

Abstract
Familial tumoral calcinosis (FTC)/hyperostosis-hyperphosphatemia syndrome (HHS) is a rare disorder caused by mutations in the genes encoding fibroblast growth factor-23 (FGF23), N-acetylgalactosaminyltransferase 3 (GALNT3), or KLOTHO. The result is functional deficiency of, or resistance to, intact FGF23 (iFGF23), causing hyperphosphatemia, increased renal tubular reabsorption of phosphorus (TRP), elevated or inappropriately normal 1,25-dihydroxyvitamin D3 (1,25D), ectopic calcifications and/or diaphyseal hyperostosis. Eight subjects with FTC/HHS were studied and treated. Clinical manifestations varied, even within families, ranging from asymptomatic to large, disabling calcifications. All subjects had hyperphosphatemia, increased TRP, and elevated or inappropriately normal 1,25D. C-terminal FGF23 was markedly elevated while iFGF23 was comparatively low, consistent with increased FGF23 cleavage. Radiographs ranged from diaphyseal hyperostosis to massive calcification. Two subjects with severe calcifications also had overwhelming systemic inflammation and elevated C-reactive protein (CRP). GALNT3 mutations were identified in 7 subjects; no causative mutation was found in the eighth. Biopsies from 4 subjects showed ectopic calcification and chronic inflammation, with areas of heterotopic ossification observed in 1 subject. Treatment with low phosphate diet, phosphate binders, and phosphaturia-inducing therapies was prescribed with variable response. One subject experienced complete resolution of a calcific mass after 13 months of medical treatment. In the 2 subjects with systemic inflammation, interleukin-1 (IL-1) antagonists significantly decreased CRP levels with resolution of calcinosis cutis and peri-lesional inflammation in one subject and improvement of overall well-being in both subjects. This cohort expands the phenotype and genotype of FTC/HHS and demonstrates the range of clinical manifestations despite similar biochemical profiles and genetic mutations. Overwhelming systemic inflammation has not been described previously in FTC/HHS; the response to IL-1 antagonists suggests that anti-inflammatory drugs may be useful adjuvants. In addition, this is the first description of heterotopic ossification reported in FTC/HHS, possibly mediated by the adjacent inflammation.
Keywords: Familial tumoral calcinosis, fibroblast growth factor 23, hyperostosis-hyperphosphatemia syndrome, hyperphosphatemia
INTRODUCTION
Hyperphosphatemic familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome (FTC/HHS) (OMIM 211900) is a rare autosomal recessive disorder caused by mutations in the genes encoding one of the following: fibroblast growth factor 23 (FGF23, 12p13.3) (1), UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase-T3 (GalNAc-T3) (GALNT3, 2q24-q31) (2), or Klotho (KL, 13q12) (3). FGF23 is produced by bone cells and is a primary regulator of phosphate homeostasis (4), with the intact hormone (iFGF23) acting via the fibroblast growth factor receptor 1 (FGFR1) along with its co-receptor Klotho to inhibit sodium phosphate co-transporter (NaPi-2a and-2c) and 25-hydroxyvitamin D 1-α-hydroxylase expression. This leads to increased renal phosphate excretion and decreased serum 1,25-dihydroxyvitamin D3 (1,25D), lowering the blood phosphate level. Intact FGF23 undergoes post-translational O-linked glycosylation by GalNAc-T3 at a pro-protein convertase cleavage site, protecting iFGF23 from cleavage into inactive fragments (5). Thus, in the absence of O-linked glycosylation, iFGF23 is readily cleaved and inactivated.
The mutations causing FTC/HHS result in relative deficiency of, or resistance to, iFGF23, which leads to hyperphosphatemia, due to increased renal tubular reabsorption of phosphate (TRP), and elevated or inappropriately normal 1,25D production, which promotes gastrointestinal absorption of phosphorus and calcium. The net effect is an increase in the calcium × phosphate product. Affected individuals develop ectopic calcifications and/or diaphyseal hyperostosis, which may manifest clinically as diaphyseal pain of the long bones and is often erroneously diagnosed as osteomyelitis (6). Characteristic dental findings of FTC/HHS are often present including short bulbous teeth, shortened roots with dilacerations, thistle-shaped dental pulps, pulp chamber and root canal obliteration, and pulp stones (7–10).
There is no standard treatment for this disease, and studies of the treatment of FTC/HHS do not exist aside from case reports or case series. Most reported therapies attempt to lower serum phosphate by instituting low phosphate diet, phosphate binders, or medications that promote renal excretion of phosphate (11–13). However, clinical response to these treatments is quite variable. Surgical debulking may be performed in subjects with functional impairment or severe pain, but is not routinely undertaken as calcinosis often recurs (14–16). In addition, post-surgical morbidity from attempted complete excision may occur. Here we present the clinical and molecular characterization of a cohort of subjects with FTC/HHS evaluated and treated with previously reported and novel therapies at the National Institutes of Health (NIH).
MATERIALS AND METHODS
Subjects
This study was approved by the Institutional Review Board of the National Institute of Dental and Craniofacial Research, NIH. Subjects were referred by their home physicians. Adult subjects or the parents of minors provided written consent to the study protocol; minor subjects > 7 years of age provided written assent. Clinical evaluations performed by subjects’ home physicians occurred prior to consenting to the NIH study, and subjects permitted use of this data for research purposes/publication upon signing informed consent. All subjects received a predefined set of evaluations that included detailed medical and family history, physical examination, dental examination, standard biochemical evaluation, detailed laboratory evaluation of calcium and phosphorus metabolism including 1,25D, intact parathyroid hormone (iPTH), TRP, iFGF23 and C-terminal FGF23 (CFGF23), genetic testing, and standard radiographs. The results of the standard evaluation were used to guide additional studies, e.g. computed tomography (CT) of symptomatic lesions, colonoscopy for evaluation of gastrointestinal luminal masses, etc. However, additional factors were considered in obtaining further studies, e.g. minimizing radiation exposure in children.
Biochemical
Testing included routine blood and urine chemistries, complete blood count (CBC), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), iPTH (electrochemiluminescence immunoassay on Roche Cobas e601 analyzer, NIH), and 1,25D (radioimmunoassay or liquid chromatography-tandem mass spectrometry, Mayo Medical Laboratories, Rochester, MN). iFGF23was measured by ELISA (Kainos Laboratories, Tokyo, Japan, or Immutopics International, San Clemente, CA), and CFGF23 was measured by ELISA (Immutopics International, San Clemente, CA). The iFGF23 assay measures only the intact protein while the CFGF23 assay reflects both the intact and C-terminal fragments.
Genetic analysis
For GALNT3 sequencing, genomic DNA was isolated from leukocytes using standard procedures. All coding exons and their adjacent intronic sequences in GALNT3 were amplified by PCR, using Multiplex PCR Kit (QIAGEN Inc., Valencia, CA). Approximately 100 ng of purified PCR products were directly sequenced from PCR primers, using Big-Dye Terminator Cycle Sequencing Kit and the ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA)(14). DNA sequencing of FGF23 from whole blood was performed in a commercial laboratory (GeneDx, Gaithersburg, MD). No subjects were tested for mutations in KLOTHO.
Imaging
Tumoral calcinosis was assessed by conventional radiography and, in some, by CT. Two subjects had magnetic resonance imaging of tumoral calcinosis, and one underwent colonoscopy. Six subjects underwent dual energy x-ray absorptiometry (DXA) (Hologic QDR 4500; Hologic Inc, Waltham, MA) of the anteroposterior (AP) spine, total hip, and 1/3 radius; sites with tumoral calcinosis were excluded. Height-adjusted Z-scores for bone mineral density (BMD) were calculated in subjects less than 20 years of age (17). Cardiac CT (Toshiba Aquillion ONE, Otawara, Japan) was performed in 5 subjects. Cardiac calcifications were quantified by using the Agatston calcium score (18). Dental radiographs were obtained in all subjects.
Treatment
All subjects received nutritional counseling and were prescribed a low phosphate diet targeting 400–900 mg/day, depending upon age. Prescription of phosphate-lowering medications was determined individually, depending on burden of calcinosis and response to previous therapies. Treatment of all subjects except FTC1 included sevelamer, a phosphate binder, prescribed with meals and snacks. All subjects but FTC3 and FTC5, who had no tumoral calcinosis, were additionally treated with acetazolamide and/or probenecid, both of which promote renal phosphate excretion. The acetazolamide was titrated up while closely monitoring the serum bicarbonate. The dose was held or decreased if the serum bicarbonate reached ≤ 16 mmol/L. The probenecid was increased until the blood uric acid level was suppressed or the maximum dose for age was reached. Aluminum hydroxide was added as an additional phosphate binder in 2 subjects, who had increased tumoral calcinosis burden with no change in blood phosphorus on sevelamer, acetazolamide and probenecid. Treatment with nicotinamide was attempted during a hospitalization in 2 subjects to assess its effect on inducing phosphaturia but was not continued upon discharge, as it did not show additional benefits. Treatment efficacy was determined by changes in serum phosphorus, TRP, and tumoral calcinosis burden. One subject with severe functional impairment underwent surgical debulking of tumoral calcinosis at NIH while others had resections performed at outside institutions.
Two subjects with systemic inflammation were treated with either the monoclonal antibody to IL-1β, canakinumab, or the recombinant IL-1 receptor antagonist, anakinra. Doses were increased as tolerated in an effort to normalize CRP levels.
RESULTS
Clinical findings
Eight subjects (FTC1-FTC8) from six families with clinical and/or biochemical features consistent with FTC/HHS were examined (Table 1). On initial evaluation, 6 subjects had tumoral calcinosis on physical examination, typically periarticular, and of various sizes in multiple locations (Figure 1). Some lesions were painful and debilitating, reducing quality of life. FTC4 had a frozen left shoulder along with decreased range of motion of the left elbow due to tumoral calcinosis, which significantly affected self-care and activities of daily living, such as toileting and feeding. FTC3 and FTC5 are siblings of significantly affected subjects (FTC2 and FTC4, respectively), yet initially had no physical exam findings despite characteristic biochemical findings on screening laboratory evaluation (Table 1). FTC3 later developed tumoral calcinosis at the elbow noted at the three-year follow-up visit.
Table 1.
Clinical, biochemical, and genetic findings in FTC/HHS cohort at initial visit
| FTC1(7) | FTC2 | FTC3α | FTC4 | FTC5β | FTC6 | FTC7 | FTC8 | ||
|---|---|---|---|---|---|---|---|---|---|
| Age at Symptom Onset |
12yr | 5.25yr | 9.9yr | 10yr | NA | 3yr | 45yr | 20mo | |
| Age at Initial NIH Visit |
36yr | 6yr | 8yr | 29yr | 19yr | 12.5yr | 56yr | 6yr | |
| Sex | F | F | F | M | M | F | F | F | |
| Clinical Findings | Calcinosis | Yes | Yes | No | Yes | No | Yes | Yes | Yes |
| Dental Findings | Yes | Yes | Yes | Yes | Yes | Yes | No | No | |
| Diaphysitis | Yes | Yes | No | Yes | No | Yes | No | Yes | |
| Treatment | ACZ, NC | SEV, ACZ, PB, AL, CAN |
SEV, ACZ, PB |
SEV, ACZ, PB, NC, AL, ANA |
NA | SEV, ACZ, PB |
SEV, ACZ, PB |
SEV, ACZ, PB |
|
| Phosphorus (Age specific normal range mg/dL) |
7.3 (2.5–4.5) |
8.8 (3.0–5.7) |
6.8 (3.0–5.7) |
5.5 (2.5–4.5) |
6.3 (2.5–4.5) |
6.6 (3.0–5.7) |
5.5 (2.5–4.5) |
7.7 (3.0–5.7) |
|
| Calcium (8.2–10 mg/dL) |
8.8 | 8.48 | 9.72 | 9.64 | 9.84 | 9.52 | 8.68 | 9.76 | |
| Calcium × Phosphorus (<55 mg2/dL2) |
64 | 75 | 66 | 53 | 62 | 63 | 48 | 75 | |
| 1,25-OH Vitamin D3 (24–86 pg/mL) |
61 | 89 | 74 | 74 | 59 | 63 | 34 | 38 | |
| Intact PTH (15–65 pg/mL) |
23 | 12 | 19 | 55 | 17 | 25 | 27 | 17 | |
| TRP (85–95%) |
96.5 | 98.2 | 98.8 | 92 | 96.6 | 96 | 91.3 | 97.3 | |
| TP/GFR (Age specific mean mg/dL(19)) |
7.05 (3.2) |
8.6 (4.4) |
6.72 (4.4) |
5.15 (3.2) |
5.31 (3.2) |
6.34 (4.4) |
4.31 (3.2) |
6.81 (4.4) |
|
| iFGF23 (22–63 pg/mL) |
17† | 34 | 39 | 8† | 39 | 38† | 55 | 77 | |
| CFGF23 (3M–17Y≤ 230, ≥18Y ≤180 RU/mL) |
1210 | 887 | 1047 | 573 | 971 | 1827 | 520 | 1031 | |
| Total FGF23/iFGF23 | 72 | 24 | 28 | 73 | 26 | 36 | 10 | 14 | |
| CRP (<4.99 mg/L) | 2.59 | 81.8 | 0.26 | 245.1 | 4.1 | 2.5 | 19.3 | 6.8 | |
| ESR (0–42 mm/hr) | 7.0 | 90.9 | 14 | 127 | NA | 7 | 70 | 32 | |
| Genetic Mutations |
GALNT3 c.1312 C>T/ c.1774 C>T |
GALNT3 c.516- 2A>T/ c.260_ 266delGG CAAA* |
GALNT3 c.516- 2A>T/ c.260_ 266delGG CAAA* |
GALNT3 c.1584_1 585 insA* |
GALNT3 c.1584_1 585 insA* |
GALNT3 c.516- 2A>T c.1524+5 G>A |
Unknown |
GALNT3 c.746_749d elTCAG* c.892 delT* |
|
Boxes indicate siblings.
FTC3 is the phenotypically unaffected sibling of FTC2.
FTC5 is the phenotypically unaffected sibling of FTC4.
Novel mutation
Kainos iFGF23 ELISA
NA: not applicable, SEV: sevelamer, ACZ: acetazolamide, PB: probenecid, AL: aluminum hydroxide, NC: nicotanimide, ANA: anakinra, CAN: canakinumab, PTH: parathyroid hormone, TRP: tubular reabsorption of phosphorus, TmP/GFR: tubular maximum reabsorption of phosphorus/glomerular filtration rate, iFGF23: intact fibroblast growth factor 23, CFGF23: c-terminal fibroblast growth factor 23, Total FGF23: iFGF23 + CFGF23, CRP: C-reactive protein, ESR: erythrocyte sedimentation rate, GALNT3: N-acetylgalactosaminyltransferase 3. To convert phosphorus to mmol/L, multiply by 0.323. To convert calcium to mmol/L, multiply by 0.25. To convert 1,25-OH Vitamin D3 to pmol/L, multiply by 2.6. For PTH pg/mL = ng/L.
Figure 1. Clinical manifestations of the FTC/HHS cohort.
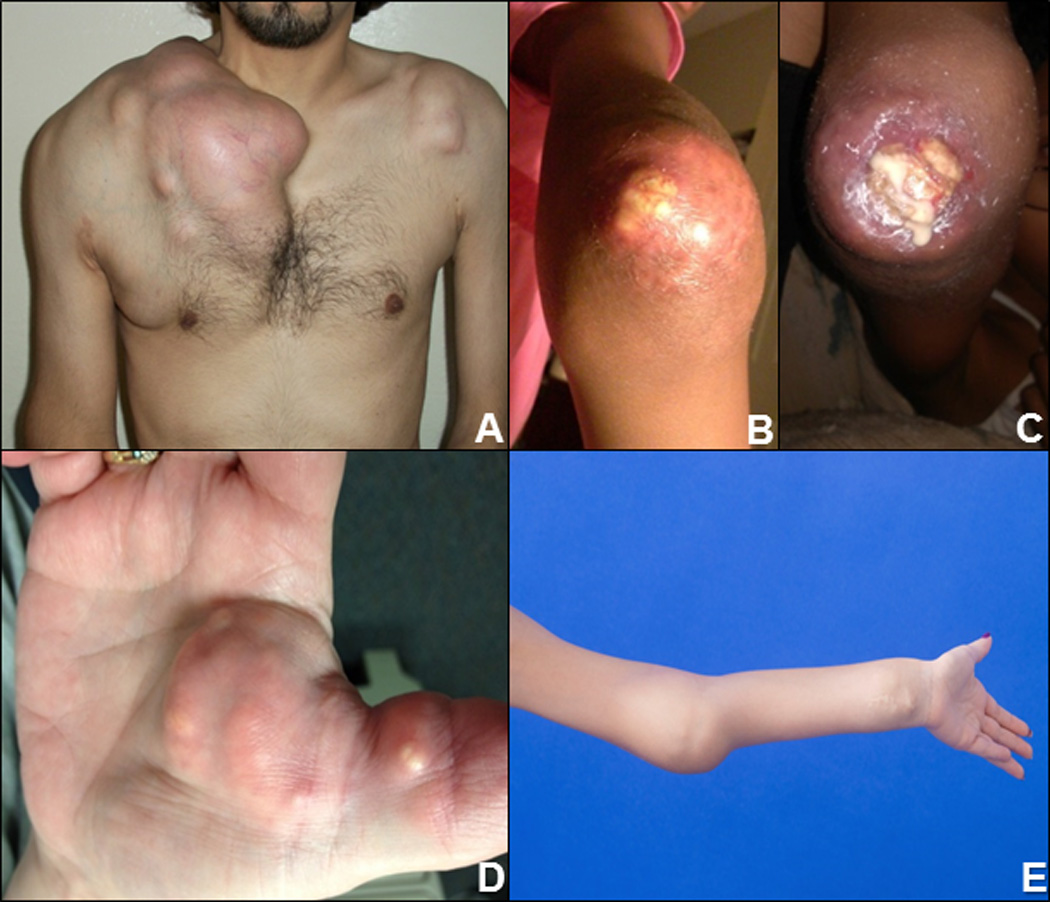
(A) Massive calcifications surrounding the right shoulder and chest in FTC4. (B&C) Tumoral calcinosis of the elbow (38), which later ulcerated and drained thick, white “milk of calcium” in FTC2. (D) Calcifications of the right palmar surface, the dominant hand, in FTC7 who was an avid bowler. (E) Tumoral calcinosis of the elbow and wrist in FTC8.
In addition to tumoral calcinosis, FTC2 had symptoms of intermittent fever as well as the onset of cutaneous calcinosis with inflammatory reaction (Figure 7B). FTC4 had no clinical manifestations of systemic inflammation other than chronic fatigue.
Figure 7. Treatment of systemic inflammation in FTC2 and FTC4.
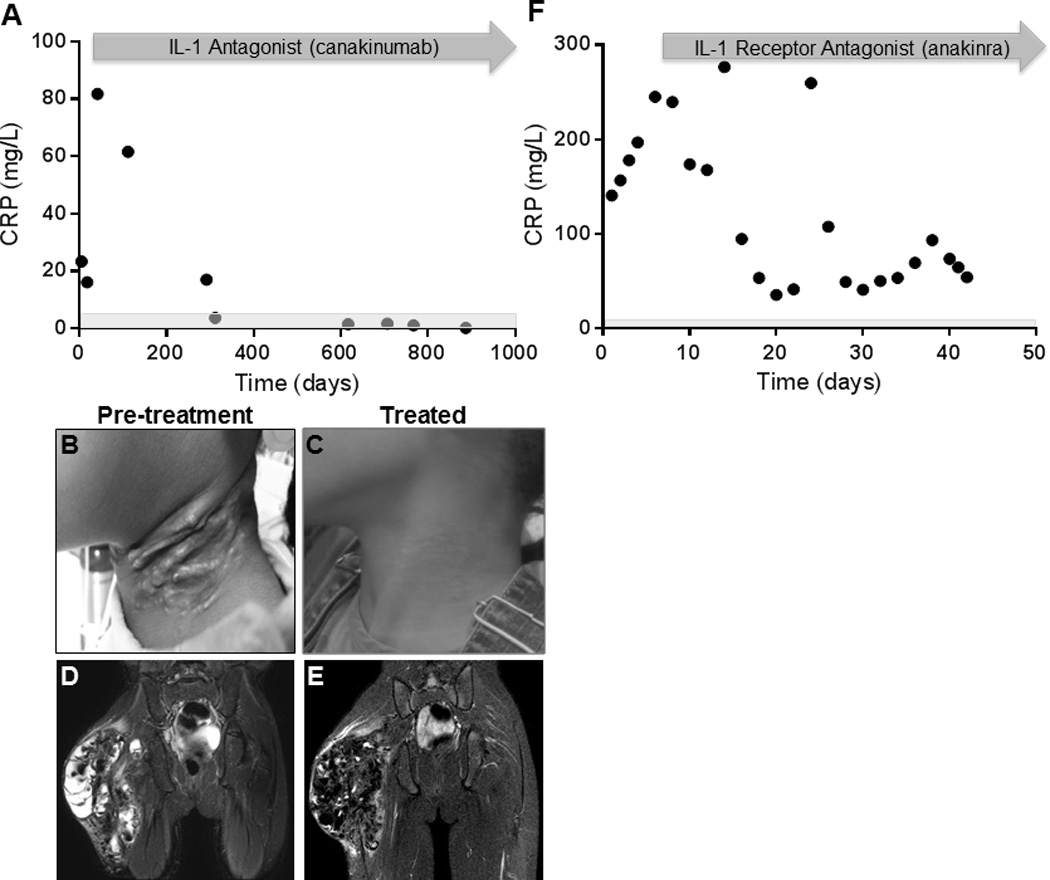
(A) In FTC2, C-reactive protein (CRP) levels normalized and (B&C) painful, erythematous, calcinosis cutis (38) resolved on canakinumab therapy. (D) Increased edema surrounding right hip tumoral calcinosis on pelvis MRI (STIR). Accounting for differences in technique (3.0 T (D) versus 1.5 T (E)), (E) there was also marked improvement in peri-lesional edema following treatment with canakinumab in FTC2. (F) Anakinra decreased CRP levels in FTC4. Rebound CRP elevation observed on Day 25 after a missed dose.
Biochemical findings
Median values are presented in text, individual values can be found in Table 1. All subjects were hyperphosphatemic with elevated TRP [96.5%] and TP/GFR [6.5 mg/dL (2.8 mmol/L)](19). The 1,25D [62 pg/mL(161.2 pmol/L)] was frankly elevated or inappropriately normal for the degree of hyperphosphatemia. Serum calcium, creatinine, and alkaline phosphatase were normal. The calcium × phosphorus product [63.5 mg2/dL2 (5.12 mmol2/L2)] was elevated in all but two patients, according to criteria defined by the National Kidney Foundation Kidney Disease Outcomes Quality Initiative (K/DOQI) guidelines (> 65 mg2/dL2 for <12Y; >55 mg2/dL2 for >12Y) (20). iPTH levels [21 pg/mL (21 ng/L)] were in the lower 1/3 of normal in all subjects, except for FTC4 who had a low 25-OH-Vitamin D3 level of 5 ng/mL (12.5 mmol/L). Consistent with increased FGF23 cleavage, iFGF23 (39 pg/mL) ranged from low to just above normal for all subjects, which is inappropriate given the hyperphosphatemia, while the CFGF23 (1001 RU/mL) was significantly elevated. The ratio of total FGF23 (iFGF23 + CFGF23) to iFGF23 was much higher in subjects with FTC/HHS (35.3 ± 24.3, mean ± SD) compared to normal controls (0.99± 0.3) (21).
Three subjects had biochemical evidence of systemic inflammation with significantly elevated CRP levels (max 81.8 mg/L in FTC2, 245.1 mg/L in FTC4, and 19.3 mg/L in FTC7; nl<4.99 mg/L) and ESR (max 90.9 in FTC2, 127 in FTC4, and 70 in FTC7; nl<25 mm/hr). FTC2 and FTC4 also had anemia consistent with inflammation-mediated chronic disease and thrombocytosis (740K in FTC2 and 490K in FTC4; nl 199–367K). FTC4 had significantly elevated ferritin levels as high as 1137 mcg/L (2555 pmol/L; nl 30–400 mcg/L). FTC2 had leukocytosis with a white blood cell count as high as 219,000. FTC6 had anemia without inflammation, which was judged to be due to nutritional iron deficiency.
Genetic analyses
Five subjects in this cohort had compound heterozygous mutations in GALNT3, three of which were novel. FTC4 and FTC5, siblings from a consanguineous family, had novel homozygous GALNT3 mutations. No mutations in FGF23 or GALNT3 were identified in subject FTC7 and KLOTHO mutation testing was not performed as her biochemical profile (no evidence of iFGF23 resistance) was not consistent with mutations in KLOTHO (Table 1).
Imaging findings
Calcinosis varied widely, ranging from none to involvement of multiple joints (Figure 2). FTC4 was the most significantly affected subject (Figure 2A, 2D, 2E) including substantial intraarticular infiltration and chondral damage of the left shoulder joint, resulting in ankylosis. FTC8 had growth plate obliteration by tumoral calcinosis with ulnar shortening. (Figure 2C).
Figure 2. Imaging of FTC/HHS cohort.
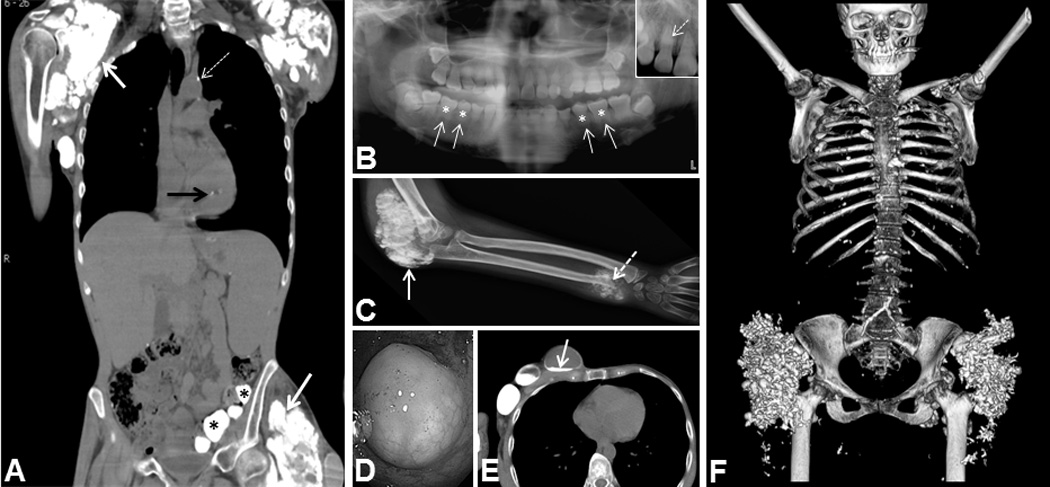
(A) CT showing calcifications in FTC4 including shoulder and hip soft tissue calcifications (white arrows), aortic calcification (dashed arrow), papillary muscle calcification (black arrow), and submucosal gut calcifications (asterisks). (B) Panoramic dental radiograph showing short, bulbous roots (solid arrows) with obliteration of dental pulp (asterisks). Inset - thistle-shaped pulp chambers and a pulp stone (dashed arrow) in FTC5. (C) Radiograph showing tumoral calcinosis of elbow (solid arrow) and ulna with growth plate invasion and destruction in FTC8 (dashed arrow). (D) Submucosal gut calcification on colonoscopy in FTC4. (E) CT showing large cystic masses in the chest wall with fluid levels containing “milk of calcium” in FTC4 (arrow). (F) CT 3-D reconstruction demonstrating extensive calcifications in bilateral hips and scattered calcifications throughout the chest in FTC7.
BMD was within 2 SD of the mean for Z-score (adults and children) and T-score (adults only) for all subjects but one at the AP spine, total hip, and 1/3 radius. FTC4, who presented with severe, longstanding inflammation, had an AP spine BMD of 0.667 gm/cm2, T-score −3.9; total hip BMD of 0.551 gm/cm2, T-score −3.2; and 1/3 radius BMD of 0.612 gm/cm2, T-score −1.4.
FTC4 (32 years old) and FTC7 (56 years old) had cardiac calcifications on CT with Agatston coronary calcium scores of 105 (95th percentile for 45-year old ethnicity-matched men) (Figure 3B) and 157 (95th percentile for age and ethnicity-matched women), respectively. FTC4, who had longstanding untreated chronic inflammation, and FTC7, who had type 2 diabetes mellitus, hypertension, hyperlipidemia, and history of 60 pack-years smoking, both had increased risk factors for development of vascular calcification independently of FTC/HHS. No cardiac calcifications were seen in the other 3 subjects studied (ages 6–36 years).
Figure 3. Cardiac CT angiography in FTC4.
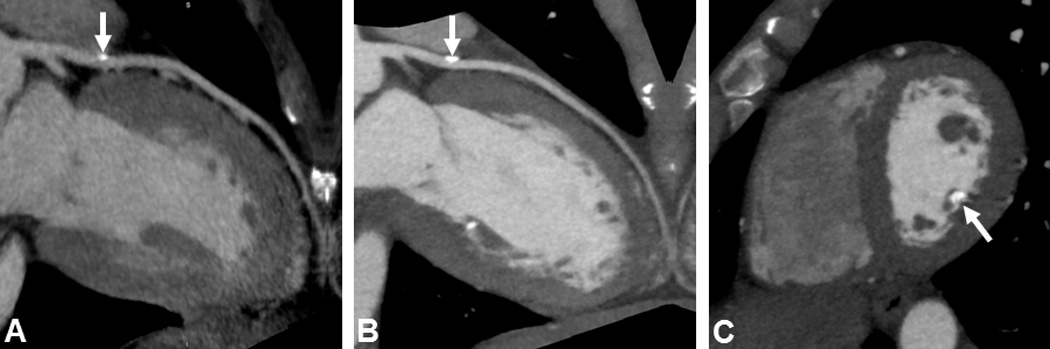
(A) Calcification of left anterior descending (LAD) artery on original scan (arrow) (Agatston score = 6). (B) Increased calcification in LAD 3 years later (arrow) (Agatston score = 105; 95th percentile). (C) Calcification of posterior medial papillary muscle (arrow) (Agatston score = 167), which remained relatively stable over 3y.
Six subjects had characteristic dental findings (Figure 2B).
Histopathology
Specimens obtained following surgical resection of calcific tumors from the right chest and shoulder of FTC4 showed histologic evidence of calcification, as expected (Figure 4A). In addition to the calcification, heterotopic ossification was evident (Figure 4B). This existed within the background of overwhelming chronic inflammation (Figure 4C). Tibia biopsy of FTC6 revealed hyperostosis with reactive woven bone which, over time, is converted to lamellar bone (Figure 5B&C).
Figure 4. Histology (H&E) and electron micrograph (EM) of tumoral calcinosis resected from FTC4.
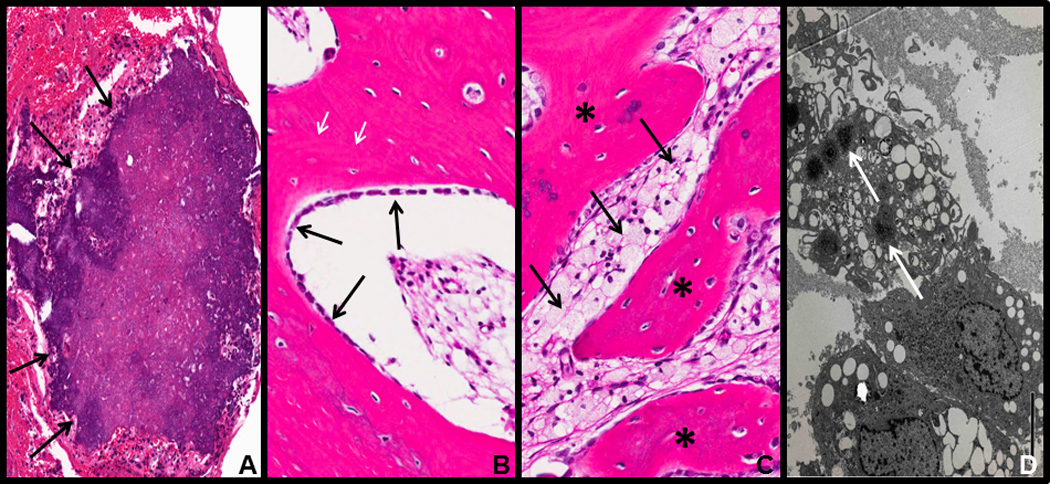
(A) Subcutaneous ectopic calcification (arrows). (B) Heterotopic ossification with osteoblasts laying down new bone (black arrows) as well as reversal lines (white arrows) indicating active bone remodeling. (C) Chronic inflammation with foamy macrophages (arrows) associated with heterotopic ossification (asterisks). (D) EM showing macrophages with stellate needle-like inclusions consistent with calcium deposition (arrows).
Figure 5. Radiographic and histopathologic features of hyperostosis.
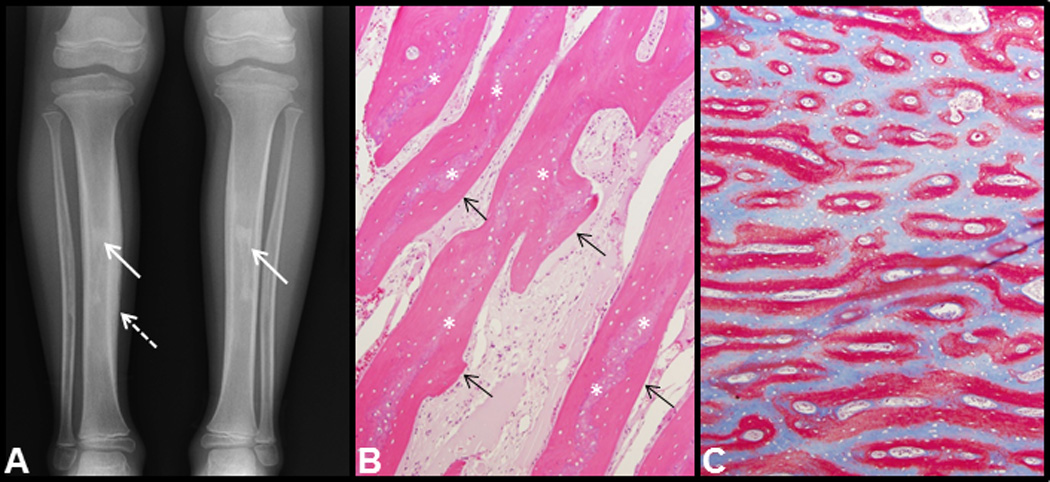
(A) Radiograph tibias/fibulas showing hyperostosis with hypermineralization of the cortical bone (dashed arrow) and patchy sclerosis (solid arrows) of the medullary cavities in FTC2. (B) H&E (100×) stained sections from tibia biopsy of FTC6 show periosteal bone with a central core of woven bone (asterisks) being replaced by lamellar bone (arrows). (C) Goldner trichrome (40×) stained undecalcified sections show woven bone in red and lamellar bone in light blue.
Treatment
Subjects had variable responses to treatment with phosphate-lowering agents. For example, after 13 months on treatment, FTC6’s serum phosphorus and TRP decreased, and the tumoral calcinosis posterior to the left elbow completely resolved on radiograph (Figure 6A). In contrast, despite optimizing medical therapy, there was no significant decrease in FTC4’s serum phosphorus or TRP (Figure 6B). Therefore, phosphate-lowering agents were discontinued in this subject prior to discharge. FTC4 underwent seven surgeries over 3 years in an effort to alleviate pain and improve ambulation.
Figure 6. Response to phosphate-lowering therapies in two subjects.
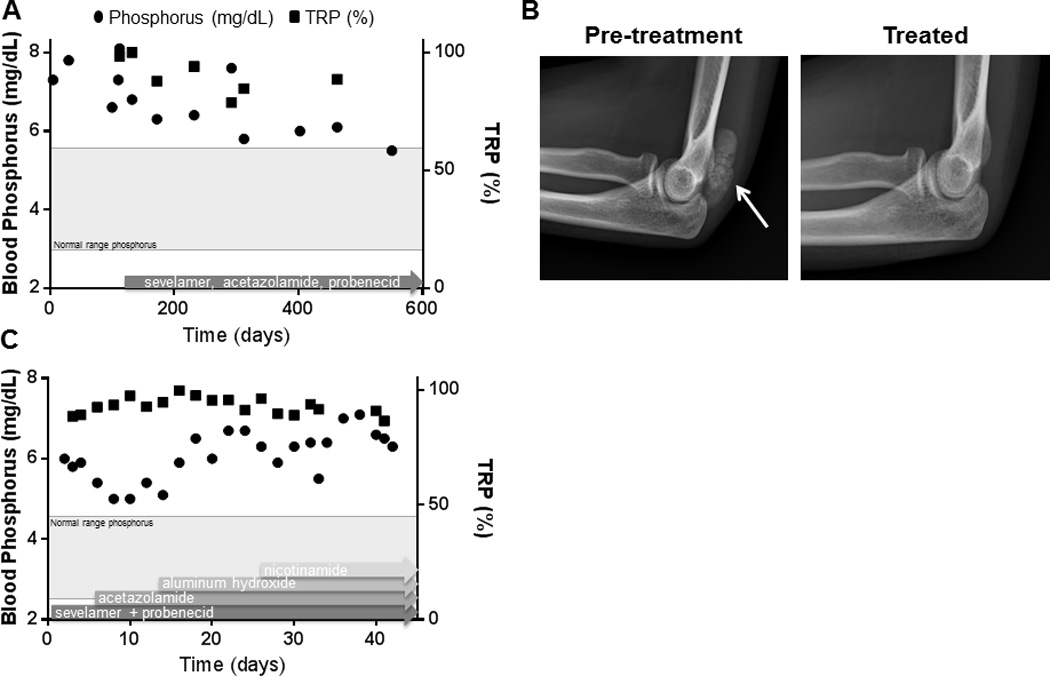
(A) After 13 months of low-phosphate diet, sevelamer, acetazolamide, and probenecid, FTC6’s serum phosphorus and TRP decreased, and (B) the tumoral calcinosis posterior to the elbow completely resolved (arrow). (C) Treatment with sevelamer, probenecid, acetazolamide, aluminum hydroxide, and nicotinamide in a stepwise approach along with a low phosphate diet during an inpatient hospitalization. Despite optimizing medical therapy, there was no significant decrease in FTC4’s serum phosphorus or TRP; to the contrary, the serum phosphorus appeared higher while on medical therapy. *Normal range for TRP is 85–95% when blood phosphorus is in the normal range.
Given the presumed increased IL-1 production by foamy macrophages seen on biopsies, inflammation in FTC4 was treated with a short course of anakinra, a recombinant IL-1 receptor antagonist. Anakinra 100 mg subcutaneous daily was started initially and then increased to 100 mg twice daily, with a decrease in CRP levels from 176 mg/L to 26.6 mg/L over 16 days. On FTC4’s subsequent admission, anakinra was restarted at 100 mg daily and increased to 200 mg daily; CRP levels declined significantly but did not normalize (Fig 7F). Treatment was continued for 4 months but then discontinued as the subject was unable to obtain the medication. Off therapy, symptoms returned and biochemical markers of inflammation increased. FTC2’s inflammatory response was initially treated with oral prednisone, topical steroids, and ibuprofen, without improvement. Therapy was changed to subcutaneous canakinumab, a monoclonal interleukin-1β (IL-1 β) antibody, at a starting dose of 2 mg/kg every 8 weeks (50 mg), and increased to 3 mg/kg every 8 weeks (100 mg), with normalization of CRP levels (Fig 7A). Because of a needle phobia, canakinumab was prescribed every eight weeks instead of once or twice daily injections of anakinra. The subject has continued on therapy for the past 2.5 years. Both subjects reported more energy, improved appetite, and an overall improved sense of well-being while on IL-1 blockade. Of note, FTC2 reported greater adherence to phosphate lowering medications during this time, which likely contributed to her improvement in symptoms.
DISCUSSION
Hyperphosphatemia due to increased renal TRP and elevated or inappropriately normal 1,25D are the biochemical hallmarks of FTC/HHS. When due to mutations in FGF23 or GALNT3, CFGF23 is significantly elevated with a low or normal iFGF23, indicating increased processing of the biologically active iFGF23. Despite the low or normal iFGF23, iFGF23 action appears blunted, suggesting that CFGF23 may competitively inhibit iFGF23 binding to FGFR1(22). Thus, FTC/HHS is the biochemical mirror of iFGF23 excess conditions, such as tumor-induced osteomalacia (23), X-linked hypophosphatemic rickets(24), and autosomal dominant hypophosphatemia(25).
The reported clinical spectrum of FTC/HHS is variable ranging from isolated eyelid calcifications to massive periarticular calcifications; this holds true in our NIH cohort. Most subjects had onset of tumoral calcinosis in childhood; however, FTC7 was unique with the development of calcifications at 45 years. Because of the late onset of tumoral calcinosis, FTC7 underwent an extensive evaluation for possible causes of ectopic calcifications including the following rheumatologic diagnoses: dermatomyositis, scleroderma, lupus, mixed connective tissue disease, and CREST syndrome; all of which were excluded. However, other theoretical environmental, acquired, or autoimmune etiologies could not be excluded. Two subjects were asymptomatic at initial evaluation despite biochemical findings similar to their respective siblings who were severely affected at an early age. Variable clinical presentation among family members with the same mutation suggests the role of genetic or environmental modifiers (i.e., trauma) in the clinical presentation. Due to the heterogeneity of this disease, systematic phenotyping and indication-directed therapy, as outlined above, is recommended.
While the distribution of calcifications has typically been reported in the skin or subcutaneous tissue, usually in periarticular locations with no joint or bony involvement, FTC4 had complete destruction of the left shoulder joint and FTC8 had destruction of the ulnar growth plate. Thus, extension into bones and/or joints is a previously unreported complication with the potential to severely affect quality of life and activities of daily living. In addition, we identified large calcifications in the submucosal lining of the large intestine, another novel location.
The greatest potential risk associated with FTC/HHS may be vascular calcification (26, 27). Two of the five subjects evaluated had coronary calcifications, with Agatston scores above the 95th percentile. As the two affected had other risk factors for cardiac calcification, the lack of cardiac calcification in other subjects may be a function of young age and other factors. Therefore, the risk of cardiac calcification associated with FTC/HHS, and its sequelae are not clear at this time.
In an effort to reduce the calcium × phosphate product, subjects were instructed to follow a low-phosphate diet. Given the ubiquity of phosphate in foods, adherence was variable. Symptomatic subjects were treated with sevelamer, a non-calcium based phosphate binder (28), to reduce intestinal absorption. Aluminum hydroxide was added as an additional phosphate binder in 2 subjects, as the potential for aluminum toxicity is low when renal function is normal. Most subjects were treated with acetazolamide, a carbonic anhydrase inhibitor, and probenecid, a uricosuric agent, both of which increase renal phosphate excretion. While the exact mechanism of probenecid is unknown, acetazolamide, induces a proximal renal tubular acidosis resulting in phosphaturia (29). Therefore, monitoring of serum bicarbonate to avoid significant acidosis is important. Recent work in the Klotho-deficient mouse, a model for FTC, suggests that acetazolamide may also prevent tissue calcification by inducing acidosis (30). In addition to its uricosuric properties, probenecid also prolongs the half-life of antibiotics. Thus, one should be cautious when prescribing other drugs such as penicillin, cephalosporins, and trimethoprim-sulfamethoxazole, which when used in combination with probenecid, can result in antibiotic toxicity. Surgery was avoided, except for severely painful or disabling lesions.
The response to medical therapy was variable amongst subjects, some of which could be attributed to nonadherence with the difficult dosing regimen. Therapy with acetazolamide and phosphate binders has previously been reported to result in complete resolution of tumoral calcinosis in two cases (11, 31) and either a decrease in size, reduced progression, or absence of new lesions (12, 32). While improvement of ectopic calcifications in rheumatologic disorders treated with probenecid has been reported (33), this is the first report of successful treatment of tumoral calcinosis in FTC/HHS with probenecid. As all of our subjects receiving probenecid were also on sevelamer and acetazolamide, it is not known if the effect was additive or coincidental. Further work is necessary to determine the benefit of probenecid in the treatment of FTC/HHS, if any, but it serves as another phosphaturic agent that may be utilized.
Two previously reported subjects had evidence of increased inflammation (31, 34). In our cohort, three of the eight subjects had overwhelming systemic inflammation. Inflammation correlated with severity of clinical disease, but there was no correlation with TRP. IL-1 blockade decreased inflammatory markers, reduced tumor burden, and led to subjective improvement in energy level, appetite, and overall well-being in two subjects. While the role of inflammation in the pathogenesis of FTC/HHS remains to be elucidated, treatment of inflammation may be important adjuvant therapy.
Histologic analyses showed ectopic calcification in a background of chronic inflammation with multiple foamy macrophages. Others have shown the presence of macrophages and multinucleated giant cells on histology from biopsies of tumoral calcinosis (12, 35), and older literature describes the presence of woven bone (36). However, there are no reports of heterotopic ossification within tumoral calcinosis lesions with mature remodeling of lamellar bone and the presence of reversal lines as seen in our cohort. We hypothesize that the chronic inflammation may contribute to recruitment/development of local progenitor cells with osteogenic potential to form ectopic bone, possibly similar to the mechanism of heterotopic ossification in fibrodysplasia ossificans progressiva (37).
This study is limited by the small number of study subjects and the individualized treatment regimens. However, this is the largest reported cohort of subjects with FTC/HHS from different families with known causative mutations in 7 subjects. Further genetic evaluation is ongoing in the subject in whom a causative mutation was not identified. While a randomized, placebo-controlled treatment trial is ideal, it is not feasible given the disorder’s rarity. In addition, interpreting the response to therapy is difficult in cases of poor adherence and lack of long-term follow-up, which occurred with some subjects. Finally, referral bias may also affect our findings. As a tertiary referral center, these subjects may be more severely affected, thus diminishing potential generalizability of the findings.
This cohort broadens the phenotype and genotype of FTC/HHS and provides insight into the efficacy of a combined treatment regimen of phosphate binders and phosphaturic agents. For the first time, we present evidence of heterotopic ossification and overwhelming systemic inflammation in subjects with FTC/HHS and improvement of these symptoms with anti-inflammatory medications. These data suggest that inflammation may play a role in the pathogenesis of this disorder and that treatment of the inflammation may be important for disease management.
Acknowledgments
The NIDCR Investigators (MSR, LCG, BAB, RIG, and MTC) receive non-salary financial support from Shire for research investigating pharmaceutical agents not discussed in this work. KEW receives royalties for licensing FGF23 to Kyowa Hakko Kirin Co., Ltd., and has funding from Eli Lilly& Co. RGM has received study support from SOBI and Novartis.
This research was supported by the Division of Intramural Research of the NIDCR, NIH, DHHS. Work performed in MJE’s lab was supported by NIH grant AR42228. We thank Brian Kirmse for patient samples and Leah R. Padgett, Austin M. Riley, and Joseph Bondi for assisting with DNA sequencing.
Footnotes
Disclosure Statement:
All other authors have no conflicts of interest.
Authors’ roles: MSR, PG, RGM, FW, TH, JDR, PSM, BA, MJ, SH, LCG, BAB, RIG, and MTC phenotyped subjects, provided treatment, and monitored subjects on treatment. SI, MJE, and KW performed DNA sequencing and genetic analyses. AM and EM assisted in histology analysis. MYC analyzed CT scans with calcium scoring. MSR prepared the manuscript, which was reviewed, edited and approved by all authors.
References
- 1.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Human molecular genetics. 2005 Feb 1;14(3):385–390. doi: 10.1093/hmg/ddi034. PubMed PMID: 15590700. [DOI] [PubMed] [Google Scholar]
- 2.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nature genetics. 2004 Jun;36(6):579–581. doi: 10.1038/ng1358. PubMed PMID: 15133511. [DOI] [PubMed] [Google Scholar]
- 3.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. Journal of musculoskeletal & neuronal interactions. 2007 Oct-Dec;7(4):318–319. PubMed PMID: 18094491. [PubMed] [Google Scholar]
- 4.Bhattacharyya N, Chong WH, Gafni RI, Collins MT. Fibroblast growth factor 23: state of the field and future directions. Trends in endocrinology and metabolism: TEM. 2012 Dec;23(12):610–618. doi: 10.1016/j.tem.2012.07.002. PubMed PMID: 22921867. Pubmed Central PMCID: 3502714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. The Journal of biological chemistry. 2006 Jul 7;281(27):18370–18377. doi: 10.1074/jbc.M602469200. PubMed PMID: 16638743. [DOI] [PubMed] [Google Scholar]
- 6.Narchi H. Hyperostosis with hyperphosphatemia: evidence of familial occurrence and association with tumoral calcinosis. Pediatrics. 1997 May;99(5):745–748. doi: 10.1542/peds.99.5.745. PubMed PMID: 9113957. [DOI] [PubMed] [Google Scholar]
- 7.Dumitrescu CE, Kelly MH, Khosravi A, Hart TC, Brahim J, White KE, et al. A case of familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2009 Jul;20(7):1273–1278. doi: 10.1007/s00198-008-0775-z. PubMed PMID: 18982401. Pubmed Central PMCID: 2692468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burkes EJ, Jr, Lyles KW, Dolan EA, Giammara B, Hanker J. Dental lesions in tumoral calcinosis. Journal of oral pathology & medicine : official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 1991 May;20(5):222–227. doi: 10.1111/j.1600-0714.1991.tb00423.x. PubMed PMID: 2066872. [DOI] [PubMed] [Google Scholar]
- 9.Gal G, Metzker A, Garlick J, Gold Y, Calderon S. Head and neck manifestations of tumoral calcinosis. Oral surgery, oral medicine, and oral pathology. 1994 Feb;77(2):158–166. doi: 10.1016/0030-4220(94)90279-8. PubMed PMID: 8139834. [DOI] [PubMed] [Google Scholar]
- 10.Krstevska A, Gale S, Blair F. Tumoral calcinosis: a dental literature review and case report. Dental update. 2012 Jul-Aug;39(6):416–418. 421. doi: 10.12968/denu.2012.39.6.416. PubMed PMID: 22928454. [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi T, Sugimoto T, Imai Y, Fukase M, Fujita T, Chihara K. Successful treatment of hyperphosphatemic tumoral calcinosis with long-term acetazolamide. Bone. 1995 Apr;16(4 Suppl):247S–250S. doi: 10.1016/8756-3282(95)00019-a. PubMed PMID: 7626311. [DOI] [PubMed] [Google Scholar]
- 12.Lammoglia JJ, Mericq V. Familial tumoral calcinosis caused by a novel FGF23 mutation: response to induction of tubular renal acidosis with acetazolamide and the non-calcium phosphate binder sevelamer. Hormone research. 2009;71(3):178–184. doi: 10.1159/000197876. PubMed PMID: 19188744. [DOI] [PubMed] [Google Scholar]
- 13.Alkhooly AZ. Medical treatment for tumoral calcinosis with eight years of follow-up: a report of four cases. Journal of orthopaedic surgery. 2009 Dec;17(3):379–382. doi: 10.1177/230949900901700328. PubMed PMID: 20065385. [DOI] [PubMed] [Google Scholar]
- 14.Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, et al. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. American journal of medical genetics Part A. 2010 Apr;152A(4):896–903. doi: 10.1002/ajmg.a.33337. PubMed PMID: 20358599. Pubmed Central PMCID: 3392654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lafferty FW, Reynolds ES, Pearson OH. Tumoral Calcinosis: A Metabolic Disease of Obscure Etiology. The American journal of medicine. 1965 Jan;38:105–118. doi: 10.1016/0002-9343(65)90164-6. PubMed PMID: 14251895. [DOI] [PubMed] [Google Scholar]
- 16.Yancovitch A, Hershkovitz D, Indelman M, Galloway P, Whiteford M, Sprecher E, et al. Novel mutations in GALNT3 causing hyperphosphatemic familial tumoral calcinosis. Journal of bone and mineral metabolism. 2011 Sep;29(5):621–625. doi: 10.1007/s00774-011-0260-1. PubMed PMID: 21347749. [DOI] [PubMed] [Google Scholar]
- 17.Zemel BS, Kalkwarf HJ, Gilsanz V, Lappe JM, Oberfield S, Shepherd JA, et al. Revised reference curves for bone mineral content and areal bone mineral density according to age and sex for black and non-black children: results of the bone mineral density in childhood study. The Journal of clinical endocrinology and metabolism. 2011 Oct;96(10):3160–3169. doi: 10.1210/jc.2011-1111. PubMed PMID: 21917867. Pubmed Central PMCID: 3200252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M, Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. Journal of the American College of Cardiology. 1990 Mar 15;15(4):827–832. doi: 10.1016/0735-1097(90)90282-t. PubMed PMID: 2407762. [DOI] [PubMed] [Google Scholar]
- 19.Alon U, Hellerstein S. Assessment and interpretation of the tubular threshold for phosphate in infants and children. Pediatric nephrology. 1994 Apr;8(2):250–251. doi: 10.1007/BF00865491. PubMed PMID: 8018507. [DOI] [PubMed] [Google Scholar]
- 20.Langman CB, Salusky IB. K/DOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Children with Chronic Kidney Disease - Foreword. Am J Kidney Dis. 2005 Oct;46(4):S6–S121. PubMed PMID: WOS:000232487400001. English. [Google Scholar]
- 21.Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, et al. Mechanism of FGF23 processing in fibrous dysplasia. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012 May;27(5):1132–1141. doi: 10.1002/jbmr.1546. PubMed PMID: 22247037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proceedings of the National Academy of Sciences of the United States of America. 2010 Jan 5;107(1):407–412. doi: 10.1073/pnas.0902006107. PubMed PMID: 19966287. Pubmed Central PMCID: 2806769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer. 2011 Jun;18(3):R53–R77. doi: 10.1530/ERC-11-0006. PubMed PMID: 21490240. Pubmed Central PMCID: 3433741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. The New England journal of medicine. 2003 Apr 24;348(17):1656–1663. doi: 10.1056/NEJMoa020881. PubMed PMID: 12711740. [DOI] [PubMed] [Google Scholar]
- 25.White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney international. 2001 Dec;60(6):2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. PubMed PMID: 11737582. [DOI] [PubMed] [Google Scholar]
- 26.Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman M, et al. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum Genet. 2005 Nov;118(2):261–266. doi: 10.1007/s00439-005-0026-8. PubMed PMID: 16151858. [DOI] [PubMed] [Google Scholar]
- 27.Shah A, Miller CJ, Nast CC, Adams MD, Truitt B, Tayek JA, et al. Severe vascular calcification and tumoral calcinosis in a family with hyperphosphatemia: a fibroblast growth factor 23 mutation identified by exome sequencing. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2014 Dec;29(12):2235–2243. doi: 10.1093/ndt/gfu324. PubMed PMID: 25378588. Pubmed Central PMCID: 4240183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahdavi H, Kuizon BD, Gales B, Wang HJ, Elashoff RM, Salusky IB. Sevelamer hydrochloride: an effective phosphate binder in dialyzed children. Pediatric nephrology. 2003 Dec;18(12):1260–1264. doi: 10.1007/s00467-003-1298-7. PubMed PMID: 14586677. [DOI] [PubMed] [Google Scholar]
- 29.Lufkin EG, Wilson DM, Smith LH, Bill NJ, DeLuca HF, Dousa TP, et al. Phosphorus excretion in tumoral calcinosis: response to parathyroid hormone and acetazolamide. The Journal of clinical endocrinology and metabolism. 1980 Apr;50(4):648–653. doi: 10.1210/jcem-50-4-648. PubMed PMID: 7364922. [DOI] [PubMed] [Google Scholar]
- 30.Leibrock CB, Alesutan I, Voelkl J, Michael D, Castor T, Kohlhofer U, et al. Acetazolamide sensitive tissue calcification and aging of klotho-hypomorphic mice. Journal of molecular medicine. 2015 Aug 27; doi: 10.1007/s00109-015-1331-x. PubMed PMID: 26307633. [DOI] [PubMed] [Google Scholar]
- 31.Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, et al. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. The Journal of clinical endocrinology and metabolism. 2006 Oct;91(10):4037–4042. doi: 10.1210/jc.2006-0305. PubMed PMID: 16868048. [DOI] [PubMed] [Google Scholar]
- 32.Finer G, Price HE, Shore RM, White KE, Langman CB. Hyperphosphatemic familial tumoral calcinosis: response to acetazolamide and postulated mechanisms. American journal of medical genetics Part A. 2014 Jun;164A(6):1545–1549. doi: 10.1002/ajmg.a.36476. PubMed PMID: 24668887. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura H, Kawakami A, Ida H, Ejima E, Origuchi T, Eguchi K. Efficacy of probenecid for a patient with juvenile dermatomyositis complicated with calcinosis. The Journal of rheumatology. 2006 Aug;33(8):1691–1693. PubMed PMID: 16881125. [PubMed] [Google Scholar]
- 34.Masi L, Beltrami G, Ottanelli S, Franceschelli F, Gozzini A, Zonefrati R, et al. Human Preosteoblastic Cell Culture from a Patient with Severe Tumoral Calcinosis-Hyperphosphatemia Due to a New GALNT3 Gene Mutation: Study of In Vitro Mineralization. Calcified tissue international. 2015 May;96(5):438–452. doi: 10.1007/s00223-015-9974-8. PubMed PMID: 25899975. [DOI] [PubMed] [Google Scholar]
- 35.Slavin RE, Wen J, Barmada A. Tumoral calcinosis--a pathogenetic overview: a histological and ultrastructural study with a report of two new cases, one in infancy. International journal of surgical pathology. 2012 Oct;20(5):462–473. doi: 10.1177/1066896912444925. PubMed PMID: 22614164. [DOI] [PubMed] [Google Scholar]
- 36.Slavin RE, Wen J, Kumar D, Evans EB. Familial tumoral calcinosis. A clinical, histopathologic, and ultrastructural study with an analysis of its calcifying process and pathogenesis. The American journal of surgical pathology. 1993 Aug;17(8):788–802. PubMed PMID: 8338191. [PubMed] [Google Scholar]
- 37.Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: diagnosis, management, and therapeutic horizons. Pediatric endocrinology reviews: PER. 2013 Jun;10(Suppl 2):437–448. PubMed PMID: 23858627. Pubmed Central PMCID: 3995352. [PMC free article] [PubMed] [Google Scholar]
- 38.Foster BL, Ramnitz MS, Gafni RI, Burke AB, Boyce AM, Lee JS, et al. Rare bone diseases and their dental, oral, and craniofacial manifestations. Journal of dental research. 2014 Jul;93(7 Suppl):7S–19S. doi: 10.1177/0022034514529150. PubMed PMID: 24700690. Pubmed Central PMCID: 4107543. [DOI] [PMC free article] [PubMed] [Google Scholar]