

Abstract
Understanding of trafficking, processing, and degradation mechanisms of amyloid precursor protein (APP) is important because APP can be processed to produce β-amyloid (Aβ), a key pathogenic molecule in Alzheimer’s disease (AD). Here, we found that APP contains KFERQ motif at its C-terminus, a consensus sequence for chaperone-mediated autophagy (CMA) or microautophagy which are another types of autophagy for degradation of pathogenic molecules in neurodegenerative diseases. Deletion of KFERQ in APP increased C-terminal fragments (CTFs) and secreted N-terminal fragments of APP and kept it away from lysosomes. KFERQ deletion did not abolish the interaction of APP or its cleaved products with heat shock cognate protein 70 (Hsc70), a protein necessary for CMA or microautophagy. These findings suggest that KFERQ motif is important for normal processing and degradation of APP to preclude the accumulation of APP-CTFs although it may not be important for CMA or microautophagy. [BMB Reports 2016;49(6): 337-342]
Keywords: Alzheimer disease (AD), β-amyloid, Chaperone mediated autophagy, Hsc70, LAMP2
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disease characterized by widespread loss of neurons and synapses, and a progressive loss of memory. Extracellular accumulation of amyloid-β (Aβ) in senile plaques is a key pathological finding in AD and is regarded as the primary causative factor of neurodegeneration (1, 2). Aβ is derived from amyloid precursor protein (APP) via intracellular proteolytic processing. APP is a type I integral membrane protein processed by α-, β- and γ-secretases. Cleavage of APP by β-secretase produces a soluble form of APP (sAPPβ) and a membrane-bound amyloidogenic 99 amino acid β-C-terminal fragment (βCTF), which is then sequentially cleaved by γ-secretase to produce a 4 kDa Aβ fragment and a 57-59 amino acid APP intracellular domain (AICD). By contrast, α-secretase cleaves APP within the Aβ region to produce sAPPα and a non-amyloidogenic 83 amino acid α-C-terminal fragment (αCTF); this processing precludes Aβ production (3, 4). In addition to Aβ toxicity (3), other APP fragments such as APP-βCTF and AICD also induce neurotoxicity (5-7). Hence, a precise understanding of APP processing is important.
Autophagy is a process whereby, cellular constituents are degraded and recycled via lysosomes. There are three major types of autophagy, namely; macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Macroautophagy is the best understood process. Here, substrates are sequestered into characteristic double-membraned vesicles, known as autophagosomes, then delivered to lysosomes for degradation (8). Microautophagy sequesters cytoplasmic material into lysosomes by direct membrane invagination of lysosomes or late endosomes (9-11). CMA is another type of lysosomal degradation whereby, substrate proteins are translocated into the lysosome by lysosome-associated membrane protein-2 (LAMP2) (12, 13).
Macroautophagy degrades various key pathogenic proteins in neurodegenerative diseases. These proteins include tau, Aβ, APP, α-synuclein and huntingtin (14-19). CMA also degrades pathogenic proteins including tau, α-synuclein and huntingtin (20-22). Microautophagy degrades cytosolic proteins by delivering them to late endosomes and it is also known to degrade pathogenic proteins (10, 11, 23); hence, all of these autophagic processes are involved in various neurodegenerative diseases (24).
Although, many pathogenic proteins including tau, α-synuclein and huntingtin are degraded by CMA (20-22), whether APP, or its cleaved products, such as CTFs or AICD, are substrates for CMA or microautophagy remains unknown. Hence, we investigated this issue and found that APP contains a KFERQ motif in its cytosolic C-terminus, and that this motif is important for APP processing.
RESULTS
The KFERQ motif is important for APP processing
We first searched for KFERQ motifs (13) in the APP sequence and identified one in the cytosolic C-terminus (Fig. 1A). Then, we generated a KFERQ-deletion construct (Fig. 1B) and expressed it in SH-SY5Y neuronal cells to investigate expression and processing profiles and to validate Hsc70 binding. Interestingly, western blots from cell lysates showed that deletion of KFERQ increased CTF expression but had little effect on the expression of full-length APP (Fig. 1C). Western blots from culture media also showed that KFERQ deletion increased the secretion of sAPPα and sAPPβ, the secreted forms of APP (Fig. 1D).
Fig. 1. KFERQ-deleted APP increases C-terminal fragment expression and APP secretion. (A) Schematic diagram of amyloid precursor protein (APP) depicting the endocytosis motif and KFERQ motif at the intracellular C-terminus. (B) Schematic diagram of wild-type APP and ΔKFERQ-APP constructs used in the experiments herein. (C) SH-SY5Y cells were transfected with wild-type APP (WT-APP)-HA or ΔKFERQAPP-HA tagged with HA. Western blots for HA using cell lysates show that ΔKFERQ-APP increased APP C-terminal fragment (CTF) protein amount. (D) SH-SY5Y cells were transfected with WT-APP-HA or ΔKFERQ-APP-HA and culture media was collected. Western blots for secreted APP show that ΔKFERQ-APP increased sAPPα and sAPPβ secretion. βactin was from cell lysates. Each data represent the mean ± S.E. from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.

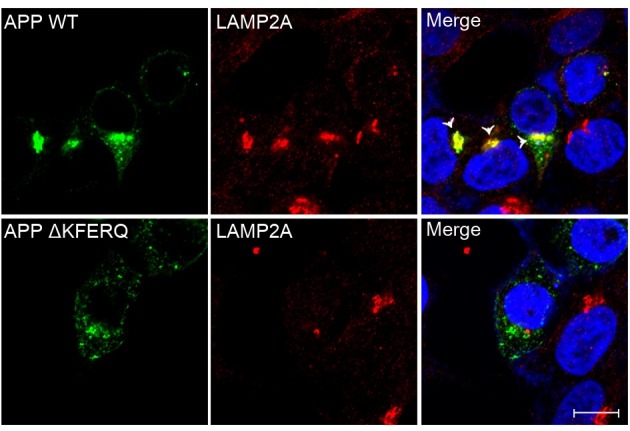
The KFERQ motif is important for targeting APP to lysosomes
SH-SY5Y neuronal cells were transfected with wild-type APP and ΔKFERQ-APP, then fixed and immunostained for LAMP2. Wild-type APP was well colocalized with LAMP2, a lysosomal marker (Fig. 2, arrowheads); however, ΔKFERQ-APP was not colocalized with LAMP2, indicating that the KFERQ motif is required for lysosomal targeting of APP.
Fig. 2. ΔKFERQ-APP exhibits less lysosomal colocalization than wild-type APP. SH-SY5Y cells were transfected with WT-APP-HA or ΔKFERQ-APP-HA, and stained with antibodies against HA (green) and lysosome-associated membrane protein-2 (LAMP2, red). ΔKFERQAPP shows less colocalization with lysosomes than WT-APP. Arrow head indicates the colocalization of WT-APP with LAMP2. Scale bar, 10 μm.
The KFERQ motif increases Hsc70 binding
Since Hsc70 binding to its substrate proteins via the KFERQ motif is necessary for both CMA and microautophagy, we investigated Hsc70 binding to wild-type APP and ΔKFERQ-APP. SH-SY5Y neuronal cells were transfected with wild-type APP and ΔKFERQ-APP, and immunoprecipitation was performed. Deletion of KFERQ did not abolish Hsc70 binding, rather increased it (Fig. 3A). Since, APP cleavage products, such as the APP-CTFs or AICD, could be substrates for CMA or microautophagy, we further generated constructs expressing αCTF, βCTF, and AICD and performed a similar experiment to the one above. Notably, CTFs and AICD bound Hsc70 (Fig. 3B, C and D). Deletion of the KFERQ motif in CTFs and AICD did not abolish binding of Hsc70, rather increased it.
Fig. 3. The KFERQ motif is not necessary for the binding of Hsc70 to APP. (A) SH-SY5Y cells were transfected with WT-APP-HA or ΔKFERQ-APP-HA. Immuno-precipitation with an anti-HA antibody shows that heat shock cognate protein-70 (Hsc70) remains bound to ΔKFERQ-APP. (B) SH-SY5Y cells were transfected with wild-type APP intracellular domain (WT-AICD)-HA or ΔKFERQ-AICD-HA. Immunoprecipitation with an anti-HA antibody shows that heat shock cognate protein-70 (Hsc70) remains bound to ΔKFERQ-AICD. (C) SH-SY5Y cells were transfected with wild-type α-C-terminal fragment (WT-αCTF)-HA or ΔKFERQ-αCTF-HA. Immunoprecipitation with an anti-HA antibody shows that Hsc70 remains bound to ΔKFERQ-αCTF. (D) SH-SY5Y cells were transfected with wild-type β-C-terminal fragment (βCTF)-HA and ΔKFERQ-βCTF-HA. Immuno-precipitation with an anti-HA antibody shows that Hsc70 remains bound to ΔKFERQ-βCTF. Each data represent the mean ± S.E. from three independent experiments. *P < 0.05, **P < 0.01.

Phosphorylated tau is increased in cells expressing KFERQ-deleted APP
Since KFERQ deletion increased APP-CTFs (Fig. 1 and 3) and we wondered whether it could have potential effects on AD pathologies, we checked phosphorylation level of tau. Western blots showed that deletion of KFERQ in APP, CTFs and AICD increased the phosphorylated tau at Ser396 (Fig. 4).
Fig. 4. Tau phosphorylation is increased by ΔKFERQ-APP. (A) SH-SY5Y cells were transfected with WT-APP-HA or ΔKFERQ-APP-HA. Western blots with an anti-Tau-pS396 antibody shows that phosphorylated tau is increased in ΔKFERQ-APP-expressing cells. (B) SH-SY5Y cells were transfected with WT-AICD-HA or ΔKFERQ-AICD-HA. Western blots with an anti-Tau-pS396 antibody shows that phosphorylated tau is increased in ΔKFERQ-AICD-expressing cells. (C) SH-SY5Y cells were transfected with WT-αCTF-HA or ΔKFERQ-αCTF-HA. Western blots with an anti-Tau-pS396 antibody shows that phosphorylated tau is increased in ΔKFERQ-αCTF-expressing cells. (D) SH-SY5Y cells were transfected with WT-βCTF-HA and ΔKFERQ-βCTF-HA. Western blots with an anti-Tau-pS396 antibody shows that phosphorylated tau is increased in ΔKFERQ-βCTF-expressing cells. Each data represent the mean ± S.E. from three independent experiments. *P < 0.05, **P < 0.01.

DISCUSSION
In the present study, we found that APP contains a KFERQ motif, typically associated with CMA and microautophagy. Deletion of this motif increased CTF levels and sAPP α/β secretion, suggesting that this sequence is important for the regulation of APP processing.
Although, many neurodegenerative disease-associated pathogenic proteins, such as tau, α-synuclein and huntingtin, are degraded by CMA (20-22), whether APP, another important pathogenic protein, or its cleaved products, such as CTFs or AICD, are a substrate for CMA or microautophagy has not previously been addressed. We found here that, APP contains a KFERQ motif in its C-terminus (Fig. 1), raising the possibilities that APP or its cleaved products could be substrates for CMA or microautophagy. Soluble cytosolic proteins are substrates for CMA (13), so AICD, which is generated from APP after γ-site cleavage, is freely exposed to the cytosol and could therefore be a potential CMA substrate. Since, organelles can be engulfed by direct invagination during microautophagy (9, 10), APP or CTFs could be potential substrates for microautophagy because of the presence of the KFERQ motif in the cytosolic domain.
Interestingly, deletion of KFERQ motif in APP increased CTF levels and sAPPα/β secretion (Fig. 2), but did not alter the level of full-length APP. This finding suggests that, trafficking and maturation of APP is normal even in the absence of KFERQ sequence at its C-terminus. After APP is endocytosed, APP is normally sorted and processed to secretase associated cleavage pathway or to degradation pathway. However, KFERQ deletion impairs its sorting and processing to degradation pathway and increased α/β-secretase cleavage, resulting in the increased sAPPα/β secretion and CTF generation, which is more supported by findings that lysosomes are not colocalized with APP in the absence of KFERQ motif (Fig. 2). It was reported that, APP-CTFs increase neurotoxicity and tau phosphorylation by glycogen synthase kinase-3β (7). Increased APP-CTFs by KFERQ deletion enhances tau phosphorylation (Fig. 4), suggesting that regulation of normal level of APP-CTF by KFERQ motif is important for the maintenance of adequate level of tau phosphorylation, which impairment could contribute to AD pathogenesis.
Substrate proteins of both CMA and microautophagy contain a KFERQ motif that is recognized and bound by heat shock cognate protein-70 (Hsc70). This is necessary for further targeting to lysosomes and late endosomes (9-13). However, we did not detect loss of Hsc70 binding to KFERQ-deleted APP, CTFs or AICD (Fig. 3), suggesting that these APP may not be substrates for CMA or microautophagy. APP-CTF is degraded by macroautophagy through interactions with LC3 via adaptor protein 2 (AP2) (15). This interaction is mediated through the AP2 recognition signal sequence, YxxΦ, which is YKFF in APP, as part of the KFERQ sequence. They also showed that phosphatidylinositol-binding clathrin assembly lymphoid myeloid leukemia protein (PICALM), a risk factor for AD identified in a genome-wide association study (25), was recruited to autophagosomes with AP2 and APP-CTF (15). This finding may also explain our observation that KFERQ deletion increased APP-CTF levels and impaired lysosomal targeting of APP (Fig. 1 and 2), because KFERQ deletion would likely abolish the interaction with AP2, resulting in impaired targeting to autophago-lysosomes.
Our findings reveal that, KFERQ motif in the C-terminus of APP is important for APP processing and degradation although it is less important for the typical CMA or microautophagy. Future studies will be necessary about whether any putative binding partners for KFERQ motif such as AP2 complex works in APP processing and how its impairments could affect AD pathogenesis.
MATERIALS AND METHODS
Plasmid constructs and antibodies
The wild-type human APP 695 construct and pcDNA5-FRT/TO-HA were kindly provided by Dr. SW Kang. The KFERQ-deficient mutant APP derivative (APPΔKFERQ) was cloned by PCR from wild-type human APP and subcloned into pcDNA5-FRT/TO-HA. As αCTF, KFERQ-deficient αCTF (αCTFΔKFERQ), βCTF, KFERQ-deficient βCTF (βCTFΔKFERQ) are secreted proteins, the APP signal sequence was added to the N-terminus of αCTF, αCTFΔKFERQ, βCTF and βCTFΔKFERQ. AICD and KFERQ-deficient AICD (AICDΔKFERQ) were also cloned by PCR amplification and inserted into pcDNA5-FRT/TO-HA. HA tags were introduced by subcloning into pcDNA5-FRT/TO-HA. All constructs were verified by DNA sequencing (CosmoGenetech, South Korea). All APP constructs were detected by western blotting and immunochemistry with rat anti-HA (Roche, Switzerland).
Cell culture analysis
SH-SY5Y cells were maintained in DMEM (Thermo, USA) supplemented with 10% fetal bovine serum (FBS) (Thermo, USA) and incubated in 5% CO2 at 37℃ as previously described (26). All experiments involving transient transfection were performed using Lipofectamine 2000 (Invitrogen, USA), according to the manufacturer’s instructions. For expression of APP constructs, cells were transiently transfected with APP or KFERQ-deleted APP constructs. Transfected cells were incubated with antibiotic-free DMEM media for 4 hours and then the media was changed to DMEM supplemented with FBS. After 24 hours, cells were analyzed by western blotting, immunochemistry or co-immunoprecipitation.
Co-immunoprecipitation and western blotting
For co-immunoprecipitation, SH-SY5Y cells transiently expressing various APP constructs (wild type, WT-APPΔKFERQ, αCTF, αCTFΔKFERQ, βCTF, βCTFΔKFERQ AICD, and AICDΔKFERQ) were lysed with lysis buffer (1% Triton X-100, 20 mM HEPESpH7.5, 150 mM NaCl, 10% glycerol, 1 mM EDTA, protease inhibitor cocktail (Calbiochem, USA) and phosphatase inhibitor cocktail (Sigma, USA) for 20 min at 4℃ as previously described with some modifications (27-29). Cell lysates were centrifuged at 10,000 g for 20 min at 4℃ to remove any insoluble material. Co-immunoprecipitation was performed using an anti-HA (Roche, Basel, Switzerland) antibody with an overnight incubation. Immunocomplexes were captured using Protein G-Sepahrose (GE Healthcare) followed by three washes with lysis buffer. The immunoprecipitated samples or 5% of the input lysates were used for immunoblotting. MG 132 (Calbiochem, USA) was used to detect AICD.
For western blotting, protein lysates from SH-SY5Y cells transfected with wt-APP, mutant constructs and other cleaved fragments were fully solubilized with 1% sodium dodecyl sulfate (SDS), 100 mM Tris, pH 8.0. Protein concentrations were measured by Bradford protein assay. Equal amounts of protein were mixed with sample buffer (62.5 mM Tris, pH 6.8, 1% SDS, 2.5% glycerol, 0.5% β-mercaptoethanol, and bromophenol blue), boiled at 100℃ for 5 min, and stored at −20℃ until use. Proteins were resolved by SDS-polyacrylamide gel electrophoresis, and subsequently transferred to polyvinylidenedi-fluoride membranes (pore size, 0.2 mm; BioRad, USA). After 1 hour incubation in blocking PBST buffer (0.1% Tween-20 in phosphate-buffered saline (PBS)), blots were incubated with primary antibodies overnight at 4℃. Blots were washed in PBST buffer, incubated with horseradish peroxidase-conjugated anti-IgG (1:5,000; Pierce, USA), and visualized using enhanced chemiluminescence reagents (Amersham, USA) and X-ray film. The primary antibodies used for western blotting were rat anti-HA (1:5,000; Roche, Switzerland), rat anti-HSC70 (1:5,000; Covance, USA), mouse anti-β-actin (1:10,000; Sigma-Aldrich, USA), mouse anti-sAPPα(1:50; IBL, Japan), and rabbit anti-sAPPβ(1:50; IBL, Japan).
Immunocytochemistry
For immunocytochemistry, SH-SY5Y cells were plated onto 12 mm coverslips (Marienfeld, Germany) as previously described (30). SH-SY5Y cells were transfected with TREM2 construct or mutant plasmids using Lipofectamine 2000. Twenty-four hours after transfection, cells were washed with PBS (140 mM NaCl, 10 mM Na2HPO4, 1.75 mM KH2PO4 in dH2O, pH7.4). Cells were fixed in 4% paraformaldehyde for 10 min. After three washes in PBS, cells were permeabilized with PBS containing 0.1% Triton X-100 for 5 min at room temperature. Cells were washed three times and blocked with PBS containing 5% bovine serum albumin for 30 min at 37℃. Cells were then washed three times and incubated overnight with rat anti-HA (1:100; Roche, Switzerland) or rabbit anti-LAMP2A (1:100; Abcam, UK) primary antibodies. After washing five times with PBS, cells were incubated with a secondary antibody coupled to TexasRed (Invitrogen, USA) or Alexa FluorⓇ 488 (Invitrogen, USA) for 60 min at 37℃. Cells were then washed five times and mounted for imaging. Images were acquired using Zeiss Axiovert 200 LSM510 confocal microscope workstation equipped with a 63x (numerical aperture, 1.4) objective.
Acknowledgments
This work was supported by the Medical Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, Information and Communications Technology, and Future Planning (2008-0062286), and the Korean Health Technology Research and Development Project, Ministry of Health & Welfare, Republic of Korea (HI13C1630).
References
- 1.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol. (2006);6:404–416. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 2.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. (2008);451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid betaprotein. J Alzheimers Dis. (2001);3:75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- 4.Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. (1987);325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 5.Choi SH, Park CH, Koo JW, et al. Memory impairment and cholinergic dysfunction by centrally administered Abeta and carboxyl-terminal fragment of Alzheimer's APP in mice. FASEB J. (2001);15:1816–1818. doi: 10.1096/fj.00-0104com. [DOI] [PubMed] [Google Scholar]
- 6.Kim HS, Park CH, Cha SH, et al. Carboxyl-terminal fragment of Alzheimer's APP destabilizes calcium homeostasis and renders neuronal cells vulnerable to excitotoxicity. FASEB J. (2000);14:1508–1517. doi: 10.1096/fj.14.11.1508. [DOI] [PubMed] [Google Scholar]
- 7.Kim HS, Kim EM, Lee JP, et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. (2003);17:1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- 8.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. (2000);290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. (2011);7:673–682. doi: 10.4161/auto.7.7.14733. [DOI] [PubMed] [Google Scholar]
- 10.Li WW, Li J, Bao JK. Microautophagy: lesserknown self-eating. Cell Mol Life Sci. (2012);69:1125–1136. doi: 10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santambrogio L, Cuervo AM. Chasing the elusive mammalian microautophagy. Autophagy. (2011);7:652–654. doi: 10.4161/auto.7.6.15287. [DOI] [PubMed] [Google Scholar]
- 12.Kaushik S, Bandyopadhyay U, Sridhar S, et al. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011;124:495–499. doi: 10.1242/jcs.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. (2012);22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar S, Rubinsztein DC. Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J. (2008);275:4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 15.Tian Y, Chang JC, Fan EY, Flajolet M, Greengard P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A. (2013);110:17071–17076. doi: 10.1073/pnas.1315110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. (2010);5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. (2011);25:219–231. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. (2003);278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 19.Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. (2013);105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperonemediated autophagy and macroautophagy in neuronal cells. J Biol Chem. (2008);283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Martinez-Vicente M, Kruger U, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. (2009);18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauer PO, Goswami A, Wong HK, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. (2010);28:256–263. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- 23.Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. (2011);20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. (2013);19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 25.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. (2009);41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho K, Cho MH, Seo JH, et al. Calpain-mediated cleavage of DARPP-32 in Alzheimer's disease. Aging Cell. (2015);14:878–886. doi: 10.1111/acel.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. (2014);10:1761–1775. doi: 10.4161/auto.29647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park YM. Oxidized LDL induces phosphorylation of non-muscle myosin IIA heavy chain in macrophages. BMB Rep. (2015);48:48–53. doi: 10.5483/BMBRep.2015.48.1.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuo J, Ma H, Cai H, Wu Y, Jiang W, Yu L. An inhibitory role of NEK6 in TGFbeta/Smad signaling pathway. BMB Rep. (2015);48:473–478. doi: 10.5483/BMBRep.2015.48.8.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JS, Ji IJ, An HJ, et al. Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus. Traffic. (2015);16:510–518. doi: 10.1111/tra.12264. [DOI] [PubMed] [Google Scholar]