièze R, Himberlin C, Randriamalala E, Preudhomme C, Jourdan E, Dombret H, Ifrah N. Dasatinib in high-risk core binding factor acute myeloid leukemia in first complete remission: a French Acute Myeloid Leukemia Intergroup trial. Haematologica. 2015;100:780–785. doi: 10.3324/haematol.2014.114884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Konopleva M, Pollyea DA, Potluri J, Chyla BJ, Busman T, McKeegan E, Salem A, Zhu M, Ricker JL, Blum W, DiNardo CD, Dunbar M, Kirby R, Falotico N, Leverson JD, Humerickhouse RA, Mabry M, Stone RM, Kantarjian HM, Letai AG. A phase 2 study of ABT-199 (GDC-0199) in patients with acute myelogenous leukaemia. Blood. 2014;124:118. [Google Scholar]
- 88.DiNardo C, Pollyea D, Pratz K, Thirman MJ, Letai A, Frattini M, Jonas B, Leverson J, Zhu M, Dunbar M, Falotico N, Kirby R, Agarwal S, Mabry M, Potluri J, Humerickhouse RA, Kantarjian HM, Konopleva M. A Phase 1b Study of venetoclax (ABT-199/GDC-0199) in combination with decitabine or azacitidine in treatment-naive patients with acute myelogenous leukemia who are ≥ to 65 years and not eligible for standard induction therapy. Blood. 2015;126:327. [Google Scholar]
- 89.Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, Poulain L, Hospital MA, Sujobert P, Joseph L, Chapuis N, Lacombe C, Moura IC, Demo S, Sarry JE, Recher C, Mayeux P, Tamburini J, Bouscary D. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126:1346–1356. doi: 10.1182/blood-2015-01-621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 91.Kojima K, Kornblau SM, Ruvolo V, Dilip A, Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N, Qiu YH, Burks JK, Kantarjian H, Shacham S, Kauffman M, Andreeff M. Prognostic impact and targeting of CRM1 in acute myeloid leukaemia. Blood. 2013;121:4166–4174. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Turner JG, Sullivan DM. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem. 2008;15:2648–2655. doi: 10.2174/092986708786242859. [DOI] [PubMed] [Google Scholar]
- 93.Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukaemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukaemia. 2013;27:66–74. doi: 10.1038/leu.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, de Revel T, Gastaud L, de Gunzburg N, Contentin N, Henry E, Marolleau JP, Aljijakli A, Rousselot P, Fenaux P, Preudhomme C, Chevret S, Dombret H, Acute Leukemia French Association Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet. 2012;379:1508–1516. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 96.Gasiorowski RE, Clark GJ, Bradstock K, Hart DNJ. Antibody therapy for acute myeloid leukaemia. Br J Haematol. 2014;164:481–495. doi: 10.1111/bjh.12691. [DOI] [PubMed] [Google Scholar]
- 97.Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, Annino L, Venditti A, Voso MT, Mazzone C, Magro D, De Fabritiis P, Muus P, Alimena G, Mancini M, Hagemeijer A, Paoloni F, Vignetti M, Fazi P, Meert L, Ramadan SM, Willemze R, de Witte T, Baron F. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: Results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34:972–979. doi: 10.1200/JCO.2015.64.0060. [DOI] [PubMed] [Google Scholar]
- 98.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, Danet-Desnoyers G, Scholler J, Grupp SA, June CH, Kalos M. Preclinical targeting of human acute myeloid leukaemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, Hoffman L, Aguilar B, Chang WC, Bretzlaff W, Chang B, Jonnalagadda M, Starr R, Ostberg JR, Jensen MC, Bhatia R, Forman SJ. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F, Galimberti S, Lopez AF, Biondi A, Bonnet D, Biagi E. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 101.Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, Lukez AP, Avigan D, Chen YB, McSweeney P, LeBoeuf NR, Rooney MS, Bowden M, Zhou CW, Granter SR, Hornick JL, Rodig SJ, Hirakawa M, Severgnini M, Hodi FS, Wu CJ, Ho VT, Cutler C, Koreth J, Alyea EP, Antin JH, Armand P, Streicher H, Ball ED, Ritz J, Bashey A, Soiffer RJ, Leukemia and Lymphoma Society Blood Cancer Research Partnership Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375:143–153. doi: 10.1056/NEJMoa1601202. [DOI] [PMC free article] [PubMed] [Google Scholar]