

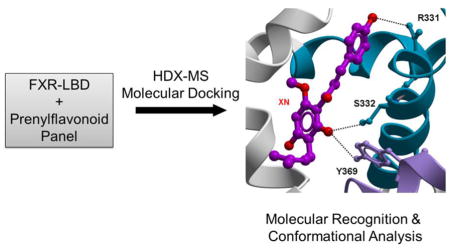
Abstract
We report on the molecular interactions of the Farnesoid X Receptor (FXR) with prenylflavonoids, an emerging class of FXR modulators. FXR is an attractive therapeutic target for mitigating metabolic syndromes (MetS) because FXR activates the inhibitory nuclear receptor, small heterodimer partner (SHP), thereby inhibiting both gluconeogenesis and de novo lipogenesis. We and others have shown that xanthohumol (XN), the principal prenylflavonoid of the hop plant (Humulus lupulus L.), is a FXR agonist based on its ability to affect lipid and glucose metabolism in vivo and to induces FXR target genes in biliary carcinoma cells and HEK293 cells. However, studies are currently lacking to rationalize the molecular mechanisms of FXR modulation by prenylflavonoids. We addressed this deficiency and report the first systematic study of FXR prenylflavonoid interactions. We combined Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) with computational studies for dissecting molecular recognition and conformational impact of prenylflavonoid interactions on the ligand binding domain (LBD) of human FXR. Four prenylflavonoids were tested: xanthohumol, a prenylated chalcone, two prenylated flavonones, namely isoxanthohumol (IX) and 8-prenylnaringenin (8PN), and a semisynthetic prenylflavonoid derivative, tetrahydroxanthohumol (TX). Enhancement of the HDX protection profile data by in silico predicted models of FXR prenylflavonoid complexes resulted in mapping of the prenylflavonoid interactions within the canonical ligand binding pocket. Our findings provide a foundation for the exploration of the chemical scaffolds of prenylated chalcones and flavanones as leads for future structure activity studies of this important nuclear receptor with potential relevance for ameliorating lipid metabolic disorders associated with obesity and MetS.
Keywords: Farnesoid X receptor, prenylflavonoids, hydrogen deuterium exchange, mass spectrometry, xanthohumol, fluorescence spectroscopy
Graphical Abstact
1. Introduction
FXR is a highly promising therapeutic target [1] for ameliorating diverse metabolic disorders because of its vital role in regulating expression of genes involved in bile acid, cholesterol and glucose homeostasis [2, 3]. Like other nuclear receptors, FXR shares the classical modular architecture of other nuclear receptors: a ligand-binding domain (LBD), a DNA-binding domain (DBD), and a connecting hinge region [4]. The LBD harbors the canonical ligand binding cavity. Ligand binding results in conformational changes that control release of the corepressor proteins and subsequent recruitment of the coactivator proteins. Multiple synthetic and semi-synthetic FXR ligands are currently under preclinical investigations. The initial results strongly suggest that FXR-targeting drugs may play future therapeutic roles in the treatment of cholestasis, dyslipidemic disorders and non-alcoholic steatohepatitis [5–8].
Bile acids function as FXR ligands [9, 10]. Natural FXR ligands have been isolated from plants and marine organisms and show promise in in vitro and preclinical studies as pharmaceutical agents for the management of metabolic diseases [11–16]. Several plant polyphenols, including dietary procyanidins and the tea catechin, epigallocatechin-3-gallate (EGCG), have been identified as FXR modulators [17–19]. Flavonoids are an emerging class of dietary modulators of nuclear receptors that show potential utilities in the management of metabolic syndrome and insulin resistance in patients with type 2 diabetes [5, 6]. As an emerging class of natural FXR modulators we chose for the current structure-function study prenylflavonoids from hops. Our panel of prenylflavonoids contained xanthohumol, a prenylated chalcone and a recognized agonist of FXR [20, 21], two prenylated flavonones, namely isoxanthohumol (IX) and 8-prenylnaringenin (8PN), and a semisynthetic prenylflavonoid derivative, tetrahydroxanthohumol (TX) (. 1).
We have shown that XN induced BSEP promoter activity, one of FXR target genes in transfected HEK293 cells. This induction occurred at a similar level as observed for chenodeoxycholic acid (CDCA) in the transfected cells [21]. Other studies also indicate that prenylflavonoids function as pharmacologically relevant modulators of FXR [22–25]: XN modulates hyperlipidemia by inhibiting expression or activity of the diacylglycerol acyltansferase [22]; XN inhibits triglyceride synthesis and secretion in HepG2 cells [23]; XN functions as an FXR agonist in a transient transfection assay, and also determination of the hepatic gene expression in XN-fed mice suggests that XN acts as FXR modulator [24]; XN and the related prenylated flavanone isoxanthohumol (IX) inhibit lipid accumulation in maturing preadipocytes, possibly partly by modulating FXR target genes [25].
HDX-MS has been successfully used to probe small molecule binding to nuclear receptors [26–28]. HDX-MS reports on changes of backbone amide hydrogen exchange rates which are highly sensitive to the structural changes, conformational flexibility, hydrogen bonding strength, and solvent accessibility of protein surfaces [29]. In this study we conducted HDX-MS for dissecting molecular recognition and conformational impact of prenylflavonoid interactions on the ligand binding domain (LBD) of human FXR. We complemented the HDX-MS studies with fluorescence quenching titration, a well-established technique for investigating protein structure and dynamics and ligand/receptor interactions [30–32]. Tryptophan fluorescence in proteins is highly sensitive to the local environment in solution and fluorescence emission spectra reflect on protein conformational transitions, subunit association, substrate binding, or denaturation [33].
The present studies provide information on the molecular recognition of prenylflavonoids by FXR-LBD and describe how prenylflavonoids modulate the conformational dynamics of FXR-LBD. Our findings provide a scientific basis for the use of XN and related prenylflavonoids as novel leads for the development of FXR–targeting therapeutics with possible application for the treatment and management of metabolic disorders.
2. Materials and Methods
2.1. Materials
Deuterium oxide (D2O, 99.9% deuterium), chenodeoxycholic acid (CDCA), and guggulsterone (GG) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA), Tocris (Ellisville, MO, USA) and ChromaDexTM Corporate (Irvine, CA, USA), respectively. All prenylflavonoids compounds were kindly provided by Dr. Fred Stevens’s group at Oregon State University and were >99% pure by HPLC. For the described experiments 10 mM stock solutions of the compounds were prepared in dimethylsulfoxide (DMSO).
2.2. Protein Expression and Purification
Protein expression and purification was performed as described previously [34]. Briefly, the protein was expressed in Escherichia coli BL 21 (DE23) pLysS with a pET 15B vector encompassing residues L193 to Q472 which were grown in 2XYT medium with antibiotics (Ampicillin 150 μg/mL, Chloramphenicol 35 μg/mL) at 37°C until absorption measurements at 600nm (A600) reached 0.6–0.8. The cells were allowed to grow for another four hours at 20°C with 0.8 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) induction to produce an N-terminal Histidine (His6)-tagged FXR LBD. Cells were harvested by centrifugation, washed and frozen at −80°C.
The frozen pellets were resuspended in pH 7.3-buffer containing 50 mM sodium phosphate, 0.5 M NaCl, 0.5 mM CHAPS, 15 mM imidazole, and 0.5 M sucrose. The cells were then sonicated, centrifuged and the supernatant saved. Isolated protein was purified by affinity chromatography on Clonetech Talon Co2+ polyhistidine affinity resins which was equilibrated with the above described buffer. The supernatant was mixed with Co2+ affinity resin and rotated at 4°C for 45 minutes to bind the His6-tagged proteins. The affinity-bound His6-tagged protein was eluted from the Co2+ resin-packed column with the above described buffer but containing 200 mM imidazole until A280 was less than 0.03. The protein fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The purest fractions were pooled and incubated with thrombin for 48 hours at 4°C to remove the His6-tag. Subsequently, the protein was additionally purified by a Co2+ resin column with and without imidazole to remove uncut His6-tagged protein and the free His6-tag. The concentration of purified FXR-LBD was determined spectrophotometrically at 280 nm, and the purity (over 95%) was judged by SDS-PAGE and mass spectrometry. The purified protein was collected and concentrated, aliquoted, snap-frozen in liquid nitrogen and stored at −80°C.
2.3. Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS)
The recombinant human FXR-LBD (15 μL, 98 μM) was equilibrated for 30 min in H2O buffer (in 50 mM sodium phosphate, 0.5 M NaCl, 0.5 mM CHAPS, 1 mM TCEP, 0.5 M sucrose, and 10% glycerol, pH 7.4) at room temperature in absence and presence of each ligand (0.5 μL, ±10 mM ligand in DMSO). The small amount of DMSO (3.3% v/v) did not affect the conformational properties of FXR-LBD as indicated by intact protein HDX data as well as by fluorescence titration data (Fig. S1). Under these conditions the bound fractions of FXR-LBD before HDX initiation is about 100% for the prenylflavonoids tested in this study [34]. To initiate the exchange-in reaction, 10-fold D2O buffer with the same composition as the above described H2O buffer was added to equilibrate the apoprotein and holo-protein solutions. The ratio of ligand to protein was kept at 3:1 as previously described [34]. The FXR LBD concentration was ~10 μM and prenylflavonoid concentration was ~30 μM in the labeling experiments. Exchange was quenched (pH 2.5, 0 C°) by adding 15 μL 0.42% chilled phosphoric acid at different reaction time points. Liquid nitrogen was used to flash-freeze all quenched samples prior to MS analysis. Experiments were performed in triplicate.
The quenched intact protein were loaded onto a Micro TrapTM C4 column (1 mm × 8 mm) and eluted with a fast 10–90% (v/v) B gradient in 12 min (solvent A: 0.1% formic acid in H2O, solvent B: 0.1% formic acid in acetonitrile, flow rate 40 μL/min). Deuterium level for the intact protein was measured using an LCT ESI-ToF mass spectrometer. HDX information of peptic peptides was obtained by using a Synapt HDMS instrument equipped with a Waters HDX module enabling online peptic digestion and “cold” LC separation. Briefly, a Poroszyme® immobilized pepsin column (2.1×30 mm) in conjunction with 0.05% aqueous formic acid at a flow rate of 100 μL/min was used for online digestion. The peptides were loaded onto an Acquity BEH C18 VanGuard Pre-column (1.7 μm, 2.1 x 5 mm) and separated using an Acquity UPLC® BEH C18 column (1.7 μm, 1.0×100 mm). The following LC gradient was applied: 8% B to 40% in 6 minutes, hold 40% B 1 minute, 40% B to 85% B in 0.5 minute, hold 85% B 1 minute, 85% to 8% in 0.5 minute, hold B at 8% for 3 minutes, total 12 minutes (using the same LC solvents as described above for intact protein analysis).
2.4. HDX Data Analysis
The deuterium numbers of the intact protein were directly obtained from the mass difference between deuterated and nondeuterated protein. Protein mass spectra were analyzed using MassLynx 4.0 program (Waters Corporation). For obtaining peptide identifications, the raw data of the undeuterated samples were analyzed in the MSE mode. ProteinLynx Global Server (PLGS) 2.4 software (Waters) was used for searching against an “in house” protein database to which the human FXR-LBD sequence was added. After database searching, a peptide list covering specific protein regions was produced. Peptides that resulted in mass spectra of high spectral quality and protein sequence coverage were chosen for the subsequently conducted HDX-MS studies. The deuterium uptake for each peptide and time point was derived by calculating the differences between the centroid of the deuterated peptide isotope distribution and the centroid of the undeuterated peptide isotope distribution. Data analysis was supported by HDX Browser software (Waters) in conjunction with HX-Express software [35].
The deuterium number of each peptide was normalized to account for differences in peptide length by applying the following equation 1:
| (1) |
Where D% is percentage of the deuterium in a peptide; m is deuterated peptide mass; m0 is undeuterated peptide mass; N is total number of the exchangeable amide hydrogens in a peptide. Because of the high reproducibility of the online pepsin UPLC setup and the comparative nature of this study we omitted to apply back exchange corrections in accord to other HDX-MS studies [36–38].
Using the measured Kd values for the prenylflavonoids compiled in Table 1 and the concentrations of FXR-LBD (10 μM) and ligand (30 μM) we estimated the fraction of bound FXR-LBD according to Shortridge et al [39] as follows: 88% for XN, 80% for IX, 81% for GG and 91% for 8-PN. The presence of a mixed population of bound and free FXR-LBD may complicate the interpretation of the HDX results. Therefore, the mass isotope pattern of each peptide at the different exchange time points were checked manually and their mass isotope distributions were evaluated (Fig. S2). We concluded that the absence of bimodal isotope distributions suggested that under the conditions used for the FXR-LBD exchange experiments in the presence of the diverse ligands HDX the fraction of ‘protein bound (holoprotein)’ was indeed the predominant population probed allowing a comparative qualitative assessment of the interaction characteristics of the prenylflavonoid-type ligands on FXR-LBD. The average relative standard deviation for deuterium content of the peptides examined was less than 5% for 542 out of 560 (97 % of all time points) and 18 out of 560 (3 % of all time points) had average relative standard deviations between 5 and 7 %. An average relative standard deviation for the deuterium content of each peptide at each labeling time point of less than 5% is consider as demonstrating high reproducibility of the peptide-level HDX-MS methodology [40].
Table 1. Stern-Volmer constant KSV, the fraction of fluorophore accessible to the quencher (fá), bimolecular quenching constants (kq), dynamic quenching constant (KD), static quenching constant (KS), and dissociation constant (Kd) of FXR-LBD-ligand systems studied.
These values were obtained from Eq. (S1) for IX and 8-PN, Eq. (S3) for GG (marked with *), Eq. (S4) for XN (marked with **), and Eq. (S2) and (S5) for all ligands (For details, see supplementary data Text S1).
| KSV × 105 (M−1) | fa | KS × 105 (M−1) | kq × 1013 (M−1S−1) | Kd (μM) | |
|---|---|---|---|---|---|
| GG* | 2.53±0.13 | 0.53±0.02 | - | 8.43±0.43 | 5.40±0.21 |
| XN** | 0.006±0.003 (KD) | - | 3.25±0.32 | 0.02±0.01 | 2.68±0.11 |
| IX | 1.15±0.02 | - | - | 3.83±0.07 | 5.92±0.25 |
| 8-PN | 1.94±0.05 | - | - | 6.47±0.17 | 2.20±0.05 |
2.5. Kinetic Modeling of Deuterium Uptake Profiles
All peptide deuterium uptake plots were fitted with a six-bin-fixed-rate-constant kinetic model, built in R software, where the sum of the square error was minimized [36]. The relationship between deuterium uptake and exchange time can be described by equation 2 because the HDX reaction follows first-order kinetics [41].
| (2) |
where D is deuterium uptake; ki is the rate constant for the exchange reaction which is fixed to six values (six bins) (10, 1, 0.1, 0.01, 0.001, and 0.0001 min−1); Ai is the amide hydrogen number in a specific bin; t is incubation time.
2.6. Fluorescence Titration Experiments
A LS 50 luminescence spectrometer (Perkin Elmer) was used to measure steady state fluorescence. The monochromator slit widths for excitation and emission were both set to 3.5 nm. For the fluorescence titrations, after each addition of ligand, the protein sample was excited at 295 nm to minimize the fluorescence contribution of tyrosine residues and the emission spectra were acquired from 305 nm to 480 nm. The ligand-FXR-LBD interactions were studied by using an intrinsic fluorescence titration method in which 400 μL of 10 μM protein (dissolved in 50 mM sodium phosphate buffer containing 0.5 M NaCl, 0.5 mM CHAPS, 1 mM TCEP, 0.5 M sucrose and 10% glycerol, pH 7.4) was titrated with 1000 μM ligand (dissolved in DMSO) until the ligand/protein ratio reached 5 to 1. Before analysis of the binding and quenching data, fluorescence spectra were corrected (Fcor) for the background fluorescence of the specific ligand FB, the instrument-dependent monochromator and photomultiplier response emission correction factor FS, and the dilution factor A: Fcor = (F0 − FB) × 100×A/FS where F0 is the measured fluorescence intensity. All measurements were performed at 25°C and repeated three times. Fluorescence quenching data analysis procedure was described in Text S1.
2.7. Molecular Docking Study
The 3D-coordinates of the FXR-LBD in complex with 3-deoxy-CDCA were retrieved from PDB ID 1OT7 [42] and energetically minimized in the internal coordinate space with Molsoft [43]. For clarity, the numbers of the relevant amino acids in PDB ID 1OT7 file, Arg328, Ser329, and Tyr366, have been adjusted as Arg331, Ser332, and Tyr369 (UniRef100_B6ZGS9). Molecular docking was run against the binding pocket of the FXR-LBD as previously reported [44]. The docking protocol was initially validated by docking 3-deoxy-CDCA into the FXR-LBD. The agonist docked with a score of −26.63 reproducing the crystallographic binding pose involving interactions with residues Arg331, Ser332, and Tyr369 [42].
3. Results
3.1. HDX-MS analysis of the intact human FXR LBD indicates molecular recognition of prenylflavonoids
We performed HDX-MS studies of the intact FXR-LBD construct to determine to what extent each of the four prenylflavonoids tested in this study would affect the global structural dynamics of the protein. The sum of the exchangeable amide hydrogens in the FXR-LBD construct is 274. In presence of prenylflavonoids the observed HDX protection pointed to an increase in structural compactness, loss of conformational flexibility, and/or decrease of global solvent accessibility upon ligand interaction (Fig. S3). For instance, comparison of the observed global deuterium levels between the apoprotein and the protein in presence of each of the prenylflavonoids indicated retardation of exchange of approximately 11±2, 13±2, 15±1, 14±2 amide hydrogens for XN, IX, 8-PN and TX, respectively, at the 60-min time point of the co-incubation experiment in the 90% D2O-solvent system (Fig. S3). The observed exchange protection let support to that the prenylflavonoids indeed directly interact with the FXR LBD and function as FXR ligands.
3.2. Peptide-level HDX protection profiles of FXR-LBD identifies the canonical ligand binding cavity as the site of prenylflavonoid interaction
Twenty peptides with high quality covering over 85% of the amino acid sequence were chosen for determining deuterium uptake levels in the different protein systems tested. Visual inspection of the exchange-in evolution of the peptide ion isotope distributions indicated that there was no indication of mixed exchange populations in the HDX-MS ligand binding studies (Fig. S2); exchange-in under the current conditions proceeded according to EX2 regime [45]. Fig. 2 summarizes the modulation of conformational dynamics for the FXR LBD for each the prenylflavonoids tested. The graphical presentations of the time course of deuterium uptake for each of the systems studied visualizes that interaction of the ligand causes retardation in deuterium uptake signifying reduced conformational mobility compared to the apoprotein. The protection against exchange-in was observed in regions that form the classical ligand binding cavity, namely regions of helices H3 (288–298), H6 (320–336) and H8 (361–375), and regions of helices H9 (388–396) and H10 (406–412).
Fig. 2. Modulation of conformational dynamics of FXR LBD by prenylflavonoids.

Time course of percent deuterium incorporation observed for apo FXR LBD in comparison to the time courses of deuterium uptake observed for co-incubation and labelling experiment of FXR LBD in presence of each of the prenylflavonoids, XN, IX, 8PN, and TX. Deuterium uptake is color-coded as indicated and mapped onto PDB 1OT7. Prenylflavonoid interactions result in gain in conformational stability in FXR-LBD reflected in lower deuterium uptake over time. Deuterium exchange data (%D) for all peptides monitored for all labeling times for FXR-LBD in presence of the different prenylflavonoids are compiled in Fig. S5. Because the N-terminal region (201–241) is not visible in the crystal structure PDB 1OT7 no color-coded HDX-MS map could be provided for this region.
Whereas, peptides encompassing the amino acid residues 201–222, 229–241, 256–268, 269–278, 337–347, and 454–465 reached maximum deuterium uptake levels of observable exchange in 30 seconds (Fig. S5). This type of deuterium exchange behavior is commonly observed for regions that are highly solvent exposed, disordered and/or conformationally highly flexible without stabilizing hydrogen bonding networks. In this context it is noteworthy that in the FXR-LBD-3-deoxy-CDCA complex crystal structure (PDB ID 1OT7) no secondary structure was assigned to the regions encompassing residues 264–266, 277–278, 339–341, 454–456, and 459–462 reflecting on the limitation of the X-ray structural analysis technique that regions with high mobility in proteins often result in large errors during data collections making any structural interpretations impossible [46].
Table S1 compiles peptide-level average differences in deuterium incorporation levels for the comparison FXR-XN vs. apoprotein, FXR-IX vs. apoprotein, FXR-8PN vs. apoprotein, FXR-TX vs. apoprotein and p-values for each comparison. Deuterium level differences of regions with p <0.001, calculated by two-way ANOVA, were considered as statistically significant [47–49]. A graphical presentation of these peptide-level average differences in deuterium incorporation levels is provided in Fig. 3. The average differences in percentage of deuterium level for the four ligand-bound FXR complexes relative to the apoprotein were overlaid onto the X-ray crystal structure of the FXR-LBD bound to 3-deoxy-CDCA (PDB 1OT7) (Fig. 3, Table S1). The comparative analysis of the peptide-level HDX protection profiles revealed that each of the prenylflavonoids induced statistically significant protection in the following regions: 288–298, 320–336, 361–375 and 406–412 (Fig. 3, Table S1). In presence of each of the prenylflavonoids, exchange in those regions was retarded indicating stabilization of the conformation and/or reduced solvent accessibility caused by ligand interaction. Three of these peptides, 288–298 (helix 3), 320–336 (helix 4–helix 5), and 361–375 (helix 7) are involved in forming the canonical ligand binding cavity in the LBD of FXR. Peptide 406–412 encompasses parts of helix 9 and is not part of the canonical ligand binding cavity. Allosteric stabilization of helix 9 upon ligand binding was also detected in our previous HDX-MS study of FXR-LBD ligand binding interactions [34]. Importantly, for all four prenylflavonoids exchange protection was observed for peptide 288–298, which is involved in the coordination of ligands as shown in two crystal structures of FXR-LBD-ligand complexes (PDB ID 1OT7 and 1OSV)[42]. Interestingly, for 8-PN and TX exchange protection was also observed for the region 348–360 (helix 6) (Fig. 3). Overall, the comparative analysis of the HDX protection profiles at the peptide level for FXR-LBD in presence of XN, IX, 8-PN and TX vs. apoprotein strongly supports the notion that prenylflavonoids are bona fide ligands of human FXR targeting the canonical ligand binding site.
Fig. 3. Comparison of the HDX protection profiles at the peptide level for FXR-LBD in presence of XN, IX, 8-PN and TX vs. apoprotein.

Left: Deuterium exchange-in percentage difference between apo and holo FXR-LBD is shown at different reaction time points (0.5, 1, 2, 5, 10, 30, and 60 min, from left to right) for each peptide. Graphical presentations were derived from data compiled in Table S1. Deuterium level differences of regions with p = <0.001, calculated by two-way ANOVA, were considered as statistically significant and these regions are marked with an asterisk. The error bars represent the average relative standard deviation of three independently conducted labeling experiments. Right: Average differences in deuterium percentage (D%) of seven time points (0.5, 1, 2, 5, 10, 30, and 60 min) mapped onto the cartoon representation of FXR-LBD-ligand complex, PDB 1OT7. NS, “not significant” and ND, “not detected”. Due to the absence of the N-terminal region (201–241) in this crystal structure no color-coding for the HDX data could be provided for this region.
For the regions that displayed retarded exchange in the presence of each of the prenylflavonoids, kinetic modeling results (Table S2) revealed that the distribution of the amide hydrogens in the bins was rearranged in presence of the prenylflavonoid. Compared to the apoprotein prenylflavonoid binding decreased the number of the amide hydrogens in the ‘fast exchange rate’ bins (k = 10 and 1 min−1) while the number of amide hydrogen that were grouped into the ‘medium’ (k = 0.1 and 0.01 min−1) and the ‘low exchange rate’ bins (k = 0.001 and 0.0001 min−1) increased (Table S2).
In contrast, regions 201–222, 229–241, 256–268, 269–278, 337–347, and 454–465 exhibited no differences in deuterium levels when the peptides derived from the holo protein systems were compared to that of the peptides derived from apoprotein (Fig. 3, Table S1). These findings are consistent with that these regions display high conformational mobility even in presence of ligand binding and, as noted above, which is in accord to that no structural information is available for the regions 264–266, 277–278, 339–341, 454–456, and 459–462 in the co-crystal structure of FXR-LBD-3-deoxy-CDCA complex (PDB ID 1OT7).
3.3. Intrinsic fluorescence studies of apo FXR-LBD and holo FXR-LBD
The FXR-LBD contains two tryptophan residues, Trp454 and Trp469, which are located in the C-terminal region of the protein (Figure 6A). In order to evaluate molecular recognition and ligand interaction of the prenylflavonoids we performed first fluorescence titration with two established ligands, CDCA, a low affinity agonist and endogenous ligand of FXR, and GG, an in vitro antagonist of FXR. We added CDCA to the protein sample and measured the changes in fluorescence intensities of the tryptophan residues (emission λmax). When excited at 295 nm, apo FXR-LBD exhibited significant intrinsic fluorescence in the range 305–480 nm with maximum emission at 343 nm (Fig. 4). Only subtle fluorescence intensity changes (±3%) were observed during the CDCA titration of FXR-LBD, suggesting the microenvironment of the tryptophan residues in FXR-LBD was either unchanged or only slightly changed consistent with a recent study [50] . In contrast, GG titration of FXR-LBD showed a concentration-dependent quenching without any changes in the emission λmax and spectral shape (Fig. 4), which suggests a direct interaction of GG with the FXR-LBD and that the tryptophan residues are involved in the binding mechanism. Under the same experimental conditions, there was a distinct decrease in the fluorescence intensity when FXR-LBD was titrated with XN and its derivatives (Fig. 4). Blue shifting of the emission were observed in the presence of XN and TX (Fig. 4), suggesting that upon ligand binding the tryptophan residues were placed in a more hydrophobic microenvironment possibly caused by exclusion of solvent at the binding sites of XN and TX. Whereas, the additions of IX and 8-PN caused red shifting, suggesting that one or more tryptophan residues in FXR-LBD were exposed to solvent to a higher degree. In addition, the isosbestic point in the spectra from the IX titration at 405 nm suggests that the presence of both bound and free IX in equilibrium [51, 52].
Fig. 6. Visualization of human FXR-LBD prenylflavonoid interactions.

A, Ribbon representation of the co-crystal structure of FXR-LBD bound to 3-deoxy-CDCA complex, PDB 1OT7, with the Trp residues at positions 454 and 469 displayed as sticks and colored in blue. Arrows mark the approximate positions of the residues, R331, S332, and Y 369, for which interactions were predicted with the prenylflavonoid ligands. The N-terminal region (201–241) is not visible in PDB 1OT7. Docking models of B, XN, C, TX, D, IX, and E, 8PN into human FXR-LBD (Molsoft ICM). Computational molecular docking predicts and identifies non-covalent interactions established between ligands and protein. Docked ligands are displayed as sticks and colored by atom type, with carbon atoms in magenta (XN), orange (TX), cyan (IX) and white (8PN); protein residues are displayed as stick with the carbon atoms colored in green. Secondary structure is displayed as ribbon. Protein-ligand hydrogen binding interactions are displayed as dashed black lines. The docking models do not encompass the N-terminal region 201–241.
Fig. 4.

Fluorescence emission spectra of FXR-LBD in the presence of different concentrations of ligands. CDCA, the endogenous ligand of FXR, and GG, an in vitro FXR antagonist, were included in the fluorescence titration studies for comparison.
3.4. Mechanism of prenylflavonoid binding to FXR-LBD and determination of dissociation constant, Kd
In fluorescence titration experiments probing ligand/receptor interaction, any observed fluorescence quenching might be due to dynamic quenching (diffusive collisions) and/or static quenching (the formation of a ground-state complex). To determine the quenching mechanism in the process of the ligand binding in the FXR-LBD, the Stern-Volmer equation (3) was used to analyze the quenching data (Fig. 5A). Of the four ligand-FXR-LBD complexes under study, the fluorescence quenching plots of IX and 8-PN were linear, indicating that the two tryptophan residues in FXR-LBD were accessible to these two ligands equally [33] and just a single type of quenching occurred, either dynamic or static [30]. For XN, the Stern-Volmer plot exhibited a slightly upward curvature (Fig. 5A) which is characteristic of a combination of both dynamic and static quenching [33]. As noted above due to the low affinity of CDCA to FXR LBD, no or minimal Trp fluorescence quenching was observed and therefore no Stern-Volmer plot can be provided. The downward curving plot (Fig. 5A) obtained for the GG titration suggested that a fraction of tryptophan residues (in this case one tryptophan residue) was shielded from GG [53]. By fitting the experimental quenching data from the GG titration to equation 5 (Fig. 5B), the fa value 0.53 was obtained, indicating that half of the tryptophan residues in FXR-LBD was accessible to GG, i.e. only one tryptophan residue was involved in the interaction between protein and ligand.
Fig. 5.

A, Stern-Volmer plots of FXR-LBD quenching by the different ligands; B, Modified Stern-Volmer plots for GG and XN quenching of FXR-LBD.
The magnitude of KSV often can be used to differentiate between dynamic and static quenching [54–59] based on the fact that if the bimolecular quenching constant kq is larger than the maximal diffusion-controlled quenching constant of 1 × 1010 M−1S−1 [33], a decrease in the fluorescence intensity is caused mainly by the formation of a non-fluorescence complex and/or the existence of a specific kind of binding interaction [33]. The kq values (Table 1) were calculated by applying Eq. S2 (Text S1), where the average lifetime τ0 of the native tryptophan fluorescence of protein was taken to be three nanoseconds [60]. The bimolecular quenching constants kq (Table 1) determined from the IX, 8-PN, and GG fluorescence titration experiments were three orders of magnitude larger than the maximal expected value of kq, suggesting that dynamic quenching was not responsible for the reduction of fluorescence intensity. Interestingly, the kq derived by KD of XN (Eq. S4) was 2×1011 M−1, 20 times larger than 1×1010 M−1S−1 (Table 1) which suggests the presence of another specific binding site located near the tryptophan residues.
The apparent dissociation constant Kd is a parameter describing how tightly the ligand binds to its receptor. The magnitude of the apparent dissociation constant for each of the prenylflavonoids was in the micromole range, implying that they are low affinity binders of the FXR-LBD, which is consistent with our mass spectrometry-based HDX data. The Kd of the GG FXR-LBD interaction was determined to be 5.4 μM, similar to the previously reported Kd value that was determined by radioligand binding assays [61].
4. Discussion
We predicted and evaluated the binding sites between prenylflavonoid molecules and the FXR-LBD by molecular docking studies (Fig. 6). The HDX profiles of the prenylflavonoid-bound FXR LBD indicated decreased deuterium incorporation in the following regions: 288–298, 320–336, and 361–375, which are all involved in forming of the FXR ligand binding cavity (Fig. 3). The docking data suggested, similar to 3-deoxy-CDCA, that the 4-hydroxyphenyl and 2-OH groups in XN and TX could interact with the side chain of residues Arg331, Ser332, and Tyr369 via hydrogen bonds (Fig. 6B and C), and the two residues Ser332 and Tyr369 could also form hydrogen bonding interactions with the carbonyl group (C=O) in IX and 8PN (Fig. 6D and E). Additionally, each prenylflavonoid molecule docked into the FXR-LBD and the scores are displayed in Table S3. Usually lower scores predict better binding. Considering the low potency exerted by agonist 3-deoxy-CDCA [62], the docking scores generated with the prenylflavonoid-type compounds (Table S3) predicted weak binding. These results also suggested that these compounds, like other FXR ligands, shared similar binding sites and were able to occupy the canonical ligand binding pocket of the FXR-LBD.
In this study, TX was used to decipher the possible effect caused by covalent modification of XN, an electrophile with the potential to alkylate nucleophilic groups in FXR LBD. The Stern-Volmer plot of FXR-LBD interaction with XN revealed the existence of both dynamic and static quenching at high ligand concentrations (Fig. 5A), which were also previously observed in the interaction of flavonoid compounds with bovine serum albumin and bovine milk xanthine oxidase [57, 58]. In addition, Kaldas et al reported that the flavonoid quercetin can covalently bind with human serum albumin (HSA) [63]. The kq value determined for XN in Table 1 implied the presence of a specific binding site in close proximity to the tryptophan residues and the formation of a ground-state complex. Importantly, MS data analysis showed that multiple XN molecules can covalently bind with FXR-LBD (Fig. S6), possibly via a Michael-type addition reaction involving the α,β-unsaturated ketone functionality of XN and the thiol(SH) functional group of a cysteine residue. Therefore, we propose that XN binds to Cys466, which is located in close proximity to Trp469, and this covalent interaction is responsible for the dynamic quenching observed in the XN-FXR-LBD complex. A comparison of the HDX protection profile (Fig. 3) showed that the binding of XN and TX resulted in exchange protection in the following regions: 288–298, 320–328, 328–336, 348–360, 361–367, and 368–375, all are part of the ligand binding pocket in FXR-LBD. Furthermore, our molecular docking study demonstrated that XN and TX were able to dock into the ligand binding pocket of the FXR-LBD with similar scores (Table S3), and that both compounds revealed the same pattern of non-covalent interactions established by the weak agonist 3-deoxy-CDCA [42]. Both ligands, similar to 3-deoxy-CDCA, interact with the side chain of residues Arg331, Ser332, and Tyr369 involving their 4-hydroxyphenyl and 2-OH groups, respectively (Fig. 6). Overall, our data suggested that covalent binding of XN to FXR-LBD has no effect on XN binding in the LBD cavity.
While X-ray crystal structures of the FXR with agonists exist, structural information for the interaction of FXR LBD with low affinity ligands is scarce. However, knowledge of the distinct modes of interaction of agonists, partial agonists and antagonists with FXR is needed for the development of new drugs that are based on the modulation of FXR as strategy for the management and treatment of metabolic disorders. In our previously HDX-MS data, we showed that apo FXR-LBD displays conformational features commonly associated with highly solvent exposed and/or disordered proteins. One may speculate that this conformational flexibility may contribute to the promiscuity of FXR [64].
In our previous work we reported the conformational dynamics of FXR-LBD induced by three well known FXR ligands that represented a diverse spectrum of chemical scaffolds, namely the high affinity agonist GW4064, the low affinity agonist CDCA, and the in vitro antagonist GG(Z) [34]. The most pronounced ligand-dependent differences in deuterium uptake for the three typical ligands were observed for the following peptides: 288–298, 320–328, 328–336, 348–360, 361–367, and 368–375 [34]. To determine to what extent the chemical scaffolds possibly govern the deuterium exchange-in profiles a hierarchical clustering analysis of the HDX profiles was performed based on the peptide-level deuterium incorporations observed for the seven ligands tested, i.e. three classical ligands and four prenylflavonoids (methodological details are provided in Text S2). Fig. 7 shows a heatmap visualizing the peptide-level deuterium incorporations observed for the seven FXR ligand systems tested. The cluster analysis revealed that the HDX profiles obtained for the four FXR-prenylflavonoid systems were more similar to each other compared to the HDX profiles obtained for the natural FXR ligand CDCA and GG(Z), both featuring a steroidal tetracyclic scaffold. The FXR-GW4064 system displayed a disparate deuterium incorporation profile in accord to that the stilbene/isoxazole scaffold of GW4064 displays a HDX profile distinct from the other two scaffolds.
Fig. 7. Hierarchical clustering analysis of the HDX data for seven FXR-LBD-bound complexes.

The peptide-level deuterium exchange-in level is color-coded from red to green indicating low (exchange-in protection) to high levels of exchange-in. “Counts” represents the pixels number (squares) of each color in the histogram based on the frequency of the pixels (squares) of each color in the heatmap. The heat map allows visualizing that the exchange-in dynamics observed for the prenylflavonoids is different from the exchange-in dynamics observed for FXR ligands with tetracyclic core skeletons. GW4064, a synthetic non-steroidal stilbene-based high affinity agonist, displays a unique FXR-LBD exchange-in dynamics different from that observed for the ligands with tetracyclic core and the prenylflavonoids.
To summarize, the current HDX-MS study demonstrates that the FXR LBD is capable of binding of prenylflavonoid-type compounds. Furthermore, our molecular docking suggest that three key functional groups in the prenylflavonoids, namely the 4-hydroxyphenyl, the 2-OH, and the carbonyl group, are important in forming hydrogen bonding interactions with the FXR-LBD and contributing to ligand binding affinity. This study hints that the chemical scaffold of the ligand will determine to a large extent the mode of interactions and in turn the conformational dynamics of FXR. The current findings provide support to the notion that the chemical scaffolds of prenylated chalcones and flavanones provide hitherto underexplored scaffolds for the development of novel bile acid receptor modulators. Future studies will need to explore the fitness of HDX-MS profiling for classifying FXR ligands according to bioactivity, i.e. agonist, partial agonist and antagonists.
5. Conclusion
In this report we describe the modulation of the conformational dynamics of the human FXR LBD by four prenylflavonoids (XN, IX, 8-PN, and TX). We provide binding and molecular recognition motifs for these prenylflavonoids based on combining findings from HDX-MS, fluorescence titration and molecular docking studies. On the basis of these findings, we conclude that prenylflavonoids are bona fide ligands of human FXR. We foresee that prenylflavonoids and possibly other dietary polyphenols [65, 66] may serve as future leads in the development of new synthetic FXR ligands for prevention and treatment of lipid and metabolic disorders. The combination of HDX-MS with computational approaches provides a promising strategy for future structure-function research of small molecule modulators of this important nuclear receptor.
Supplementary Material
Fig. 1.

Chemical structures of the prenylflavonoids used in the present study.
Highlights.
Solution-phase HDX dynamics of FXR LBD interactions with prenylflavonoids
Fluorescence spectroscopy support interaction of prenylflavonoids with FXR LBD
Molecular docking reveals site of interactions of prenylflavonoids with FXR LBD
Prenylflavonoids interacts with the canonical site of the FXR LBD
Prenylflavonoids are bona fide ligands of FXR
Acknowledgments
This study was supported in part by a National Institutes of Health grant (S10RR025628, P30ES000210, R01AT009168), Research Equipment Reserve Funds provided by Oregon State University (Corvallis, OR) and Waters Corporation.
Abbreviations
- FXR
farnesoid X receptor
- HDX-MS
hydrogen deuterium exchange mass spectrometry
- MetS
metabolic syndromes
- SHP
small heterodimer partner
- BSEP
bile salt export pump
- LBD
ligand binding domain
- XN
xanthohumol
- IX
isoxanthohumol
- 8-PN
8-prenylnarigenin
- TX
tetrahydroxanthohumol
- DBD
DNA-binding domain
- CDCA
chenodeoxycholic acid
- GG
guggulsterone
- IPTG
isopropyl-β-D-thiogalactopyranoside
- DMSO
dimethylsulfoxide
- HSA
human serum albumin
- PLGS
Protein Lynx Global Server
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mencarelli A, Fiorucci S. FXR an emerging therapeutic target for the treatment of atherosclerosis. J Cell Mol Med. 2010;14:79–92. doi: 10.1111/j.1582-4934.2009.00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 3.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem. 2003;278:104–110. doi: 10.1074/jbc.M209505200. [DOI] [PubMed] [Google Scholar]
- 5.Westin S, Heyman RA, Martin R. FXR, a therapeutic target for bile acid and lipid disorders. Mini Rev Med Chem. 2005;5:719–727. doi: 10.2174/1389557054553802. [DOI] [PubMed] [Google Scholar]
- 6.Fiorucci S, Rizzo G, Donini A, Distrutti E, Santucci L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol Med. 2007;13:298–309. doi: 10.1016/j.molmed.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Crawley ML. Farnesoid X receptor modulators: a patent review. Expert Opin Ther Pat. 2010;20:1047–1057. doi: 10.1517/13543776.2010.496777. [DOI] [PubMed] [Google Scholar]
- 8.Merk D, Steinhilber D, Schubert-Zsilavecz M. Medicinal chemistry of farnesoid X receptor ligands: from agonists and antagonists to modulators. Future Med Chem. 2012;4:1015–1036. doi: 10.4155/fmc.12.47. [DOI] [PubMed] [Google Scholar]
- 9.Sepe V, Festa C, Renga B, Carino A, Cipriani S, Finamore C, Masullo D, Del Gaudio F, Monti MC, Fiorucci S, Zampella A. Insights on FXR selective modulation. Speculation on bile acid chemical space in the discovery of potent and selective agonists. Scientific reports. 2016;6:19008. doi: 10.1038/srep19008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 11.De Marino S, Ummarino R, D'Auria MV, Chini MG, Bifulco G, Renga B, D'Amore C, Fiorucci S, Debitus C, Zampella A. Theonellasterols and conicasterols from Theonella swinhoei. Novel marine natural ligands for human nuclear receptors. J Med Chem. 2011;54:3065–3075. doi: 10.1021/jm200169t. [DOI] [PubMed] [Google Scholar]
- 12.Sepe V, Bifulco G, Renga B, D'Amore C, Fiorucci S, Zampella A. Discovery of sulfated sterols from marine invertebrates as a new class of marine natural antagonists of farnesoid-X-receptor. J Med Chem. 2011;54:1314–1320. doi: 10.1021/jm101336m. [DOI] [PubMed] [Google Scholar]
- 13.Choi H, Hwang H, Chin J, Kim E, Lee J, Nam SJ, Lee BC, Rho BJ, Kang H. Tuberatolides, potent FXR antagonists from the Korean marine tunicate Botryllus tuberatus. J Nat Prod. 2011;74:90–94. doi: 10.1021/np100489u. [DOI] [PubMed] [Google Scholar]
- 14.Di Leva FS, Festa C, D'Amore C, De Marino S, Renga B, D'Auria MV, Novellino E, Limongelli V, Zampella A, Fiorucci S. Binding mechanism of the farnesoid X receptor marine antagonist suvanine reveals a strategy to forestall drug modulation on nuclear receptors. Design, synthesis, and biological evaluation of novel ligands. J Med Chem. 2013;56:4701–4717. doi: 10.1021/jm400419e. [DOI] [PubMed] [Google Scholar]
- 15.Sepe V, Ummarino R, D'Auria MV, Taglialatela-Scafati O, Marino SD, D'Amore C, Renga B, Chini MG, Bifulco G, Nakao Y, Fusetani N, Fiorucci S, Zampella A. Preliminary structure-activity relationship on theonellasterol, a new chemotype of FXR antagonist, from the marine sponge Theonella swinhoei. Mar Drugs. 2012;10:2448–2466. doi: 10.3390/md10112448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin K, Chin J, Hahn D, Lee J, Hwang H, Won DH, Ham J, Choi H, Kang E, Kim H, Ju MK, Nam SJ, Kang H. Sterols from a soft coral, Dendronephthya gigantea as farnesoid X-activated receptor antagonists. Steroids. 2012;77:355–359. doi: 10.1016/j.steroids.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 17.Del Bas JM, Ricketts ML, Vaque M, Sala E, Quesada H, Ardevol A, Salvado MJ, Blay M, Arola L, Moore DD, Pujadas G, Fernandez-Larrea J, Blade C. Dietary procyanidins enhance transcriptional activity of bile acid-activated FXR in vitro and reduce triglyceridemia in vivo in a FXR-dependent manner. Mol Nutr Food Res. 2009;53:805–814. doi: 10.1002/mnfr.200800364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heidker RM, Caiozzi GC, Ricketts ML. Dietary procyanidins selectively modulate intestinal farnesoid X receptor-regulated gene expression to alter enterohepatic bile acid recirculation: elucidation of a novel mechanism to reduce triglyceridemia. Mol Nutr Food Res. 2016;60:727–736. doi: 10.1002/mnfr.201500795. [DOI] [PubMed] [Google Scholar]
- 19.Li G, Lin W, Araya JJ, Chen T, Timmermann BN, Guo GL. A tea catechin, epigallocatechin-3-gallate, is a unique modulator of the farnesoid X receptor. Toxicol Appl Pharmacol. 2012;258:268–274. doi: 10.1016/j.taap.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nozawa H. Xanthohumol, the chalcone from beer hops (Humulus lupulus L.), is the ligand for farnesoid X receptor and ameliorates lipid and glucose metabolism in KK-A(y) mice. Biochem Biophys Res Commun. 2005;336:754–761. doi: 10.1016/j.bbrc.2005.08.159. [DOI] [PubMed] [Google Scholar]
- 21.Campbell BSY, Zhen J, Fantacone ML, Esfandiari E, Yang L, Maier CS, Stevens JF, Gombart AF. Induction of human cathelicidin antimicrobial peptide gene expression by xanthohumol via the farnesoid X receptor. Molecular Nutrition and Food Research. 2016 under revision. [Google Scholar]
- 22.Tabata N, Ito M, Tomoda H, Omura S. Xanthohumols, diacylglycerol acyltransferase inhibitors, from Humulus lupulus. Phytochemistry. 1997;46:683–687. doi: 10.1016/s0031-9422(97)00157-x. [DOI] [PubMed] [Google Scholar]
- 23.Casaschi A, Maiyoh GK, Rubio BK, Li RW, Adeli K, Theriault AG. The chalcone xanthohumol inhibits triglyceride and apolipoprotein B secretion in HepG2 cells. J Nutr. 2004;134:1340–1346. doi: 10.1093/jn/134.6.1340. [DOI] [PubMed] [Google Scholar]
- 24.Nozawa H. Xanthohumol, the chalcone from beer hops (Humulus lupulus L.), is the ligand for farnesoid X receptor and ameliorates lipid and glucose metabolism in KK-A(y) mice. Biochem Biophys Res Commun. 2005;336:754–761. doi: 10.1016/j.bbrc.2005.08.159. [DOI] [PubMed] [Google Scholar]
- 25.Yang JY, Della-Fera MA, Rayalam S, Baile CA. Effect of xanthohumol and isoxanthohumol on 3T3-L1 cell apoptosis and adipogenesis. Apoptosis. 2007;12:1953–1963. doi: 10.1007/s10495-007-0130-4. [DOI] [PubMed] [Google Scholar]
- 26.Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert review of proteomics. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass spectrometry reviews. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 28.Balasubramaniam D, Komives EA. Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochimica et biophysica acta. 2013;1834:1202–1209. doi: 10.1016/j.bbapap.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan X, Maier CS. Hydrogen/deuterium exchange mass spectrometry. Methods Mol Biol. 2009;492:255–271. doi: 10.1007/978-1-59745-493-3_15. [DOI] [PubMed] [Google Scholar]
- 30.Silva D, Cortez CM, Cunha–Bastos J, Louro SR. Methyl parathion interaction with human and bovine serum albumin. Toxicol Lett. 2004;147:53–61. doi: 10.1016/j.toxlet.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Chao PW, Chow CS. Monitoring aminoglycoside-induced conformational changes in 16S rRNA through acrylamide quenching. Bioorg Med Chem. 2007;15:3825–3831. doi: 10.1016/j.bmc.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakkialakshmi S, Chandrakala D. A spectroscopic investigations of anticancer drugs binding to bovine serum albumin. Spectrochim Acta A Mol Biomol Spectrosc. 2012;88:2–9. doi: 10.1016/j.saa.2011.10.076. [DOI] [PubMed] [Google Scholar]
- 33.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer Science+Business Media, LLC; Boston, MA: 2006. SpringerLink (Online service) [Google Scholar]
- 34.Yang L, Broderick D, Jiang Y, Hsu V, Maier CS. Conformational dynamics of human FXR-LBD ligand interactions studied by hydrogen/deuterium exchange mass spectrometry: insights into the antagonism of the hypolipidemic agent Z-guggulsterone. Biochim Biophys Acta. 2014;1844:1684–1693. doi: 10.1016/j.bbapap.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 36.Sperry JB, Huang RY, Zhu MM, Rempel DL, Gross ML. Hydrophobic Peptides Affect Binding of Calmodulin and Ca as Explored by H/D Amide Exchange and Mass Spectrometry. Int J Mass Spectrom. 2011;302:85–92. doi: 10.1016/j.ijms.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker CH, Morgan CR, Rand KD, Engen JR, Jorgenson JW, Stafford DW. A Conformational Investigation of Propeptide Binding to the Integral Membrane Protein gamma-Glutamyl Carboxylase Using Nanodisc Hydrogen Exchange Mass Spectrometry. Biochemistry. 2014;53:1511–1520. doi: 10.1021/bi401536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang RY, Wen J, Blankenship RE, Gross ML. Hydrogen-deuterium exchange mass spectrometry reveals the interaction of Fenna-Matthews-Olson protein and chlorosome CsmA protein. Biochemistry. 2012;51:187–193. doi: 10.1021/bi201620y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shortridge MD, Hage DS, Harbison GS, Powers R. Estimating protein-ligand binding affinity using high-throughput screening by NMR. J Comb Chem. 2008;10:948–958. doi: 10.1021/cc800122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G, Iacob RE, Engen JR. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug discovery today. 2014;19:95–102. doi: 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 42.Mi LZ, Devarakonda S, Harp JM, Han Q, Pellicciari R, Willson TM, Khorasanizadeh S, Rastinejad F. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell. 2003;11:1093–1100. doi: 10.1016/s1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 43.Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, Liu Y, Gavrilova O, Patterson AD, Gonzalez FJ. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun. 2015;6:10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perkins A, Phillips JL, Kerkvliet NI, Tanguay RL, Perdew GH, Kolluri SK, Bisson WH. A Structural Switch between Agonist and Antagonist Bound Conformations for a Ligand-Optimized Model of the Human Aryl Hydrocarbon Receptor Ligand Binding Domain. Biology. 2014;3:645–669. doi: 10.3390/biology3040645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia RA, Pantazatos D, Villarreal FJ. Hydrogen/deuterium exchange mass spectrometry for investigating protein-ligand interactions. Assay and drug development technologies. 2004;2:81–91. doi: 10.1089/154065804322966342. [DOI] [PubMed] [Google Scholar]
- 46.Acharya KR. the advantages and limitations of protein crystal structure. Trends Pharmacol Sci. 2005;26:10–14. doi: 10.1016/j.tips.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 47.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Garcia-Ordonez RD, Pascal BD, Burris TP, Dodge JA, Griffin PR. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure. 2010;18:1332–1341. doi: 10.1016/j.str.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, Griffin PR. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 50.Han KC, Kim JH, Kim KH, Kim EE, Seo JH, Yang EG. Identification of farnesoid X receptor modulators by a fluorescence polarization-based interaction assay. Anal Biochem. 2010;398:185–190. doi: 10.1016/j.ab.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 51.Moreno F, Gonzalez-Jimenez J. Binding of the Promen fluorescent probe to human serum albumin: a fluorescence spectroscopic study. Chemico-biological interactions. 1999;121:237–252. doi: 10.1016/s0009-2797(99)00111-8. [DOI] [PubMed] [Google Scholar]
- 52.Gasymov OK, Abduragimov AR, Glasgow BJ. A simple model-free method for direct assessment of fluorescent ligand binding by linear spectral summation. Journal of fluorescence. 2014;24:231–238. doi: 10.1007/s10895-013-1290-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehrer SS. Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 54.Wang Z, Tan X, Chen D, Yue Q, Song Z. Study on the binding behavior of lysozyme with cephalosporin analogues by fluorescence spectroscopy. Journal of fluorescence. 2009;19:801–808. doi: 10.1007/s10895-009-0477-8. [DOI] [PubMed] [Google Scholar]
- 55.Kandagal PB, Ashoka S, Seetharamappa J, Shaikh SM, Jadegoud Y, Ijare OB. Study of the interaction of an anticancer drug with human and bovine serum albumin: spectroscopic approach. J Pharm Biomed Anal. 2006;41:393–399. doi: 10.1016/j.jpba.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 56.Ding F, Liu W, Liu F, Li ZY, Sun Y. A study of the interaction between malachite green and lysozyme by steady-state fluorescence. Journal of fluorescence. 2009;19:783–791. doi: 10.1007/s10895-009-0475-x. [DOI] [PubMed] [Google Scholar]
- 57.Papadopoulou A, Green RJ, Frazier RA. Interaction of flavonoids with bovine serum albumin: a fluorescence quenching study. J Agric Food Chem. 2005;53:158–163. doi: 10.1021/jf048693g. [DOI] [PubMed] [Google Scholar]
- 58.Rasoulzadeh F, Jabary HN, Naseri A, Rashidi MR. Fluorescence quenching study of quercetin interaction with bovine milk xanthine oxidase. Spectrochim Acta A Mol Biomol Spectrosc. 2009;72:190–193. doi: 10.1016/j.saa.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 59.Cui FL, Fan J, Li JP, Hu ZD. Interactions between 1-benzoyl-4-p-chlorophenyl thiosemicarbazide and serum albumin: investigation by fluorescence spectroscopy. Bioorg Med Chem. 2004;12:151–157. doi: 10.1016/j.bmc.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 60.Lakowicz JR, Weber G. Quenching of fluorescence by oxygen. A probe for structural fluctuations in macromolecules. Biochemistry. 1973;12:4161–4170. doi: 10.1021/bi00745a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burris TP, Montrose C, Houck KA, Osborne HE, Bocchinfuso WP, Yaden BC, Cheng CC, Zink RW, Barr RJ, Hepler CD, Krishnan V, Bullock HA, Burris LL, Galvin RJ, Bramlett K, Stayrook KR. The hypolipidemic natural product guggulsterone is a promiscuous steroid receptor ligand. Mol Pharmacol. 2005;67:948–954. doi: 10.1124/mol.104.007054. [DOI] [PubMed] [Google Scholar]
- 62.Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nuclear receptor signaling. 2010;8:e005. doi: 10.1621/nrs.08005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaldas MI, Walle UK, van der Woude H, McMillan JM, Walle T. Covalent binding of the flavonoid quercetin to human serum albumin. J Agric Food Chem. 2005;53:4194–4197. doi: 10.1021/jf050061m. [DOI] [PubMed] [Google Scholar]
- 64.Hsu CW, Zhao J, Huang R, Hsieh JH, Hamm J, Chang X, Houck K, Xia M. Quantitative high-throughput profiling of environmental chemicals and drugs that modulate farnesoid X receptor. Scientific reports. 2014;4:6437. doi: 10.1038/srep06437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Del Bas Josep Maria, Ricketts Marie-Louise, Vaqué Montserrat, Sala Esther, Quesada Helena, Ardevol Anna, Josepa Salvadó M, Blay Mayte, Arola Lluís, Moore David D, Pujadas Gerard, JF-L, Bladé C. Dietary procyanidins enhance transcriptional activity of bile acid-activated FXR in vitro and reduce triglyceridemia in vivo in a FXR-dependent manner. Mol Nutr Food Res. 2009;53:805–814. doi: 10.1002/mnfr.200800364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li G, Lin W, Araya JJ, Chen T, Timmermann BN, Guo GL. A tea catechin, epigallocatechin-3-gallate, is a unique modulator of the farnesoid X receptor. Toxicol Appl Pharmacol. 2012;258:268–274. doi: 10.1016/j.taap.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.