

Abstract
Study Objectives:
Emerging evidence suggests a role for sleep in contributing to the progression of Alzheimer disease (AD). Slow wave sleep (SWS) is the stage during which synaptic activity is minimal and clearance of neuronal metabolites is high, making it an ideal state to regulate levels of amyloid beta (Aβ). We thus aimed to examine relationships between concentrations of Aβ42 in the cerebrospinal fluid (CSF) and measures of SWS in cognitively normal elderly subjects.
Methods:
Thirty-six subjects underwent a clinical and cognitive assessment, a structural MRI, a morning to early afternoon lumbar puncture, and nocturnal polysomnography. Correlations and linear regression analyses were used to assess for associations between CSF Aβ42 levels and measures of SWS controlling for potential confounders. Resulting models were compared to each other using ordinary least squared linear regression analysis. Additionally, the participant sample was dichotomized into “high” and “low” Aβ42 groups to compare SWS bout length using survival analyses.
Results:
A significant inverse correlation was found between CSF Aβ42 levels, SWS duration and other SWS characteristics. Collectively, total SWA in the frontal lead was the best predictor of reduced CSF Aβ42 levels when controlling for age and ApoE status. Total sleep time, time spent in NREM1, NREM2, or REM sleep were not correlated with CSF Aβ42.
Conclusions:
In cognitively normal elderly, reduced and fragmented SWS is associated with increases in CSF Aβ42, suggesting that disturbed sleep might drive an increase in soluble brain Aβ levels prior to amyloid deposition.
Citation:
Varga AW, Wohlleber ME, Giménez S, Romero S, Alonso JF, Ducca EL, Kam K, Lewis C, Tanzi EB, Tweardy S, Kishi A, Parekh A, Fischer E, Gumb T, Alcolea D, Fortea J, Lleó A, Blennow K, Zetterberg H, Mosconi L, Glodzik L, Pirraglia E, Burschtin OE, de Leon MJ, Rapoport DM, Lu S, Ayappa I, Osorio RS. Reduced slow-wave sleep is associated with high cerebrospinal fluid Aβ42 levels in cognitively normal elderly. SLEEP 2016;39(11):2041–2048.
Keywords: Alzheimer disease, amyloid beta, sleep, prevention, elderly
Significance.
Sleep problems are common in people who have Alzheimer disease (AD), but they may also be an indicator of early disease prior to the development of clinical symptoms. As of yet, it is unknown whether poor sleep quality affects the development of AD or vice-versa. For a long time, experts believed AD was wounding centers of the brain responsible for sleep regulation, but recent research suggests the link between sleep and AD may be more complicated. Our evidence indicates that deep sleep reduces production/cleanses the brain of toxins that form amyloid plaques, a hallmark of AD. To establish whether lack of sleep leaves the brain vulnerable to AD, future work should be directed at studying this process in humans.
INTRODUCTION
The “Amyloid Cascade Hypothesis” posits that the deposition of amyloid beta (Aβ) in the brain is the initiating pathological event in Alzheimer disease (AD).1 Several studies have provided evidence that Aβ dynamics are influenced by sleep. In transgenic mice, Aβ levels are higher in the interstitial fluid during wakefulness and lower during sleep, while sleep deprivation increases Aβ concentrations and accelerates plaque deposition.2 In humans, cerebrospinal fluid (CSF) Aβ42 also exhibits a diurnal pattern, with the lowest levels occurring in the morning.3 This CSF Aβ42 physiological morning decrease is attenuated by total sleep deprivation.4 All these findings suggest that sleep may play a unique role in AD by resetting soluble Aβ42 to lower levels; however, the precise regulation of this diurnal pattern is not well understood.
Aβ production is thought to be neuronal activity-dependent, and plaque deposition preferentially targets brain regions with high neuronal and large-scale synchronous activity.5 During sleep, the brain remains predominantly active with preservation of cortico-cortical connectivity during light sleep, i.e., non-rapid eye movement (NREM) sleep stages 1–2. However, there is a reduction in fronto-parietal connectivity that occurs with increasing depth of sleep, to the point of being significantly reduced at deep sleep, i.e., slow wave sleep (SWS) or NREM stage 3 (NREM3).6 During rapid eye movement (REM) sleep, brain activation becomes more frequent.7 In this study, we assessed the effect of SWS characteristics on morning CSF Aβ42 levels in a group of normal elderly. We hypothesized that conserved SWS would be associated with low CSF Aβ42 levels, while disrupted SWS would be associated with high Aβ42 levels. Given that reduced CSF Aβ42 levels or SWS duration can be associated with advanced age,8 the apolipoprotein E-4 (ApoE4) allele,8 sex,9 lower education,10 and sleep disordered breathing (SDB),11 we also tested the extent to which the variation in CSF Aβ42 predicted by SWS were influenced by these factors.
METHODS
Study Design and Participants
Among a pool of elderly participating in NIH-supported longitudinal studies, this study included a sub-set of 41 subjects that agreed to undergo nocturnal polysomnography (NPSG). Subjects were recruited from multiple community sources in NYC as previously described,11 individuals with conditions that could affect brain structure or function were excluded.
Procedures
Subjects received the standardized Uniform Data Set II diagnostic assessment at the NYU Center for Brain Health (CBH).12 In addition, subjects had laboratory examinations and underwent a structural MRI, a morning to early afternoon lumbar puncture (LP) and a NPSG. The interval between polysomnography and CSF collection was of 6.7 ± 7.5 months. All subjects were cognitively normal (Clinical Dementia Rating [CDR] = 0). CSF samples were processed as described pre -viously.11 CSF was analyzed for Aβ42, phosphorylated tau at threonine181 (P-tau) and total tau (T-Tau) using commercially available enzyme-linked immunosorbent assay (ELISA) kits (Fujirebo, Ghent, Belgium), CSF Aβ40 was analyzed using commercially available high sensitivity ELISA kits (Merck, Darmstadt, Germany).
All subjects received structural volumetric magnetic resonance imaging (MRI) scans as part of the parent NIH studyies on a 1.5T (GE, USA) or 3T (Siemens, Germany) system using standardized procedures.13,14 These scans were obtained to rule out MRI evidence of intracranial mass and white matter disease prior to performing the LPs. No subjects were excluded due to these abnormalities. In view of recent evidence suggesting that frontal atrophy is associated with reduced SWS in normal elderly,15,16 we measured cortical volumes from MPRAGE sequences using the FreeSurfer toolkit17 and computed gray matter volumes to create a medial prefrontal cortex (mPFC) region of interest (ROI) using the following bilateral ROIs: caudal anterior cingulate cortex, medial orbitofrontal cortex, rostral anterior cingulate cortex and superior frontal gyrus.18 Resulting ROIs were adjusted (residualized) to their intracranial volume using linear regression.
Sleep recordings were performed using American Academy of Sleep Medicine (AASM) guidelines.19 They consisted of 6 electroencephalogram (EEG) channels (F3, F4, C3, C4, O1, and O2), 2 electro-oculographic (EOG) leads, and one chin electromyo-graphic channel. Visual scoring of recordings, total sleep time (TST) and sleep duration in minutes were determined according to AASM criteria.19 Respiratory events were scored using AASM criteria as described previously.20 AHI4% was defined as the sum of all apneas and hypopneas with ≥ 4% desaturation divided by TST in hours. AHIall was defined as the sum of apneas and hypopneas (3% or arousal) divided by TST in hours.
Sleep studies were first scored in 30-s epochs.19 NREM REM cycles were defined according to the criteria of Feinberg and Floyd21 starting with NREM2 and containing at least 15 minutes of NREM2 or NREM3 followed by a REM episode of at least 5 minutes. EEG signals, acquired with a sampling frequency of 256Hz, were then segmented into 5-s epochs. Power spectra of artifact-free epochs were computed using the fast-Fourier transform and matched with the 5-s sleep scores. Slow wave activity (SWA) was calculated using the average power density in the 0.5–4.0 Hz range of F4, C3 and O2 full-night EEG recordings. Changes in SWA were evaluated using area under the curve (AUC) for each NREM sleep cycle and for the full night. To account for individual differences in the occurrence and duration of sleep cycles, NREM episodes were first subdivided into 24 equal segments and then averaged.22 Where appropriate, group comparisons of SWS characteristics were performed using the median CSF Aβ42 value to divide the sample into two equal sized (high/low) Aβ42 groups.
Covariates used in statistical analyses were age, gender, ApoE4, years of education, CSF biomarkers (Aβ42, P-Tau, T-Tau), SWS duration, percent of TST spent in SWS (%SWS), mean SWS bout length, total SWA, SWA in NREM cycles 1–4, and mPFC volume.
Statistical Analyses
Logarithm transformation was applied to normalize right skewed variables (CSF Aβ42, SWA in F4, C3 and O2) prior to analysis. We first assessed the effect of SWS duration and other SWS characteristics on morning CSF Aβ42 levels in the entire group using correlation analyses. We then used ordinary least squared linear regression to evaluate the associations between the SWS characteristic with the highest correlation coefficient and CSF Aβ42, using Aβ42 as the dependent variable. Age, ApoE4, sex, and years of education were included as covariates only if they improved the R2 and adjusted R2 for the model. The best fitting model was then replicated with each of the other SWS characteristics. On a final step we compared the resulting models looking at percent increase in R2 for each model.
Finally, we analyzed mean SWS bout length after dichotomizing the sample into “low Aβ42” and “high Aβ42” groups using the median CSF Aβ42 (536.9 pg/mL). A sleep bout of any particular sleep stage was defined as the duration of consecutive 30-s epochs of sleep scored as that stage, terminated by 1 or more epochs scored as another stage, including wake. A bootstrap-based analysis that accounted for the number of sleep bouts contributed by each subject was performed to determine a cumulative duration probability distribution for each sleep stage (REM, NREM1, NREM2, and SWS). In order to remain consistent across all subjects and, at the same time, retain a sufficient number of data points, for those subjects with more than the median number of bouts, bouts were randomly sampled up to the median number. Log rank tests were used to subjects with “low Aβ42” vs. “high Aβ42” on the survival curves derived from the sampling procedure. This procedure was repeated 1,000 times. At each step of the iteration, a P value was estimated from the history of prior P values, yielding an increasingly stable result as the number of iterations increased.
RESULTS
Healthy, Cognitively Normal Group with Low Overall Risk for AD
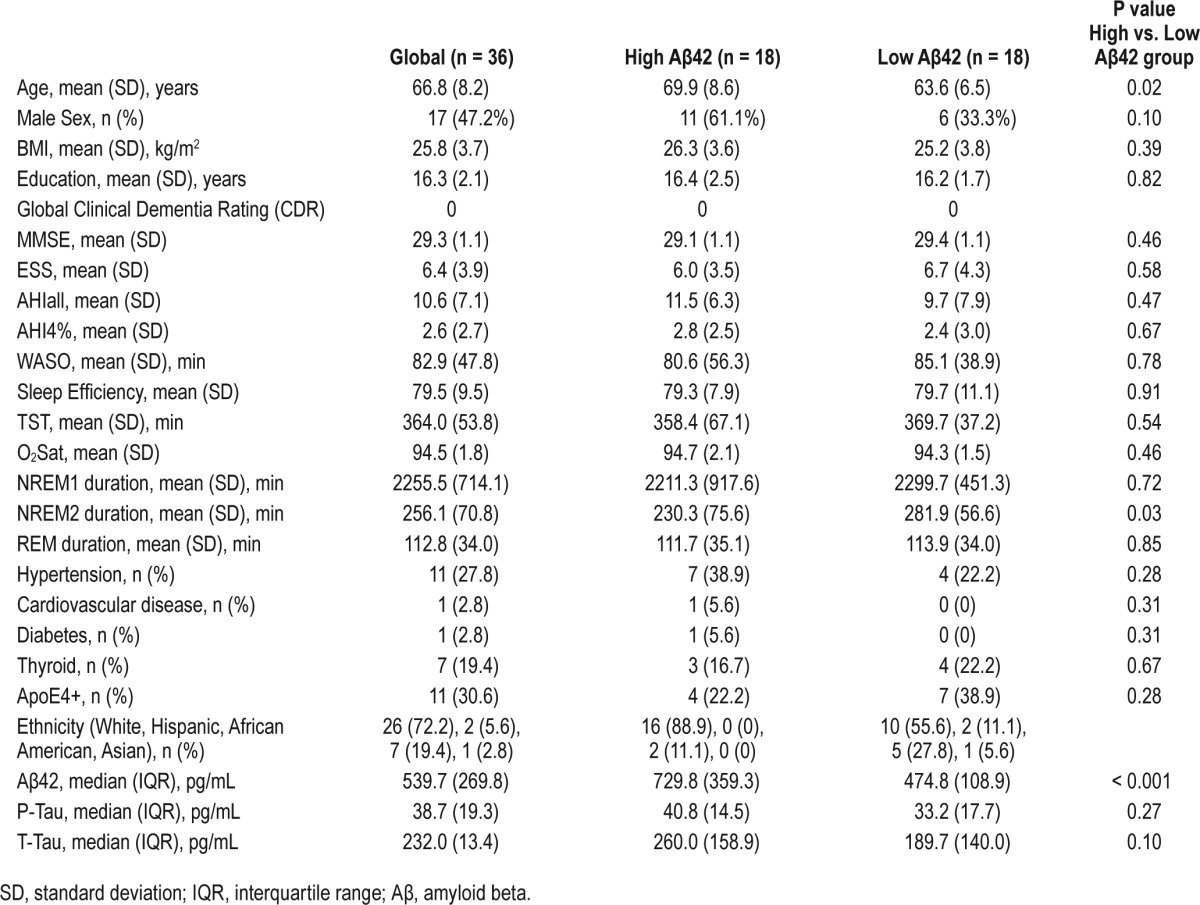
Forty-one eligible participants completed all study procedures. Five were excluded: 3 due to moderate to severe SDB (AHI4% ≥ 15), 1 due to fragmented sleep with TST < 3 hours during the NPSG, and 1 due to significant alcohol consumption prior to the NPSG. Demographic, cognitive, and health characteristics of the remaining 36 participants are shown in Table 1 (“Global”). Results are reported as mean ± SD. Overall, it was a sample of mostly non-obese (BMI 25.8 ± 3.7 kg/m2), highly educated (16.3 ± 2.1 years of education), elderly (age 66.9 ± 8.3 years), in good general health. Table 1 displays median values of CSF Aβ42, T-Tau, and P-Tau levels. Only one subject had CSF Aβ42 levels in the AD range (below 369.53 pg/mL), suggestive of possible cerebral Aβ deposition (cutoff based on ROC analysis from the NYU CBH cohort). Overall, it was a healthy group with low overall risk for AD.
Table 1.
Sociodemographic, clinical, and CSF data of study group.
Sleep Characteristics
Table 1 also summarizes sleep architecture characteristics. Only 5 subjects had mild SDB (AHI4% = 5–14.99), while the majority of subjects had normal breathing during sleep (AHI4% < 5). Epworth Sleepiness Scale scores did not suggest the presence of daytime sleepiness in our sample (value 6.4 ± 3.9). Table 2 shows the SWS characteristics of our sample. SWS duration was inversely correlated with age (r = −0.36, P < 0.05) and wake after sleep onset (WASO) (r = −0.51, P < 0.05), and positively correlated with sleep efficiency (r = 0.39, P < 0.05), but was not associated with BMI; AHI4%, AHIall, mean O2Sat during sleep, TST, NREM1, NREM2, or REM duration; or with mPFC volume. As expected,9,23 SWS duration was higher in females than in males even after controlling for age and WASO (F1-33 = 4.5, P < 0.05; females: 76.8 ± 27.1 min, males: 47.4 ± 30.2 min). Mean time of LP was 11:54 ± 01:08 hours. Time of LP was not associated with CSF Aβ42 levels (rho = −0.2, n.s).
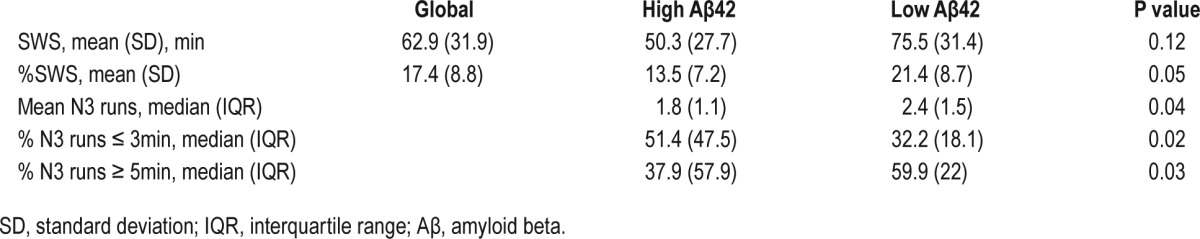
Table 2.
Slow wave sleep characteristics of study group.
We calculated home sleep schedules using self-reported sleep logs and clinical interviews. Subjective sleep times were used in our analyses because an objective measure of sleep (actigraphy) was not available in all subjects. Home bedtime was obtained by calculating the average bedtime from the sleep logs rounded to the nearest quarter hour. For participants without a sleep log, mean bedtime was assessed using the following question “At what time do you usually fall asleep?” In the lab, participants were allowed to fall asleep when they wanted to. Median in-lab sleep onset time was 23:13:35 and median home sleep time was 23:00:00. We used a Wilcoxon Signed Ranks to compare home and in lab sleep times. Participant sleep times in the lab were later than sleep times at home (Z = −2.0, P < 0.05). However, although statistically significant, a 13-min difference is likely not functionally relevant in terms of the circadian fluctuation of CSF Aβ42.
Effects of SWS Duration and Power on CSF Aβ42 Levels
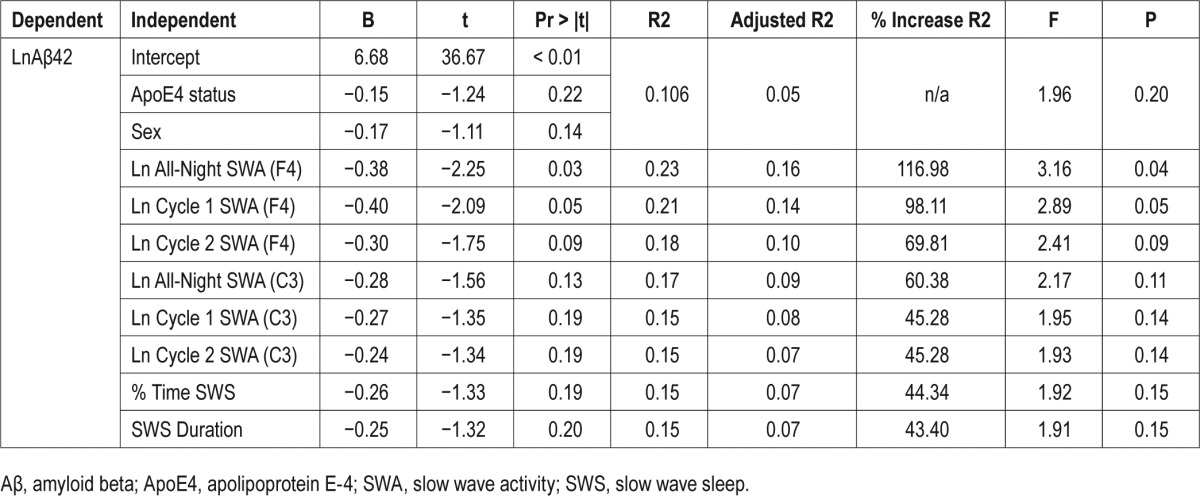
We first examined whether SWS duration correlated with CSF Aβ42 levels. There was a significant inverse correlation between CSF Aβ42 and SWS duration (r = −0.35, P < 0.05) (Figure 1), %SWS (r = −0.36, P < 0.05), total SWA in F4 (r = −0.45, P < 0.01) (Figure 1), SWA in cycle 1 in F4 (r = −0.41, P < 0.05), and SWA in cycle 2 in F4 (r = −0.38, P < 0.05). Similar but weaker inverse correlations were found between CSF Aβ42 and SWA in the C3 channels (Table 4) and there were no associations with SWA in the O2 channels. CSF Aβ42 was not correlated with the duration of other sleep stages or TST. We then repeated the analyses with CSF Aβ40 levels. We observed no significant correlation between CSF Aβ40 and SWS duration (r = −0.13, n.s), %SWS (r = −0.08, n.s.), total SWA in F4 (r = −0.24, n.s), SWA in cycle 1 in F4 (r = −0.19, n.s) or SWA in cycle 2 in F4 (r = −0.16, n.s). Nonetheless, the associations were consistently inverse, as with CSF Aβ42. Using OLS, the best prediction model for CSF Aβ42 included total SWA in F4, sex and ApoE4 status (Table 3). Based on % increase in R2, total SWA in F4 reduced the variation in Aβ42 by 116.98%, compared to the model that only included sex and ApoE4. The next best sleep predictors for CSF Aβ42 were SWA in cycle 1 in F4, followed by SWA in cycle 2 in F4, which increased the R2 by 98.11% and 69.81%, respectively (Table 3). SWS duration was not associated with levels of CSF P-Tau or T-Tau. Time of LP was not associated with CSF Aβ42.
Figure 1.

Scatter plots of natural log of Aβ42 and F4 slow wave activity (full night) and slow wave sleep duration.
Table 4.
Pearson correlation with LnAβ42.
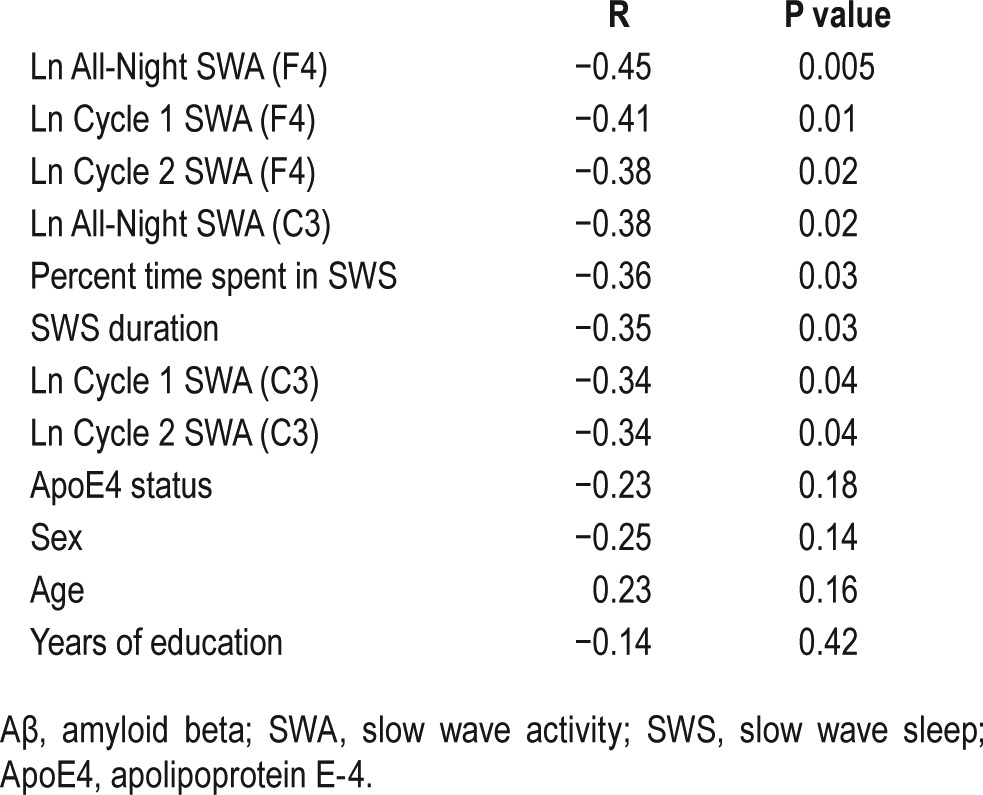
Table 3.
Linear regression analysis.
Effects of SWS Continuity in “Low Aβ42” and “High Aβ42” Groups on CSF Aβ42 Levels
There were no clinical differences between “high” and “low” Aβ42 groups except for age, which was lower in the ‘low Aβ42’ (F1-35 = 4.5, P < 0.05; 70.2 ± 9.0 vs. 63.7 ± 6.3years) (Table 1). The cumulative duration probability distribution for SWS was significantly left-shifted in the “high Aβ42” compared to “low Aβ42” subjects (P < 0.01), indicating that SWS was more fragmented and occurred in shorter bouts in high Aβ42 subjects. Conversely, the cumulative duration probability distribution for NREM2 was significantly right-shifted in the high Aβ42 compared to low Aβ42 subjects (P < 0.01), indicating NREM2 sleep was less fragmented and occurred in longer bouts in high Aβ42 subjects. There were no significant differences in the duration probability distribution of NREM1 or REM sleep between groups.
While the cumulative duration probability distribution reflects sleep continuity across groups, individual measures of sleep stage continuity can be represented by stage mean bout length (with longer length reflecting increased sleep consolidation) and percent of runs of sleep lasting less than 3 minutes (with a higher percentage reflecting decreased sleep consolidation). Based on the results of the survival analysis, we examined the correlation between CSF Aβ42 levels and measures of NREM2 and SWS continuity. Across all subjects, there were no significant correlations between CSF Aβ42 levels and NREM2 continuity variables. On the other hand, we observed a significant inverse correlation between CSF Aβ42 levels and SWS mean bout length (r = −0.37, P < 0.05) and a significant positive correlation between CSF Aβ42 levels and percent of runs of SWS less than 3 minutes (r = 0.42, P = 0.01). Using a partial correlation to control for age, we showed a continued significant positive association between CSF Aβ42 levels and percent of runs of SWS less than 3 minutes (r = 0.36, P < 0.05), suggesting that controlling for age had little effect on the strength of the relationship between these variables. A partial correlation controlling for age demonstrated a reduced strength of association between CSF Aβ42 levels and SWS mean bout length (r = −0.31, P = 0.069), suggesting that age may have some mediating effect on this relationship.
Effects of Mild SDB on the Effect of SWS on CSF Aβ42 Levels
To conclude, although most subjects in our group had no significant SDB, based on our prior work showing positive associations between severity of SDB and CSF AD biomarkers,11 we repeated the above analysis controlling for AHI4% and AHIall. While controlling for AHI4% did not significantly modify the associations (data not shown), controlling for AHIall increased the strength of the associations between CSF Aβ42 levels and SWS duration (r = −0.38, P < 0.05), %SWS (r = −0.39, P < 0.05), total SWA in F4 (r = −0.46, P < 0.01), SWA in cycle 1 in F4 (r = −0.47, P < 0.01), and SWA in cycle 2 in F4 (r = −0.43, P = 0.01).
DISCUSSION
Understanding the relation between sleep and Aβ might present important opportunities for therapy to delay the onset of AD. Although soluble Aβ42 levels may be influenced by several factors in the elderly, changes in sleep common in late-life such as age-dependent loss of SWS and increased incidence of insomnia and sleep apnea24 could lead to relative high brain soluble Aβ42 levels in the stages prior to amyloid deposition. Our findings provide a link between diminished SWS duration, continuity, and frontal SWA with high CSF Aβ42 in a normal aging group.
Evidence from both human and animal models suggests that Aβ production is neuronal activity-dependent, following a diurnal pattern wherein peak levels occur during periods of activity and decline during sleep.12–14 Aβ production is thus postulated to decrease predominantly during SWS due to the decreased neuronal activity observed in this sleep stage. In view of our results, a decrease in CSF Aβ42 would occur in periods of sleep with high SWA in the frontal lobes, although these relationships were also observed in the central electrodes and were present in all NREM sleep cycles.
We did not observe significant associations between measures of slow wave sleep and CSF Aβ40, a phenomenon possibly influenced by the age of our cohort. Significant inverse associations between SWS and CSF Aβ40 have been found previously in a cohort containing younger middle aged (53.2 ± 5.7) individuals.25 In any case, the consistent inverse associations between SWS and Aβ42 remain of special interest as this peptide appears to have the greatest propensity to deposit into insoluble plaques.
In a recent human study, nadirs in lumbar CSF Aβ42 levels occurred at 10 AM, 6 hours later than the peak sleep time (4 AM), after which most SWS has occurred and after which sleep is predominated by stages NREM1-2 and REM.3 The timing of the LPs in the current study occurred within a roughly 2-hour window. Therefore, it is possible that some of the variance observed is related to different timing of the LPs, however we did not find a significant correlation between LP time and CSF Aβ42 levels. Attenuation of the Aβ diurnal pattern with age has been previously described in a study in which circadian amplitudes were approximately 2 times higher for both Aβ40 and Aβ42 in the younger healthy group.3 This may be explained by a relative increase in neuronal activity following disturbed sleep with advanced age, possibly reflecting chronic age-related loss of SWS amongst other factors.
Synaptic downscaling during sleep is thought to be necessary to counter waking activity synaptic potentiation and associated growth, which would otherwise exceed available resources of energy and space.26 This synaptic homeostasis theory proposes that most downscaling is achieved during SWS. Given that synaptic activity is thought to increase CSF Aβ concentrations, and SWS is a stage of sleep where there is a decrease in brain connectivity and a global downscaling, SWS may therefore be the stage that is most responsible for the morning after sleep decreases in CSF Aβ42.27 An additional possible mechanism involves sleep's putative role in the clearance of brain metabolites, including Aβ.28 Fragmented SWS would reduce egress of Aβ out of the brain, leaving higher concentrations in the brain interstitial fluid that is ultimately reflected in CSF concentrations, as suggested by the fact that controlling for AHIall,29,30 which has an excellent correlation with NPSG EEG defined arousals31 and is a good measure of SDB-related sleep fragmentation in mild SDB, increased the strength of the associations.
It bears noting that the functional significance of elevated CSF Aβ42 levels is not established. Although it makes intuitive sense that higher concentrations of CSF Aβ42 would foster its aggregation, longitudinal studies of how CSF concentrations of Aβ42 change over time, particularly as cognitively normal subjects progress to dementia, has not been carefully studied. While mouse models show early increases in Aβ before late decreases,32 there is also longitudinal evidence of preclinical elevations in CSF Aβ42 prior to decreases in human subjects who were not carrying mutations in APP, PSEN1, or PSEN2 genes33 and cross sectional evidence of preclinical CSF Aβ42 elevations in cognitively normal elderly in early pre-symptomatic stages of the disease and ApoE4 negative status.34,35 While it has been demonstrated that CSF levels of Aβ42 are about 50% of control levels when compared to age-matched subjects without AD,36 it remains to be determined how universal a period of elevated CSF Aβ42 in humans is prior to decline as our data suggests.
Limitations of this study are that the interval between polysomnography and CSF collection was of 6.7 ± 7.5 months, the fact that LPs occurred within a roughly 2-hour window and that sleep times in the lab were slightly later than sleep times reported at home. Although neither SWS nor CSF Aβ42 change markedly over this short duration in normal subjects, we nonetheless recognize this may have introduced variability. Additionally, measurement of SWS itself may have been affected by the equipment required for its recording. However, most subjects completed home sleep monitoring as part of existing studies prior to in-lab polysomnography such that some level of acclimation to the recording equipment was likely.
Our results cannot define the causal relationship between reduced SWS and high CSF Aβ42. Although we favor the model in which age-dependent long term disruption of SWS promotes higher Aβ42, disturbed sleep may alternatively be a consequence of accumulated extracellular Aβ42 early in the progression of AD pathology rather than a key event in AD pathogenesis. The sleep-wake changes described in APP-PS1 mice may be due to induced changes in synaptic activity and excitability occurring in brain regions affected by amyloid deposition.37 In humans, a recent study found that individuals diagnosed with AD had fewer neurons than controls in the intermediate nucleus, a brain region that is thought to promote sleep by inhibiting wake-promoting brain regions.38 Additionally, the impairment in sleep-dependent declarative memory consolidation observed in subjects with high amyloid load was found to be mediated by the loss in frontal SWA,39 suggesting that changes in SWS lie downstream of Aβ42 deposition. Because these observations are not mutually exclusive, the interaction between a loss of SWS and Aβ42 may perpetuate a positive feedforward-cycle. Amyloid deposition may damage neurons responsible for generating slow waves, further disturbing sleep and elevating Aβ42 levels during wakefulness,13 until a certain degree of amyloid burden is reached that captures and prevents the transport of soluble Aβ42 from the brain to the lumbar-space CSF.40
CONCLUSIONS
Irrespective of an effect of amyloid on sleep, the potential of an effect of SWS on soluble Aβ42 is exciting because it raises the possibility that modifying SWS can slow AD progression. The mechanism by which SWS can be modified may take many forms. In older subjects with SDB, treatment with CPAP improves sleep architecture including SWS.41 In older subjects without SDB, transcranial magnetic stimulation42 and existing medications43,44 may increase or trigger SWS and reduce Aβ42 production. Whether such interventions affect Aβ42 metabolism and/or disease progression remains to be tested, but our current findings support further investigation.
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by grants from: NIH/NIA/NHLBI R01HL118624, R01HL111724, R21AG049348, R01AG035137, R01AG032554, R01AG022374, R01AG13616 and R01AG1210; Foundation for Research in Sleep Disorders and CTSI UL1TR000038; R01HL118624 1R21AG049348-01, the American Sleep Medicine Foundation Junior Faculty Award, the Leon Levy Foundation. CIBER-BBN is an initiative of the Instituto de Salud Carlos III, Spain. This work has been partially supported by the Ministry of Economy and Competitiveness (MINECO), Spain, under contract DPI2014-59049-R. Additional support is acknowledged from the philanthropy of Mr. James B. Kuhn. Dr. Ayappa has received research support from Fisher & Paykel Healthcare; holds multiple US and foreign patents covering techniques and analysis algorithms for the diagnosis of obstructive sleep apnea hypopnea syndrome (OSAHS) and techniques for administering continuous positive airway pressure (CPAP). Several of these have been licensed to Fisher & Paykel Healthcare and Advanced Brain Monitoring. Dr. Rapoport has received research support from Fisher & Paykel Healthcare, Ventus Medical, and speaking and consulting engagements for Fisher & Paykel Healthcare. Dr. Rapoport. holds multiple US and foreign patents covering techniques and analysis algorithms for the diagnosis of OSAHS and techniques for administering CPAP. Several of these have been licensed to Biologics, Fisher & Paykel Healthcare, Advanced Brain Monitoring, and Tyco (Health C'Aire). Dr. de Leon serves on the external advisory board of Roche Pharmaceuticals and holds patents issued through NYU related to the image analysis of PET and MRI scans. Dr. Blennow has served at Advisory Boards for Roche Diagnostic, IBL International, Eli Lilly, and has served on the speakers bureau of Fujirebio Europe and Lundbeck. He also has received research support from the Swedish Research Council, grant #14002. Drs. Blennow and Zetterberg are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based company at the University of Gothenburg. The other authors have indicated no financial conflicts of interest. Drs. Varga, Burschtin, Rapoport, and Ayappa are currently affiliated with Mount Sinai Integrative Sleep Center, Division of Pulmonary Critical Care, and Sleep Medicine, Icahn School of Medicine at Mount Sinai, New York, NY.
ACKNOWLEDGMENTS
The authors are indebted to the study subjects for their patience, and for their participation in and contribution to the research. The authors acknowledge contributions to patient recruitment and data collection by Ms. Margo Miller, Ms. Kimberly Clay, Mr. Michael Yablon, Ms. Christine Grosso, and Ms. Gabriella Petrongolo. They also thank Dr. Tracy Butler and Dr. Pauline McHugh for their assessment of research subjects.
REFERENCES
- 1.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang Y, Potter R, Sigurdson W, et al. Effects of age and amyloid deposition on abeta dynamics in the human central nervous system. Arch Neurol. 2012;69:51–8. doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71:971–7. doi: 10.1001/jamaneurol.2014.1173. [DOI] [PubMed] [Google Scholar]
- 5.Ovsepian SV, O'Leary VB. Neuronal activity and amyloid plaque pathology: an update. J Alzheimers Dis. 2015;49:13–9. doi: 10.3233/JAD-150544. [DOI] [PubMed] [Google Scholar]
- 6.McGinty D, Szymusiak R. Keeping cool: a hypothesis about the mechanisms and functions of slow-wave sleep. Trends Neurosci. 1990;13:480–7. doi: 10.1016/0166-2236(90)90081-k. [DOI] [PubMed] [Google Scholar]
- 7.Steriade M, Amzica F, Contreras D. Synchronization of fast (30-40 Hz) spontaneous cortical rhythms during brain activation. J Neurosci. 1996;16:392–417. doi: 10.1523/JNEUROSCI.16-01-00392.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peskind ER, Li G, Shofer J, et al. Age and apolipoprotein E4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol. 2006;63:936–9. doi: 10.1001/archneur.63.7.936. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda N, Honma H, Kohsaka M, et al. Gender difference of slow wave sleep in middle aged and elderly subjects. Psychiatry Clin Neurosci. 1999;53:151–3. doi: 10.1046/j.1440-1819.1999.00508.x. [DOI] [PubMed] [Google Scholar]
- 10.Almeida RP, Schultz SA, Austin BP, et al. Effect of cognitive reserve on age-related changes in cerebrospinal fluid biomarkers of Alzheimer disease. JAMA Neurol. 2015;72:699–706. doi: 10.1001/jamaneurol.2015.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osorio RS, Ayappa I, Mantua J, et al. The interaction between sleep-disordered breathing and apolipoprotein E genotype on cerebrospinal fluid biomarkers for Alzheimer's disease in cognitively normal elderly individuals. Neurobiol Aging. 2014;35:1318–24. doi: 10.1016/j.neurobiolaging.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21:249–58. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 13.Glodzik L, Rusinek H, Brys M, et al. Framingham cardiovascular risk profile correlates with impaired hippocampal and cortical vasoreactivity to hypercapnia. J Cereb Blood Flow Metab. 2011;31:671–9. doi: 10.1038/jcbfm.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glodzik L, Mosconi L, Tsui W, et al. Alzheimer's disease markers, hypertension, and gray matter damage in normal elderly. Neurobiology Aging. 2011;33:1215–27. doi: 10.1016/j.neurobiolaging.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varga AW, Ducca EL, Kishi A, et al. Effects of aging on slow-wave sleep dynamics and human spatial navigational memory consolidation. Neurobiol Aging. 2016;42:142–9. doi: 10.1016/j.neurobiolaging.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mander BA, Rao V, Lu B, et al. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat Neurosci. 2013;16:357–64. doi: 10.1038/nn.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yau PL, Kang EH, Javier DC, Convit A. Preliminary evidence of cognitive and brain abnormalities in uncomplicated adolescent obesity. Obesity (Silver Spring) 2014;22:1865–71. doi: 10.1002/oby.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–80. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 19.Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8:597–619. doi: 10.5664/jcsm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osorio RS, Ayappa I, Mantua J, et al. The interaction between sleep-disordered breathing and apolipoprotein E genotype on cerebrospinal fluid biomarkers for Alzheimer's disease in cognitively normal elderly individuals. Neurobiol Aging. 2013;35:1318–24. doi: 10.1016/j.neurobiolaging.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinberg I, Floyd TC. Systematic trends across the night in human sleep cycles. Psychophysiology. 1979;16:283–91. doi: 10.1111/j.1469-8986.1979.tb02991.x. [DOI] [PubMed] [Google Scholar]
- 22.Aeschbach D, Borbely AA. All-night dynamics of the human sleep EEG. J Sleep Res. 1993;2:70–81. doi: 10.1111/j.1365-2869.1993.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 23.Wauquier A, van SB, Lagaay AM, Kemp B, Kamphuisen HA. Ambulatory monitoring of sleep-wakefulness patterns in healthy elderly males and females (greater than 88 years): the “Senieur” protocol. J Am Geriatr Soc. 1992;40:109–14. doi: 10.1111/j.1532-5415.1992.tb01928.x. [DOI] [PubMed] [Google Scholar]
- 24.Lucey BP, Bateman RJ. Amyloid-beta diurnal pattern: possible role of sleep in Alzheimer's disease pathogenesis. Neurobiol Aging. 2014;35S2:S29–34. doi: 10.1016/j.neurobiolaging.2014.03.035. [DOI] [PubMed] [Google Scholar]
- 25.Ju YS, Finn MB, Sutphen CL, et al. Obstructive sleep apnea decreases central nervous system-derived proteins in the cerebrospinal fluid. Ann Neurol. 2016;80:154–9. doi: 10.1002/ana.24672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Born J, Feld GB. Sleep to upscale, sleep to downscale: balancing homeostasis and plasticity. Neuron. 2012;75:933–5. doi: 10.1016/j.neuron.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosselet J, Ayappa I, Norman RG, Krieger AC, Rapoport DM. Classification of sleep-disordered breathing. Am J Respir Crit Care Med. 2001;163:398–405. doi: 10.1164/ajrccm.163.2.9808132. [DOI] [PubMed] [Google Scholar]
- 30.Hosselet JJ, Norman RG, Ayappa I, Rapoport DM. Detection of flow limitation with a nasal cannula/pressure transducer system. Am J Respir Crit Care Med. 1998 May;157(5 Pt 1):1461–7. doi: 10.1164/ajrccm.157.5.9708008. [DOI] [PubMed] [Google Scholar]
- 31.Ayappa I, Norman RG, Suryadevara M, Rapoport DM. Comparison of limited monitoring using a nasal-cannula flow signal to full polysomnography in sleep-disordered breathing. Sleep. 2004;27:1171–9. doi: 10.1093/sleep/27.6.1171. [DOI] [PubMed] [Google Scholar]
- 32.Maia LF, Kaeser SA, Reichwald J, et al. Increased CSF Abeta during the very early phase of cerebral Abeta deposition in mouse models. EMBO Mol Med. 2015;7:895–903. doi: 10.15252/emmm.201505026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alcolea D, Martinez-Lage P, Sanchez-Juan P, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology. 2015;85:626–33. doi: 10.1212/WNL.0000000000001859. [DOI] [PubMed] [Google Scholar]
- 35.Osorio RS, Pirraglia E, Gumb T, et al. Imaging and cerebrospinal fluid biomarkers in the search for Alzheimer's disease mechanisms. Neurodegener Dis. 2014;13:163–5. doi: 10.1159/000355063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer's disease. Neurotherapeutics. 2004;1:213–25. doi: 10.1602/neurorx.1.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roh JH, Huang Y, Bero AW, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med. 2012;4:150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim AS, Ellison BA, Wang JL, et al. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain. 2014;137(Pt 10):2847–61. doi: 10.1093/brain/awu222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mander BA, Marks SM, Vogel JW, et al. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18:1051–7. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 41.Verma A, Radtke RA, VanLandingham KE, King JH, Husain AM. Slow wave sleep rebound and REM rebound following the first night of treatment with CPAP for sleep apnea: correlation with subjective improvement in sleep quality. Sleep Med. 2001;2:215–23. doi: 10.1016/s1389-9457(00)00069-1. [DOI] [PubMed] [Google Scholar]
- 42.Bellesi M, Riedner BA, Garcia-Molina GN, Cirelli C, Tononi G. Enhancement of sleep slow waves: underlying mechanisms and practical consequences. Front Syst Neurosci. 2014;8:208. doi: 10.3389/fnsys.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walsh JK. Enhancement of slow wave sleep: implications for insomnia. J Clin Sleep Med. 2009;5(2 Suppl):S27–32. [PMC free article] [PubMed] [Google Scholar]
- 44.Walsh JK, Hall-Porter JM, Griffin KS, et al. Enhancing slow wave sleep with sodium oxybate reduces the behavioral and physiological impact of sleep loss. Sleep. 2010;33:1217–25. doi: 10.1093/sleep/33.9.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]