

Abstract
Oncogenic fusions in TRK family receptor tyrosine kinases have been identified in several cancers and can serve as therapeutic targets. We identified ETV6-NTRK3, MYO5A-NTRK3 and MYH9-NTRK3 fusions in Spitz tumours and demonstrate that NTRK3 fusions constitutively activate the MAPK, PI3K and PLCγ1 pathways in melanocytes. This signalling was inhibited by DS-6051a, a small molecule inhibitor of NTRK1/2/3 and ROS1. NTRK3 fusions expand the range of oncogenic kinase fusions in melanocytic neoplasms and offer targets for a small subset of melanomas for which currently no targeted options exist.
Keywords: NTRK3 fusion, Spitz tumour, melanoma, kinase inhibitor, Spitz nevus, atypical Spitz tumour, spitzoid melanoma, oncogene, genetics
Introduction
Constitutive activation of signalling pathways controlling cell growth and proliferation by activating mutations in dominant oncogenes such BRAF, NRAS, KIT, GNAQ or GNA11 is found in the majority of melanocytic tumours [1–5]. These mutations tend to occur early during progression and are almost always mutually exclusive with each other. While inactivating mutations in NF1 have emerged as an additional genetic alteration[6], a subset of melanoma remains ‘wild-type’ despite exhaustive studies sequencing the coding regions of genes [7,8]. Recently, we and others described recurrent rearrangements of kinases as a novel class of oncogenic alterations in this subset of melanocytic neoplasms [9–12]. In some cases, the presence of a fusion kinase was associated with copy number transitions within the kinase gene with relative gain of the 3′ portion of the gene. The transitions result from duplication of the DNA fragment that encodes the 3′ portion of the kinase gene and/or loss of the other fragment that carries the 5′ portion of the kinase gene.
In our clinical practice, we perform array comparative genomic hybridization (aCGH) as an adjunct to the histopathologic diagnosis of difficult to classify melanocytic tumours. In our database of copy number profiles we noticed cases with copy number transitions within the NTRK3 locus on chromosome 15q with a relative gain of the NTRK3 kinase domain suggesting the presence of an activating NTRK3 fusion kinase in these cases.
Here, we report ETV6-NTRK3 fusions in melanocytic tumours as well as novel NTRK3 fusions with MYO5A and MYH9. These kinase fusions appear in a mutually exclusive pattern with previously identified melanoma oncogenes, are constitutively active, and may serve as therapeutic targets for a subset of melanomas.
Materials and Methods
Study Population
We analysed the copy number profiles of 1,202 melanocytic neoplasms, for which array comparative genomic hybridization (aCGH) was performed as part of the diagnostic assessment at the UCSF Dermatopathology Service of the Departments of Dermatology and Pathology at the University of California, San Francisco over a 36 month period from 2010 through 2013. The majority of these cases are ‘borderline’ or ambiguous tumours with histopathological features that overlap those of melanocytic nevi and melanoma, for which aCGH was performed as an ancillary diagnostic test. The final diagnosis was determined by review of histopathologic features and aCGH results. The diagnostic criteria for Spitz nevi, atypical Spitz tumour and spitzoid melanoma used are those detailed by LeBoit and Massi [13]. The study was approved by the Committee on Human Research (11-07951) and was conducted according to the Declaration of Helsinki. Human tissues were obtained with informed consent during the course of clinical care and a waiver of consent was granted for use of archival material in this study.
DNA extraction
Areas of tumour were microdissected from 20 μm thick sections of formalin-fixed, paraffin-embedded (FFPE) tumour. After deparaffinization by washing with SafeClear and ethanol, DNA was extracted by phenol/chloroform extraction.
Array comparative genomic hybridization (aCGH)
This was carried out using 500–1000 ng of genomic DNA on Agilent 4×180k microarrays (Agilent, Santa Clara, CA). The raw microarray images were processed with Agilent Feature Extraction software, and analysed using Nexus Copy Number Software version 7.0 (Biodiscovery, El Segundo, CA). Segmentation was performed requiring at least 10 probes per segment and copy number status was defined by log2ratio as follows: gain >= 0.2, amplification >=0.7, loss <= −0.17, homozygous loss <= −0.75. Cases demonstrating copy number transitions with relative copy number increase of the 3′ portion of NTRK3 with sufficient residual DNA or tissue were selected for sequencing. Given the resolution of our aCGH platform, we included copy number transitions within 50 kb of NTRK3.
DNA sequencing and analysis
Multiplex library preparation was performed using the Ovation Ultralow Library System (NuGEN, San Carlos, CA, p/n 0331-32) or Nextflex (Bioo Scientific, Austin, TX, p/n No. 5140-53) according to the manufacturer’s specifications, with up to 200 ng of sample DNA. Hybridization-capture of pooled libraries was performed using custom-designed bait libraries (Nimblegen SeqCap EZ Choice, p/n 06588786001) spanning ~1.8 Mb of the genome including the exons of BRAF, NRAS, HRAS, KIT, GNAQ and intron 14 of NTRK3 and introns 3 and 4 of ETV6 (supplementary information, Tables S1–S4). The target intervals cover mostly exonic but also some intronic and untranslated regions of 293 (version 1) or 365 target genes (version 2). The target genes were curated to comprise common cancer genes with particular relevance to melanoma.
Captured libraries were sequenced as paired-end 100bp reads on a HiSeq 2000 or HiSeq 2500 instrument (Illumina). Sequence reads were mapped to the reference human genome (hg19) using the Burrows-Wheeler aligner (BWA) [14]. Recalibration of reads and variant calling were performed using the Genome Analysis Toolkit (GATK) [15]. Coverage and sequencing statistics were determined using Picard CalculateHsMetrics and Picard CollectInsertSizeMetrics [16] (Supplementary Table S5). Variant annotation was performed with Annovar [17]. For fusion detection, read pairs with one or more reads unaligned, insert sizes greater than 1000 bp, or with soft clipping of at least one read were extracted and re-aligned using BWA-SW [18] and used as input to CREST [19]. Structural variants predicted by CREST were reviewed by visual inspection in the Integrative Genomics Viewer [20]. We predicted the resulting fusion transcripts by joining the exon directly upstream from the genomic breakpoint with the exon directly downstream. Predicted protein sequences were then determined from the predicted transcripts.
RNA sequencing and analysis
RNA was extracted from FFPE tumours after microdissection using the Qiagen RNEasy FFPE kit (p/n 73504) following the manufacturer’s protocol. Double stranded cDNA was synthesized with the NEBNext mRNA Library Prep Reagent Set for Illumina (New England Biolabs E6100S) with up to 250 ng of input total RNA. Hybrid selection of indexed, adaptor-ligated libraries was performed using the cDNA Kinome hybridization kit with 612 transcripts of kinases and kinase-related genes (Agilent SureSelect Human Kinome Kit, 5190-4801). Sequencing was performed on the HiSeq 2500 instrument (Illumina) with paired-end 100bp reads (Supplementary Table S2). Sequence reads were mapped to the reference human genome (hg19) using Tophat and fusions were identified by Tophat Fusion in combination with manual review [21,22].
Confirmation by RT-PCR
Total RNA extracted from FFPE tissue sections was reverse transcribed with random hexamer primers using an NEBNext RNA First Strand Synthesis module. RT-PCR amplification was done for 32 cycles using Platinum Taq DNA polymerase (Invitrogen) with positive and negative controls using primer pairs described in Supplementary Table S6. The PCR products were resolved on a 2% agarose-TBE gel, stained with SYBR safe (Invitrogen), and visualized under UV illumination.
Plasmid construction
The cDNA of the ETV6-NTRK3 and MYO5A-NTRK3 fusions were generated by 30 cycles of overlap PCR using Addgene plasmid #23901 and sequences corresponding to the N-terminal portions of ETV6 and MYO5A (synthesized by Genewiz, South Plainfield, NJ) with Platinum Pfx DNA polymerase (Invitrogen). Subsequent cloning into pENTR was performed with the D-TOPO kit (ThermoFisher Scientific) following the manufacturer’s instructions. Subsequently, the cDNA was cloned by LR Clonase II (Life Technologies) into the pLenti6.3/TO/V5-Dest backbone (Life Technologies).
Generation of stably transduced cell lines
Melan-a cells were generously provided by Dr. Dorothy C. Bennett (St. George’s Hospital, University of London, London, UK) [23]and maintained in glutamine-containing RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum, 200 nM of 12-O-tetradecanoylphorbol-13-acetate (TPA), penicillin (100 units/mL) and streptomycin (50 mg/mL). 293FT cells were purchased from Life Technologies and maintained in DME-H21 medium containing 10% heat inactivated fetal bovine serum, minimal essential media (MEM) Non-Essential Amino Acids (0.1 mM), sodium pyruvate (1 mM), penicillin (100 units/mL) and streptomycin (50 mg/mL).
Lentiviruses were produced by transfecting 293FT cells with plasmid DNA pLenti6.2-GFP or pLenti6.3/TO/V5-Dest backbone with pCMV-VSV-G and pCMV delta R8.2 using JetPrime (Polyplus-transfection, Illkirch). Melan-a cells were transduced by infection with lentivirus in the presence of 10 μg/ml of polybrene (Santa Cruz Biotechnology).
Drug studies
The selective ROS1 and NTRK1/2/3 inhibitor DS-6051a was a gift from Daiichi Sankyo. Transduced melan-a cells were treated with glutamine-containing RPMI-1640 with drug (1 μM) or without for 4 h before collection of lysates.
Western Blotting
Antibodies: anti-phospho-Erk (#9101), anti-pan NTRK (C17F1), anti-NTRK3 (sc117), anti-phospho AKT (Ser473) (#9271), anti-AKT (#9272), anti-phospho-PLCγ1 (Tyr783) (#2821), and anti-PLCγ1 (#2822), from Cell Signaling Technologies, anti-HSP60 (sc-1722) and anti-ERK (sc154) from Santa Cruz Biotechnology. Cell lysates were prepared in RIPA buffer supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). Equal amounts of protein (30 μg), as measured by bicinchoninic (BCA) protein assay, were resolved in 4–12% Bis-Tris NuPage gradient gels (Life Technologies) and transferred electrophoretically to a polyvinylidene difluoride 0.45 μM pore membrane. Membranes were blocked in 5% non-fat milk in Tris-buffered saline with Tween 20 (TBST) before being incubated overnight at 4° C with the primary antibodies (1:1000 dilution in 5% non-fat milk in TBST). Signal detection was achieved by incubation of the membrane in enhanced chemiluminescence (ECL) solution (Millipore) and exposure of autoradiography film. The full blots are shown in supplementary Figure S12.
Results
Identification of NTRK3 kinase fusions
Of 1202 cases of difficult to classify melanocytic tumours for which aCGH was performed for clinical purposes, 22 (1.8%) demonstrated copy number transitions in NTRK3 with a net gain of the 3′ portion of NTRK3 (Supplementary Table S7–8). Of these cases, 10 demonstrated a gain of the 3′ end of NTRK3, 9 demonstrated a loss of the 5′ end of NTRK3, and 3 demonstrated both gain of the 3′ end and loss of the 5′ end of NTRK3. Amplification of any portion of NTRK3 was not observed.
Leftover material was available for 12 of these 22 cases and was subjected to kinome RNA sequencing and/or targeted DNA sequencing of ~ 300 melanoma and other cancer-related genes, including all exons of NTRK3, intron 14 of NTRK3, and introns 3 and 4 of ETV6 (n=10). We identified NTRK3 fusions in 8 cases (75%), with the 5′ fusion partner ETV6 (chr12p13) in four cases, MYO5A (chr15q21) in 3 cases, and MYH9 (chr22q12) in 1 case. For 5 cases (those with available RNA), the NTRK3 fusions were confirmed by RT-PCR, (Supplementary Figure S1). Activating mutations in BRAF, NRAS, HRAS, GNAQ, GNA11 or fusions in other kinases were not identified in tumours with NTRK3 fusions.
In all cases with NTRK3 fusions, the predicted or sequenced fusion transcript was in frame and contained the kinase domain of NTRK3 (Figure 1). The ETV6-NTRK3 fusion transcripts were comprised of the first 5 exons of ETV6 (NM_001987) followed by exons 15 – 20 of NTRK3 (NM-001012338), similar to what has been commonly observed in other tumour types due to the reciprocal translocation t(12;15)(p13;q25)[24–26]. The MYH9-NTRK3 fusion found in one case arose from a breakpoint within exon 32 of MYH9 (NM_002473), joining it to intron 13 of NTRK3. This resulted in an insertion of 22 nt of NTRK3 intron 13 into the coding sequence, adding 7 amino acids. Two different MYO5A-NTRK3 fusion genes were identified in three other cases. Fusion transcripts joining exon 1–32 of MYO5A (NM_001142495) to exon 13–20 of NTRK3 were present in cases 6 and 8. In case 7, transcriptome sequencing revealed two fusion transcripts, spanning MYO5A through exon 23 followed by NTRK3 exon 11–20 in one and exons 12–20 in the other. This likely reflects a fusion gene with a genomic breakpoint in intron 10 of NTRK3, with alternative splicing of the fusion transcript resulting from skipping of exon 11 of NTRK3, which encodes 9 amino acids. Among the four known transcript variants of NTRK3 described in the NCBI database, exon 11 is skipped in transcript variant 4 (NM_001243101).
Figure 1. Structural details of NTRK3 fusions.
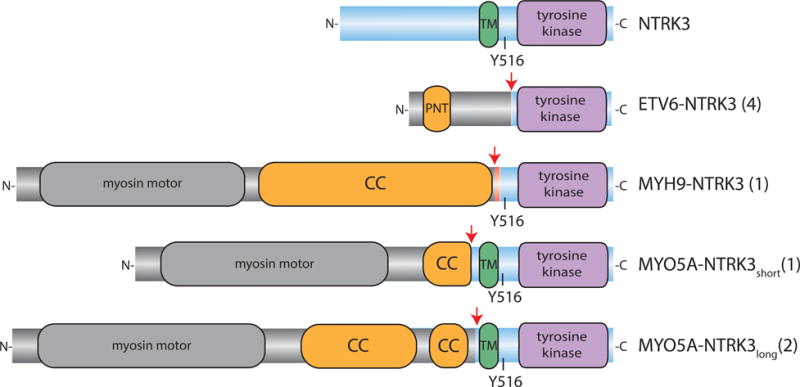
The protein domain structure of NTRK3 is shown at the top with the N-terminal portion to the left and the transmembrane (TM, green) and tyrosine kinase (purple) domains highlighted. Below, the protein domain structure of the NTRK3 fusions identified are shown. Red arrows denote the fusion junctions. The blue backbone denotes the contribution from NTRK3 and the grey backbone denotes the contribution from the N-terminal partner. The domains contributed by the N-terminal partners that mediate dimerization of the native proteins (coiled-coil CC, sterile alpha motif SAM) are coloured orange. The red backbone in MYH9-NTRK3 represents the novel sequence contributed by NTRK3 intron 13.
Three of four cases with ETV6-NTRK3 fusions had copy number profiles suggesting non-reciprocal translocations with loss of distal chromosome 15q (Cases 1–3, Supplementary Figure S2). The remaining case with an ETV6-NTRK3 fusion had a copy number profile compatible with reciprocal translocation followed by duplication of the fragment encoding the NTRK3 kinase domain (Case 4). MYO5A resides ~35 Mb upstream from NTRK3 on chromosome 15. Given that the genes are in the same orientation, MYO5A-NTRK3 fusion could arise from tandem duplication. The copy number profile of case 8 suggests tandem duplication and loss of the unaffected chromosome 15 (Supplementary Figures S2 and S3). Two other cases in our cohort, from which material was not available for sequencing analysis, demonstrated a gain on chromosome 15 starting within MYO5A and ending within NTRK3, thus suggesting the possibility of a recurrent tandem duplication of the region between MYO5A and NTRK3 (Supplementary Figure S3).
MYO5A and MYH9 are both myosin genes that contribute coiled coil domains to their respective NTRK3 fusion genes. These coiled coil domains dimerize the native myosin proteins. NTRK3 fusions genes with MYH9 and MYO5A arose from breakpoints in introns of NTRK3 upstream from intron 14, setting them apart from ETV6-NTRK3 fusions. As a consequence, the novel MYH9- and MYO5A-NTRK3 fusions contain, in addition to the kinase domain, the critical tyrosine 516 of NTRK3, which is a site for SHC interaction, and in other instances also contained the transmembrane domain of NTRK3 (Figure 1).
Functional validation and fusion kinase inhibition
To determine whether MYO5A-NTRK3 is constitutively active and which downstream signalling pathways are activated by ETV6-NTRK3 and MYO5A-NTRK3 fusions in the melanocytic context, we expressed full-length NTRK3, ETV6-NTRK3 and MYO5A-NTRK3 constructs in immortalized mouse melanocytes (Melan-a) [23] (Figure 2) and in 293FT cells (Supplementary Figure S4). NTRK3 demonstrated little to no signalling in the absence of its ligand NT-3, whereas both ETV6- and MYO5A-NTRK3 fusions constitutively activated the mitogen-activated protein kinase (MAPK), phosphoinositol-3 kinase (PI3K), and the phospholipase C gamma 1 (PLCγ-1) pathways (Figure 2). Treatment with DS-6051a (an inhibitor of ROS1 and NTRK1/2/3), at 1 μM for 4 h completely abrogated MAPK and PLCγ-1 signalling and significantly diminished PI3K signalling (Figure 2).
Figure 2. NTRK3 fusions constitutively activate oncogenic signalling pathways and are inhibited by the small molecule inhibitor DS-6051a.
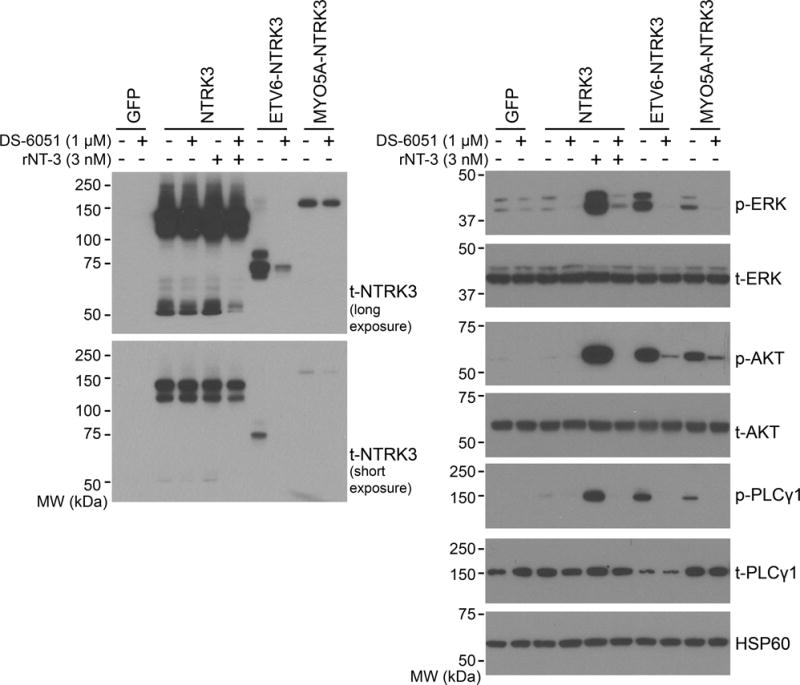
Melan-a cells (immortalized mouse melanocytes) were transduced with GFP, full-length NTRK3, ETV6-NTRK3 or MYO5A-NTRK3 expression constructs. A pan-NTRK antibody detected the expressed fusion proteins at their predicted molecular mass of 75 kDa and 174 kDa (left panels). In the absence of its ligand, expression of full-length NTRK3 did not result in significant increase of p-ERK, p-AKT, or p-PLCγ1 as compared to GFP transduced control, but these pathways were activated in the presence of recombinant neurotrophin 3 (rNT-3) at 3 nM (right panels). Expression of the fusions resulted in increased p-ERK, p-AKT, and p-PLCγ1 as compared to GFP-transduced control in the absence of rNT-3 (right panels). Signalling downstream of NTRK3 activation was significantly inhibited by DS-6051a (1 μM) with only p-AKT level exhibiting residual increase above the GFP-transduced control.
Clinical characteristics of melanocytic tumours with NTRK3 fusions
The median age of patients with melanocytic tumours harbouring NTRK3 fusions was 10 years, ranging from 2–72 years. Six of 8 patients (75%) were younger than 18 years (Table 1). The 72-year-old patient recalled the tumour had been present for over 30 years without change. In all cases, the tumours were spitzoid with epithelioid to spindled melanocytes with increased amounts of cytoplasm as compared to the small melanocytes in ‘common’ or conventional nevi [13]. The histopathological diagnoses ranged from probable, or unusual, Spitz nevus in 2 (25%) cases to atypical/ambiguous Spitz tumour for the remaining 6 (75%) cases. The tumours showed dermal involvement, often in form of large dermal nests or nodules, in 7 of 8 cases. (Figure 3, Supplementary Figures S5–S11). The Spitz designation indicates the presence of neoplastic melanocytes, with Spitz nevi often have distinct architectural features such as epidermal hyperplasia and clefting around nests of melanocytes in the epidermis. spitzoid features are not very reproducibly assessed, particularly in metastatic lesions in which architectural features are lacking.
Table 1.
Clinical characteristics of cases with NTRK3 fusions.
| Case | Age (yr) | Sex | Site | Clinical description | Diagnosis | Breslow depth (mm) | Follow-up | NTRK3 fusion |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | F | leg | 7×6 mm pink papule. Possible Spitz nevus, xanthogranuloma, mastocytoma. | inflamed Spitz nevus | >1.4 | not available | ETV6-NTRK3 |
| 2 | 6 | F | cheek | Spitz nevus or keratotic nevus, less likely verruca vulgaris | atypical Spitz tumour | 2.6 | SLN with deposit in 1/2 nodes. Completion lymphadenectomy with 0/33 nodes involved. No recurrence (3.5 yr) | ETV6-NTRK3 |
| 3 | 7 | M | cheek | not available | atypical Spitz tumour | 1.2 | not available | ETV6-NTRK3 |
| 4 | 10 | F | earlobe | nevus, possibly with central pyogenic granuloma | atypical Spitz tumour | >1.1 | Complete excision, no recurrence (4.5 yr) | ETV6-NTRK3 |
| 5 | 10 | M | cheek | pilomatricoma? | Spitz nevus | 4.2 | not available | MYH9-NTRK3 |
| 6 | 16 | F | scalp | new nevus? | atypical Spitz tumour | 1.5 | Complete excision, no recurrence (3 yr) | MYO5A-NTRK3 |
| 7 | 41 | F | chest | haemangioma or dysplastic nevus | atypical Spitz tumour | >1 | not available | MYO5A-NTRK3 |
| 8 | 72 | F | forehead | present for over 30 yr without change | atypical Spitz tumour | >1.1 | Complete excision, no recurrence (35 mo) | MYO5A-NTRK3 |
Figure 3. Case 5.
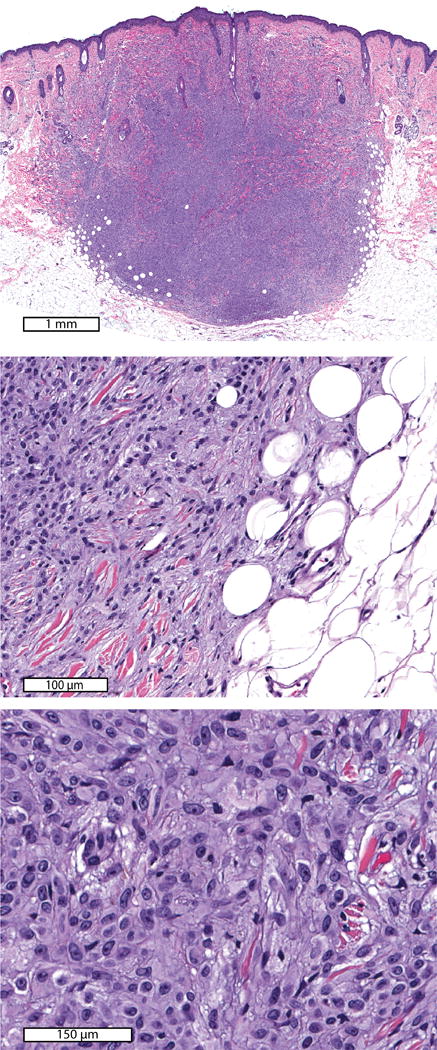
Spitz nevus with MYH9-NTRK3 fusion on the cheek of a 10-year-old boy. Low power view (top panel) demonstrates a dermal nodule that fills the dermis and extends into the superficial subcutis. The neoplastic melanocytes intercalate between adipocytes and collagen fibres (middle panel) and contain moderate amounts of cytoplasm (bottom panel).
Clinical follow-up of at least 3 years was available for half of the patients, and there were no reported recurrences. Sentinel lymph node biopsy was performed in one case and revealed involvement of one of the two biopsied lymph nodes. The patient underwent a completion lymphadenectomy, and no involvement of additional lymph nodes was found. In this case, the portions of chromosome 7 and 18 in addition to regions of the chromosomes harbouring ETV6 and NTRK3 (chromosomes 12 and 15) showed copy loss (Supplementary Figure S2). There has been no evidence of recurrence after 3.5 years of follow-up.
Discussion
Receptor tyrosine kinase fusions involving ALK, MET, NTRK1, RET and ROS1 are present in significant fraction of Spitz tumours [10,12,27], but are rare in non-acral cutaneous melanoma as demonstrated by the Cancer Genome Atlas (TCGA) [28]. In acral melanoma, a rare and genetically distinct subtype of melanoma, ALK gene rearrangements are present in ~7% by fluorescence in situ hybridization [29], and the frequency of other fusions is unknown. Activating fusions of the serine/threonine kinase BRAF are present in 7% of Spitz tumours and 1% of non-acral cutaneous melanoma in the TCGA [10,12].
In this study, we identified ETV6-NTRK3, MYO5A-NTRK3 and MYH9-NTRK3 fusions in melanocytic tumours. NTRK3 fusion kinases were identified in 0.7% of our cohort of 1,202 diagnostically challenging melanocytic tumours by sequencing cases with copy-number transitions within NTRK3. Our approach likely underestimates the frequency of NTRK3 fusions in our cohort as almost half of the cases with copy number transitions in NTRK3 did not have available material for sequencing and, based on the fact that we identified NTRK3 fusions in two-thirds of samples sequenced, may have harboured additional cases with NTRK3 fusions. In addition, our approach does not detect any fusions that were not associated with a copy number change in NTRK3 that can be identified by aCGH. In other tumours, ETV6-NTRK3 fusions commonly arise by reciprocal translocations and the same may be true for melanocytic tumours. NTRK3 fusions expand the range of kinase fusions observed in melanocytic neoplasms and similar to other kinase fusions, occur in a mutually exclusive pattern with other oncogenes including BRAF and NRAS activating mutations and other kinase fusions.
NTRK3 belongs to the Trk family of neurotrophin receptors (which consists of NTRK1, NTRK2, and NTRK3). They are transmembrane receptor tyrosine kinases that mediate response to one or more of four neurotrophins. In the developing nervous system Trk receptor signalling regulates cell fate, proliferation and axon and dendrite patterning. Trk receptors influence synaptic strength and plasticity in the adult nervous system[30]. Melanocytes share a common neuroectodermal origin with the nervous system and Trk receptors are expressed in normal melanocytes and melanoma cell lines[31]. Melanoma cell lines respond to Trk receptor signalling with increased proliferation and migration[31].
NTRK fusions are present at a low frequency in tumours of the central nervous system (high-grade astrocytoma, adult glioblastoma, and paediatric high-grade glioma). In melanocytic tumours, NTRK1 fusions are present in 23% of spitzoid melanocytic tumours, spanning a spectrum from benign to malignant [12]. A single NTRK3 fusion, ETV6-NTRK3, was detected in 1 (0.2%) of 374 cutaneous melanomas in the cancer genome atlas (TCGA) [28].
ETV6-NTRK3 was first identified in 1998 in a series of three congenital fibrosarcomas [25]. Constitutive MAPK and PI3K pathway signalling is required for ETV6-NTRK3 to transform fibroblasts [32]. ETV6-NTRK3 signalling requires dimerization, which is mediated by the pointed or sterile alpha motif (SAM) domain of ETV6 [33]. ETV6-NTRK3 is present in the majority of secretory breast carcinomas [26], congenital infantile fibrosarcomas [25], cellular type congenital mesoblastic nephromas [34], and mammary analogue secretory carcinomas [35]. ETV6-NTRK3 fusions have been identified at low frequency in additional cancer types. In thyroid cancer, the prevalence of ETV6-NTRK3 fusion is increased to 14.5% when there is a history of radiation exposure [36]. ETV6-NTRK3 fusions have also been identified in rare cases of acute myelogenous leukaemia, chronic eosinophilic leukaemia, gastrointestinal stromal tumour, inflammatory myofibroblastic tumour, paediatric high grade glioma, head and neck squamous cell carcinoma and metastatic colorectal carcinoma [28,37–42]. Additional NTRK3 fusions have been reported (LYN-NTRK3, RBPMS-NTRK3, BTBD1-NTRK3), but to our knowledge none are recurrent [28,41].
We identified the known ETV6-NTRK3 fusion in Spitz tumours and the previously undescribed MYO5A-NTRK3 and MYH9-NTRK3 fusions. The tumours in our series were classified as probably benign or of low malignant potential, and only one had evidence of metastatic dissemination to the sentinel lymph nodes. Taken together with a prior report of ETV6-NTRK3 fusion in a BRAF and NRAS wild-type primary melanoma in the TCGA [28] our findings suggest NTRK3 fusions are oncogenes in melanocytic neoplasia that arise early in the course of progression, in a mutually exclusive pattern with other MAPK activating melanoma oncogenes. Both MYO5A and MYH9 contribute coiled-coiled dimerization domains to the fusion protein. The MYO5A-NTRK3 fusion was recurrent, occurring in 3 cases in our series. MYO5A has been reported as an N-terminal fusion partner with ROS1 in a spitzoid melanocytic tumour and MYH9 as a fusion partner with ALK in anaplastic large cell lymphoma [12,43]. We demonstrate in the melanocytic context that both ETV6-NTRK3 and MYO5A-NTRK3 fusion proteins constitutively signal through the MAPK, AKT and PLCγ1 pathways. ETV6-NTRK3 is effectively inhibited by small molecule ATP-competitive kinase inhibitors including crizotinib and cabozantinib [37,44,45]. We demonstrate that the downstream signalling of both ETV6-NTRK3 and MYO5A-NTRK3 are inhibited by DS-6051a, a selective ATP-competitive small molecule inhibitor of NTRK3 and ROS1. Additional drug sensitivity studies using melanoma cell lines with endogenous fusions of NTRK3 are required to determine the most active compounds in melanoma. Those cell lines are currently not available.
NTRK3 fusions are promising drug-targets as multiple NTRK inhibitors are in early phase clinical trials. A metastatic mammary analogue secretory carcinoma with an ETV6-NTRK3 fusion responded clinically to entrectinib, a selective inhibitor of NTRK1/2/3, ALK and ROS1. The cancer relapsed and the recurrence had a mutation in ETV6-NTRK3, validating the fusion kinase as therapeutic target [46]. ETV6-NTRK3, MYO5A-NTRK3 and MYH9-NTRK3 are newly described NTRK3 fusions in melanocytic tumours, and additional studies to validate methods for clinical detection, identify their prevalence in melanoma in need of systemic treatment, and determine the clinical impact of NTRK3 inhibitors will advance our understanding of NTRK3 fusions in melanoma.
Supplementary Material
Figure S1. Confirmation of fusion transcripts by RT-PCR.
Figure S10. Case 7. Atypical Spitz tumour on the chest of a 41 year old woman.
Figure S11. Case 8. Atypical Spitz tumour on the forehead of a 72 year old woman.
Figure S12. Full western blots corresponding to Figure 3.
Figure S2. Copy number profiles for tumours with NTRK3 fusions.
Figure S3. MYO5A-NTRK3 fusion may arise from tandem duplication.
Figure S4. Expression of NTRK3, ETV6-NTRK3 (EN) and MYO5A-NTRK3 (MN) in 293FT cells.
Figure S5. Case 1. Inflamed Spitz nevus on the leg of a 2 year old girl.
Figure S6. Case 2. Atypical Spitz tumour on the cheek of a 6 year old girl.
Figure S7. Case 3. Atypical Spitz tumour on the cheek of a 7 year old boy.
Figure S8. Case 4. Atypical Spitz tumour on the earlobe of a 10 year old girl.
Figure S9. Case 6. Atypical Spitz tumour on the scalp of a 16 year old girl.
Table S1. Sequencing QC metrics.
Table S2. RT-PCR primers.
Table S3. Clinicopathological features of cases with relative gain of the 3′ end of NTRK3.
Table S4. Target genes on the Sequencing Panel version 1.
Table S5. Bait Intervals for version 1 of targeted sequencing platform (hg19).
Table S6. Target genes on the Sequencing Panel version 2.
Table S7. Bait Intervals for version 2 of targeted sequencing platform (hg19).
Table S8. GEO submission file for array data.
Acknowledgments
This work was funded by grants from Daiichi-Sankyo (to B.C.B), the National Institutes of Health P01 CA025874 (to Meenhard Herlyn), the Dermatology Foundation (to I. Y) and the Melanoma Research Alliance (to I. Y. and to B.C.B) and the Melanoma Research Foundation (to I. Y.). We thank Sonia Mirza and Jingly Weier for assistance with DNA extraction and aCGH, and Arthur Delance for assistance with DNA and cDNA sequencing library preparation. Drs. John Abernethy, Duane Barber, Carrie Davis, Garth Fraga, Chris Nebesio, Sheila Roumpf, Jeffrey Wagner and Simon Warren provided clinical follow-up information.
Footnotes
Conflict of interest statement:
K.N. and T.I. are employees of Daiichi Sankyo, Co., Ltd. The remaining authors declare no conflict of interest.
Statement of Author Contributions:
Project planning and experimental design: I. Y., M.K.T, K.N., T.I., T.B., B.C.B.; sample collection and classification: P.E.L., T.H.M.; aCGH: S.S.V.; preparation of DNA sequencing libraries: A.G.; preparation of RNA and sequencing libraries A.J.S.; sequence data analysis: I.Y., A.H.S.; generation of the translocation constructs: M.K.T.; RT-PCR confirmation: M.K.T.; in vitro experiments including viral transfections and western blotting: M.K.T.; manuscript writing: I.Y., M.K.T., B.C.B.; review of final manuscript: all authors.
SUPPLEMENTARY INFORMATION ONLINE
The following supplementary information may be found in the online version of this article:
References
- 1.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002 Jun 27;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 3.Curtin JA, Busam K, Pinkel D, et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2006 Sep 10;24:4340–6. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 4.Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009 Jan 29;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in Uveal Melanoma. N Engl J Med. 2010;363:2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in Cutaneous Melanoma Is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014 Apr 15;74:2340–50. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012 Sep;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012 Jul 20;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010 Jul;16:793–8. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Botton T, Yeh I, Nelson T, et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res. 2013 Nov 1;26:845–51. doi: 10.1111/pcmr.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutchinson KE, Lipson D, Stephens PJ, et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res Off J Am Assoc Cancer Res. 2013 Dec 15;19:6696–702. doi: 10.1158/1078-0432.CCR-13-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014 Jan 20;5:3116. doi: 10.1038/ncomms4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massi G, LeBoit PE. Histological Diagnosis of Nevi and Melanoma. 2nd. Springer; Heidelberg: 2014. pp. 481–510. [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2009 Jul 15;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 Sep 1;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broad Institute. Picard [Internet] Available from: http://broadinstitute.github.io/picard/
- 17.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010 Sep 1;38:e164–e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2010 Mar 1;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Mullighan CG, Easton J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011 Aug;8:652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013 Mar 1;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009 May 1;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett DC, Cooper PJ, Hart IR. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer. 1987;39:414–8. doi: 10.1002/ijc.2910390324. [DOI] [PubMed] [Google Scholar]
- 24.De Braekeleer E, Douet-Guilbert N, Morel F, et al. ETV6 fusion genes in hematological malignancies: A review. Leuk Res. 2012 Aug;36:945–61. doi: 10.1016/j.leukres.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 25.Knezevich SR, McFadden DE, Tao W, et al. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998 Feb;18:184–7. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- 26.Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002 Nov;2:367–76. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 27.Yeh I, Botton T, Talevich E, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun. 2015;6:7174. doi: 10.1038/ncomms8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun [Internet] 2014 Sep 10; doi: 10.1038/ncomms5846. [cited 2014 Oct 21];5. Available from: http://www.nature.com/ncomms/2014/140910/ncomms5846/full/ncomms5846.html. [DOI] [PMC free article] [PubMed]
- 29.Niu H-T, Zhou Q-M, Wang F, et al. Identification of anaplastic lymphoma kinase break points and oncogenic mutation profiles in acral/mucosal melanomas. Pigment Cell Melanoma Res. 2013 Sep;26:646–53. doi: 10.1111/pcmr.12129. [DOI] [PubMed] [Google Scholar]
- 30.Huang EJ, Reichardt LF. Trk Receptors: Roles in Neuronal Signal Transduction *. Annu Rev Biochem. 2003;72:609–42. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 31.Truzzi F, Marconi A, Lotti R, et al. Neurotrophins and Their Receptors Stimulate Melanoma Cell Proliferation and Migration. J Invest Dermatol. 2008;128:2031–40. doi: 10.1038/jid.2008.21. [DOI] [PubMed] [Google Scholar]
- 32.Tognon C, Garnett M, Kenward E, et al. The Chimeric Protein Tyrosine Kinase ETV6-NTRK3 Requires both Ras-Erk1/2 and PI3-Kinase-Akt Signaling for Fibroblast Transformation. Cancer Res. 2001 Dec 15;61:8909–16. [PubMed] [Google Scholar]
- 33.Tognon CE, Mackereth CD, Somasiri AM, et al. Mutations in the SAM domain of the ETV6-NTRK3 chimeric tyrosine kinase block polymerization and transformation activity. Mol Cell Biol. 2004 Jun;24:4636–50. doi: 10.1128/MCB.24.11.4636-4650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knezevich SR, Garnett MJ, Pysher TJ, et al. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998 Nov 15;58:5046–8. [PubMed] [Google Scholar]
- 35.Skálová A, Vanecek T, Sima R, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol. 2010 May;34:599–608. doi: 10.1097/PAS.0b013e3181d9efcc. [DOI] [PubMed] [Google Scholar]
- 36.Leeman-Neill RJ, Kelly LM, Liu P, et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer. 2014 Mar 15;120:799–807. doi: 10.1002/cncr.28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenca M, Rossi S, Polano M, et al. Transcriptome sequencing identifies ETV6–NTRK3 as a gene fusion involved in GIST. J Pathol. 2016 Mar 1;238:543–9. doi: 10.1002/path.4677. [DOI] [PubMed] [Google Scholar]
- 38.Kralik JM, Kranewitter W, Boesmueller H, et al. Characterization of a newly identified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn Pathol. 2011 Mar 15;6:19. doi: 10.1186/1746-1596-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forghieri F, Morselli M, Potenza L, et al. Chronic eosinophilic leukaemia with ETV6-NTRK3 fusion transcript in an elderly patient affected with pancreatic carcinoma. Eur J Haematol. 2011 Apr 1;86:352–5. doi: 10.1111/j.1600-0609.2011.01576.x. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto H, Yoshida A, Taguchi K, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology. 2016 Jul 1;69:72–83. doi: 10.1111/his.12910. [DOI] [PubMed] [Google Scholar]
- 41.Project the SJCRH-WUPCG. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014 May;46:444–50. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hechtman JF, Zehir A, Yaeger R, et al. Identification of Targetable Kinase Alterations in Patients with Colorectal Carcinoma That are Preferentially Associated with Wild-Type RAS/RAF. Mol Cancer Res MCR. 2016 Mar;14:296–301. doi: 10.1158/1541-7786.MCR-15-0392-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamant L, Gascoyne RD, Duplantier MM, et al. Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer. 2003 Aug;37:427–32. doi: 10.1002/gcc.10232. [DOI] [PubMed] [Google Scholar]
- 44.Taipale M, Krykbaeva I, Whitesell L, et al. Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Nat Biotechnol. 2013 Jul;31:630–7. doi: 10.1038/nbt.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts KG, Li Y, Payne-Turner D, et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N Engl J Med. 2014 Sep 11;371:1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drilon A, Li G, Dogan S, et al. What hides behind the MASC: Clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC) Ann Oncol. 2016 Feb 15;5:920–6. doi: 10.1093/annonc/mdw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Confirmation of fusion transcripts by RT-PCR.
Figure S10. Case 7. Atypical Spitz tumour on the chest of a 41 year old woman.
Figure S11. Case 8. Atypical Spitz tumour on the forehead of a 72 year old woman.
Figure S12. Full western blots corresponding to Figure 3.
Figure S2. Copy number profiles for tumours with NTRK3 fusions.
Figure S3. MYO5A-NTRK3 fusion may arise from tandem duplication.
Figure S4. Expression of NTRK3, ETV6-NTRK3 (EN) and MYO5A-NTRK3 (MN) in 293FT cells.
Figure S5. Case 1. Inflamed Spitz nevus on the leg of a 2 year old girl.
Figure S6. Case 2. Atypical Spitz tumour on the cheek of a 6 year old girl.
Figure S7. Case 3. Atypical Spitz tumour on the cheek of a 7 year old boy.
Figure S8. Case 4. Atypical Spitz tumour on the earlobe of a 10 year old girl.
Figure S9. Case 6. Atypical Spitz tumour on the scalp of a 16 year old girl.
Table S1. Sequencing QC metrics.
Table S2. RT-PCR primers.
Table S3. Clinicopathological features of cases with relative gain of the 3′ end of NTRK3.
Table S4. Target genes on the Sequencing Panel version 1.
Table S5. Bait Intervals for version 1 of targeted sequencing platform (hg19).
Table S6. Target genes on the Sequencing Panel version 2.
Table S7. Bait Intervals for version 2 of targeted sequencing platform (hg19).
Table S8. GEO submission file for array data.