

Abstract
Corosolic acid (CA), a triterpenoid compound isolated from Lagerstroemia speciosa L. (Banaba) leaves, exerts anti-inflammatory effects by regulating phosphorylation of interleukin receptor- associated kinase (IRAK)-2 via the NF-κB cascade. However, the protective effect of CA against endotoxic shock has not been reported. LPS (200 ng/mL, 30 min) induced phosphorylation of IRAK-1 and treatment with CA (10 μM) significantly attenuated this effect. In addition, CA also reduced protein levels of NLRP3 and ASC which are the main components of the inflammasome in BMDMs. LPS-induced inflammasome assembly through activation of IRAK-1 was down-regulated by CA challenge. Treatment with Bay11-7082, an inhibitor of IκB-α, had no effect on CA-mediated inhibition of IRAK-1 activation, indicating that CA-mediated attenuation of IRAK-1 phosphorylation was independent of NF-κB signaling. These results demonstrate that CA ameliorates acute inflammation in mouse BMDMs and CA may be useful as a pharmacological agent to prevent acute inflammation. [BMB Reports 2016; 49(5): 276-281]
Keywords: Acute inflammation, Corosolic acid, Inflammasome, IRAK-1, Macrophages
INTRODUCTION
Acute inflammation is the body’s local response to any infection or injury (1) and is the first line of defense against infection or injury and involves the release of immune mediators and vasodilation in the area of infection (2). The process of acute inflammation occurs within a short duration in response to infection or injury and after the immune response is evoked, it is generally restored (3, 4). Clinically, endotoxin shock (e.g. sepsis or septic shock) occurs when acute inflammation persists for a prolonged period.
Toll-like receptors (TLRs) are a major recognition and signaling component of the mammalian host defense that activate nuclear factor kappa B (NF-κB), mitogen-activated protein (MAP) kinases, and interferon (IFN) response factor proteins (5, 6). Nucleotide-binding oligomerization domain-like receptors (NLRs) are located in the cytosol and are stimulated by microbial inducers like bacterial toxins (7, 8). The inflammasome complex assembly stimulates secretion of pro-inflammatory cytokines, such as interleukin (IL)-1β and 18 through activation and cleavage of caspase-1 (9). In a recent study, two different mechanisms to activate NLRP3 inflammasome assembly have been identified. One is the early phase of inflammasome induced by TLR signaling through the TLR-signaling molecule IL-1 receptor-associated kinase (IRAK-1), a serine/threonine kinase. And another pathway is the late phase of acute inflammasome activation regulated by IRAK-2 and responses to activation of the NLRP3 signaling (10).
Mouse IRAK family members IRAK-1, 2, 3, and 4 are expressed in the liver, kidneys, and testis, where they play key roles in multiple signaling pathways involved in TLR signaling and pro-inflammatory cytokine secretion (11, 12). IRAK proteins contain a proST domain, an N-terminal death domain (DD), a central conserved kinase domain, and a C-terminal domain (13). IRAK-1 is phosphorylated by TLR4 signaling activated by the MyD88-IRAK-4 axis and functions in IL-1β signaling by recruiting the IκB kinase complex (14). Phosphorylated IRAK-1 combines with tumor necrosis factor (TNF) receptorassociated factor 6 (TRAF6) to induce stimulation of NF-κB, c-Jun N-terminal kinase (JNK), and signal transducer and activator of transcription 3 (ATF3) (15, 16).
Corosolic acid (CA), a natural pentacyclic triterpenoid acid isolated from Lagerstroemia speciosa L. (Banaba), possesses antiatherosclerotic, antihyperlipidemic, antioxidant, anti-inflammatory, antidiabetic, antifungal, antiviral, and antineoplastic activities (17-24). The effects of CA are thought to be mediated by peroxisome proliferator activator receptor (PPAR), mitogenactivated protein kinase, NF-κB, and other signal transduction factors (25). However, the precise mechanisms underlying the effects of CA against acute inflammation remain unclear, hampering the development of effective drugs based on CA for clinical use. In the present study, CA was found to ameliorate acute inflammation by suppressing phosphorylation and transcription of IRAK-1 in mouse bone marrow-derived macrophages (BMDM).
RESULTS
CA confers resistance to proinflammatory toxic shock
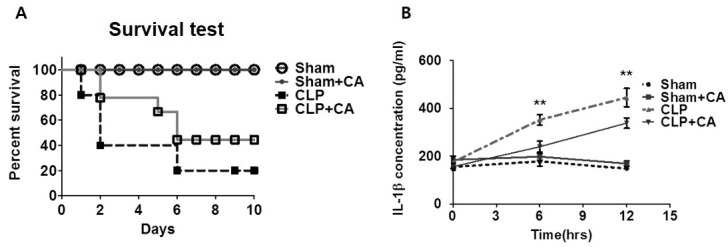
Macrophages play a pivotal role in the innate immune response to pathogen challenge. Because CA inhibits NF-kB signaling, we examined the hypothesis that CA might have a protective effect against septic shock by using an in vivo model system to evaluate responses to endogenous pathogen engagement. Survival was monitored for 10 days after cecal ligation and puncture (CLP) surgery was performed. Survival curves showed that the survival rate of the CLP group was significantly decreased in comparison with that of the sham and sham+CA mice groups (controls); however, the survival rates of the two control groups were not significantly different. Strikingly, on day 10, mortality was decreased by 50% in the CLP+CA mice group relative to the group that received no treatment in addition to CLP, indicating the potential of CA to prevent CLP-induced sepsis (Fig. 1A). After CLP surgery, survival was correlated with serum levels of pro-inflammatory cytokine IL-1β (Fig. 1B). After CA administration, serum IL-1β levels were significantly reduced in comparison with those of the group that received no treatment in addition to CLP. Taken together, these results suggest that CA reduces susceptibility to endotoxin shock in an in vivo model of sepsis.
Fig. 1. Effect of CA on mice subjected to cecal ligation and puncture (CLP) surgery and inflammasome assembly. (A) Survival of mice after CLP with or without CA (2 μg/kg). Mortality of Sham (n = 14), Sham+CA (n = 14), CLP (n = 14), and CLP+CA mice (n = 15) was monitored for 10 days after CLP surgery. (B) IL-1β secretion in vivo. Serum samples were collected 0, 6, and 12 h after surgery. The IL-1β concentration was measured by ELISA. The survival rate was estimated by the Kaplan-Meier method and compared by using the log-rank test. **P < 0.01. CLP versus CLP+CA, Sham, sham laparotomy; CLP, one puncture in the cecum; CLP+CA, one puncture in the cecum and CA injection.
CA regulates expression and phosphorylation of IRAK-1
LPS-induced inflammatory responses stimulate proinflammatory cytokine secretion by increasing IRAK activity through phosphorylation (26). Therefore, to measure the effect of CA on IL-1β secretion, enzyme-linked immunosorbent assays (ELISA) were performed to measure pro-inflammatory cytokine secretion from BMDMs. As shown in Fig. 2A, IL-1β secretion was markedly increased after LPS treatment; however, LPS-stimulated IL-1β expression was significantly decreased after CA treatment. Because infection-induced IL-1β secretion in macrophages requires activation of the NLRP3-ASC inflammasome complex, we performed immunocytochemistry to localize NLRP3 and ASC. While the number of inflammasome complex-positive cells in the LPS group was increased, the population of inflammasome complex-positive cells in the CA and LPS+CA groups was low (Fig. 2B), which suggested a weak inflammatory response to CA at the protein and mRNA levels. These results demonstrated that LPS treatment for a short duration induced inflammasome complex formation, which was inhibited by CA (Fig. 2B). These data demonstrate that CA ameliorated acute inflammation by inhibiting inflammasome complex formation.
Fig. 2. CA regulates inflammasome assembly and IRAK-1 phosphorylation in BMDMs. BMDMs were isolated from mice and exposed to LPS (200 ng/mL) and/or CA (6 μM) for 30 min. (A) IL-1β secretion in vitro. BMDMs were treated with LPS and/or CA for 6 h and supernatants were collected. IL-1β secretion was measured by ELISA. (B) CA prevents inflammasome assembly. Immunocytochemistry for the inflammasome complex. BMDMs were treated with LPS and/or CA for 30 min, after which the treated cells were stained with fluorescence-labeled antibodies. Fluorescence was detected using confocal laser microscopy and LSM 3 EXCITER software. qPCR was used to measure mRNA levels of NLRP3 (C), IRAK1 (D), IRAK2 (E), and IRAK4 (F). (G) Protein samples were assessed by immunoblotting with the indicated antibodies. Graphs illustrate the mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.01 versus the relevant control group.
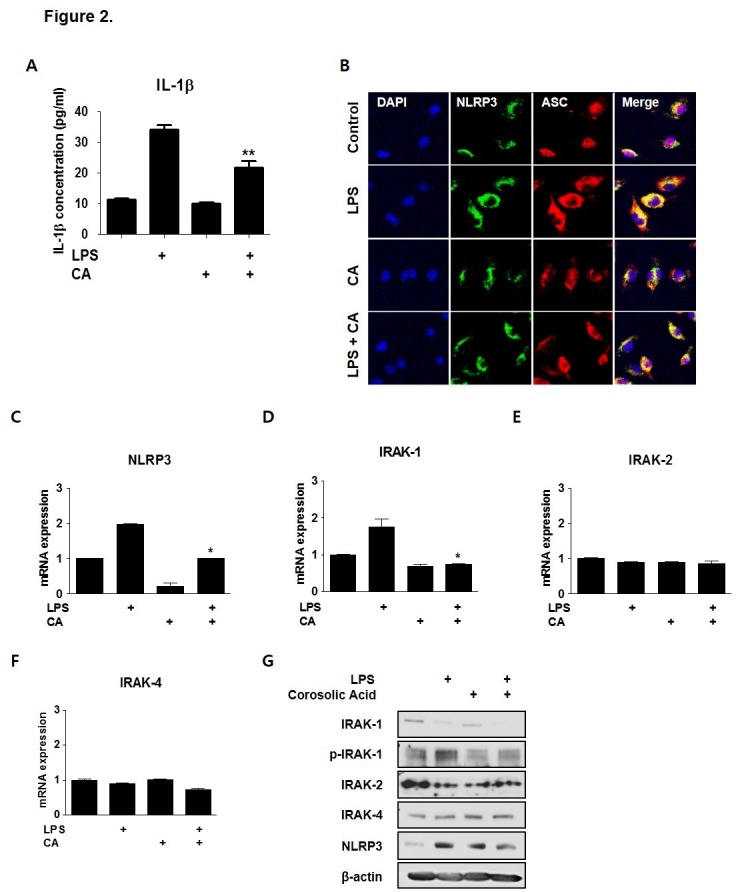
To investigate the involvement of molecular targets of CA in LPS-induced inflammation, after BMDMs were treated with LPS and CA for 30 min to induce acute inflammation, which expression levels of transcripts encoding TLR4 signaling proteins, including IRAK-1, IRAK-2, and IRAK-4 were measured by qPCR. The NLRP3 and IRAK-1 expression levels in the LPS-treated group were increased 2-fold in comparison with those in the control group. CA markedly decreased LPS-induced up-regulation of NLRP3 and IRAK-1 by approximately 40% (Fig. 2C and D). In particular, IRAK1 expression was increased rapidly following LPS treatment. IRAK-2 and IRAK-4 expression levels were unaffected after the 30-min LPS treatment (Fig. 2E and F). These data indicated that CA may be involved in transcriptional regulation of IRAK-1. Next, to assess whether CA modulates IRAK phosphorylation, IRAK-1 phosphorylation level was detected by immunoblotting. Interestingly, LPS drastically reduced LPS-mediated IRAK-1 phosphorylation, but this effect was attenuated by CA. Based on this data, we inferred that CA regulates IRAK-1 phosphorylation. The level of NLRP3 protein, which was elevated by LPS, decreased after treatment with CA during acute inflammation (Fig. 2G). This is the first study to demonstrate CA-induced inhibition of IRAK-1 phosphorylation in acute inflammation.
Effect of CA on LPS-mediated TLR4 signaling
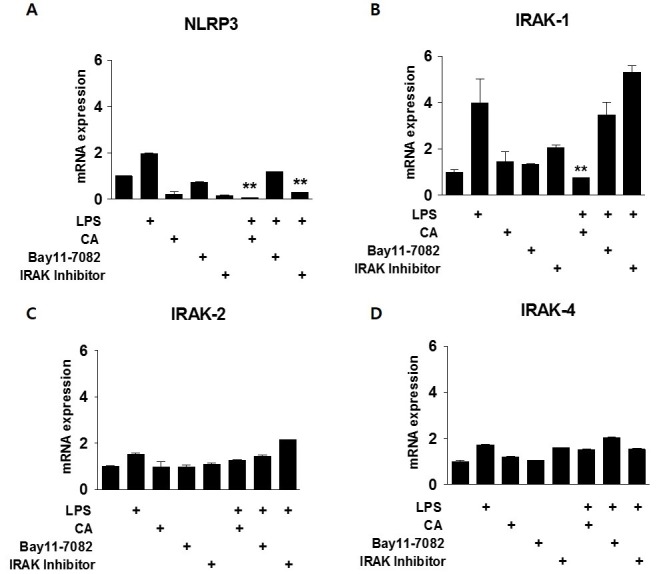
To further address the early-phase anti-inflammatory effects of CA, we investigated the dependence of the anti-inflammatory effect of CA on NF-κB signaling. Mouse BMDMs were incubated with the indicated concentrations of Bay11-7082 (an inhibitor of IκB-α) and IRAK inhibitor as an inhibitor of both IRAK-1and IRAK-4, followed by stimulation with LPS and/or CA for 30 min to induce inflammation (Fig. 3). LPS treatment increased NLRP3 expression by more than 2-fold in comparison with that of the control cells. The effect of LPS on NLRP3 expression was attenuated by treatment with CA or the IRAK inhibitor, but not by Bay11-7082 treatment (Fig. 3A). CA attenuated LPS-induced IRAK-1 expression by approximately 75%, but Bay11-7082 and the IRAK inhibitor had no effect, suggesting that CA-induced inhibition of IRAK-1 expression may independent of NF-κB signaling (Fig. 3B). IRAK-2 and -4 expression levels were not affected by exposure to LPS or CA (Fig. 3C and D).
Fig. 3. Relationship between CA and NF-kB signaling in acute inflammation. BMDMs were isolated from mice and exposed to LPS (200 ng/mL) and/or CA (6 μM), Bay11-7082 (10 μM), or an IRAK inhibitor (2.5 μM) for 30 min. qPCR was used to measure mRNA levels of NLRP3 (A), IRAK1 (B), IRAK2 (C), and IRAK4 (D). *P < 0.05, **P < 0.01 versus the relevant control group.
Anti-inflammatory effect of CA on acute inflammation
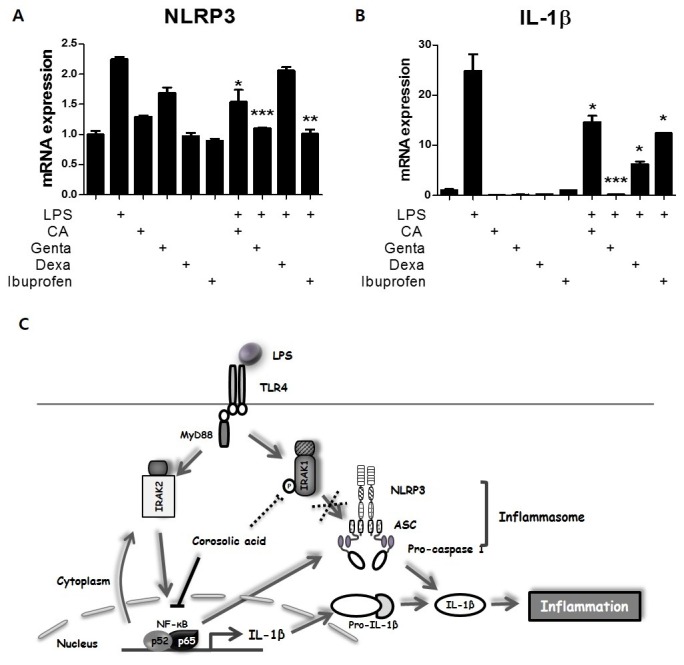
In order to further explore the anti-inflammatory effect of CA, mouse BMDMs were treated for 30 min with gentamicin (an antibiotic), ibuprofen (a nonsteroidal anti-inflammatory drug), or dexamethasone (a steroidal anti-inflammatory agent). LPS-induced expression of NLRP3, a target gene of NF-κB, was 2-fold greater than that of the control cells; however, LPS-induced NLRP3 expression was decreased by approximately 30% after CA treatment and by approximately 40% after treatment with gentamicin and ibuprofen. Dexamethasone treatment did not affect NLRP3 expression (Fig. 4A). LPS up-regulated expression of IL-1β, another NF-κB target, by 25-fold; however, this effect was decreased by approximately 40% following treatment with CA and ibuprofen, by 50% following treatment with dexamethasone, and by more than 90% following treatment with gentamicin (Fig. 4B). These findings demonstrate the therapeutic potential of CA as an anti-inflammatory drug.
Fig. 4. Effect of CA on LPS-mediated inflammation. (A-B) Effect of CA as an anti-inflammatory drug. BMDMs were isolated from mice and exposed to LPS (200 ng/mL) and/or gentamicin (10 μM), dexamethasone (10 μM), ibuprofen (10 μM), or CA (6 μM) for 30 min. qPCR was used to measure mRNA levels of NLRP3 and IL-1β. (C) Schematic summary of the role of CA in regulating early-phage inflammation. Graphs illustrate the mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus the relevant control group.
DISCUSSION
In the present study, we describe the mechanism underlying NF-κB-independent regulation of acute inflammation by CA. We investigated new targets of CA in the context of LPS-induced acute inflammation. Although the anti-inflammatory effect of CA has been reported (17), the cellular mechanism underlying this effect has not been fully elucidated. Our results elucidated the mechanism underlying CA-mediated regulation of acute inflammation in vitro and in vivo (Fig. 4C).
LPS stimulates the production of cytokines and other inflammatory mediators by inflammatory cells, and is generally regarded as a key initiating factor in the pathogenesis of septic shock. Studies have demonstrated that LPS induces cellular responses by activating signaling molecules belonging to the TLR family (27). Here, we treated BMDMs with LPS for 30 min to induce acute inflammation. Similar to the results of a previous study (20), the inflammatory cytokine expression of the CA-treated group was significantly attenuated in comparison with that of the group treated with LPS alone. In the priming stimulation to induce NLRP3 inflammasome activation, TLR signaling connects to NLRP3 sequentially bypassing NF-κB signaling (10, 28). LPS-dependent TLR activation is directly implicated in acute NLRP3 inflammasome activation through the MyD88-IRAK4-IRAK1 signaling network (29). The early priming inflammasome assembly differs from the late primingmediated signaling. Therefore, IRAK-1 may play a pivotal role in acute inflammation appears to control inflammasome complex assembly and activation (30). We also showed that LPS-induced acute inflammation increased expression of IRAK-1, but not IRAK-2. Although the total protein level of IRAK-1 was decreased, LPS increased IRAK-1 phosphorylation, which was attenuated by CA treatment. Furthermore, immunocytochemical staining of the inflammasome complex assembly revealed that LPS-induced complex formation was inhibited by CA treatment in the early period of inflammation. While a previous report showed that IRAK-1 phosphorylation stimulated inflammatory cytokine release by inducing inflammasome complex assembly (10), our results suggested that CA abolished inflammasome complex formation through inhibition of LPS-induced IRAK-1 phosphorylation. The precise mechanism underlying the regulation of inflammasome complex formation by IRAK-1 phosphorylation remains to be elucidated.
Based on previous studies that demonstrated different regulatory scenarios for chronic and acute inflammation (30), we sought to determine whether the anti-inflammatory effects of CA were mediated via an NF-κB-dependent pathway. In a chronic inflammatory condition, exposure of LPS-pretreated BMDMs to CA down-regulated expression of NF-κB target genes NLRP3 and IL-1β. Similar results were observed when cells were treated with LPS and Bay11-7082, an NF-κB inhibitor, indicating that the effects of CA were mediated by NF-κB signaling. Similar results were observed upon treatment with an inhibitor of IRAK, which is an upstream signaling molecule of LPS-induced activation of TLR4, which ultimately activates NF-κB. According to the available literature, acute inflammation occurs independently of NF-κB. However, based on our findings, specifically data on IRAK-2 and NLRP3 expression after treatment with Bay11-7082 and IRAK inhibitors in cells treated with LPS for 30 min, showed that acute inflammatory reactions are not completely NF-κB-independent. Rather, NF-κB has a role in the regulation of target gene expression during acute inflammation. Importantly, IRAK-1 expression did not decrease after treatment with Bay11-7082 or an IRAK inhibitor, implicating the role of another transcription factor in LPS-induced acute inflammation. CA inhibited this unknown transcription factor, this regulating IRAK-1 gene expression. In TLR signaling, IRAK-1 is phosphorylated by IRAK-4 through the MyD88 pathway (31). Here, we did not clearly address how the phosphorylation of IRAK-1 changes without IRAK-4 being affected. And we need to investigate the process of IRAK-1 phosphorylation modulation by CA as a further study.
Because CA regulated IRAK-1 activity in an acute inflammation condition known to be NF-κB-independent, we hypothesized inhibitory effects of CA on endotoxin shock response or septic shock. Indeed, the administration of CA had a significant protective effect against CLP-induced septic animal model. And also IL-1β secretion levels at 6 and 12 h after CLP surgery were reduced in CLP plus CA group compared to CLP group. These findings suggest that CA reduces mortality in polymicrobial sepsis through inhibition of IRAK-1 phosphorylation.
In conclusion, our study indicates that the CA plays a role in inhibitory effect on an acute inflammation. Furthermore, we identified a novel pathway through which CA regulates IRAK-1 phosphorylation via an NF-κB-independent pathway. These results propose the possibility of using CA as a medication for acute inflammatory diseases, such as sepsis and endotoxin shock.
MATERIALS AND METHODS
Chemicals and reagents
Fetal bovine serum (FBS) and Dulbecco’s modified eagle’s medium (DMEM) media for cell culture were purchased from Hyclone. LPS (lipopolysaccharide), Corosolic acid (PubChem CID : 6918774), Bay11-7082 (PubChem CID : 24891842), and Interleukin-1 Receptor-Associated-kinase 1/4 inhibitor (PubChem CID : 11983295) were purchased from Sigma-Aldrich and dissolved in the recommended solvents. Antibodies against IRAK-1 and IRAK-4 were purchased from Cell Signaling Technology; anti-IRAK-2 and ASC were purchased from Novus Biologicals, Inc.; phospho-IRAK-1 was purchased from Bioworld Technology; β-actin was purchased from Santa Cruz Biotechnology.
Animal studies
8-week-old male C57BL/6 mice (Jung-Ang Experimental Animals, Seoul, Republic of Korea) were used in the experiment. All animal experiments were performed in accordance with the rules and regulations of the Institutional Animal Use and Care Committee (IAUCC), Keimyung University School of Medicine (KM-2012-24R).
Isolation and culture of primary mouse bone marrow-derived macrophages
Bone marrow-derived macrophages (BMDM) were isolated from C57BL/6 as previously described (32). Briefly, the C57BL/6 mice were sacrificed by cervical dislocation and femurs were dissected free of adherent tissue. Mouse bone marrow cells were flushed from femurs and tibias. Separated bone marrow cells were cultured in DMEM media, containing 10% FBS, 1% penicillin/streptomycin, and 20% L929 cells conditioning media for 7 days to differentiate into BMDM.
Real-time PCR
Total RNA was isolated from mouse primary hepatocytes and livers using the Trizol method (Invitrogen). cDNA was synthesized using a SuperScript III First-strand cDNA synthesis kit (Invitrogen) and used for qPCR with Roche LightcyclerⓇ Real-Time PCR systems (Roche Applied Science, Indianapolis, IN, USA). All data were normalized to ribosomal L32 expression. The following primer sets for mouse were used: mouse NF-κB, forward, 5’-GTGCAGTGTCTTGGCTTTCT-3’; reverse, 5’-CGGAGGGACAGCAGTAACAAC-3’; mouse IL-1β, forward, 5’-ATG AGAGCATCCAGCTTCAA-3’, reverse, 5’-TGAAGGAAAAGA AGGTGCTC-3’; mouse NLRP3, forward, 5’-ATTACCCGCCC GAGAAAGG-3’, reverse, 5’-CATGAGTGTGGCTAGATCCA AG-3’; mouse ASC, forward, 5’-ACTATCTGGAGTCGTATGG CTT-3’, reverse, 5’-CTGGTCCACAAAGT GTCCTGT-3’; mouse IRAK-1, forward, 5’-CCTTCAGAGAGGCTAGCTGTACC-3’, reverse, 5’-ACTTTGACCTCTGAGTCTGAGGG-3’, mouse IRAK-2, forward, 5’-AGC ACAGCCATCCACCAG-3’, reverse, 5’-GATCTCAATTTTCCATGAAGTCT-3’; IRAK-4, forward, 5’-CATACGCAACCTTAATGTGGGG-3’, reverse, 5’-GGAACTGAT TGTATCTGTCGTCG-3’; mouse L32, forward, 5’-ACATTTGCCCTGAATGTGGT-3’; reverse, 5’-ATCCTCTTGCCCTGATCCTT-3’.
Western blot analyses
Mouse BMDM were isolated and processed according to a method described previously (33). The membranes were probed with indicated antibodies, and then developed using an enhanced chemiluminescent Western blot detection kit (Amersham Bioscience, Piscataway, NJ, USA). The intensities of bands were calculated using ImageJ, verifying for non-saturation and subtracting background. Values are expressed as the integrals (target area density) of each band (normalized to total indicated protein band).
Immunocytochemistry analysis
8-chamber culture dish was subjected to either control medium or LPS and/or CA contained medium were fixed in 4% buffered paraformaldehyde in PBS. Fixed cells were permeabilized and incubated in blocking solution (1% BSA and 0.1% Triton-X in PBS) at room temperature for 1 h before overnight incubation at 4℃ with primary antibodies for NLRP3 and ASC. Following incubation with primary antibodies, the cells were incubated with the appropriate Alexa Fluor-conjugated secondary antibodies (Life Technologies) for 1 h at room temperature. Following secondary antibody incubation, chamber slides were sealed with fluorescent mounting medium containing DAPI (molecular probes) on glass slides. Images were acquired using a confocal laser scanning microscopy (Carl Zeiss).
Cecal ligation and puncture model
Cecal ligation and puncture (CLP) surgery was used to induce acute septic peritonitis (34). Blood samples were taken from the infraorbital plexus after subcutaneous injections with CA. The blood was spun down for 10 min in a centrifuge at 1,000 × g and 4℃ after drawing. The serum was stored at −80℃ until it was assayed. Animal survival data were evaluated by the Kaplan–Meier survival analysis and compared by using the log-rank test. A probability value of 0.01 or less was considered statistically significant.
IL-1β ELISA assay
BMDMs were cultured and stimulated with LPS and/or CA supernatants collected after 6 h. The release of IL-1β was analyzed by ELISA (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instruction.
Statistical analysis
Results are presented as mean ± standard deviation of at least three separate experiments. Differences between groups were detected by one-way analysis of variance or a paired Student’s t-test. Differences were considered statistically significant at P < 0.05.
Acknowledgments
This study was supported by grants of the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120864), the Korea New Faculty, Korea Research Foundation, Republic of Korea (NRF-2013R1A1A1006606) and the Korea Research Foundation, Medical Research Center (2014R1A5A2010008) to S.S.I.
References
- 1.Kushner I. The phenomenon of the acute phase response. Ann N Y Acad Sci. (1982);389:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- 2.Sherwood ER, Toliver-Kinsky T. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesthesiol. (2004);18:385–405. doi: 10.1016/j.bpa.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Kim KK, Jin SH, Lee BJ. Herpes virus entry mediator signaling in the brain is imperative in acute inflammation-induced anorexia and body weight loss. Endocrinol Metab (Seoul) (2013);28:214–220. doi: 10.3803/EnM.2013.28.3.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. (2008);3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akira S. Toll-like receptors and innate immunity. Adv Immunol. (2001);78:1–56. doi: 10.1016/s0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Janeway C., Jr Innate immune recognition: mechanisms and pathways. Immunol Rev. (2000);173:89–97. doi: 10.1034/j.1600-065X.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- 7.Chen G, Shaw MH, Kim YG, Nunez G. NODlike receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. (2009);4:365–398. doi: 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
- 8.Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012);13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. (2009);10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin KM, Hu W, Troutman TD, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. (2014);111:775–780. doi: 10.1073/pnas.1320294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trofimova M, Sprenkle AB, Green M, Sturgill TW, Goebl MG, Harrington MA. Developmental and tissue-specific expression of mouse pelle-like protein kinase. J Biol Chem. (1996);271:17609–17612. doi: 10.1074/jbc.271.30.17609. [DOI] [PubMed] [Google Scholar]
- 12.Uematsu S, Sato S, Yamamoto M, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. (2005);201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. (2010);80:1981–1991. doi: 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 14.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. (1996);271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 15.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. (2004);431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 16.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. (2002);2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 17.Chen H, Yang J, Zhang Q, Chen LH, Wang Q. Corosolic acid ameliorates atherosclerosis in apolipoprotein E-deficient mice by regulating the nuclear factor-kappaB signaling pathway and inhibiting monocyte chemoattractant protein-1 expression. Circ J. (2012);76:995–1003. doi: 10.1253/circj.CJ-11-0344. [DOI] [PubMed] [Google Scholar]
- 18.Bai N, He K, Roller M, et al. Active compounds from Lagerstroemia speciosa, insulin-like glucose uptake-stimulatory/inhibitory and adipocyte differentiation-inhibitory activities in 3T3-L1 cells. J Agric Food Chem. (2008);56:11668–11674. doi: 10.1021/jf802152z. [DOI] [PubMed] [Google Scholar]
- 19.Priya TT, Sabu MC, Jolly CI. Free radical scavenging and anti-inflammatory properties of Lagerstroemia speciosa (L). Inflammopharmacology. (2008);16:182–187. doi: 10.1007/s10787-008-7002-6. [DOI] [PubMed] [Google Scholar]
- 20.Aguirre MC, Delporte C, Backhouse N, et al. Topical anti-inflammatory activity of 2alpha-hydroxy pentacyclic triterpene acids from the leaves of Ugni molinae. Bioorg Med Chem. (2006);14:5673–5677. doi: 10.1016/j.bmc.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 21.Judy WV, Hari SP, Stogsdill WW, Judy JS, Naguib YM, Passwater R. Antidiabetic activity of a standardized extract (Glucosol) from Lagerstroemia speciosa leaves in Type II diabetics. A dose-dependence study. J Ethnopharmacol. (2003);87:115–117. doi: 10.1016/S0378-8741(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 22.Sato J, Goto K, Nanjo F, Kawai S, Murata K. Antifungal activity of plant extracts against Arthrinium sacchari and Chaetomium funicola. J Biosci Bioeng. (2000);90:442–446. doi: 10.1016/S1389-1723(01)80016-5. [DOI] [PubMed] [Google Scholar]
- 23.Choi HJ, Bae EY, Song JH, Baek SH, Kwon DH. Inhibitory effects of orobol 7-O-D-glucoside from banaba (Lagerstroemia speciosa L.) on human rhinoviruses replication. Lett Appl Microbiol. (2010);51:1–5. doi: 10.1111/j.1472-765X.2010.02845.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee MS, Cha EY, Thuong PT, Kim JY, Ahn MS, Sul JY. Down-regulation of human epidermal growth factor receptor 2/neu oncogene by corosolic acid induces cell cycle arrest and apoptosis in NCI-N87 human gastric cancer cells. Biol Pharm Bull. (2010);33:931–937. doi: 10.1248/bpb.33.931. [DOI] [PubMed] [Google Scholar]
- 25.Shim KS, Lee SU, Ryu SY, Min YK, Kim SH. Corosolic acid stimulates osteoblast differentiation by activating transcription factors and MAP kinases. Phytother Res. (2009);23:1754–1758. doi: 10.1002/ptr.2843. [DOI] [PubMed] [Google Scholar]
- 26.Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. (2011);187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Means TK, Golenbock DT, Fenton MJ. The biology of Toll-like receptors. Cytokine Growth Factor Rev. (2000);11:219–232. doi: 10.1016/S1359-6101(00)00006-X. [DOI] [PubMed] [Google Scholar]
- 28.Diano S. Role of reactive oxygen species in hypothalamic regulation of energy metabolism. Endocrinol Metab (Seoul) (2013);28:3–5. doi: 10.3803/EnM.2013.28.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. (2010);465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pauls E, Nanda SK, Smith H, Toth R, Arthur JS, Cohen P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J Immunol. (2013);191:2717–2730. doi: 10.4049/jimmunol.1203268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci U S A. (2002);99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilchrist M, Thorsson V, Li B, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. (2006);441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 33.Im SS, Kim MY, Kwon SK, et al. Peroxisome proliferator-activated receptor {alpha} is responsible for the up-regulation of hepatic glucose-6-phosphatase gene expression in fasting and db/db Mice. J Biol Chem. (2011);286:1157–1164. doi: 10.1074/jbc.M110.157875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayala A, Perrin MM, Kisala JM, Ertel W, Chaudry IH. Polymicrobial sepsis selectively activates peritoneal but not alveolar macrophages to release inflammatory mediators (interleukins-1 and -6 and tumor necrosis factor). Circ Shock. (1992);36:191–199. [PubMed] [Google Scholar]