

Abstract
GPR88, an orphan receptor richly expressed in the striatum, is implicated in a number of basal ganglia-associated disorders. In order to elucidate the functions of GPR88, an in vivo probe appropriate for CNS investigation is required. We previously reported that 2-PCCA was able to modulate GPR88-mediated cAMP production through a Gαi-coupled pathway. Early structure–activity relationship (SAR) studies suggested that the aniline moiety of 2-PCCA is a suitable site for diverse modifications. Aimed at elucidating structural requirements in this region, we have designed and synthesized a series of analogues bearing a variety of substituents at the phenyl ring of the aniline moiety. Several compounds (e.g., 5j, 5o) showed improved or comparable potency, but have lower lipophilicity than 2-PCCA (clogP 6.19). These compounds provide the basis for further optimization to probe GPR88 in vivo functions. Computational studies confirmed the SAR trends and supported the notion that 4′-substituents on the biphenyl ring exit through a largely hydrophobic binding site to the extracellular loop.
Keywords: Orphan GPR88, 2-PCCA, SAR, molecular modeling
Graphical Abstract
The G protein-coupled receptors (GPCRs) constitute the largest family of membrane proteins and mediate most cellular responses to hormones and neurotransmitters. GPCRs also play important roles in disease pathogenesis and account for more than 30% of targets of marketed drugs.1 Analysis of the human genome revealed more than 800 GPCR sequences, of which around 140 GPCRs, excluding olfactory receptors, are still classified as orphan receptors for which endogenous ligands are unknown.2 The complex biology and potential for drug therapy within this class provide strong incentives for small molecule probe development to enable modulation of individual receptors and facilitate elucidation of their biological functions.3
GPR88 is an orphan receptor highly expressed in the CNS, with particularly robust expression in the striatum throughout the dorsal and ventral areas.4 Transcriptional profiling studies have revealed Gpr88 gene expression is altered by conditions related to Parkinson’s disease,4c schizophrenia,5 bipolar disorder,6 depression,7 and drug addiction.8 Interestingly, both receptor up- and down-regulation have been observed depending on the treatment and brain region. For example, in dopamine-depleted striatum, Gpr88 expression was differentially regulated in striatonigral and striatopallidal medium spinal neurons (MSNs), and L-DOPA treatment normalized Gpr88 expression levels.4c Gpr88 gene expression was up-regulated in the prefrontal cortex following methamphetamine exposure6b and down-regulated in the amygdala following treatment with the NMDA receptor antagonist MK-801.5
More direct evidence of GPR88 function was obtained from GPR88 knockout studies. GPR88 knockout mice demonstrated disrupted prepulse inhibition of startle response, a phenotype of schizophrenia, and exhibited D2 receptor hypersensitivity, which were normalized to control levels by antipsychotic drug administration.9 In another study, the GPR88 knockout animals exhibited increased locomotion, poor motor coordination and impaired cue-based learning.10 Examination of MSNs electrophysiology in brain slices revealed decreased tonic GABAergic inhibition and increased glutamatergic excitation, which promoted enhanced firing rates observed in vivo.10 In addition, GPR88 re-expression normalized these impaired behaviors and electrophysiological properties, indicating that GPR88 dysfunction may contribute to abnormal behaviors observed in basal ganglia-associated disorders such as Parkinson’s disease and schizophrenia. Interestingly, a recent study showed that the GPR88-deficient mice performed better in spatial tasks and reduced levels of anxiety, indicating GPR88 may also play a role in cognitive and anxiety disorders.11
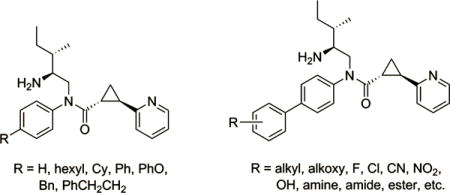
Despite emerging pharmacological implications of GPR88, the signaling mechanisms and biological functions of this orphan receptor are still largely unknown due to the lack of potent and selective native or synthetic ligands. We previously reported that 2-PCCA [(1R*,2R*)-2-(pyridin-2-yl)-cyclopropanecarboxylic acid ((2S,3S)-2-amino-3-methylpentyl)-(4′-propylbiphenyl-4-yl)-amide (1); Figure 1] was able to modulate GPR88-mediated cAMP production through a Gαi− coupled pathway.12 Unfortunately, 2-PCCA has a high calculated lipophilicity (clogP 6.19) and was reported to be a P-glycoprotein substrate, which might limit its effectiveness as a tool.13 In order to elucidate the functions of GPR88, an improved in vivo agonist probe appropriate for CNS investigation is required. Early structure–activity relationship (SAR) studies suggest that the aniline moiety of 2-PCCA is a suitable site for diverse modifications.12,13 Aimed at elucidating SAR requirements in this region, we have designed and synthesized a series of analogues (4a–g, Figure 1 and 5a–aa, Figure 2) bearing a variety of substituents at the phenyl ring of the aniline moiety. In this paper, we report the SAR and computational studies of these compounds with the goal of improving potency and lowering the lipophilicity.
Figure 1.
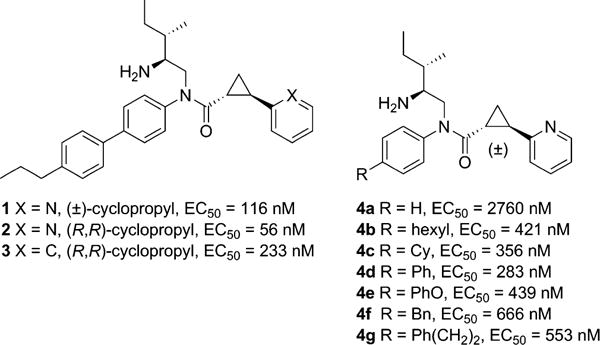
Structures of 2-PCCA (1), (1R,2R)-2-PCCA (2), and analogues 3, 4a–g.
Figure 2.
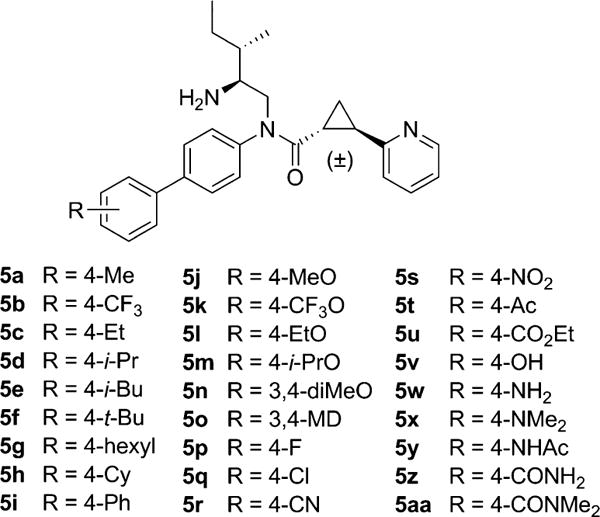
Structure of 2-PCCA analogues 5a–aa.
RESULTS AND DISCUSSION
Chemistry
The overall approach to the synthesis followed methods detailed in our earlier work.12 Synthesis of the designed compounds 4a–g is outlined in Scheme 1. Reductive amination of aldehyde 7, prepared by Dess-Martin oxidation of commercially available (−)-(2S,3S)-N-Boc-2-amino-3-methyl-1-pentanol, with 4-substituted aniline 6a–h afforded amine 8a–h in 46–77% yields. Amide formation with the acid chloride of racemic (±)-trans-2-(pyridin-2-yl)-cyclopropanecarboxylic acid gave 9a–h in 32–72% yields. Removal of the Boc protecting group of 9b–h with 4 M HCl in dioxane provided target compounds 4a–g in 83–97% yields.
Scheme 1.
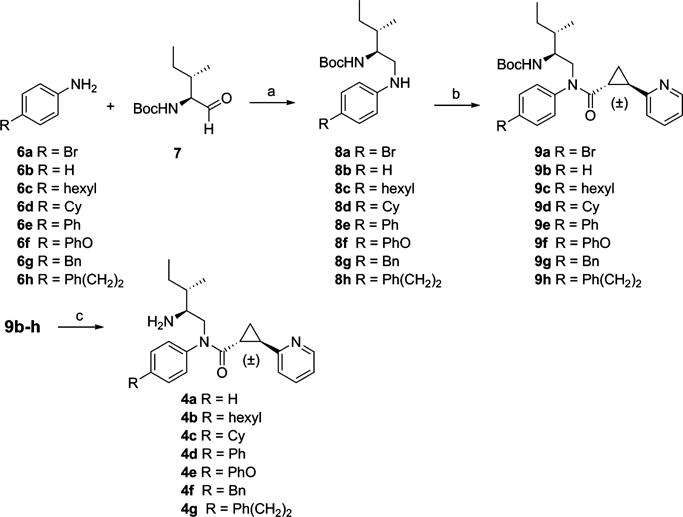
aReagents: (a) NaBH(OAc)3, 1,2-dichloroethane, rt, overnight; (b) (±)-trans-2-(pyridin-2-yl)cyclopropanecarboxylic acid/oxalyl chloride/DCM/40 °C/2 h, concentrated, then 8a–h/Et3N/DCM, rt, overnight; (c) 4 M HCl/dioxane, DCM, rt, 6 h.
Synthesis of the biphenyl derivatives 5a–aa is illustrated in Scheme 2. Suzuki coupling of the bromo compound 9a with an appropriate arylboronic acid under microwave conditions gave intermediates 10a–aa in the range of 51–81% yields. Deprotection of the Boc group furnished 5a–aa in 86–100% yields. Target compounds 4a–g and 5a–aa were determined to be 1:1 mixture of (1R,2R)- and (1S,2S)-enantiomers, differentiating at the configuration of the trans-substituted cyclopropane ring, by 1H NMR and HPLC analyses.
Scheme 2.
aReagents: (a) arylboronic acid, Pd(dppf)Cl2·DCM, K3PO4, DME/H2O (3:1), microwave, 160 °C, 6 min; (b) 4 M HCl/dioxane, DCM, rt, 6 h.
Biological Results and SAR Studies
We previously demonstrated that 2-PCCA (1, Figure 1) inhibited isoproterenol-stimulated cAMP accumulation with EC50 = 911 nM in HEK293 cells stably expressing the human GPR88 receptor and the GloSensor–22F cAMP construct, indicating that GPR88 is coupled to the Gαi subunits.12 Recently, Bi et al. reported that the (1R,2R)-enantiomer of 2-PCCA exhibited an EC50 of 3.1 nM in a HTRF (Cisbio Bioassays) cAMP assay.13 The discrepancy between the EC50 values of 2-PCCA is possibly due to the sensitivity of the two different assay systems. In order to facilitate the SAR study and discovery of potent GPR88 ligands, a reliable and sensitive assay is needed. Thus, we created a Chinese Hamster Ovary (CHO) cell line stably expressing the PPLS-HA-GPR88 construct and measured cAMP levels using the Lance assay platform (PerkinElmer). A preprolactin leader sequence (PPLS) and an influenza hemagglutinin (HA) tag were used in the construct to facilitate membrane expression and immunostaining of GPR88 in CHO cells. In the stable PPLS-HA-GPR88 CHO cells, 2-PCCA and its pure enantiomer 2 had EC50 values of 116 and 56 nM, respectively, shown in Figure 1. The phenyl analogue 3 possessed an EC50 of 233 nM.
Early SAR studies suggest that the aniline moiety of 2-PCCA is a suitable site for optimization.12,13 It appears that the phenyl ring of the aniline moiety can tolerate substitution at the 4-position, in some cases leading to improved potency. In order to gain additional information about the structural requirements in this region for obtaining high potency ligands with possibly improved drug-like properties (e.g., clogP < 5), we have synthesized a series of analogues (4a–g, Figure 1 and 5a–aa, Figure 2) and tested their agonist activity in the Lance cAMP assay. These compounds bear a variety of substituents at the phenyl ring of the aniline moiety to systematically evaluate the effects of size, polarity, lipophilicity, and steric and electronic tolerance. The unsubstituted analogue 4a was significantly less potent than 2-PCCA (2760 vs 116 nM, Figure 1). Addition of a hexyl group at the 4-position of the phenyl ring led to 4b, which regained most of the potency (EC50 = 421 nM), indicating a hydrophobic pocket may be present in the binding site to interact with this region. Replacing the hexyl group with cyclohexyl (4c) or phenyl (4d) further improved the EC50 to 283 nM. However, moving the distal phenyl group in 4d away from the aniline phenyl ring by replacement with phenoxy (4e), benzyl (4f), or phenethyl (4g) resulted in deteriorated activity. These findings suggested that an aromatic stacking interaction between the biphenyl moiety and the receptor might contribute to the agonist activity of 2-PCCA.
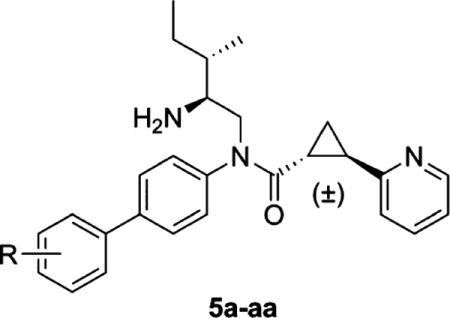
Substituent effect of the distal phenyl ring of 4d was next examined and the results are summarized in Table 1. Consistent with the previous findings,12 the 4-position of the distal phenyl ring tolerated small to medium sized alkyl substitutions with the ethyl analogue 5c (EC50 = 85 nM) being the most potent compound and the bulk t-butyl 5f (EC50 = 515 nM) being the least potent compound in the series. Interestingly, the large alkyl substitutions, such as hexyl (5g), cyclohexyl (5h) and phenyl (5i), were also well tolerated giving good to moderate (EC50 = 136–289 nM) activity. This provided further evidence to support the previous hypothesis13 that the 4′-substitution at the biphenyl group likely extends into a hydrophobic pocket.
Table 1.
Structures and Activities of Compounds 5a–aa
| |||||
|---|---|---|---|---|---|
| compda | R | pEC50 (EC50, nM)b | compda | R | pEC50 (EC50, nM)b |
| 1 | 4-Pr | 6.94 ± 0.11 (116) | 5n | 3,4-diMeO | 6.04 ± 0.01 (917) |
| 4d | H | 6.55 ± 0.03 (283) | 5o | 3,4-MD | 6.69 ± 0.09 (204) |
| 5a | 4-Me | 6.98 ± 0.09 (104) | 5p | 4-F | 5.67 ± 0.04 (268) |
| 5b | 4-CF3 | 6.73 ± 0.06 (187) | 5q | 4-Cl | 6.67 ± 0.07 (213) |
| 5c | 4-Et | 7.07 ± 0.01 (85) | 5r | 4-CN | 6.71 ± 0.05 (197) |
| 5d | 4-i-Pr | 6.59 ± 0.07 (260) | 5s | 4-NO2 | 6.56 ± 0.06 (274) |
| 5e | 4-i-Bu | 6.57 ± 0.02 (271) | 5t | 4-Ac | 6.45 ± 0.05 (351) |
| 5f | 4-t-Bu | 6.29 ± 0.07 (515) | 5u | 4-CO2Et | 6.59 ± 0.09 (257) |
| 5g | 4-hexyl | 6.87 ± 0.11 (136) | 5v | 4-OH | 5.54 ± 0.06 (2860) |
| 5h | 4-Cy | 6.76 ± 0.10 (176) | 5w | 4-NH2 | 5.95 ± 0.02 (1120) |
| 5i | 4-Ph | 6.54 ± 0.03 (289) | 5x | 4-NMe2 | 6.53 ± 0.04 (294) |
| 5j | 4-MeO | 7.02 ± 0.08 (96) | 5y | 4-NHAc | 8500c |
| 5k | 4-CF3O | 6.86 ± 0.04 (139) | 5z | 4-CONH2 | NAc |
| 5l | 4-EtO | 7.09 ± 0.02 (82) | 5aa | 4-CONMe2 | 5.48 ± 0.05 (3320) |
| 5m | 4-i-PrO | 7.11 ± 0.09 (77) | |||
All compounds were tested as the HCl salt.
pEC50 values are means ± standard error of at least three independent experiments performed in duplicate.
EC50 > 10 μM, mean of two independent experiments.
Improvement of the potency of 4d was also observed by adding small alkoxy groups at the 4-position (5j–m). Attachment of an additional methoxy group at the 3-position of 5j led to the 3,4-dimethoxy analogue 5n, resulting in approximately 10-fold loss of activity (96 nM vs 917 nM). The loss in potency of 5n was probably due to the limited steric tolerance proximal to the distal phenyl ring, as evidenced in 5f. Somewhat surprisingly, the 3,4-methylenedioxy analogue 5o possessed good activity with an EC50 of 204 nM. The electron-withdrawing groups (5b, 5p–u) were well tolerated at the 4-postion, with EC50 values ranging from 187 nM to 351 nM. However, substituents containing a hydrogen-bond donor (5v and 5w) led to a significant decrease in activity. The dimethylamino group (5x) had an improved potency compared to the amino group in 5w (294 vs 1120 nM), suggesting that both basicity and lipophilicity of the substituents at the 4-postion may have a marked influence on the activity. Finally, substitution at the 4-position with an amide group (5y, 5z, and 5aa) gave the least activity in the series.
Calculated physiochemical properties such as lipophilicity (clogP), topological polar surface area (TPSA), and derived values such as logBB are useful indicators of a compound’s potential to penetrate the brain. In general, CNS drugs have clogP in the range of 2–4,14 TPSA less than 76 Å2,15 and logBB greater than −1.16 These molecular descriptors were calculated for 2-PCCA, as well as selected compounds 5a, 5c, 5j, 5l, 5m, and 5o (Table 2). 2-PCCA has the highest clogP value of 6.19. All other calculated compounds have clogP greater than 4, among which 5o has the lowest clogP = 4.41. 2-PCCA and all of the analogues, except 5o, have TPSA less than 76 Å2. Even 5o has a TPSA = 77.68 Å2, just above the recommended threshold (a TPSA cutoff of 90 Å2 has also been suggested for CNS drugs17). All compounds have logBB values greater than −1, predicting potential brain penetration.
Table 2.
Calculated Physiochemical Properties
| compd | EC50 (nM) | clogPa | TPSA (Å2)a | logBB |
|---|---|---|---|---|
| 1 | 116 | 6.19 | 59.22 | 0.20 |
| 5a | 104 | 5.30 | 59.22 | 0.07 |
| 5c | 85 | 5.74 | 59.22 | 0.14 |
| 5j | 96 | 4.63 | 68.45 | −0.17 |
| 5l | 82 | 4.99 | 68.45 | −0.12 |
| 5m | 77 | 5.40 | 68.45 | −0.05 |
| 5o | 204 | 4.41 | 77.68 | −0.34 |
cLogP and TPSA were calculated using Instant JChem 5.4.0 (ChemAxon Ltd.).
QSAR Analysis
In a complementary effort to assess the SAR, we computed a series of properties of the substituents and analyzed their importance in explaining the observed variations in ln EC50 values. We computed optimized geometries of the hydrogen capped substituted biphenyl fragments using MOPAC718 and then computed shape/steric (Sterimol parameters/globularity),19 polar surface area (PSA), volume, solvent accessible area, total hydrophobicity (TotalHyd) and HOMO/LUMO frontier orbital energetics using these semi-empirical results in addition to GRAPHA assessments of the varied substituted biphenyl fragments.20 LogP and Molar refractivity and number of rotatable bonds were computed using OpenBabel 2.3.2.21 Isotropic polarizabilities of the substituents were computed using GAMESS-UK22 at 6-31G** B3LYP DFT optimized geometries.
The covariance matrix of all the descriptors was computed to facilitate exploring QSAR employing descriptors with reasonable “orthogonality” in variance. In this way we maximized the linear independence of the descriptors and sought to explain the ln EC50 variations via the QSAR description with the smallest number of possible terms while probing multiple facets of each of the substitutions. While many models were explored in an attempt to find explanations for the ln EC50’s in terms of all the computed metrics, we focused on a buildup of three models and examined at each stage the impact of an additional descriptor.
Table 3 reports the best (model C) of three successive models, while the Supporting Information contains the description and statistics for the other two models A and B upon which model C was based. In the model C, ln EC50 ∼ −1.0 L + 1.7 TotalHyd +0.04 V_S + 0.1 PSA − 0.9LogP + 1.1 B4 + 8.0. The significance of both the “metric” within the given QSAR model as well as the ANOVA computations allowed one to decide on the significance of any improvement in the multivariate-linear least-squares with model expansion. The model table provides the coefficients of the metrics in the model QSAR equation, the standard error (an error level of the model computed from the sums-of-squares of deviations from the theoretical fit and the number of cases), the t-value (the t-statistic providing an indication of whether the coefficient is different than zero), and Pr (the p-value for the hypothesis test for which the t-value is the test statistic). As shown in Table 3, the p-value was <0.05 and indicated significance. At a 95% confidence limit, the model had significance in explaining the variations of ln EC50 in terms of the physiochemical descriptors probed. The R2 value illustrated reasonable correlations in the fit described by the theoretical QSAR equation to the observed ln EC50 value, and this coefficient of determination showed that the variables employed provided a 68% explanation for the variations in ln EC50. Figure 3 shows a plot of the predicted versus the experimental ln EC50 values.
Table 3.
QSAR Model Descriptiona
| model C: ln EC50 ∼ L + TotalHyd + V_S + PSA + logP + B4
| ||||
|---|---|---|---|---|
| descriptor | Estimate coefficients | std error | t-value | Pr (>|t|) |
| intercept | 8.05770 | 1.68999 | 4.768 | 5.23 × 10−5 |
| L | −1.01903 | 0.46050 | −4.587 | 8.58 × 10−5 |
| TotalHyd | 1.70887 | 0.46050 | 3.711 | 0.000907 |
| V_S | 0.04528 | 0.01001 | 4.523 | 0.000102 |
| PSA | 0.14556 | 0.02630 | 5.535 | 6.43 × 10−6 |
| logP | −0.88637 | 0.37052 | −2.392 | 0.023701 |
| B4 | 1.11513 | 0.41208 | 2.706 | 0.011461 |
Residual standard error = 0.7779 on 28 degrees of freedom, multiple R2 = 0.6797, adjusted R2 = 0.6111, F-statistic = 9.903 on 6 and 28 degrees of freedom, p-value = 7.063 × 10−6.
Figure 3.
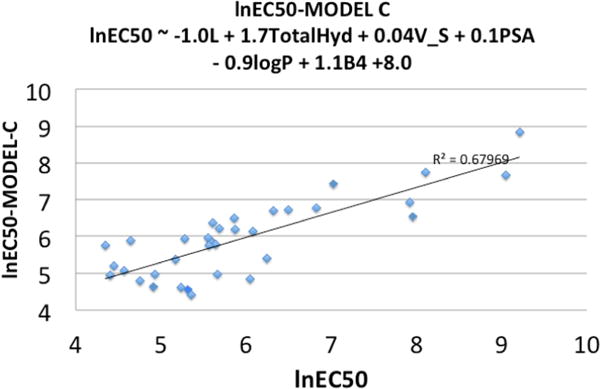
Plot of experimental ln EC50 vs model C ln EC50.
The model C contains six physiochemical descriptors. PSA, logP, and total hydrophobicity are common features used in the assessment of drug candidates, but the L- and B4-Sterimol (L and B4, respectively) and the vibrational thermochemical (V_S) descriptors are much less well-known. An example of the L- and B4-Sterimol metrics for two ligands 5j and 5o is shown in Figure 4. The L-Sterimol axis is the longest dimension spanning the molecular surface of the substituent. The B4-Sterimol descriptor is orthogonal to this L-dimension and provides a metric of a width of the substituent to the L-axis. Such orthogonal descriptor captures the width of the binding pocket orthogonal to the long axis of the substituent.
Figure 4.
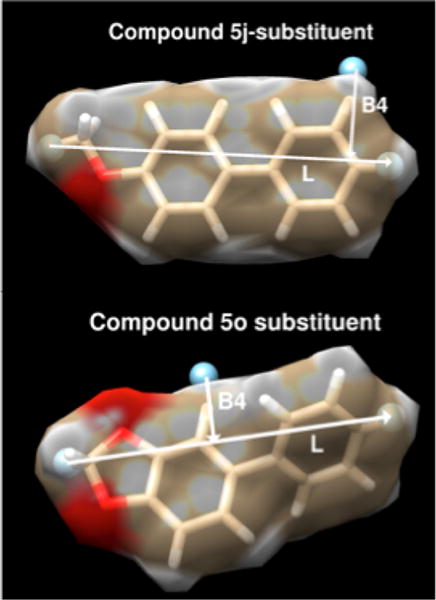
Illustrations of the L- and B4-Sterimol metrics for 5j and 5o.
The vibrational parameter (V_S) is a descriptor that captures a dynamical aspect of the ligand and possibly indicates a propensity for cooperative binding. While differential binding to the orthosteric site of a GPCR is not likely to result in significant receptor structural changes near the orthosteric site, small changes in a ligand’s architecture may result in differential stabilization of activated conformations of the GPCR. The effect of the local fluctuations in a portion of a ligand substituent may bias larger cooperative structural changes at the intracellular side to conformations constituting an “active state”.23,24 Small changes in a ligand’s dynamics may in this way alter ionic locks/loop and helical structure at the intracellular region that modifies interactions with GPCR partners (e.g., G proteins, β-arrestin). While such a rationale for V_S remains speculative, there was a plausible connection of the vibrational parameter to the ln EC50 in our QSAR model.
Docking Analysis of (1R,2R)-2-PCCA
While the QSAR model compared reasonably with the qualitative facets of the SAR deductions at the aniline moiety, we sought to discover if a homology model, based on existing crystallographic templates, would give useful information about the receptor binding site characteristics for this region. The focus of this limited structural exploration was to ascertain whether the QSAR is consistent with structural principles/orthosteric ligand binding site based on the docking and MMGBSA (Molecular Mechanics Generalized Born Surface Area)25 scoring analysis of (1R,2R)-2-PCCA.
Sequence alignments of human GPR88 with selected GPCR templates of known structure were performed using a BLOSUM62 matrix. While no GPCR crystal structures with high or moderate sequence identity (>40% by our own experience) to GPR88 have yet been solved, the β1-adrenergic receptor (PDB: 5A8E) has a 26% sequence identity to GPR88 deduced from a pairwise alignment. This template also has one of the highest sequence identities in the alignment of 9 GPCRs with GPR88 (∼20%, see the Supporting Information). The β1-adrenergic receptor template was used to construct an initial backbone model of GPR88 employing Sali’s MODELER v9.11,26 refining the backbone model “energetically” using simulated annealing with topological constraints. We next employed SCWRL27 rotamer exploration to find nonclashing side chain orientations for nonconserved residues with conserved residues between the target and template employing conservation assessments shown in the alignment. Conserved residue side chain from the β1-adrenergic receptor template were initially retained in the SCWRL rotamer exploration. This initial level model was then energy minimized using AMBER1228 to provide a homology model for docking and free-energy scoring.
We docked (1R,2R)-2-PCCA using both Autodock VINA29 with AMBER12 MMGBSA rescoring as well as GLIDE-SP (Schrödinger)30 followed by Prime-MMGBSA rescoring. As shown in Figure 5, the 4′-propylbiphenyl group in (1R,2R)-2-PCCA pointed out of the orthosteric site into the extracellular loop region. The residue character along the binding site region housing the biphenyl is quite hydrophobic. The initial GPR88 homology model appears to be consistent with the results of QSAR analysis in that the long-spatial axis, parametrized at the L-Sterimol, is an important facet as is the overall hydrophobicity. The model is not, at this stage, predictive of ln EC50. Refinement of the model using pure (R,R)-diastereomers is currently under investigation.
Figure 5.
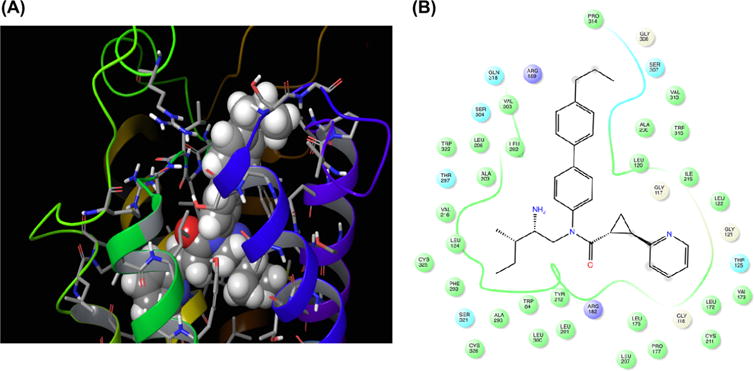
Examination of an early stage homology model of GPR88 with (1R,2R)-2-PCCA lowest MMGBSA scored pose in 3D (A) and LIGPLOT (B) format. Note the largely hydrophobic residues around the biphenyl scaffold portion indicated by the green line.
CONCLUSIONS
In summary, we have designed and synthesized a series of 2-PCCA analogues bearing a variety of substituents at the phenyl ring of the aniline moiety to determine the SAR in this region. The target compounds were evaluated in a Lance cAMP assay using stable PPLS-HA-GPR88 CHO cells. SAR studies suggested that there is a hydrophobic pocket in the GPR88 binding site interacting with the aniline region of 2-PCCA. In addition, the 4′-position of the biphenyl group was well tolerated with alkyl, alkoxy and electron-withdrawing groups, but less tolerated with the substituents having a hydrogen-bond donor. Several compounds had a slightly improved or comparable potency, but lower calculated lipophilicity than 2-PCCA. Combined with calculated TPSA and logBB values, compound 5j may provide the basis for further optimization to develop an in vivo probe for GPR88 functional studies.
Exploration of both a computational QSAR and an initial homology model and docking of (1R,2R)-2-PCCA supported the SAR conclusions. The findings of statistically sound models including both L- and B4-Sterimol parameters in the QSAR were consistent with the positioning of the 4′-substituted biphenyl encased in a largely hydrophobic GPR88 homology model binding site.
METHODS
Chemistry
General Methods
Melting points were determined using a MEL-TEMP II capillary melting point apparatus and are uncorrected. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained on a Bruker Avance DPX-300 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) with reference to internal solvent. 13C NMR data of diastereomeric mixtures were not reported due to the complicity of the spectra. Mass spectra (MS) were run on a PerkinElmer Sciex API 150 EX mass spectrometer. HRMS spectra were run on a Waters Synapt G2 HDMS Q-TOF mass spectrometer, using electrospray ionization in positive ion mode. Analytical thin-layer chromatography (TLC) was carried out using EMD silica gel 60 F254 TLC plates. TLC visualization was achieved with a UV lamp or in an iodine chamber. Flash column chromatography was done on a CombiFlash Companion system using Isco prepacked silica gel columns. Unless otherwise stated, reagent-grade chemicals were obtained from commercial sources and were used without further purification. All moisture- and air-sensitive reactions and reagent transfers were carried out under dry nitrogen. Synthesis and characterization of compounds 1–3, 5a–c, 5e, 5h, 5p, 5q, and 5t have been previously reported.12 All synthesized compounds were ≥95% pure as determined by HPLC analyses (see the Supporting Information).
tert-Butyl {(2S,3S)-1-[(4-Bromophenyl)amino]-3-methylpentan-2-yl}carbamate (8a)
To a solution of (−)-(2S,3S)-N-Boc-2-amino-3-methyl-1-pentanol (2.17 g, 10.0 mmol) in water-saturated CH2Cl2 (10 mL) at room temperature was added Dess-Martin reagent (8.90 g, 21.0 mmol) and the reaction was stirred for 1 h. Additional water-saturated CH2Cl2 (5 mL) was added every 15 min during the reaction time. The mixture was diluted with Et2O (100 mL) and poured into a solution of Na2S2O3 (17 g) in 80% saturated NaHCO3 (100 mL). After stirring for 10 min, the layers were separated and the aqueous layer was extracted with Et2O (100 mL). The combined organic layers were washed with ice-cold saturated NaHCO3 (30 mL) and water (30 mL). The solution was dried (Na2SO4) and concentrated under reduced pressure to give the crude aldehyde 7. To a solution of 4-bromoaniline (6a) (1.72 g, 10.0 mmol) in dichloroethane (60 mL) was added the above crude aldehyde, followed by NaBH(OAc)3 (4.24 g, 20.0 mmol). The mixture was stirred at room temperature overnight. Saturated NaHCO3 (20 mL) was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were washed with brine (3 × 30 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–30% EtOAc in hexanes afforded 8a (2.78 g, 75%) as a white solid: mp 103–105 °C; 1H NMR (300 MHz; CDCl3) δ 7.24 (d, J = 9.0 Hz, 2H), 6.45 (d, J = 9.0 Hz, 2H), 4.52 (d, J = 9.0 Hz, 1H), 4.20 (br s, 1H), 3.82−3.65 (m, 1H), 3.30−3.14 (m, 1H), 3.05−2.89 (m, 1H), 1.60−1.47 (m, 1H), 1.44 (s, 9H), 1.23−1.10 (m, 1H), 0.95 (d, J = 6.0 Hz, 3H), 0.95 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 154.9, 145.7, 130.1, 112.4, 106.9, 77.9, 52.9, 45.0, 35.7, 26.6, 23.6, 13.8, 9.9; MS (ESI) m/z 371.3 [M + H]+ (79Br), 373.3 [M + H]+ (81Br).
tert-Butyl [(2S,3S)-1-Phenylamino-3-methylpentan-2-yl]-carbamate (8b)
The procedure for 8a was followed using 140 mg (1.5 mmol) of 6b to give 336 mg (77%) of 8b as a white solid: mp 77–79 °C; 1H NMR (300 MHz; CDCl3) δ 7.16 (t, J = 7.5 Hz, 2H), 6.69 (t, J = 7.5 Hz, 1H), 6.59 (d, J = 9.0 Hz, 2H), 4.57 (d, J = 9.0 Hz, 1H), 4.13 (br s, 1H), 3.82−3.68 (m, 1H), 3.26 (dd, J = 12.0, 3.0 Hz, 1H), 3.02 (t, J = 10.5 Hz, 1H), 1.68−1.46 (m, 1H), 1.45 (s, 9H), 1.28−1.10 (m, 1H), 0.98−0.89 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 156.6, 148.3, 129.2, 117.4, 112.8, 79.5, 54.7, 46.6, 37.5, 28.4, 25.4, 15.5, 11.7; MS (ESI) m/z 293.3 [M + H]+.
tert-Butyl {(2S,3S)-1-[(4-Hexylphenyl)amino]-3-methylpentan-2-yl}carbamate (8c)
The procedure for 8a was followed using 355 mg (2.0 mmol) of 6c to give 526 mg (70%) of 8c as an oil: 1H NMR (300 MHz; CDCl3) δ 6.98 (d, J = 9.0 Hz, 2H), 6.53 (d, J = 9.0 Hz, 2H), 4.52 (d, J = 9.0 Hz, 1H), 3.86 (br s, 1H), 3.80−3.66 (m, 1H), 3.30−3.18 (m, 1H), 3.06−2.90 (m, 1H), 2.48 (t, J = 6.0 Hz, 2H), 1.70−1.53 (m, 2H), 1.44 (s, 9H), 1.38−1.10 (m, 8H), 1.00−0.80 (m, 9H); 13C NMR (75 MHz; CDCl3) δ 156.5, 146.5, 131.8, 129.1, 112.8, 79.4, 54.8, 46.7, 37.5, 35.1, 31.9, 31.8, 29.0, 28.4, 25.4, 22.7, 15.5, 14.1, 11.7; MS (ESI) m/z 377.3 [M + H]+.
tert-Butyl {(2S,3S)-1-[(4-Cyclohexylphenyl)amino]-3-methylpentan-2-yl}carbamate (8d)
The procedure for 8a was followed using 263 mg (1.5 mmol) of 6d to give 345 mg (61%) of 8d as a white solid: mp 108–110 °C; 1H NMR (300 MHz; CDCl3) δ 7.02 (d, J = 9.0 Hz, 2H), 6.55 (d, J = 9.0 Hz, 2H), 4.53 (d, J = 9.0 Hz, 1H), 4.00 (br s, 1H), 3.80−3.66 (m, 1H), 3.32−3.20 (m, 1H), 3.10−2.90 (m, 1H), 2.46−2.31 (m, 1H), 1.90−1.70 (m, 5H), 1.70−1.10 (m, 16H), 0.98−0.90 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 156.5, 146.5, 137.3, 127.5, 122.8, 79.4, 54.8, 46.7, 43.7, 37.4, 34.8, 28.4, 27.1, 26.3, 25.4, 15.5, 11.7; MS (ESI) m/z 375.2 [M + H]+.
tert-Butyl [(2S,3S)-1-Biphenylamino-3-methylpentan-2-yl]-carbamate (8e)
The procedure for 8a was followed using 545 mg (2.5 mmol) of 6e to give 560 mg (76%) of 8e as a white solid: mp 98− 100 °C; 1H NMR (300 MHz; CDCl3) δ 7.56−7.50 (m, 2H), 7.46−7.34 (m, 4H), 7.28−7.21 (m, 1H), 6.66 (d, J = 9.0 Hz, 2H), 4.47 (d, J = 9.0 Hz, 1H), 4.18 (br s, 1H), 3.81−3.71 (m, 1H), 3.32−3.22 (m, 1H), 3.10−2.96 (m, 1H), 1.68−1.46 (m, 1H), 1.45 (s, 9H), 1.30−1.10 (m, 1H), 0.98 (d, J = 9.0 Hz, 3H), 0.95 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 156.7, 148.1, 141.4, 130.2, 128.7, 127.6, 126.6, 126.1, 133.0, 79.6, 54.8, 46.6, 37.5, 28.5, 25.5, 15.6, 11.7; MS (ESI) m/z 369.4 [M + H]+.
tert-Butyl {(2S,3S)-1-[(4-Phenoxyphenyl)amino]-3-methylpentan-2-yl}carbamate (8f)
The procedure for 8a was followed using 370 mg (2.0 mmol) of 6f to give 350 mg (46%) of 8f as an oil: 1H NMR (300 MHz; CDCl3) δ 7.32−7.25 (m, 2H), 7.05−6.98 (m, 1H), 6.96−6.86 (m, 4H), 6.59 (d, J = 9.0 Hz, 2H), 4.56 (d, J = 9.0 Hz, 1H), 4.02 (br s, 1H), 3.81−3.72 (m, 1H), 3.31−3.20 (m, 1H), 3.06−2.95 (m, 1H), 1.66−1.50 (m, 1H), 1.45 (s, 9H), 1.26−1.10 (m, 1H), 1.02−0.90 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 159.3, 156.7, 147.5, 145.3, 129.9, 121.9, 121.3, 117.1, 113.7, 79.4, 54.8, 46.9, 37.5, 28.5, 25.4, 15.6, 11.7; MS (ESI) m/z 385.4 [M + H]+.
tert-Butyl {(2S,3S)-1-[(4-Benzylphenyl)amino]-3-methylpentan-2-yl}carbamate (8g)
The procedure for 8a was followed using 275 mg (1.5 mmol) of 6g to give 390 mg (68%) of 8g as an off-white solid: mp 65−66 °C; 1H NMR (300 MHz; CDCl3) δ 7.30−7.15 (m, 5H), 6.99 (d, J = 9.0 Hz, 2H), 6.54 (d, J = 9.0 Hz, 2H), 4.54 (d, J = 9.0 Hz, 1H), 3.95 (br s, 1H), 3.87 (s, 2H), 3.83−3.70 (m, 1H), 3.30−3.18 (m, 1H), 3.06−2.95 (m, 1H), 1.65−1.50 (m, 1H), 1.44 (s, 9H), 1.28−1.10 (m, 1H), 1.01−0.90 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 156.5, 146.6, 142.1, 130.0, 129.7, 128.8, 128.3, 125.8, 113.0, 79.5, 54.7, 46.8, 41.1, 37.5, 28.4, 25.4, 15.5, 11.7; MS (ESI) m/z 383.5 [M + H]+.
tert-Butyl {(2S,3S)-1-[(4-Phenethylphenyl)amino]-3-methylpentan-2-yl}carbamate (8h)
The procedure for 8a was followed using 290 mg (1.5 mmol) of 6h to give 325 mg (55%) of 8h as a white solid: mp 55–57 °C; 1H NMR (300 MHz; CDCl3) δ 7.30−7.10 (m, 5H), 6.99 (d, J = 9.0 Hz, 2H), 6.59 (d, J = 9.0 Hz, 2H), 4.62 (d, J = 9.0 Hz, 1H), 3.95 (br s, 1H), 3.80−3.68 (m, 1H), 3.25 (dd, J = 12.0, 3.0 Hz, 1H), 3.02 (t, J = 10.5 Hz, 1H), 2.90−2.76 (, 4H), 1.65−1.50 (m, 1H), 1.44 (s, 9H), 1.23−1.10 (m, 1H), 0.98−0.88 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 156.6, 145.9, 142.1, 131.4, 129.2, 128.5, 128.3, 125.8, 113.5, 79.5, 54.6, 47.2, 38.3, 37.5, 37.1, 28.4, 25.4, 15.5, 11.7; MS (ESI) m/z 397.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{4-Bromophenyl-[(1R*,2R*)-2-(pyridin-2-yl)-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9a)
To a solution of (±)-trans-2-(pyridin-2-yl)-cyclopropanecarboxylic acid (0.40 g, 2.0 mmol) in CH2Cl2 (20 mL) at room temperature was added oxalyl chloride (0.35 mL, 4.0 mmol) and DMF (50 μL). The mixture was stirred at 40 °C for 2 h, then cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (20 mL) and treated with 8a (0.74 g, 2.0 mmol) and Et3N (1.1 mL, 8.0 mmol). The resulting solution was stirred at room temperature overnight. Saturated NaHCO3 (10 mL) was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (3 × 20 mL), dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0–25% EtOAc in hexanes afforded 9a (0.74 g, 72%, 1:1 diastereomeric mixture) as a light yellow foam: 1H NMR (300 MHz; CDCl3) δ 8.30 (d, J = 6.0 Hz, 1H), 7.58−7.34 (m, 3H), 7.22−6.98 (m, 4H), 4.96 (d, J = 9.0 Hz, 1H), 4.45−4.25 (m, 1H), 3.80−3.78 (m, 1H), 3.21−3.06 (m, 1H), 2.71−2.62 (m, 0.5H), 2.58−2.48 (m, 0.5H), 1.98−1.86 (m, 1H), 1.75−1.62 (m, 1H), 1.60−1.35 (m, 3H), 1.45 and 1.40 (2s, 9H), 1.15−1.02 (m, 1H), 0.92−0.80 (m, 6H); MS (ESI) m/z 516.7 [M + H]+ (79Br), 518.6 [M + H]+ (81Br).
tert-Butyl [(2S,3S)-1-{Phenyl-[(1R*,2R*)-2-(pyridin-2-yl)-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9b)
The procedure for 9a was followed using 102 mg (0.35 mmol) of 8b to give 85 mg (56%) of 9b (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.26 (d, J = 6.0 Hz, 1H), 7.54−7.46 (m, 1H), 7.38−7.10 (m, 6H), 7.03−6.96 (m, 1H), 4.96 (t, J = 7.5 Hz, 1H), 4.48−4.32 (m, 1H), 3.81−3.62 (m, 1H), 3.22−3.08 (m, 1H), 2.72−2.62 (m, 0.5H), 2.58−2.48 (m, 0.5H), 2.00−1.88 (m, 1H), 1.72−1.48 (m, 4H), 1.46 and 1.42 (2s, 9H), 1.15−1.00 (m, 1H), 0.91−0.78 (m, 6H); MS (ESI) m/z 438.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{4-Hexylphenyl-[(1R*,2R*)-2-(pyridin-2-yl)-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9c)
The procedure for 9a was followed using 188 mg (0.5 mmol) of 8c to give 140 mg (54%) of 9c (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.28−8.22 (m, 1H), 7.53−7.45 (m, 1H), 7.20−6.92 (m, 6H), 5.10 (t, J = 9.0 Hz, 1H), 4.45−4.31 (m, 1H), 3.80−3.60 (m, 1H), 3.18−3.06 (m, 1H), 2.70−2.61 (m, 0.5H), 2.60−2.47 (m, 2.5H), 2.00−1.89 (m, 1H), 1.71−1.30 (m, 24H), 1.20−1.02 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 522.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{4-Cyclohexylphenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9d)
The procedure for 9a was followed using 131 mg (0.35 mmol) of 8d to give 104 mg (57%) of 9d (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.30−8.20 (m, 1H), 7.52−7.45 (m, 1H), 7.20−6.92 (m, 6H), 5.12 (dd, J = 12.0, 9.0 Hz, 1H), 4.46−4.30 (m, 1H), 3.80−3.60 (m, 1H), 3.20−3.06 (m, 1H), 2.70−2.60 (m, 0.5H), 2.58−2.47 (m, 1.5H), 2.00−1.65 (m, 7H), 1.55−1.20 (m, 17H), 1.18−1.00 (m, 1H), 0.92−0.78 (m, 6H); MS (ESI) m/z 520.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{Biphenyl-[(1R*,2R*)-2-(pyridin-2-yl)-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9e)
The procedure for 9a was followed using 184 mg (0.5 mmol) of 8e to give 130 mg (51%) of 9e (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.29−8.23 (m, 1H), 7.60−7.15 (m, 11H), 7.01−6.95 (m, 1H), 5.11 (t, J = 9.0 Hz, 1H), 4.50−4.37 (m, 1H), 3.86−3.68 (m, 1H), 3.26−3.17 (m, 1H), 2.75−2.68 (m, 0.5H), 2.60−2.51 (m, 0.5H), 2.10−1.98 (m, 1H), 1.75−1.67 (m, 0.5H), 1.66−1.58 (m, 0.5H), 1.57−1.40 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.20−1.03 (m, 1H), 0.93−0.80 (m, 6H); MS (ESI) m/z 514.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{4-Phenoxyphenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9f)
The procedure for 9a was followed using 130 mg (0.34 mmol) of 8f to give 57 mg (32%) of 9f (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.33−8.27 (m, 1H), 7.52−7.43 (m, 1H), 7.40−7.30 (m, 2H), 7.22−7.10 (m, 4H), 7.02−6.78 (m, 5H), 5.08 (t, J = 9.0 Hz, 1H), 4.55−4.40 (m, 1H), 3.82−3.66 (m, 1H), 3.20−3.02 (m, 1H), 2.68−2.60 (m, 0.5H), 2.52−2.43 (m, 0.5H), 2.02−1.90 (m, 1H), 1.75−1.45 (m, 4H), 1.44 and 1.41 (2s, 9H), 1.18−1.00 (m, 1H), 0.92−0.80 (m, 6H); MS (ESI) m/z 530.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{4-Benzylphenyl-[(1R*,2R*)-2-(pyridin-2-yl)-cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9g)
The procedure for 9a was followed using 100 mg (0.26 mmol) of 8g to give 65 mg (47%) of 9g (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.30−8.26 (m, 1H), 7.60−7.52 (m, 1H), 7.38−6.95 (m, 11H), 5.06 (t, J = 9.0 Hz, 1H), 4.48−4.30 (m, 1H), 3.92 and 3.88 (2s, 2H), 3.80−3.66 (m, 1H), 3.20−3.05 (m, 1H), 2.70−2.62 (m, 0.5H), 2.50−2.42 (m, 0.5H), 1.98−1.90 (m, 1H), 1.76−1.46 (m, 4H), 1.44 and 1.42 (2s, 9H), 1.20−1.02 (m, 1H), 0.93−0.82 (m, 6H); MS (ESI) m/z 528.8 [M + H]+.
tert-Butyl [(2S,3S)-1-{4-Phenethylphenyl-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (9h)
The procedure for 9a was followed using 139 mg (0.35 mmol) of 8h to give 96 mg (51%) of 9h (1:1 diastereomeric mixture) as an oil: 1H NMR (300 MHz; CDCl3) δ 8.30−8.25 (m, 1H), 7.55−7.46 (m, 1H), 7.30−6.96 (m, 11H), 5.10 (dd, J = 9.0, 3.0 Hz, 1H), 4.46−4.32 (m, 1H), 3.80−3.62 (m, 1H), 3.20−3.06 (m, 1H), 2.90−2.80 (m, 4H), 2.71−2.632 (m, 0.5H), 2.56−2.47 (m, 0.5H), 1.98−1.90 (m, 1H), 1.72−1.47 (m, 4H), 1.46 and 1.42 (2s, 9H), 1.16−1.02 (m, 1H), 0.90−0.78 (m, 6H); MS (ESI) m/z 528.8 [M + H]+.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(phenyl-4-yl)amide (4a)
A solution of 9b (85 mg, 0.19 mmol) and 4 M HCl in dioxane (2 mL) in CH2Cl2 (5 mL) was stirred at room temperature for 6 h. The solvent was removed under reduced pressure. The resulting residue was triturated with hexanes to give 4a dihydrochloride (76 mg, 96%, 1:1 diastereomeric mixture) as an off-white solid: 1H NMR (300 MHz; CD3OD) δ 8.66−8.56 (m, 1H), 8.40−8.28 (m, 1H), 7.85−7.74 (m, 1H), 7.61−7.33 (m, 6H), 4.40 (dd, J = 15.0, 9.0 Hz, 0.5H), 4.29 (dd, J = 15.0, 9.0 Hz, 0.5H), 3.78−3.56 (m, 1.5H), 3.39−3.30 (m, 0.5H), 3.08−2.99 (m, 0.5H), 2.98−2.89 (m, 0.5H), 2.11−1.84 (m, 2H), 1.82−1.58 (m, 2H), 1.40−1.10 (m, 2H), 1.00−0.76 (m, 6H); HRMS (ESI) calcd for C21H27N3O [M + H]+: 338.2227. Found: 338.2241.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-hexylphenyl-4-yl)amide (4b)
The procedure for 4a was followed using 135 mg (0.26 mmol) of 9c to give 121 mg (95%) of 4b dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.26−8.16 (m, 1H), 7.70−7.56 (m, 1H), 7.28−7.08 (m, 6H), 4.32−4.16 (m, 1H), 3.70−3.58 (m, 1H), 3.36−3.20 (m, 1H), 2.66−2.50 (m, 3H), 1.92−1.80 (m, 1H), 1.78−1.48 (m, 4H), 1.48−1.10 (m, 9H), 1.00−0.76 (m, 9H); HRMS (ESI) calcd for C27H39N3O [M + H]+: 422.3166. Found: 422.3170.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-cyclohexylphenyl-4-yl)amide (4c)
The procedure for 4a was followed using 104 mg (0.2 mmol) of 9d to give 93 mg (95%) of 4c dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.63−8.52 (m, 1H), 8.38−8.24 (m, 1H), 7.85−7.72 (m, 1H), 7.60−7.48 (m, 1H), 7.48−7.20 (m, 4H), 4.36 (dd, J = 15.0, 9.0 Hz, 0.5H), 4.25 (dd, J = 15.0, 9.0 Hz, 0.5H), 3.78−3.56 (m, 1.5H), 3.40−3.30 (m, 0.5H), 3.04−2.95 (m, 0.5H), 2.94−2.83 (m, 0.5H), 2.60−2.42 (m, 1H), 2.10−2.00 (m, 1H), 1.98−1.52 (m, 7H), 1.52−1.05 (m, 8H), 1.00−0.76 (m, 6H); HRMS (ESI) calcd for C27H37N3O [M + H]+: 420.3009. Found: 420.3022.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(biphenyl-4-yl)amide (4d)
The procedure for 4a was followed using 125 mg (0.24 mmol) of 9e to give 115 mg (97%) of 4d dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.28−8.21 (m, 1H), 7.72−7.52 (m, 5H), 7.48−7.32 (m, 5H), 7.30−7.23 (m, 1H), 7.19−7.10 (m, 1H), 4.40−4.22 (m, 1H), 3.80−3.66 (m, 1H), 3.40−3.22 (m, 1H), 2.70−2.53 (m, 1H), 2.05−1.90 (m, 1H), 1.84−1.62 (m, 2H), 1.50−1.15 (m, 3H), 0.99 and 0.97 (2d, J = 6.0 Hz, 3H), 0.90−0.80 (m, 3H); HRMS (ESI) calcd for C27H31N3O [M + H]+: 414.2540. Found: 414.2541.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-phenoxyphenyl-4-yl)amide (4e)
The procedure for 4a was followed using 50 mg (0.09 mmol) of 9f to give 46 mg (97%) of 4e dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.60−8.46 (m, 1H), 8.32−8.20 (m, 1H), 7.78−7.64 (m, 1H), 7.60−7.20 (m, 5H), 7.12−7.02 (m, 1H), 7.00−6.76 (m, 4H), 4.32 (dd, J = 15.0, 9.0 Hz, 0.5H), 4.13 (dd, J = 15.0, 9.0 Hz, 0.5H), 3.68−3.40 (m, 1.5H), 3.35−3.25 (m, 0.5H), 3.02−2.90 (m, 0.5H), 2.88−2.78 (m, 0.5H), 2.08−1.54 (m, 4H), 1.42−1.08 (m, 2H), 0.98−0.68 (m, 6H); HRMS (ESI) calcd for C27H31N3O [M + H]+: 430.2489. Found: 430.2505.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-benzylphenyl-4-yl)amide (4f)
The procedure for 4a was followed using 65 mg (0.12 mmol) of 9g to give 51 mg (83%) of 4f dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.52−8.46 (m, 1H), 8.22−8.10 (m, 1H), 7.70−7.60 (m, 1H), 7.50−7.32 (m, 3H), 7.30−7.06 (m, 7H), 4.32 (dd, J = 15.0, 9.0 Hz, 0.5H), 4.21 (dd, J = 15.0, 9.0 Hz, 0.5H), 3.92 and 3.90 (2s, 2H), 3.75−3.65 (m, 1.5H), 3.38−3.26 (m, 0.5H), 3.00−2.90 (m, 0.5H), 2.88−2.80 (m, 0.5H), 2.08−1.58 (m, 4H), 1.42−1.12 (m, 2H), 0.98−0.72 (m, 6H); HRMS (ESI) calcd for C28H33N3O [M + H]+: 428.2696. Found: 428.2711.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-phenethylphenyl-4-yl)amide (4g)
The procedure for 4a was followed using 96 mg (0.18 mmol) of 9h to give 87 mg (95%) of 4g dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.66−8.56 (m, 1H), 8.38−8.25 (m, 1H), 7.82−7.70 (m, 1H), 7.58−7.42 (m, 1H), 7.40−7.30 (m, 2H), 7.30−7.05 (m, 7H), 4.42 (dd, J = 15.0, 9.0 Hz, 0.5H), 4.27 (dd, J = 15.0, 9.0 Hz, 0.5H), 3.75−3.56 (m, 1.5H), 3.38−3.28 (m, 0.5H), 3.06−2.96 (m, 0.5H), 2.96−2.78 (m, 4.5H), 2.10−1.82 (m, 2H), 1.80−1.58 (m, 2H), 1.40−1.10 (m, 2H), 1.00−0.70 (m, 6H); HRMS (ESI) calcd for C29H35N3O [M + H]+: 442.2853. Found: 442.2859.
tert-Butyl [(2S,3S)-1-{(4′-Methylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10a)
A mixture of 9a (30 mg, 0.058 mmol), 4-methylphenylboronic acid (12 mg, 0.087 mmol), Pd(dppf)-Cl2•CH2Cl2 (4.35 mg, 0.0058 mmol) and K3PO4 (38 mg, 2.7 mmol) in dimethoxyethane (1 mL) and water (0.3 mL) was heated in a sealed vessel by microwave irradiation at 160 °C for 6 min. The resulting mixture was poured into 1 N NaOH solution (5 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 20% EtOAc in hexanes afforded 10a (20 mg, 65%) as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.29−8.22 (m, 1H), 7.58−7.38 (m, 5H), 7.37−7.16 (m, 5H), 7.02−6.92 (m, 1H), 5.13−5.06 (m, 1H), 4.48−4.37 (m, 1H), 3.83−3.68 (m, 1H), 3.24−3.12 (m, 1H), 2.73−2.65 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.39 (s, 3H), 2.08−1.96 (m, 1H), 1.76−1.55 (m, 1H), 1.54−1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.17−1.00 (m, 1H), 0.92−0.78 (m, 6H); MS (ESI) m/z 528.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Trifluoromethylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10b)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 17 mg (0.087 mmol) of 4-trifluoromethylphenylboronic acid to give 25 mg (74%) of 10b as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 7.72−7.40 (m, 7H), 7.38−7.16 (m, 3H), 7.02−6.93 (m, 1H), 5.13−5.02 (m, 1H), 4.50−4.38 (m, 1H), 3.84−3.65 (m, 1H), 3.28−3.15 (m, 1H), 2.76−2.65 (m, 0.5H), 2.61−2.48 (m, 0.5H), 2.08−1.96 (m, 1H), 1.76−1.55 (m, 1H), 1.54−1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.18−1.00 (m, 1H), 0.94−0.78 (m, 6H); MS (ESI) m/z 582.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Ethylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10c)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 13 mg (0.087 mmol) of 4-ethylphenylboronic acid to give 25 mg (80%) of 10c as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.23 (m, 1H), 7.58−7.38 (m, 5H), 7.37−7.15 (m, 5H), 7.04−6.95 (m, 1H), 5.15−5.05 (m, 1H), 4.50−4.37 (m, 1H), 3.85−3.68 (m, 1H), 3.25−3.14 (m, 1H), 2.76−2.66 (m, 2.5H), 2.60−2.48 (m, 0.5H), 2.10−1.96 (m, 1H), 1.76−1.54 (m, 1H), 1.53−1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.28 (t, J = 7.5 Hz, 3H), 1.17−1.00 (m, 1H), 0.93−0.78 (m, 6H); MS (ESI) m/z 542.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Isopropylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10d)
The procedure for 10a was followed using 68 mg (0.13 mmol) of 9a and 34 mg (0.2 mmol) of 4-isopropylphenylboronic acid to give 50 mg (69%) of 10d as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.26−8.20 (m, 1H), 7.54−7.38 (m, 5H), 7.35−7.12 (m, 5H), 7.03−6.92 (m, 1H), 5.14−5.05 (m, 1H), 4.48−4.35 (m, 1H), 3.88−3.68 (m, 1H), 3.26−3.17 (m, 1H), 3.02−2.88 (m, 1H), 2.72−2.66 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.10−1.95 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.29 (d, J = 6.0 Hz, 6H), 1.18−1.02 (m, 1H), 0.95−0.82 (m, 6H); MS (ESI) m/z 556.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Isobutylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10e)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 16 mg (0.087 mmol) of 4-isobutylphenylboronic acid to give 23 mg (77%) of 10e as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.23 (m, 1H), 7.60−7.38 (m, 5H), 7.34−7.15 (m, 5H), 7.04−6.95 (m, 1H), 5.14−5.05 (m, 1H), 4.50−4.36 (m, 1H), 3.86−3.68 (m, 1H), 3.24−3.14 (m, 1H), 2.74−2.65 (m, 0.5H), 2.60−2.46 (m, 2.5H), 2.10−1.82 (m, 2H), 1.76−1.54 (m, 1H), 1.53−1.36 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.16−1.00 (m, 1H), 0.98−0.78 (m, 12H); MS (ESI) m/z 570.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-tert-butylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10f)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 16 mg (0.087 mmol) of 4-tert-butylphenylboronic acid to give 20 mg (61%) of 10f as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.20 (m, 1H), 7.58−7.45 (m, 7H), 7.30−7.12 (m, 3H), 7.00−6.92 (m, 1H), 5.12−5.04 (m, 1H), 4.45−4.33 (m, 1H), 3.82−3.66 (m, 1H), 3.25−3.15 (m, 1H), 2.72−2.65 (m, 0.5H), 2.60−2.48 (m, 0.5H), 2.08−1.95 (m, 1H), 1.75−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.36 (s, 9H), 1.16−1.02 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 570.8 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-hexylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10g)
The procedure for 10a was followed using 68 mg (0.13 mmol) of 9a and 42 mg (0.2 mmol) of 4-hexylphenylboronic acid to give 40 mg (51%) of 10g as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.21 (m, 1H), 7.58−7.40 (m, 5H), 7.36−7.15 (m, 5H), 7.05−6.94 (m, 1H), 5.16−5.08 (m, 1H), 4.50−4.38 (m, 1H), 3.88−3.68 (m, 1H), 3.27−3.15 (m, 1H), 2.76−2.50 (m, 3H), 2.10−1.95 (m, 1H), 1.72−1.22 (m, 12H), 1.47 and 1.43 (2s, 9H), 1.18−1.02 (m, 1H), 0.98−0.80 (m, 9H); MS (ESI) m/z 599.0 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Cyclohexylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10h)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 18 mg (0.087 mmol) of 4-cyclohexylphenylboronic acid to give 28 mg (81%) of 10h as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.21 (m, 1H), 7.58−7.40 (m, 5H), 7.32−7.12 (m, 5H), 7.02−6.92 (m, 1H), 5.15−5.05 (m, 1H), 4.50−4.36 (m, 1H), 3.88−3.68 (m, 1H), 3.26−3.13 (m, 1H), 2.75−2.64 (m, 0.5H), 2.60−2.48 (m, 1.5H), 2.05−1.71 (m, 7H), 1.69−1.25 (m, 8H), 1.47 and 1.43 (2s, 9H), 1.17−1.00 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 596.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Phenylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10i)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 18 mg (0.087 mmol) of 4-biphenylboronic acid to give 22 mg (65%) of 10i as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 7.70−7.42 (m, 10H), 7.42−7.16 (m, 5H), 7.03−6.95 (m, 1H), 5.15−5.05 (m, 1H), 4.50−4.38 (m, 1H), 3.88−3.68 (m, 1H), 3.30−3.18 (m, 1H), 2.78−2.65 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.10−1.98 (m, 1H), 1.80−1.40 (m, 4H), 1.48 and 1.44 (2s, 9H), 1.20−1.02 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 590.4 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Methoxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10j)
The procedure for 10a was followed using 68 mg (0.13 mmol) of 9a and 30 mg (0.2 mmol) of 4-methoxyphenylboronic acid to give 52 mg (74%) of 10j as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.25 (m, 1H), 7.56−7.36 (m, 5H), 7.32−7.16 (m, 3H), 7.04−6.93 (m, 3H), 5.15−5.06 (m, 1H), 4.50−4.37 (m, 1H), 3.85 (s, 3H), 3.84−3.68 (m, 1H), 3.28−3.12 (m, 1H), 2.76−2.68 (m, 0.5H), 2.60−2.52 (m, 0.5H), 2.10−1.98 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.02 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 544.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Trifluoromethoxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10k)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 18 mg (0.087 mmol) of 4-trifluoromethoxyphenylboronic acid to give 18 mg (52%) of 10k as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 7.58−7.40 (m, 5H), 7.38−7.16 (m, 5H), 7.04−6.96 (m, 1H), 5.10−5.03 (m, 1H), 4.50−4.38 (m, 1H), 3.82−3.68 (m, 1H), 3.28−3.16 (m, 1H), 2.76−2.68 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.08−1.94 (m, 1H), 1.75−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.05 (m, 1H), 0.96−0.82 (m, 6H); MS (ESI) m/z 598.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Ethoxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10l)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 14 mg (0.087 mmol) of 4-ethoxyphenylboronic acid to give 22 mg (68%) of 10l as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.20 (m, 1H), 7.55−7.40 (m, 5H), 7.30−7.12 (m, 3H), 7.02−6.92 (m, 3H), 5.12−5.05 (m, 1H), 4.48−4.38 (m, 1H), 4.15−4.04 (m, 2H), 3.86−3.68 (m, 1H), 3.26−3.15 (m, 1H), 2.76−2.68 (m, 0.5H), 2.60−2.52 (m, 0.5H), 2.08−1.96 (m, 1H), 1.78−1.40 (m, 4H), 1.47 and 1.42 (2s, 9H), 1.20−1.04 (m, 1H), 0.94−0.82 (m, 9H); MS (ESI) m/z 558.8 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Isopropoxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10m)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 16 mg (0.087 mmol) of 4-isopropoxyphenylboronic acid to give 20 mg (60%) of 10m as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.22 (m, 1H), 7.54−7.38 (m, 5H), 7.32−7.15 (m, 3H), 7.05−6.92 (m, 3H), 5.15−5.06 (m, 1H), 4.68−4.55 (m, 1H), 4.50−4.38 (m, 1H), 3.86−3.68 (m, 1H), 3.25−3.15 (m, 1H), 2.76−2.68 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.10−1.96 (m, 1H), 1.77−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.37 (d, J = 6.0 Hz, 6H), 1.20−1.05 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 572.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{(3′,4′-Dimethoxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10n)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 16 mg (0.087 mmol) of 3,4-dimethoxyphenylboronic acid to give 19 mg (57%) of 10n as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.20 (m, 1H), 7.55−7.40 (m, 2H), 7.32−6.90 (m, 8H), 5.15−5.08 (m, 1H), 4.50−4.38 (m, 1H), 3.96 (s, 3H), 3.33 (s, 3H), 3.85−3.68 (m, 1H), 3.26−3.15 (m, 1H), 2.78−2.68 (m, 0.5H), 2.62−2.53 (m, 0.5H), 2.10−1.98 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.03 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 574.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{(3′,4′-Methylenedioxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10o)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 14 mg (0.087 mmol) of 3,4-methylenedioxyphenylboronic acid to give 20 mg (62%) of 10o as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 7.56−7.33 (m, 3H), 7.32−7.15 (m, 3H), 7.08−6.92 (m, 3H), 6.90−6.85 (m, 1H), 6.01 (s, 2H), 5.10−5.04 (m, 1H), 4.50−4.38 (m, 1H), 3.85−3.68 (m, 1H), 3.26−3.16 (m, 1H), 2.76−2.66 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.10−1.98 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.42 (2s, 9H), 1.20−1.02 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 558.7 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Fluorobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10p)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 12 mg (0.087 mmol) of 4-fluorophenylboronic acid to give 20 mg (74%) of 10p as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 7.56−7.48 (m, 4H), 7.30−7.18 (m, 6H), 7.02−6.92 (m, 1H), 5.12−5.05 (m, 1H), 4.52−4.38 (m, 1H), 3.82−3.65 (m, 1H), 3.25−3.12 (m, 1H), 2.76−2.65 (m, 0.5H), 2.61−2.48 (m, 0.5H), 2.08−1.95 (m, 1H), 1.78−1.55 (m, 1H), 1.54−1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.18−1.00 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 532.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Chlorobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10q)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 14 mg (0.087 mmol) of 4-chlorophenylboronic acid to give 22 mg (74%) of 10q as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.21 (m, 1H), 7.55−7.36 (m, 6H), 7.30−7.15 (m, 4H), 7.02−6.95 (m, 1H), 5.10−5.00 (m, 1H), 4.50−4.35 (m, 1H), 3.80−3.62 (m, 1H), 3.26−3.15 (m, 1H), 2.76−2.65 (m, 0.5H), 2.61−2.48 (m, 0.5H), 2.08−1.95 (m, 1H), 1.75−1.55 (m, 1H), 1.54−1.37 (m, 3H), 1.47 and 1.42 (2s, 9H), 1.16−1.00 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 548.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Cyanobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10r)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 13 mg (0.087 mmol) of 4-cyanophenylboronic acid to give 20 mg (64%) of 10r as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.20 (m, 1H), 7.76−7.42 (m, 7H), 7.40−7.16 (m, 3H), 7.02−6.92 (m, 1H), 5.10−5.00 (m, 1H), 4.50−4.38 (m, 1H), 3.80−3.64 (m, 1H), 3.26−3.14 (m, 1H), 2.76−2.66 (m, 0.5H), 2.61−2.52 (m, 0.5H), 2.08−1.95 (m, 1H), 1.75−1.40 (m, 4H), 1.47 and 1.42 (2s, 9H), 1.18−1.00 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 539.3 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Nitrobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10s)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 15 mg (0.087 mmol) of 4-nitrophenylboronic acid to give 21 mg (65%) of 10s as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.35−8.21 (m, 3H), 7.75−7.44 (m, 5H), 7.40−7.16 (m, 3H), 7.02−6.95 (m, 1H), 5.08−4.98 (m, 1H), 4.50−4.38 (m, 1H), 3.80−3.62 (m, 1H), 3.28−3.18 (m, 1H), 2.76−2.66 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.08−1.94 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.42 (2s, 9H), 1.17−1.00 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 559.4 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Acetylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10t)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 14 mg (0.087 mmol) of 4-acetylphenylboronic acid to give 25 mg (76%) of 10t as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.21 (m, 1H), 8.06−7.98 (m, 2H), 7.68−7.48 (m, 5H), 7.38−7.15 (m, 3H), 7.02−6.93 (m, 1H), 5.10−5.00 (m, 1H), 4.50−4.36 (m, 1H), 3.80−3.62 (m, 1H), 3.28−3.15 (m, 1H), 2.75−2.65 (m, 0.5H), 2.64 (s, 3H), 2.61−2.48 (m, 0.5H), 2.08−1.95 (m, 1H), 1.76−1.55 (m, 1H), 1.54−1.37 (m, 3H), 1.47 and 1.43 (2s, 9H), 1.16−0.98 (m, 1H), 0.92−0.78 (m, 6H); MS (ESI) m/z 557.2 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Ethoxycarbonylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10u)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 17 mg (0.087 mmol) of 4-ethoxycarbonylphenylboronic acid to give 22 mg (65%) of 10u as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 8.16−8.08 (m, 2H), 7.66−7.48 (m, 5H), 7.38−7.16 (m, 3H), 7.02−6.95 (m, 1H), 5.10−5.02 (m, 1H), 4.50−4.36 (m, 3H), 3.85−3.66 (m, 1H), 3.28−3.16 (m, 1H), 2.75−2.65 (m, 0.5H), 2.62−2.50 (m, 0.5H), 2.10−1.98 (m, 1H), 1.76−1.37 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.02 (m, 1H), 0.95−0.81 (m, 9H); MS (ESI) m/z 586.5 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Hydroxybiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10v)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 25 mg (0.087 mmol) of 4-(tert-butoxycarboxy)phenylboronic acid to give 22 mg (72%) of 10v as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.52 (br s, 1H), 8.28−8.18 (m, 1H), 7.60−7.50 (m, 1H), 7.38−7.10 (m, 7H), 7.08−7.00 (m, 1H), 7.76−6.68 (m, 2H), 5.26−5.18 (m, 1H), 4.52−4.40 (m, 1H), 3.82−3.68 (m, 1H), 3.20−3.08 (m, 1H), 2.78−2.68 (m, 0.5H), 2.62−2.52 (m, 0.5H), 2.15−2.08 (m, 1H), 1.76−1.40 (m, 4H), 1.51 and 1.46 (2s, 9H), 1.20−1.02 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 530.9 [M + H]+.
tert-Butyl [(2S,3S)-1-{[4′-(tert-Butoxycarbonylamino)biphenyl-4-yl]-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10w)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 21 mg (0.087 mmol) of 4-(tert-butoxycarbonylamino)phenylboronic acid to give 22 mg (60%) of 10w as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.20 (m, 1H), 7.52−7.40 (m, 7H), 7.30−7.12 (m, 3H), 7.02−6.94 (m, 1H), 6.68 (s, 1H), 5.12−5.06 (m, 1H), 4.48−4.38 (m, 1H), 3.82−3.67 (m, 1H), 3.25−3.15 (m, 1H), 2.76−2.68 (m, 0.5H), 2.58−2.48 (m, 0.5H), 2.08−1.98 (m, 1H), 1.77−1.40 (m, 4H), 1.53 (s, 9H), 1.47 and 1.43 (2s, 9H), 1.20−1.02 (m, 1H), 0.95−0.80 (m, 6H); MS (ESI) m/z 629.8 [M + H]+.
tert-Butyl [(2S,3S)-1-{[4′-(Dimethylamino)biphenyl-4-yl]-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10x)
The procedure for 10a was followed using 68 mg (0.13 mmol) of 9a and 35 mg (0.2 mmol) of 4-(dimethylamino)phenylboronic acid to give 52 mg (72%) of 10x as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.22 (m, 1H), 7.55−7.36 (m, 5H), 7.30−7.12 (m, 3H), 7.02−6.96 (m, 1H), 6.84−6.76 (m, 2H), 5.18−5.10 (m, 1H), 4.50−4.38 (m, 1H), 3.88−3.70 (m, 1H), 3.26−3.16 (m, 1H), 2.99 (s, 6H), 2.76−2.68 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.10−1.98 (m, 1H), 1.72−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.02 (m, 1H), 0.98−0.82 (m, 6H); MS (ESI) m/z 557.8 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Acetamidobiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10y)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 15.6 mg (0.087 mmol) of 4-acetamidophenylboronic acid to give 18 mg (54%) of 10y as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.20 (m, 1H), 7.96−7.80 (m, 1H), 7.62−7.34 (m, 7H), 7.30−7.15 (m, 3H), 7.02−6.96 (m, 1H), 5.18−5.08 (m, 1H), 4.50−4.38 (m, 1H), 3.82−3.66 (m, 1H), 3.28−3.15 (m, 1H), 2.76−2.67 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.18 (s, 3H), 2.08−1.95 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.02 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 571.6 [M + H]+.
tert-Butyl [(2S,3S)-1-{(4′-Carbamoylbiphenyl-4-yl)-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]-carbamate (10z)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 14 mg (0.087 mmol) of 4-carbamoylphenylboronic acid to give 18 mg (56%) of 10z as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.30−8.21 (m, 1H), 7.92−7.86 (m, 2H), 7.66−7.46 (m, 5H), 7.40−7.18 (m, 3H), 7.05−6.98 (m, 1H), 6.16 (br s, 1H), 5.80 (br s, 1H), 5.10−5.05 (m, 1H), 4.50−4.38 (m, 1H), 3.82−3.64 (m, 1H), 3.28−3.16 (m, 1H), 2.76−2.68 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.08−1.96 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.42 (2s, 9H), 1.20−1.02 (m, 1H), 0.95−0.81 (m, 6H); MS (ESI) m/z 558.1 [M + H]+.
tert-Butyl [(2S,3S)-1-{[4′-(Dimethylcarbamoyl)biphenyl-4-yl]-[(1R*,2R*)-2-(pyridin-2-yl)cyclopropanecarbonyl]amino}-3-methylpentan-2-yl]carbamate (10aa)
The procedure for 10a was followed using 30 mg (0.058 mmol) of 9a and 17 mg (0.087 mmol) of 4-(dimethylcarbamoyl) phenylboronic acid to give 20 mg (56%) of 10aa as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CDCl3) δ 8.28−8.22 (m, 1H), 7.60−7.38 (m, 7H), 7.32−7.12 (m, 3H), 7.02−6.95 (m, 1H), 5.10−5.00 (m, 1H), 4.50−4.38 (m, 1H), 3.82−3.65 (m, 1H), 3.28−3.16 (m, 1H), 3.14 (s, 3H), 3.04 (s, 3H), 2.75−2.66 (m, 0.5H), 2.60−2.50 (m, 0.5H), 2.08−1.96 (m, 1H), 1.76−1.40 (m, 4H), 1.47 and 1.43 (2s, 9H), 1.20−1.02 (m, 1H), 0.96−0.80 (m, 6H); MS (ESI) m/z 585.8 [M + H]+.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-methylbiphenyl-4-yl)amide (5a)
The procedure for 4a was followed using 20 mg (0.038 mmol) of 10a and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 5a dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.55 (m, 1H), 8.42−8.30 (m, 1H), 7.88−7.40 (m, 8H), 7.30−7.20 (m, 2H), 4.50−4.40 (m, 0.5H), 4.40−4.28 (m, 0.5H), 3.88−3.56 (m, 1H), 3.48−3.38 (m, 1H), 3.12−3.05 (m, 0.5H), 3.05−2.96 (m, 0.5H), 2.37 (s, 3H), 2.22−2.10 (m, 1H), 2.10−1.88 (m, 1H), 1.87−1.60 (m, 2H), 1.48−1.15 (m, 2H), 1.06−0.80 (m, 6H); HRMS (ESI) calcd for C28H33N3O [M + H]+: 428.2696. Found: 428.2701.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-trifluoromethylbiphenyl-4-yl)amide (5b)
The procedure for 4a was followed using 20 mg (0.034 mmol) of 10b and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 5b dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70−8.58 (m, 1H), 8.42−8.30 (m, 1H), 7.90−7.50 (m, 10H), 4.55−4.40 (m, 0.5H), 4.40−4.26 (m, 0.5H), 3.90−3.60 (m, 1H), 3.50−3.38 (m, 1H), 3.15−3.05 (m, 0.5H), 3.05−2.92 (m, 0.5H), 2.25−2.10 (m, 1H), 2.10−1.92 (m, 1H), 1.92−1.62 (m, 2H), 1.50−1.15 (m, 2H), 1.08−0.80 (m, 6H); HRMS (ESI) calcd for C28H30F3N3O [M + H]+: 482.2414. Found: 482.2416.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-ethylbiphenyl-4-yl)amide (5c)
The procedure for 4a was followed using 20 mg (0.037 mmol) of 10c and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 5c dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70−8.58 (m, 1H), 8.45−8.30 (m, 1H), 7.88−7.45 (m, 8H), 7.32−7.20 (m, 2H), 4.52−4.40 (m, 0.5H), 4.38−4.28 (m, 0.5H), 3.88−3.56 (m, 1H), 3.48−3.35 (m, 1H), 3.15−3.05 (m, 0.5H), 3.05−2.95 (m, 0.5H), 2.80−2.60 (m, 2H), 2.25−2.10 (m, 1H), 2.10−1.90 (m, 1H), 1.87−1.60 (m, 2H), 1.50−1.10 (m, 5H), 1.08−0.80 (m, 6H); HRMS (ESI) calcd for C29H35N3O [M + H]+: 442.2853. Found: 442.2867.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-isopropylbiphenyl-4-yl)amide (5d)
The procedure for 4a was followed using 45 mg (0.08 mmol) of 10d and 2 mL of 4 M HCl in dioxane to give 40 mg (95%) of 5d dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.60−8.50 (m, 1H), 8.30−8.18 (m, 1H), 7.70−7.48 (m, 8H), 7.36−7.28 (m, 2H), 4.48−4.22 (m, 1H), 3.83−3.62 (m, 1H), 3.46−3.38 (m, 1H), 3.06−2.88 (m, 2H), 2.20−2.10 (m, 1H), 2.00−1.84 (m, 1H), 1.84−1.72 (m, 1H), 1.72−1.60 (m, 1H), 1.50−1.20 (m, 2H), 1.28 (d, J = 9.0 Hz, 6H), 1.05−0.80 (m, 6H); HRMS (ESI) calcd for C30H37N3O [M + H]+: 456.3009. Found: 456.3014.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-isobutylbiphenyl-4-yl)amide (5e)
The procedure for 4a was followed using 20 mg (0.035 mmol) of 10e and 1 mL of 4 M HCl in dioxane to give 17 mg (90%) of 5e dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.58 (m, 1H), 8.40−8.28 (m, 1H), 7.92−7.43 (m, 8H), 7.30−7.20 (m, 2H), 4.50−4.40 (m, 0.5H), 4.38−4.25 (m, 0.5H), 3.88−3.60 (m, 1H), 3.50−3.38 (m, 1H), 3.13−3.05 (m, 0.5H), 3.05−2.90 (m, 0.5H), 2.51 (d, J = 6.0 Hz, 2H), 2.25−2.10 (m, 1H), 2.05−1.60 (m, 4H), 1.50−1.20 (m, 2H), 1.10−0.76 (m, 12H); HRMS (ESI) calcd for C31H39N3O [M + H]+: 470.3166. Found: 470.3178.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-tert-butylbiphenyl-4-yl)amide (5f)
The procedure for 4a was followed using 20 mg (0.035 mmol) of 10f and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 5f dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.58 (m, 1H), 8.42−8.30 (m, 1H), 7.88−7.46 (m, 10H), 4.50−4.38 (m, 0.5H), 4.38−4.22 (m, 0.5H), 3.88−3.60 (m, 1H), 3.46−3.38 (m, 1H), 3.14−3.05 (m, 0.5H), 3.05−2.94 (m, 0.5H), 2.22−2.08 (m, 1H), 2.08−1.90 (m, 1H), 1.88−1.65 (m, 2H), 1.50−1.20 (m, 2H), 1.35 (s, 9H), 1.05−0.80 (m, 6H); HRMS (ESI) calcd for C31H39N3O [M + H]+: 470.3166. Found: 470.3178.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-hexylbiphenyl-4-yl)amide (5g)
The procedure for 4a was followed using 35 mg (0.058 mmol) of 10g and 1 mL of 4 M HCl in dioxane to give 30 mg (90%) of 5g dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.52−8.42 (m, 1H), 8.22−8.10 (m, 1H), 7.65−7.32 (m, 8H), 7.25−7.10 (m, 2H), 4.40−4.28 (m, 0.5H), 4.28−4.16 (m, 0.5H), 3.75−3.55 (m, 1H), 3.36−3.25 (m, 1H), 2.98−2.86 (m, 0.5H), 2.86−2.68 (m, 0.5H), 2.60−2.45 (m, 2H), 2.10−1.95 (m, 1H), 1.90−1.60 (m, 2H), 1.60−1.45 (m, 3H), 1.40−1.05 (m, 8H), 0.95−0.68 (m, 9H); HRMS (ESI) calcd for C33H43N3O [M + H]+: 498.3479. Found: 498.3496.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-cyclohexylbiphenyl-4-yl)amide (5h)
The procedure for 4a was followed using 20 mg (0.034 mmol) of 10h and 1 mL of 4 M HCl in dioxane to give 19 mg (98%) of 5h dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.55−8.40 (m, 1H), 8.15−8.00 (m, 1H), 7.75−7.40 (m, 8H), 7.35−7.20 (m, 2H), 4.48−4.20 (m, 1H), 3.88−3.68 (m, 1H), 3.42−3.35 (m, 1H), 2.98−2.88 (m, 1H), 2.68−2.48 (m, 1H), 2.20−2.00 (m, 1H), 1.98−1.70 (m, 7H), 1.66−1.10 (m, 8H), 1.05−0.78 (m, 6H); HRMS (ESI) calcd for C33H41N3O [M + H]+: 496.3322. Found: 496.3323.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-phenylbiphenyl-4-yl)amide (5i)
The procedure for 4a was followed using 22 mg (0.037 mmol) of 10i and 1 mL of 4 M HCl in dioxane to give 20 mg (95%) of 5i dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70−8.58 (m, 1H), 8.42−8.30 (m, 1H), 7.88−7.52 (m, 12H), 7.50−7.30 (m, 3H), 4.52−4.40 (m, 0.5H), 4.40−4.25 (m, 0.5H), 3.90−3.60 (m, 1H), 3.38−3.48 (m, 1H), 3.16−3.06 (m, 0.5H), 3.06−2.96 (m, 0.5H), 2.26−2.10 (m, 1H), 2.10−1.90 (m, 1H), 1.90−1.68 (m, 2H), 1.52−1.20 (m, 2H), 1.00−0.80 (m, 6H); HRMS (ESI) calcd for C33H35N3O [M + H]+: 490.2853. Found: 490.2870.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-methoxybiphenyl-4-yl)amide (5j)
The procedure for 4a was followed using 36 mg (0.066 mmol) of 10j and 2 mL of 4 M HCl in dioxane to give 32 mg (94%) of 5j dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.60−8.52 (m, 1H), 8.32−8.20 (m, 1H), 7.76−7.48 (m, 8H), 7.05−6.96 (m, 2H), 4.46−4.26 (m, 1H), 3.83 (s, 3H), 3.80−3.62 (m, 1H), 3.45−3.32 (m, 1H), 3.10−2.98 (m, 0.5H), 2.98−2.88 (m, 0.5H), 2.22−2.06 (m, 1H), 2.00−1.82 (m, 1H), 1.82−1.58 (m, 2H), 1.46−1.15 (m, 2H), 1.05−0.75 (m, 6H); HRMS (ESI) calcd for C28H33N3O2 [M + H]+: 444.2646. Found: 444.2655.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-trifluoromethoxybiphenyl-4-yl)amide (5k)
The procedure for 4a was followed using 18 mg (0.03 mmol) of 10k and 1 mL of 4 M HCl in dioxane to give 16 mg (94%) of 5k dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.56 (m, 1H), 8.45−8.30 (m, 1H), 7.90−7.56 (m, 8H), 7.42−7.30 (m, 2H), 4.52−4.40 (m, 0.5H), 4.38−4.22 (m, 0.5H), 3.90−3.60 (m, 1H), 3.48−3.38 (m, 1H), 3.15−3.06 (m, 0.5H), 3.06−2.95 (m, 0.5H), 2.22−2.08 (m, 1H), 2.08−1.90 (m, 1H), 1.90−1.64 (m, 2H), 1.50−1.16 (m, 2H), 1.06−0.78 (m, 6H); HRMS (ESI) calcd for C28H30F3N3O2 [M + H]+: 498.2363. Found: 498.2385.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-ethoxybiphenyl-4-yl)amide (5l)
The procedure for 4a was followed using 22 mg (0.039 mmol) of 10l and 1 mL of 4 M HCl in dioxane to give 21 mg (100%) of 5l dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70−8.58 (m, 1H), 8.42−8.30 (m, 1H), 7.88−7.76 (m, 1H), 7.76−7.38 (m, 7H), 7.02−6.90 (m, 2H), 4.52−4.40 (m, 0.5H), 4.38−4.24 (m, 0.5H), 4.07 (q, J = 6.0 Hz, 2H), 3.86−3.55 (m, 1H), 3.46−3.33 (m, 1H), 3.15−3.05 (m, 0.5H), 3.05−2.92 (m, 0.5H), 2.25−2.08 (m, 1H), 2.08−1.90 (m, 1H), 1.90−1.60 (m, 2H), 1.50−1.16 (m, 5H), 1.08−0.78 (m, 6H); HRMS (ESI) calcd for C29H35N3O2 [M + H]+: 458.2802. Found: 458.2812.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-isopropoxybiphenyl-4-yl)amide (5m)
The procedure for 4a was followed using 20 mg (0.035 mmol) of 10m and 1 mL of 4 M HCl in dioxane to give 19 mg (100%) of 5m dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70−8.55 (m, 1H), 8.40−8.30 (m, 1H), 7.90−7.40 (m, 8H), 7.05−6.90 (m, 2H), 4.70−4.58 (m, 1H), 4.55−4.38 (m, 0.5H), 4.38−4.22 (m, 0.5H), 3.85−3.60 (m, 1H), 3.46−3.38 (m, 1H), 3.16−3.05 (m, 0.5H), 3.05−2.94 (m, 0.5H), 2.25−2.08 (m, 1H), 2.08−1.90 (m, 1H), 1.90−1.64 (m, 2H), 1.50−1.20 (m, 2H), 1.33 (d, J = 6.0 Hz, 6H), 1.06−0.80 (m, 6H); HRMS (ESI) calcd for C30H37N3O2 [M + H]+: 472.2959. Found: 472.2957.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(3′,4′-dimethoxybiphenyl-4-yl)amide (5n)
The procedure for 4a was followed using 19 mg (0.033 mmol) of 10n and 1 mL of 4 M HCl in dioxane to give 18 mg (100%) of 5n dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.58 (m, 1H), 8.46−8.32 (m, 1H), 7.90−7.78 (m, 1H), 7.78−7.53 (m, 5H), 7.20−7.10 (m, 2H), 7.08−6.98 (m, 1H), 4.52−4.40 (m, 0.5H), 4.40−4.25 (m, 0.5H), 3.89 (s, 3H), 3.86 (s, 3H), 3.80−3.62 (m, 1H), 3.46−3.35 (m, 1H), 3.15−3.06 (m, 0.5H), 3.06−2.96 (m, 0.5H), 2.25−2.10 (m, 1H), 2.10−1.90 (m, 1H), 1.90−1.65 (m, 2H), 1.50−1.18 (m, 2H), 1.05−0.80 (m, 6H); HRMS (ESI) calcd for C29H35N3O3 [M + H]+: 474.2751. Found: 474.2763.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(3′,4′-methylenedioxybiphenyl-4-yl)amide (5o)
The procedure for 4a was followed using 20 mg (0.036 mmol) of 10o and 1 mL of 4 M HCl in dioxane to give 19 mg (100%) of 5o dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.56 (m, 1H), 8.42−8.28 (m, 1H), 7.85−7.73 (m, 1H), 7.73−7.50 (m, 5H), 7.12−7.00 (m, 2H), 6.92−6.82 (m, 1H), 5.99 (s, 2H), 4.50−4.35 (m, 0.5H), 4.34−4.20 (m, 0.5H), 3.85−3.60 (m, 1H), 3.45−3.32 (m, 1H), 3.12−3.02 (m, 0.5H), 3.02−2.90 (m, 0.5H), 2.22−2.08 (m, 1H), 2.08−1.88 (m, 1H), 1.88−1.60 (m, 2H), 1.50−1.15 (m, 2H), 1.06−0.78 (m, 6H); HRMS (ESI) calcd for C28H31N3O3 [M + H]+: 458.2438. Found: 458.2458.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-fluorobiphenyl-4-yl)amide (5p)
The procedure for 4a was followed using 20 mg (0.038 mmol) of 10p and 1 mL of 4 M HCl in dioxane to give 18 mg (94%) of 5p dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.70−8.58 (m, 1H), 8.45−8.35 (m, 1H), 7.90−7.50 (m, 8H), 7.25−7.10 (m, 2H), 4.52−4.40 (m, 0.5H), 4.40−4.25 (m, 0.5H), 3.90−3.60 (m, 1H), 3.52−3.40 (m, 1H), 3.16−3.06 (m, 0.5H), 3.05−2.92 (m, 0.5H), 2.25−2.10 (m, 1H), 2.10−1.92 (m, 1H), 1.92−1.60 (m, 2H), 1.50−1.18 (m, 2H), 1.10−0.80 (m, 6H); HRMS (ESI) calcd for C27H30FN3O [M + H]+: 432.2446. Found: 432.2453.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-chlorobiphenyl-4-yl)amide (5q)
The procedure for 4a was followed using 20 mg (0.036 mmol) of 10q and 1 mL of 4 M HCl in dioxane to give 18 mg (96%) of 5q dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.72−8.58 (m, 1H), 8.45−8.35 (m, 1H), 7.90−7.55 (m, 8H), 7.55−7.40 (m, 2H), 4.55−4.42 (m, 0.5H), 4.40−4.25 (m, 0.5H), 3.90−3.60 (m, 1H), 3.50−3.40 (m, 1H), 3.18−3.07 (m, 0.5H), 3.06−2.96 (m, 0.5H), 2.25−2.10 (m, 1H), 2.08−1.90 (m, 1H), 1.90−1.68 (m, 2H), 1.58−1.12 (m, 2H), 1.10−0.80 (m, 6H); HRMS (ESI) calcd for C27H30ClN3O [M + H]+: 448.2150. Found: 448.2162.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-cyanobiphenyl-4-yl)amide (5r)
The procedure for 4a was followed using 20 mg (0.037 mmol) of 10r and 1 mL of 4 M HCl in dioxane to give 18 mg (95%) of 5r dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.65−8.55 (m, 1H), 8.36−8.25 (m, 1H), 7.90−7.50 (m, 10H), 4.50−4.38 (m, 0.5H), 4.38−4.20 (m, 0.5H), 3.85−3.60 (m, 1H), 3.45−3.28 (m, 1H), 3.12−3.02 (m, 0.5H), 3.02−2.92 (m, 0.5H), 2.22−2.06 (m, 1H), 2.06−1.90 (m, 1H), 1.90−1.60 (m, 2H), 1.58−1.15 (m, 2H), 1.05−0.80 (m, 6H); HRMS (ESI) calcd for C28H30N4O [M + H]+: 439.2492. Found: 439.2506.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-nitrobiphenyl-4-yl)amide (5s)
The procedure for 4a was followed using 21 mg (0.037 mmol) of 10s and 1 mL of 4 M HCl in dioxane to give 19 mg (95%) of 5s dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.65−8.55 (m, 1H), 8.45−8.20 (m, 2H), 7.95−7.55 (m, 9H), 4.50−4.30 (m, 1H), 3.85−3.60 (m, 1H), 3.45−3.35 (m, 1H), 3.12−3.02 (m, 0.5H), 3.02−2.95 (m, 0.5H), 2.25−2.10 (m, 1H), 2.10−1.90 (m, 1H), 1.89−1.60 (m, 2H), 1.50−1.15 (m, 2H), 1.10−0.82 (m, 6H); HRMS (ESI) calcd for C27H30N4O3 [M + H]+: 459.2391. Found: 459.2404.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-acetylbiphenyl-4-yl)amide (5t)
The procedure for 4a was followed using 20 mg (0.036 mmol) of 10t and 1 mL of 4 M HCl in dioxane to give 17 mg (90%) of 5t dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.35−8.22 (m, 1H), 8.05−7.92 (m, 2H), 7.90−7.75 (m, 1H), 7.72−7.60 (m, 4H), 7.52−7.40 (m, 2H), 7.35−7.22 (m, 2H), 4.38−4.16 (m, 1H), 3.80−3.55 (m, 1H), 3.38−3.20 (m, 1H), 2.80−2.60 (m, 1H), 2.54 (s, 3H), 2.00−1.88 (m, 1H), 1.78−1.60 (m, 2H), 1.50−1.10 (m, 3H), 0.95−0.70 (m, 6H); HRMS (ESI) calcd for C29H33N3O2 [M + H]+: 456.2646. Found: 456.2658.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-ethoxycarbonylbiphenyl-4-yl)amide (5u)
The procedure for 4a was followed using 22 mg (0.038 mmol) of 10u and 1 mL of 4 M HCl in dioxane to give 21 mg (100%) of 5u dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.65−8.48 (m, 1H), 8.35−8.18 (m, 1H), 8.15−8.10 (m, 2H), 7.85−7.50 (m, 8H), 4.50−4.20 (m, 3H), 3.85−3.62 (m, 1H), 3.45−3.35 (m, 1H), 3.12−3.00 (m, 0.5H), 3.00−2.90 (m, 0.5H), 2.20−2.03 (m, 1H), 2.03−1.55 (m, 3H), 1.45−1.12 (m, 5H), 1.08−0.78 (m, 6H); HRMS (ESI) calcd for C30H35N3O3 [M + H]+: 486.2751. Found: 486.2774.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-hydroxybiphenyl-4-yl)amide (5v)
The procedure for 4a was followed using 22 mg (0.042 mmol) of 10v and 1 mL of 4 M HCl in dioxane to give 21 mg (100%) of 5v dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.55 (m, 1H), 8.40−8.28 (m, 1H), 7.95−7.82 (m, 1H), 7.82−7.38 (m, 7H), 6.88−6.68 (m, 2H), 4.48−4.36 (m, 0.5H), 4.36−4.22 (m, 0.5H), 3.82−3.65 (m, 1H), 3.45−3.32 (m, 1H), 3.12−3.02 (m, 0.5H), 3.02−2.92 (m, 0.5H), 2.20−2.06 (m, 1H), 2.06−1.88 (m, 1H), 1.88−1.60 (m, 2H), 1.50−1.15 (m, 2H), 1.05−0.80 (m, 6H); HRMS (ESI) calcd for C27H31N3O2 [M + H]+: 430.2489. Found: 430.2508.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-aminobiphenyl-4-yl)amide (5w)
The procedure for 4a was followed using 22 mg (0.035 mmol) of 10w and 1 mL of 4 M HCl in dioxane to give 20 mg (95%) of 5w trihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.61−8.55 (m, 1H), 8.40−8.30 (m, 1H), 7.84−7.47 (m, 10H), 4.52−4.42 (m, 0.5H), 4.40−4.28 (m, 0.5H), 3.80−3.65 (m, 1H), 3.42−3.32 (m, 1H), 3.12−3.00 (m, 0.5H), 3.00−2.93 (m, 0.5H), 2.22−2.08 (m, 1H), 2.08−1.88 (m, 1H), 1.88−1.60 (m, 2H), 1.50−1.16 (m, 2H), 1.06−0.80 (m, 6H); HRMS (ESI) calcd for C27H32N4O [M + H]+: 429.2649. Found: 429.2661.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-[4′-(dimethylamino)biphenyl-4-yl]amide (5x)
The procedure for 4a was followed using 46 mg (0.083 mmol) of 10x and 2 mL of 4 M HCl in dioxane to give 40 mg (86%) of 5x trihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.55−8.45 (m, 1H), 8.25−8.12 (m, 1H), 7.70−7.40 (m, 10H), 4.42−4.28 (m, 0.5H), 4.28−4.15 (m, 0.5H), 3.82−3.65 (m, 1H), 3.55 (s, 6H), 3.32−3.22 (m, 1H), 3.02−2.90 (m, 0.5H), 2.90−2.82 (m, 0.5H), 2.10−1.95 (m, 1H), 1.95−1.50 (m, 3H), 1.45−1.10 (m, 2H), 0.95−0.70 (m, 6H); HRMS (ESI) calcd for C29H36N4O [M + H]+: 457.2962. Found: 457.2973.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-acetamidobiphenyl-4-yl)amide (5y)
The procedure for 4a was followed using 18 mg (0.032 mmol) of 10y and 1 mL of 4 M HCl in dioxane to give 17 mg (99%) of 5y dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.58 (m, 1H), 8.45−8.30 (m, 1H), 7.85−7.50 (m, 10H), 4.50−4.38 (m, 0.5H), 4.38−4.20 (m, 0.5H), 3.82−3.65 (m, 1H), 3.55−3.35 (m, 1H), 3.12−3.02 (m, 0.5H), 3.02−2.90 (m, 0.5H), 2.15 (s, 3H), 2.15−1.96 (m, 4H), 1.50−1.16 (m, 2H), 1.05−0.80 (m, 6H); HRMS (ESI) calcd for C29H34N4O2 [M + H]+: 471.2755. Found: 471.2768.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-(4′-carbamoylbiphenyl-4-yl)amide (5z)
The procedure for 4a was followed using 18 mg (0.032 mmol) of 10z and 1 mL of 4 M HCl in dioxane to give 17 mg (99%) of 5z dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.68−8.55 (m, 1H), 8.45−8.30 (m, 1H), 8.00−7.50 (m, 10H), 4.50−4.38 (m, 0.5H), 4.38−4.22 (m, 0.5H), 3.85−3.65 (m, 1H), 3.46−3.35 (m, 1H), 3.12−3.02 (m, 0.5H), 3.02−2.92 (m, 0.5H), 2.25−2.10 (m, 1H), 2.10−1.90 (m, 1H), 1.90−1.55 (m, 2H), 1.50−1.16 (m, 2H), 1.08−0.80 (m, 6H); HRMS (ESI) calcd for C28H32N4O2 [M + H]+ : 457.2598. Found: 457.2616.
(1R*,2R*)-2-(Pyridin-2-yl)cyclopropanecarboxylic Acid [(2S,3S)-2-Amino-3-methylpentyl]-[4′-(dimethylcarbamoyl)biphenyl-4-yl]-amide (5aa)
The procedure for 4a was followed using 20 mg (0.034 mmol) of 10aa and 1 mL of 4 M HCl in dioxane to give 19 mg (100%) of 5aa dihydrochloride as a 1:1 diastereomeric mixture: 1H NMR (300 MHz; CD3OD) δ 8.86−8.55 (m, 1H), 8.50−8.25 (m, 1H), 8.00−7.40 (m, 10H), 4.50−4.38 (m, 0.5H), 4.38−4.20 (m, 0.5H), 3.85−3.65 (m, 1H), 3.40−3.25 (m, 1H), 3.20−2.95 (m, 7H), 2.25−2.10 (m, 1H), 2.10−1.60 (m, 3H), 1.50−1.16 (m, 2H), 1.08−0.80 (m, 6H); HRMS (ESI) calcd for C30H36N4O2 [M + H]+ : 485.2911. Found: 485.2931.
Pharmacology
Materials
Cell culture materials were purchased from Fisher SSI. Forskolin was purchased from Sigma-Aldrich. An expression plasmid containing a preprolactin leader sequence (PPLS) and an influenza hemagglutanin (HA) tag fused in frame upstream of the human GPR88 cDNA was prepared in a modified pcDNA3.1+mammalian expression vector by GenScript (Piscataway, NJ). Midi-prep DNA was prepared and the PPLS-HA-GPR88 construct was verified by sequencing.
Lance cAMP Assay Using Stable PPLS-HA-GPR88 CHO Cells
A stable PPLS-HA-GPR88 CHO cell line was created by overexpressing the PPLS-HA-GPR88 construct in CHO cells. These cells were maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS), 100 units each of penicillin and streptomycin, and 400 μg/mL Geneticin (for antibiotic resistance). PerkinElmer’s Lance Ultra kit (TRF0262) was used to detect cAMP accumulation. Stimulation buffer containing 1× Hank’s balanced salt solution (HBSS), 5 mM HEPES, 0.1% BSA stabilizer, and 0.5 mM final IBMX was prepared and titrated to 7.4 at room temperature. Serial dilutions of the test compounds (5 μL) and 300 nM forskolin (5 μL), both prepared at 4× the desired final concentration in 2% DMSO/stimulation buffer, were added to a 96-well white 1/2 area microplate (PerkinElmer). A cAMP standard curve was prepared at 4× the desired final concentration in stimulation buffer, and 5 μL was added to the assay plate. Cells were lifted with versene and spun at 270g for 10 min. The cell pellet was resuspended in stimulation buffer and 4000 cells (10 μL) were added to each well except wells containing the cAMP standard curve. After incubating for 30 min at RT, Eu-cAMP tracer and uLIGHT-anti-cAMP working solutions were added per the manufacturer’s instructions. After incubation at RT for 1 h, the TR-FRET signal (ex 337 nm) was read on a CLARIOstar multimode plate reader (BMG Biotech, Cary NC).
Data Analysis
The TR-FRET signal (665 nm) was converted to fmol cAMP by interpolating from the standard cAMP curve. Fmol cAMP was plotted against the log of compound concentration and data were fit to a three-parameter logistic curve to generate EC50 values (Prism, version 6.0, GraphPad Software, Inc., San Diego, CA).
Computational Studies
Property Evaluation
Properties of fragments in the SAR substitution region were evaluated using semiempirical QM (MOPAC718) models at optimized geometries using this semiempirical Hamiltonian and with isotropic polarizabilities computed at B3LYP hybrid functional optimized geometries employing GAMESS-UK.22 Postcomputation analysis of the results was done with GRAPHA, a utility that extracts QM-energetic, electrostatic, hydrophobic, frontier-orbital energetic, Sterimol, shape, polar–non-polar surface area, volume, and thermodynamic descriptors.20 Substituted biphenyls were used in the computations, capping the substituent at the biphenyl end where it attaches to the central N in the ligands with a hydrogen.
Fragment/2D/3D QSAR
Properties compiled were analyzed to develop quantitative structure relationships in an effort to ascertain the key physiochemical properties modulating GPR88 ln EC50 values as a function of substitution. Multivariate least-squares approaches embodied in the R PROJECT software for Statistical Computing (http://www.r-project.org).
Homology Modeling
Multiple and pairwise sequence alignments were made using the human sequence of GPR88 and sequences of known GPCR crystallographic template with their chimeric stabilizing proteins removed. Alignments were performed using Blosum scoring matrices employing CLUSTALX. An initial level homology model was developed using β1-adrenergic receptor template (PDB: 5A8E) first employing Sali’s MODELER v9.1126 to obtain an initial backbone model through simulated annealing with topological constraints and employing the CHARMM potential function and then using SCWRL27 to predict the rotameric side chain states of nonconserved residues in the adrenergic receptor/GPR88 alignment. AMBER12 was then used to energy minimized the structure using the Simmerling modified AMBER99 potential function and SANDER.
VINA/GLIDE Docking and MMGBSA Scoring
(1R,2R)-2-PCCA was docked with both Autodock VINA29 and GLIDE-SP30 (Schrödinger) during an evaluation period. The Autodock VINA poses were MMGBSA rescored using AMBER12 GAFF+AMBER99SB potential functions and SANDER optimization, including SQM28 deduced electrostatic charges using scripted workflow developed at RTI for parallelized CPU/GPU implementation analogous to literature reports,31 while GLIDE-SP/XP-Prime-MMGBSA30,32 results were used to deduce the lowest free energy poses using that methodology. The low-free energy poses of these two approaches were largely self-consistent and indicated a common mode of binding of (1R,2R)-2-PCCA at the level of exploration of this early stage model.
Supplementary Material
Acknowledgments
We thank Tiffany Langston, Taylor Rosa, and Dr. Elaine Gay for their valuable technical assistance. We also thank Dr. Rangan Maitra for valuable discussions during the course of this study.
Funding
We are grateful to National Institute of Mental Health, National Institutes of Health, U.S. (Grant MH103708 to C.J.) for the financial support of this research.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.6b00182.
Copies of HPLC results of 4a–g and 5a–aa, QSAR models A and B description, sequence alignment of GPR88 with selected GPCRs and β1-adrenergic receptor templates, PDB coordinates for GPR88 homology model (PDF)
Author Contributions
C.J. designed the studies, performed the synthesis and analyzed the data, A.M.D. performed the biological assays and analyzed the biological data, D.L.H. performed computational studies, and B.E.B. analyzed the biological data. All authors wrote the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discovery. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 2.Armbruster BN, Roth BL. Mining the receptorome. J Biol Chem. 2005;280:5129–5132. doi: 10.1074/jbc.R400030200. [DOI] [PubMed] [Google Scholar]
- 3.Civelli O, Reinscheid RK, Zhang Y, Wang Z, Fredriksson R, Schioth HB. G protein-coupled receptor deorphanizations. Annu Rev Pharmacol Toxicol. 2013;53:127–146. doi: 10.1146/annurev-pharmtox-010611-134548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Mizushima K, Miyamoto Y, Tsukahara F, Hirai M, Sakaki Y, Ito T. A novel G-protein-coupled receptor gene expressed in striatum. Genomics. 2000;69:314–321. doi: 10.1006/geno.2000.6340. [DOI] [PubMed] [Google Scholar]; (b) Ghate A, Befort K, Becker JA, Filliol D, Bole-Feysot C, Demebele D, Jost B, Koch M, Kieffer BL. Identification of novel striatal genes by expression profiling in adult mouse brain. Neuroscience. 2007;146:1182–1192. doi: 10.1016/j.neuroscience.2007.02.040. [DOI] [PubMed] [Google Scholar]; (c) Massart R, Guilloux JP, Mignon V, Sokoloff P, Diaz J. Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur J Neurosci. 2009;30:397–414. doi: 10.1111/j.1460-9568.2009.06842.x. [DOI] [PubMed] [Google Scholar]; (d) Van Waes V, Tseng KY, Steiner H. GPR88–a putative signaling molecule predominantly expressed in the striatum: Cellular localization and developmental regulation. Basal Ganglia. 2011;1:83–89. doi: 10.1016/j.baga.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Becker JA, Befort K, Blad C, Filliol D, Ghate A, Dembele D, Thibault C, Koch M, Muller J, Lardenois A, Poch O, Kieffer BL. Transcriptome analysis identifies genes with enriched expression in the mouse central extended amygdala. Neuroscience. 2008;156:950–965. doi: 10.1016/j.neuroscience.2008.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuoka T, Tsunoda M, Sumiyoshi T, Takasaki I, Tabuchi Y, Seo T, Tanaka K, Uehara T, Itoh H, Suzuki M, Kurachi M. Effect of MK-801 on gene expressions in the amygdala of rats. Synapse. 2008;62:1–7. doi: 10.1002/syn.20455. [DOI] [PubMed] [Google Scholar]
- 6.(a) Brandish PE, Su M, Holder DJ, Hodor P, Szumiloski J, Kleinhanz RR, Forbes JE, McWhorter ME, Duenwald SJ, Parrish ML, Na S, Liu Y, Phillips RL, Renger JJ, Sankaranarayanan S, Simon AJ, Scolnick EM. Regulation of gene expression by lithium and depletion of inositol in slices of adult rat cortex. Neuron. 2005;45:861–872. doi: 10.1016/j.neuron.2005.02.006. [DOI] [PubMed] [Google Scholar]; (b) Ogden CA, Rich ME, Schork NJ, Paulus MP, Geyer MA, Lohr JB, Kuczenski R, Niculescu AB. Candidate genes, pathways and mechanisms for bipolar (manic-depressive) and related disorders: an expanded convergent functional genomics approach. Mol Psychiatry. 2004;9:1007–1029. doi: 10.1038/sj.mp.4001547. [DOI] [PubMed] [Google Scholar]
- 7.Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, Bilbe G, Hoyer D, Bartfai T. Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol Psychiatry. 2007;12:167–189. doi: 10.1038/sj.mp.4001897. [DOI] [PubMed] [Google Scholar]
- 8.Befort K, Filliol D, Ghate A, Darcq E, Matifas A, Muller J, Lardenois A, Thibault C, Dembele D, Le Merrer J, Becker JA, Poch O, Kieffer BL. Mu-opioid receptor activation induces transcriptional plasticity in the central extended amygdala. Eur J Neurosci. 2008;27:2973–2984. doi: 10.1111/j.1460-9568.2008.06273.x. [DOI] [PubMed] [Google Scholar]
- 9.Logue SF, Grauer SM, Paulsen J, Graf R, Taylor N, Sung MA, Zhang L, Hughes Z, Pulito VL, Liu F, Rosenzweig-Lipson S, Brandon NJ, Marquis KL, Bates B, Pausch M. The orphan GPCR, GPR88, modulates function of the striatal dopamine system: a possible therapeutic target for psychiatric disorders? Mol Cell Neurosci. 2009;42:438–447. doi: 10.1016/j.mcn.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Quintana A, Sanz E, Wang W, Storey GP, Guler AD, Wanat MJ, Roller BA, La Torre A, Amieux PS, McKnight GS, Bamford NS, Palmiter RD. Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat Neurosci. 2012;15:1547–1555. doi: 10.1038/nn.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meirsman AC, Le Merrer J, Pellissier LP, Diaz J, Clesse D, Kieffer BL, Becker JA. Mice lacking GPR88 show motor deficit, improved spatial learning, and low anxiety reversed by delta opioid antagonist. Biol Psychiatry. 2016;79:917–927. doi: 10.1016/j.biopsych.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin C, Decker AM, Huang XP, Gilmour BP, Blough BE, Roth BL, Hu Y, Gill JB, Zhang XP. Synthesis, pharmacological characterization, and structure-activity relationship studies of small molecular agonists for the orphan GPR88 receptor. ACS Chem Neurosci. 2014;5:576–587. doi: 10.1021/cn500082p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bi Y, Dzierba CD, Fink C, Garcia Y, Green M, Han J, Kwon S, Kumi G, Liang Z, Liu Y, Qiao Y, Zhang Y, Zipp G, Burford N, Ferrante M, Bertekap R, Lewis M, Cacace A, Westphal RS, Kimball D, Bronson JJ, Macor JE. The discovery of potent agonists for GPR88, an orphan GPCR, for the potential treatment of CNS disorders. Bioorg Med Chem Lett. 2015;25:1443–1447. doi: 10.1016/j.bmcl.2015.02.038. [DOI] [PubMed] [Google Scholar]
- 14.Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther. 2007;322:205–213. doi: 10.1124/jpet.107.121525. [DOI] [PubMed] [Google Scholar]
- 15.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem Neurosci. 2012;3:50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark DE, Pickett SD. Computational methods for the prediction of ‘drug-likeness’. Drug Discovery Today. 2000;5:49–58. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
- 17.Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J Med Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 18.Stewart JJ. MOPAC: A semiempirical molecular orbital program. J Comput-Aided Mol Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 19.(a) Verloop A. The STERIMOL Approach to Drug Design. Marcel Dekker; New York: 1987. [Google Scholar]; (b) Verloop A, Tipker J. QSAR in Drug Design and Toxicology. Vol. 97. Elsevier; New York: 1987. [Google Scholar]
- 20.Harris D, Clayton T, Cook J, Sahbaie P, Halliwell RF, Furtmüller R, Huck S, Sieghart W, DeLorey TM. Selective influence on contextual memory: Physiochemical properties associated with selectivity of benzodiazepine ligands at GABAA receptors containing the a5 subunit. J Med Chem. 2008;51:3788–3803. doi: 10.1021/jm701433b. [DOI] [PubMed] [Google Scholar]
- 21.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guest MF, Bush IJ, van Dam HJJ, Sherwood P, Thomas JMH, van Lenthe JH, Havenith RWA, Kendrick J. The GAMESS-UK electronic structure package: algorithms, developments and applications. Mol Phys. 2005;103:719–747. [Google Scholar]
- 23.McCammon J, Lee C, Northrup S. Sidechain rotational isomerization in proteins: a mechanism involving gating and transient packing defects. J Am Chem Soc. 1983;105:2232–2237. [Google Scholar]
- 24.Dawson MI, Harris DL, Liu G, Hobbs PD, Lange CW, Jong L, Bruey-Sedano N, James SY, Zhang XK, Peterson VJ, Leid M, Farhana L, Rishi AK, Fontana JA. Antagonist analogue of 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN) family of apoptosis inducers that effectively blocks AHPN-induced apoptosis but not cell-cycle arrest. J Med Chem. 2004;47:3518–3536. doi: 10.1021/jm030524k. [DOI] [PubMed] [Google Scholar]
- 25.(a) Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, Mangano TJ, Deshpande DA, Jiang A, Penn RB, Jin J, Koller BH, Kenakin T, Shoichet BK, Roth BL. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015;527:477–483. doi: 10.1038/nature15699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Heifetz A, Storer RI, McMurray G, James T, Morao I, Aldeghi M, Bodkin MJ, Biggin PC. Application of an integrated GPCR SAR-modeling platform to explain the activation selectivity of human 5-HT2C over 5-HT2B. ACS Chem Biol. 2016;11:1372–1382. doi: 10.1021/acschembio.5b01045. [DOI] [PubMed] [Google Scholar]
- 26.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 27.Dunbrack RL., Jr Comparative modeling of CASP3 targets using PSI-BLAST and SCWRL. Proteins Suppl. 1999;3:81–87. doi: 10.1002/(sici)1097-0134(1999)37:3+<81::aid-prot12>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 28.Case DA, Darden T, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Götz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER. Vol. 12. University of California; San Francisco: 2012. [Google Scholar]
- 29.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Wong SE, Lightstone FC. Toward fully automated high performance computing drug discovery: a massively parallel virtual screening pipeline for docking and molecular mechanics/generalized Born surface area rescoring to improve enrichment. J Chem Inf Model. 2014;54:324–337. doi: 10.1021/ci4005145. [DOI] [PubMed] [Google Scholar]
- 32.Greenidge PA, Kramer C, Mozziconacci JC, Sherman W. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J Chem Inf Model. 2014;54:2697–2717. doi: 10.1021/ci5003735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.