

Abstract
Background
In order to perform selective C-H functionalization upon visible light irradiation, Ru(II)-diimine functionalized P450 heme enzymes have been developed. The sL407C-1 enzyme containing the Ru(bpy)2PhenA (bpy = 2,2′-bipyridine and PhenA = 5-acetamido-1,10-phenanthroline) photosensitizer (1) covalently attached to the non-native single cysteine L407C of the P450BM3 heme domain mutant, displays high photocatalytic activity in the selective C-H bond hydroxylation of several substrates.
Methods
A combination of X-ray crystallography, site-directed mutagenesis, transient absorption measurements and enzymatic assays was used to gain insights into its photocatalytic activity and electron transfer pathway.
Results
The crystal structure of the sL407C-1 enzyme was solved in the open and closed conformations revealing a through-space electron transfer pathway involving highly conserved, F393 and Q403, residues. Several mutations of these residues (F393A, F393W or Q403W) were introduced to probe their roles in the overall reaction. Transient absorption measurements confirm rapid electron transfer as heme reduction is observed in all four hybrid enzymes. Compared to the parent sL407C-1, photocatalytic activity was negligible in the dF393A-1 enzyme while 60% increase in activity with total turnover numbers of 420 and 90% product conversion was observed with the dQ403W-1 mutant.
Conclusions
In the sL407C-1 enzyme, the photosensitizer is ideally located to rapidly deliver electrons, using the naturally occurring electron transfer pathway, to the heme center in order to activate molecular dioxygen and sustain photocatalytic activity.
General Significance
The results shed light on the design of efficient light-driven biocatalysts and the approach can be generalized to other members of the P450 superfamily.
Keywords: cytochrome P450, electron transfer, hybrid P450 BM3 enzymes, crystal structure, enzyme catalysis, photocatalytic activity
Graphical abstract
1. Introduction
The superfamily of cytochrome P450 heme thiolate enzymes has received considerable attention due to their biological importance and the chemical reactions that they carry out in the regio- and stereoselective functionalization of unactivated C-H bonds.[1–3] These enzymes use two reducing equivalents to activate molecular dioxygen at the heme center and produce a high-valent Fe(IV)-oxo porphyrin radical species responsible for the C-H bond functionalization via a rebound mechanism.[4] In most P450 enzymes, the mechanism is initiated by substrate binding, which displaces the water bound to the ferric resting state resulting in a low-to-high spin conversion and a higher reduction potential to facilitate the first electron injection.[1] The necessary electrons are supplied in successive steps and in a timely manner by the redox partners, reductases, in order to achieve efficient oxygen activation and minimize the formation of reactive oxygen species from uncoupling.
In order to circumvent the use of redox partners and perform P450 reactions upon visible light excitation, our laboratory has developed hybrid P450 enzymes containing a Ru(II)-diimine photosensitizer covalently attached to the heme domain of various P450 enzymes.[5, 6] This approach stems from the extensive use of these photosensitizers in long range electron transfer studies in metalloproteins and their applications in the light driven activation of small molecules with several enzymes.[7] In the hybrid P450 enzyme, the photosensitizer has been designed to deliver electrons to the heme domain upon visible light irradiation and sustain photocatalytic activity without the need for reductase and cofactor. The covalent attachment of the photosensitizer enables intramolecular electron transfers and provides control over its position relative to the heme center. The sL407C-1 hybrid enzyme, containing the Ru(bpy)2PhenA photosensitizer, 1, (bpy = 2,2′-bipyridine, PhenA = 5-acetamido-1,10-phenanthroline) attached to the non-native single cysteine residue (L407C) in the P450 BM3 heme domain, showed high photocatalytic activity in the selective hydroxylation of various substrates.[8, 9] The high total turnover numbers and initial reaction rates suggested efficient coupling between electron delivery and oxygen activation at the heme center. We have then employed a combination of X-ray crystallography, site-directed mutagenesis, transient absorption measurements and enzymatic assays to gain some insights into the electron transfer pathway and the photocatalytic activity. The sL407C-1 enzyme was crystallized following reported conditions and the structures were solved in the open and closed conformations. The closed conformation contains a tightly bound substrate, N-palmitoylglycine (NPG) in the active site channel. In both conformations, the tertiary protein fold is not affected by the presence of the photosensitizer and the only noticeable contacts are C-H---π interactions between the ancillary ligands on the photosensitizer and several protein residues. A through-space electron transfer pathway was identified involving the F393 and Q403 residues, highly conserved among the P450 superfamily. These residues are known to participate in the natural electron transfer pathway in the P450 BM3 holoenzyme.[10] In order to probe individually the role of these residues, we introduced additional single point mutations in the sL407C-1 hybrid enzyme. Accordingly, we generated three doubly mutated hybrid enzymes, dF393A-1, dF393W-1 and dQ403W-1, containing the photosensitizer 1 attached to the L407C residue with the additional F393A, F393W or Q403W mutations, respectively. The F393A and F393W mutations in the holoenzyme were shown to alter the heme redox potential, electron transfer rate and stability of the oxy-ferrous adduct.[11–14] The novel Q403W mutation was sought to enhance the electronic communication between the photosensitizer and the heme via the staggering of multiple aromatic rings. We determine the kinetics of the first electron transfer using transient absorption measurements as heme reduction is observed in the four hybrid enzymes. A colorimetric assay compatible with the light-driven process was used to assess the photocatalytic activity of the double mutants relative to the sL407C-1.[15] Photocatalytic activity is negligible in the dF393A-1 enzyme while a 60% increase in activity with total turnover numbers of 420 and 90% product conversion is observed in the dQ403W-1 enzyme.
2. Materials and methods
2.1. Mutagenesis, Expression, and Purification of hybrid P450 BM3 mutants
The mutants of the P450 BM3 heme domain were generated as described previously.[15] The following primers were used to create the desired mutations: F393A 5′-GCGTTTAAACCGGCTGGAAACGGTCAG-3′; F393W 5′-GCGTTTAAACCGTGGGGAAACGGTC AG-3′; Q403W 5′-CGTGTATCGGTTGGCAGTTCGCTTGC-3′. The underlined regions indicate the section of the oligonucleotides modified from the sL407C sequence to produce the codon change required for the desired amino acid substitutions. The proteins were then expressed, purified and labeled at the L407C position with the Ru(bpy)2PhenA (1) photosensitizer to yield the sL407C-1 and the doubly mutated dF393A-1, dF393W-1, dQ403W-1 hybrid enzymes. An additional purification step by fast protein liquid chromatography using a Superose 12 column produced homogeneous sL407C-1 enzyme for crystallographic studies.
2.2. Crystallization
The hybrid P450 BM3 enzyme, sL407C-1, was crystallized by the sitting drop method at 4 °C using reported procedures.[16] Sitting drops were prepared by adding 2 μL of mother liquor to 2 μL of 15 mg/ml enzyme. N-palmitoylglycine (NPG) was used as preferred substrate for crystallization.[17] The NPG-bound crystals were obtained by crystallizing purified substrate bound enzyme while the open conformation structure with a DMSO bound to the heme resulted from cocrystallization experiments. High quality crystals were obtained by microseeding techniques. Crystals were formed in both cases after 2 days. Crystals were immersed in cryoprotectant (glycerol), before being mounted on a nylon loop and flash-cooled in liquid nitrogen.
2.3. Data Collection, Structure Elucidation, and Refinement
The data used for refinement were collected at the Swiss Light Source (SLS). Crystals were cooled at 100 K, and diffraction data were collected in oscillations of 0.2°. Data processing were carried out with the program XDS.[18] Both structures were solved via molecular replacement. The crystal structures, PDB ID: 1JPZ[17] and 3NPL[19], were used as the starting model for the DMSO- and NPG-bound structures, respectively. Data collection and final refinement statistics are given in Table 1. The atomic coordinates and structure factors for both crystal structures have been deposited in the Protein Data Bank with codes 5JTD and 5JQ2 for the DMSO and NPG-bound structures, respectively.
Table 1.
Statistics of data collection, refinement and validation
| DMSO bound | NPG bound | |
|---|---|---|
| Data collectiona | ||
| Space group | P 21 | P 21 21 21 |
| Unit-cell parameters (Å, °) | a = 58.7, b= 145.4, c = 62.9 β = 97.1 |
a = 59.9, b= 112.5, c = 156.4 |
| Radiation source | PXII-X10SA, SLS | PXII-X10SA, SLS |
| Wavelength (Å) | 1.0 | 1.0 |
| Temperature (K) | 100 | 100 |
| Resolution range (Å) | 47.36 - 1.4 (1.5-1.4) | 47.54 – 1.9 (2.0-1.9) |
| No. of observed reflections | 613594 (63761) | 418751 (60114) |
| No. of unique reflections | 183724 (22601) | 83423 (11822) |
| Multiplicity | 3.3 (2.8) | 5.0 (5.0) |
| Wilson B factor (Å2) | 17.3 | 24.7 |
| I/σ (I) | 13.2 (1.3) | 10.5 (1.4) |
| Rmerge (%) | 4.9 (92.6) | 12.9 (110.4) |
| CC (1/2) (%) | 99.9 (48.5) | 99.7 (57.3) |
|
| ||
| Refinement | ||
| Resolution range used for refinement | 47.36 - 1.50 | 47.54 - 2.0 |
| No. of reflections | 153091 | 68037 |
| Observed (%) | 96.7 | 99.1 |
| Reflections in test set (%) | 5.0 | 5.0 |
| R work/R free (%) | 17.3/ 19.8 | 17.4/ 21.8 |
| Reflections in test set (%) | 5.0 | 5.0 |
| RMSD bond/angles (Å))/(°) | 0.011/ 1.6 | 0.015/ 1.8 |
| Average B factor (Å2) | 23 | 29.0 |
|
| ||
| Ramachandran plot | ||
| Favored (%) | 97.04 | 96.17 |
| Additional allowed (%) | 2.49 | 3.72 |
| Number of outliers | 1 | 1 |
| PDB accession code | 5JTD | 5JQ2 |
Values in parentheses are for the highest resolution shell.
2.4. Transient Absorption measurements
All measurements were conducted at the Beckman Institute Laser Resources Center at Caltech. A degassed solution of 10 μM of each hybrid enzyme in Tris buffer (25 mM, pH = 8.2) containing 100 mM diethyldithiocarbamate was exposed to laser excitation (3mJ/pulse) as described elsewhere.[20] For the substrate bound experiments, 15 μL of a N-palmitoylglycine solution (1 mM) in DMSO was added to the protein solution prior to degassing. UV-vis spectra were collected before and after degassing to ensure complete substrate binding. Two sets of signal amplifiers were used to cover the range from 10 ns to 0.1 s. Kinetics traces were collected from 390 to 440 nm, every 10 nm. Single wavelength kinetics traces were fitted to a sum of exponential functions as described elsewhere[19] to produce the observed intramolecular electron transfer rate constants summarized in Table 2 and the lines of best fit in black (Fig. 2 and S2).
Table 2.
Kinetic data for the four hybrid enzymes
| dF393A-1 | dF393W-1 | sL407C-1 | dQ403W-1 | |
|---|---|---|---|---|
| reduction rate (kET) | ||||
| SFb | n.d.a | 1.20x105 ± 0.06 s−1 | 1.49x105 ± 0.02 s−1 | 2.02x105 ± 0.01s−1 |
| SB | 1.04x105 ± 0.02 s−1 | 6.25x104 ± 0.01 s−1 | 7.32x104 ± 0.04 s−1 | 1.45x105 ± 0.02s−1 |
| kcat (eq.min−1), | < 2 | 21 | 36c | 44 |
| Km (μM) | 11.3 | 5.0 | 11.0c | 9.5 |
| TTNd | <5 | 192 | 230 | 420 |
could not be determined as the SF-dF393A-1 was unstable under the laser experiments leading to the SB-F393A-1 species;
SF: Substrate free and N-palmitoylglycine bound (SB);
value determined previously[15]
TTN: total turnover numbers as nmol of products / nmol of enzymes at the end of the reaction.
Fig. 2.

Transient absorption traces for the dQ403W-1 recorded from 390 to 440 nm and spanning 10 ns to 0.1 s showing the formation of the reduced heme species noticeable at 390 and 400 nm in the substrate free conformation (A) and at 410–440 nm in the NPG bound form (B) with the lines of best fit in black. Panels C and D show the difference spectra for the actual ΔOD data points at 100 μs versus the theoretical difference spectra generated from original Fe(II) and Fe(III) spectra for the low spin substrate free (C) and high spin NPG bound (D).
2.5. Photocatalytic reactions
For the photocatalytic reactions, a solution of 1 μM hybrid enzyme in Tris buffer (25 mM, pH = 8.2) containing 100 mM diethyldithiocarbamate and 375 μM of the chromogenic substrate, p-nitrophenoxyhexadecanoic acid (16-pNCA), was irradiated for 90 min. The reaction was then quenched by the addition of 10 μL of 6 M NaOH and the absorbance of the product was read at 405 nm using a molar absorbance coefficient of 13,200 M−1cm−1. The enzyme kinetics parameters were determined as previously reported.[15]
3. Results
3.1. Crystal Structures of the open and closed conformations
High quality protein crystals were obtained using reported conditions and micro seeding techniques.[17] The open conformation structure was solved at 1.5 Å resolution and contained two molecules in the asymmetric unit, as did the substrate bound structure, which was solved at 2.0 Å (See Table 1). N-palmitoylglycine (NPG) was used as substrate due to its tight binding to the heme domain. It has also been found in the substrate channel of several P450 BM3 heme domain structures.[17, 21, 22] The two molecules in the asymmetric unit of the open conformation structure contain a DMSO molecule bound to the ferric ion via the sulfur atom with a d(Fe-S) distance of 2.35 Å. The structures are identical to the Hi-DMSO[23] (PDB ID: 2J1M) with rmsd of 0.16 Å using the structurally invariant residues described by Haines et al.[17]
For the NPG bound structures, the most noticeable difference between the two molecules in the asymmetric unit is the mode of binding of the substrate. In molecule A, the NPG conformation is identical to the one observed in the 1JPZ structure with rmsd of 0.18 Å.[17] The carboxylate group of the NPG in chain A is hydrogen bonded to the amide backbones of Q73 and A74 with Van der Waals contact of 2.8 Å, and the amide carbonyl group is hydrogen bonded to Y51 with a close contact of 2.6 Å. The molecule B, however, matches the 4KPA structure (rmsd of 0.21 Å) where the NPG molecule forms a strong hydrogen bond with the R47 residue as noted previously.[21] In all four molecules, the photosensitizer could be identified in the difference map and modeled as covalently attached to the L407C residue. The distance between the two metal centers, d(Ru-Fe) is 19 Å (See Fig. 1).
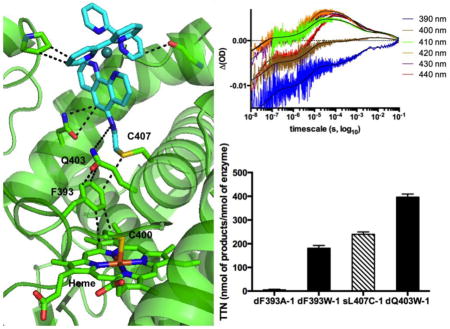
Fig. 1.

A) Surface representation of the sL407C-1 hybrid enzyme with contour map at 1.6 Å around the photosensitizer; B) Superposition of the DMSO (green) and N-palmitoylglycine bound (blue) conformation showing the photosensitizer in stick representation; C) Close-up of the interactions between the photosensitizer (blue) and several protein residues.
Close examination of the photosensitizer environment reveals strong interactions involving aromatic C-H bonds on the photosensitizer with several protein residues (See Fig. 1C). The methylene groups of the proline 382 side chain form edge-to-face interactions with one of the bipyridine rings of the photosensitizer with a distance of 3.8 Å. Interestingly, motion of the 310-helix containing the P382 is noticed in several molecules with up to 2.1 Å motion while still maintaining the C-H---π interactions (See Fig. S1). The second bipyridine ring of the photosensitizer is also involved in a strong C-H---π interaction with the carbonyl backbone of the K309 residue with a short distance of 2.9 Å. The remaining ancillary phenanthroline ligand is involved in a bifurcated interaction involving the N319 side chain and a C-H bond on the phenyl ring with distances of 3.2 and 3.6 Å, respectively (See Fig. 1C).
The sulfur of the non-native single cysteine at the 407 position is 4.1 Å away from the F393 phenyl ring and the photosensitizer amide linkage is 6 Å from the Q403 residue (See Fig. 1C). Subsequently, the Q403 residue is involved in a strong face-to-face interaction with the F393, having only 3.4 Å between the carbonyl carbon of the Q403 residue and the phenyl ring of the 393 residue. The F393 residue participates in an edge-to-face C-H---π interaction with the pyrrole ring of the heme, with a distance of 3.8 Å (See Fig 1C).
3.2. Electron transfer studies
We used transient absorption measurements to monitor, under flash quench reductive conditions, the heme reduction of the low-spin (LS) and NPG bound high-spin (HS) ferric heme species in the four hybrid enzymes (sL407C-1, dF393A-1, dF393W-1, dQ403W-1). The typical transient absorption traces are shown in Fig. 2 and S2. Traces were recorded from 390 to 440 nm every 10 nm and span from 10 ns to 0.1 s. A highly reductive [Ru]+ species is photogenerated within the first 100 ns from the biomolecular quenching of the Ru(II)* excited state with diethyldithiocarbamate (DTC) according to Scheme 1. For the substrate free (SF, Fig. 2A), a new feature is observed within 100 μs as noted in the 390 and 400 nm traces with concomitant signal bleach in the 410 and 420 nm traces. For the NPG bound (Fig. 2B), a new feature is observed in the 410–440 nm traces while the signal disappearance is mainly detected in the 390 nm trace.
Scheme 1.
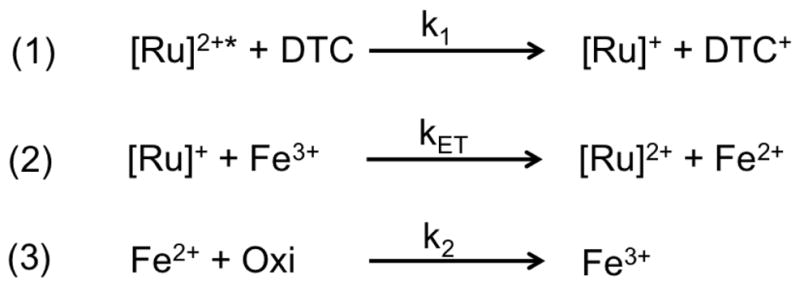
Proposed scheme for the reduction of the ferric species by the photogenerated [Ru]+ in the hybrid enzymes. Oxidizing species, Oxi, such as [DTC]+ or dioxygen traces are involved in the back electron transfer (3).
Transient absorption data points taken at 100 μs for the six different wavelengths were plotted against the theoretical difference spectra generated from the actual ferric and ferrous spectra (Fig 2C.,D. and Fig. S2–S3). The spectrum for the reduced Fe(II) heme was generated spectroelectrochemically following reported procedures and is identical for both species (See Fig. S3).[24] This unambiguously confirms that heme reduction was achieved in all four mutants for both low and high spin species. Additionally, the traces were fitted to a set of differential equations[19, 20] to extract the intramolecular rate constants for heme reduction (kET in Scheme 1) and their values are summarized in Table 2. The lines of best fit are shown in black (Fig. 2 and S2).
3.3. Photocatalytic activity of the four mutants
We used a colorimetric assay compatible with the light-driven conditions[15] to determine the Michaelis-Menten parameters and total turnover numbers (TTN) at the end of the reaction for the four hybrid enzymes in the hydroxylation of the long chain fatty acid analog, 16-pNCA, upon visible light-irradiation. The data are summarized in Table 2 and Figure S4. The dF393A-1 mutant showed minimal photocatalytic activity, while the dF393W-1 shows a kcat of 21 eq/min and TTN of 192, a 20% decrease in activity compared to the values determined for the sL407C-1.[15] The kcat for the dQ403W-1 is 42 eq/min and the TTN values are 60% higher (420) than those observed for the sL407C-1.
4. Discussion
The consensus P450 mechanism involves two successive one-electron transfer (ET) steps from the electron providing reductase to the heme center and the formation of several well-characterized intermediates.[25] The first electron transfer gated by substrate binding is thought to be rate determining in most P450 enzymes, however, other steps such as the second electron transfer step, dioxygen binding or product release can then become the rate limiting step.[10] In addition, the sequential delivery of electrons needs to be well orchestrated in order to achieve high catalytic efficiency and minimize the formation of reactive oxygen species via uncoupled pathways.
In order to circumvent the need for the reductase and cofactor, several alternative approaches to deliver electrons and activate P450 heme domains have emerged. These include the use of fusion P450 proteins,[26] surrogate oxygen atom donors[27, 28], direct chemical[29, 30] and electrochemical reductions[31] as well as light-activated approaches[32–36]. The main challenges still reside in the timely delivery of the electrons to achieve high activity and limit the unproductive pathways [37, 38]. The light-driven approach used in the hybrid P450 enzymes[5, 6, 8, 9, 15] is taking advantages of the photochemical properties of Ru(II)-diimine photosensitizers and the extensive electron transfer studies in metalloproteins.[7]
Despite the relevance of P450 enzymes and their importance for biotechnological applications, structural information on cytochrome P450 heme domains interacting with their redox partners remains scarce. To date, only three crystal structures have been solved with the heme domain in close contact with its redox partner [39–41]. An early structure from the Poulos group shows a proteolytically cleaved FMN domain, one component of the reductase, interacting with the P450 BM3 heme domain.[40] Recent molecular dynamics (MD) simulations using the crystal structure as starting point identified putative electron transfer pathways from the FMN cofactor to the heme.[42, 43]
Crystal structures of the hybrid enzymes in the open and closed conformations were solved at 1.5 and 2.0 Å resolution, respectively. This expands the crystallographically characterized structures of Ru(II)-diimine modified P450 BM3 enzyme[19] and metalloproteins.[44, 45] The X-ray structures confirm the covalent attachment of the Ru(II) photosensitizer to the non-native L407C residue as previously established using various techniques.[7] The photosensitizer resides in a bowl-shaped cavity on the proximal side of the heme domain (See Fig. 1A) where the reductase is binding. The structures reveal minimal perturbation of the heme domain by the presence of the photosensitizer as both structures are identical to previously reported crystal structures of the wild type heme domain. The only noticeable contacts involve the aromatic rings of the photosensitizer in strong C-H---π interactions with several protein residues especially the side chain of the proline 382. There is mounting evidence that C-H---π interactions involving either aliphatic or aromatic C-H bonds play a determining role in stabilizing tertiary structures and folding of proteins and in molecular recognition.[46]
In the current hybrid enzyme structures, the photosensitizer is found in the same location as the FMN cofactor determined from MD calculations.[40, 43] The d(Fe-Ru) distance is 19 Å well within the range for electron transfer tunneling [44] and is 5 Å shorter than in the tK97C-1 structure previously reported.[19] To support the fast electron transfer rate constants (Table 2), a through-space pathway is preferred rather than a through-bond pathway via the protein backbone (C407 to C400). Based on the close distances highlighted in Fig. 1C, the through-space pathway originating at the phenanthroline ring of the photosensitizer involves the pair of Q403/F393 residues en route to the porphyrin edge. While the F393 residue is highly conserved among the P450 superfamily and has been the subject of several studies, [11–14] the Q403 residue has not been investigated much despite the fact that is in very close contact (3.4 Å) with the F393 residue. It is worth noting that the 403 residue is also highly conserved among P450 heme domains, especially human P450 enzymes, with the occurrence is either Q or E at this position in more than two thirds of the sequences (See Fig. S5).
In order to probe the role of the F393 and Q403 residues, we generated three additional mutants of the sL407C-1 hybrid enzyme and studied their electron transfer properties and photocatalytic activity. These new hybrid enzymes include the doubly mutated dF393A-1, dF393W-1 and dQ403W-1. In the holoenzyme, the F393A and F393W mutations were shown to alter the heme redox potential as well as the stability of the oxy-ferrous intermediate.[11–14] Introduction of the tryptophan residue at the 403 position was sought to enhance the electronic communication between the photosensitizer and the heme via the staggering of multiple aromatic rings. Pecht and coworkers showed in the extensively studied electron transfer protein azurin that introduction of the V31W mutation in close contact with the native W48 resulted in an electron transfer rate increase [47]. It was proposed that the aromatic residues in successive positions could act as an extended relay enhancing the electronic coupling between the electron donor and acceptor.
The electron transfer kinetics and photocatalytic activity were determined for the four hybrid enzymes and the data are summarized in Table 2. The transient absorption measurements confirm rapid electron transfer from the photosensitizer upon visible light excitation as heme reduction is observed in all hybrid enzyme mutants within 100 μs after the laser pulse. Under flash quench reductive conditions according to Scheme 1, a highly reductive [Ru]+ species, with an estimated reduction potential of −1.28 V, is photogenerated within 100 ns (See Fig. 2). Successively, new spectral features are observed within 100 μs as shown in Fig. 2A and B. The generated difference spectra from Figure 2C and D unambiguously confirm that the [Ru]+ species is able to rapidly reduce the substrate free and substrate bound species. The rate constants for heme reduction range from 5x104 to 2x105 s−1 with lower values for the substrate bound high spin species compared to the substrate free low spin species. These values are in good agreement with the ones predicted from the semi-classical Marcus equation. [48] For example, the rate constant for the reduction of the LS heme ferric species (E° = −427 mV vs SHE) [14] by the photogenerated [Ru]+ species with an estimated potential of −1.28 V vs NHE is predicted to be 2.2x105 s−1 considering the 19 Å distance between the two redox centers, a reorganization energy of 0.8 eV and a distance decay constant (β) of 1.1 Å−1 [44, 45]. The heme reduction rate constants in the hybrid enzymes are three orders of magnitude higher than those observed for the holoenzyme, typically ranging around 200 s−1.[14] The high driving force of the photogenerated [Ru]+ enables the rapid reduction of the starting LS ferric species and thus potentially alleviating the gating system of substrate binding towards the functionalization of non-natural substrates with minimal low-to-high spin conversion. There are only few variants of the P450 BM3 holoenzyme that are able to reduce the low-spin ferric spin with the same rate constant as the high-spin ferric species[10]. Such variants as KT2[49] or I401P[50] show high catalytic activity towards a wide range of non-natural substrates and display first electron transfer rate constant at least an order of magnitude higher than the turnover rate.
The photocatalytic activity of the doubly mutated enzymes was compared to the sL407C-1 using a colorimetric assay compatible with the light-driven reaction conditions. As noted previously with P450 BM3 holoenzyme variants,[10] the differences in kcat among the four hybrid enzymes (Table 2) correlate with the differences in the first electron transfer rate constants, except for the dF393A-1 mutant. Negligible photocatalytic activity was observed despite the photosensitizer still able to deliver electrons to the heme domain (Table 2, Fig. S2). The lack of photocatalytic activity reflects high level of uncoupling, most likely in the second electron delivery as proposed for the holoenzyme mutant.[14] Meanwhile, introduction of the non-native Trp in the dQ403W-1 mutant resulted in a 60% increase in photocatalytic activity due to a better electronic coupling between the photosensitizer and the heme domain as reflected in the electron transfer rate constants (Table 2).
5. Conclusion
The crystallographic data reveals that the photosensitizer is found in the same location as the reductase FMN cofactor and at a distance of 19 Å to the heme. In addition, the presence of the Ru(II)-diimine photosensitizer does not alter the protein fold as minimal interactions with the heme domain are noticed. The proposed through space electron transfer pathway in the hybrid enzyme involves highly conserved F393 and Q403 residues, which are known to participate in the natural electron transfer pathway. The spectroscopic measurements confirm the ability of the photosensitizer to rapidly deliver electrons to the heme domain and the enzymatic data supports the important role of these residues in the photocatalytic activity of the hybrid enzymes. Altogether, the data indicate that the photosensitizer in the sL407C-1 hybrid enzyme is ideally located to deliver electrons to the heme center using the naturally occurring electron transfer pathway to sustain photocatalytic activity. This approach could then be generalized to other members of the P450 superfamily using the identified electron transfer pathway.
Supplementary Material
Highlights.
Hybrid P450 BM3 enzyme crystal structures in the open and closed conformations.
ET rate constant three orders of magnitude greater than in the holoenzyme
Single point mutation (Q403W) results in 60% increase in photocatalytic activity.
Acknowledgments
L.E.C. gratefully acknowledges financial support by the National Institute of Health through Grant number SC3 GM095415. J.S. would like to thank the NSF RUMBA program at SJSU. D.G. thanks financial support by the National Institute of Health through Grant number GM41049-27. L.E.C. is thankful for the use of facilities at the Beckman Institute Laser Resource Center and to the director Jay R. Winkler for helpful discussion. We thank Prof. Ilme Schlichting for help with the protein crystallization and data collection. Diffraction data were collected on beamline X10SA at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland. We thank the PXII staff for their support in setting up the beamline. M.Kl. gratefully acknowledges support and funding by the Max Planck Society.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 2.Urlacher VB, Girhard M. Cytochrome P450 monooxygenases: an update on perspectives for synthetic application. Trends Biotechnol. 2012;30:26–36. doi: 10.1016/j.tibtech.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Girvan HM, Munro AW. Applications of microbial cytochrome P450 enzymes in biotechnology and synthetic biology. Curr Opin Chem Biol. 2016;31:136–145. doi: 10.1016/j.cbpa.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Rittle J, Green MT. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 5.Tran NH, Huynh N, Chavez G, Nguyen A, Dwaraknath S, Nguyen TA, Nguyen M, Cheruzel L. A series of hybrid P450 BM3 enzymes with different catalytic activity in the light-initiated hydroxylation of lauric acid. J Inorg Biochem. 2012;115:50–56. doi: 10.1016/j.jinorgbio.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato M, Lam Q, Bhandarkar M, Banh T, Heredia J, Cheruzel AUL. Selective C–H bond functionalization with light-driven P450 biocatalysts. C R Chimie. 2016 In press. [Google Scholar]
- 7.Lam Q, Kato M, Cheruzel L. Ru(II)-diimine functionalized metalloproteins: From electron transfer studies to light-driven biocatalysis. Biochim Biophys Acta. 2015 doi: 10.1016/j.bbabio.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato M, Nguyen D, Gonzalez M, Cortez A, Mullen SE, Cheruzel LE. Regio- and stereoselective hydroxylation of 10-undecenoic acid with a light-driven P450 BM3 biocatalyst yielding a valuable synthon for natural product synthesis. Bioorg Med Chem. 2014;22:5687–5691. doi: 10.1016/j.bmc.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran NH, Nguyen D, Dwaraknath S, Mahadevan S, Chavez G, Nguyen A, Dao T, Mullen S, Nguyen TA, Cheruzel LE. An Efficient Light-Driven P450 BM3 Biocatalyst. J Am Chem Soc. 2013;135:14484–14487. doi: 10.1021/ja409337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitehouse CJC, Bell SG, Wong LL. P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Ost TW, Schelvis JP. Phe393 mutants of cytochrome P450 BM3 with modified heme redox potentials have altered heme vinyl and propionate conformations. Biochemistry. 2004;43:1798–1808. doi: 10.1021/bi034920g. [DOI] [PubMed] [Google Scholar]
- 12.Ost TW, Munro AW, Mowat CG, Taylor PR, Pesseguiero A, Fulco AJ, Cho AK, Cheesman MA, Walkinshaw MD, Chapman SK. Structural and spectroscopic analysis of the F393H mutant of flavocytochrome P450 BM3. Biochemistry. 2001;40:13430–13438. doi: 10.1021/bi010717e. [DOI] [PubMed] [Google Scholar]
- 13.Ost TW, Miles CS, Munro AW, Murdoch J, Reid GA, Chapman SK. Phenylalanine 393 exerts thermodynamic control over the heme of flavocytochrome P450 BM3. Biochemistry. 2001;40:13421–13429. doi: 10.1021/bi010716m. [DOI] [PubMed] [Google Scholar]
- 14.Ost TW, Clark J, Mowat CG, Miles CS, Walkinshaw MD, Reid GA, Chapman SK, Daff S. Oxygen activation and electron transfer in flavocytochrome P450 BM3. J Am Chem Soc. 2003;125:15010–15020. doi: 10.1021/ja035731o. [DOI] [PubMed] [Google Scholar]
- 15.Lam Q, Cortez A, Nguyen TT, Kato M, Cheruzel L. Chromogenic nitrophenolate-based substrates for light-driven hybrid P450 BM3 enzyme assay. J Inorg Biochem. 2015 doi: 10.1016/j.jinorgbio.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Clark JP, Miles CS, Mowat CG, Walkinshaw MD, Reid GA, Simon NDA, Chapman SK. The role of Thr268 and Phe393 in cytochrome P450BM3. J Inorg Biochem. 2006;100:1075–1090. doi: 10.1016/j.jinorgbio.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 17.Haines DC, Tomchick DR, Machius M, Peterson JA. Pivotal role of water in the mechanism of P450BM-3. Biochemistry. 2001;40:13456–13465. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- 18.Kabsch W. Xds. Acta Crystallogr D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ener ME, Lee YT, Winkler JR, Gray HB, Cheruzel L. Photooxidation of cytochrome P450-BM3. Proc Natl Acad Sci USA. 2010;107:18783–18786. doi: 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warren JJ, Herrera N, Hill MG, Winkler JR, Gray HB. Electron Flow through Nitrotyrosinate in Pseudomonas aeruginosa Azurin. J Am Chem Soc. 2013;135:11151–11158. doi: 10.1021/ja403734n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Catalano J, Sadre-Bazzaz K, Amodeo GA, Tong L, McDermott A. Structural Evidence: A Single Charged Residue Affects Substrate Binding in Cytochrome P450 BM-3. Biochemistry. 2013;52:6807–6815. doi: 10.1021/bi4000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fasan R, Meharenna YT, Snow CD, Poulos TL, Arnold FH. Evolutionary History of a Specialized P450 Propane Monooxygenase. J Mol Biol. 2008;383:1069–1080. doi: 10.1016/j.jmb.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuper J, Wong TS, Roccatano D, Wilmanns M, Schwaneberg U. Understanding a mechanism of organic cosolvent inactivation in heme monooxygenase P450BM-3. J Am Chem Soc. 2007;129:5786–5788. doi: 10.1021/ja067036x. [DOI] [PubMed] [Google Scholar]
- 24.Daff SN, Chapman SK, Turner KL, Holt RA, Govindaraj S, Poulos TL, Munro AW. Redox control of the catalytic cycle of flavocytochrome P-450 BM3. Biochemistry. 1997;36:13816–13823. doi: 10.1021/bi971085s. [DOI] [PubMed] [Google Scholar]
- 25.Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT. Reactive Intermediates in Cytochrome P450 Catalysis. J Biol Chem. 2013;288:17074–17081. doi: 10.1074/jbc.R113.473108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munro AW, Girvan HM, McLean KJ. Cytochrome P450 - redox partner fusion enzymes. Biochim Biophys Acta -Gen Subjects. 2007;1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Cirino PC, Arnold FH. A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew Chem Int Ed. 2003;42:3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- 28.Chen MM, Coelho PS, Arnold FH. Utilizing Terminal Oxidants to Achieve P450-Catalyzed Oxidation of Methane. Adv Synth Catal. 2012;354:964–968. [Google Scholar]
- 29.Schwaneberg U, Appel D, Schmitt J, Schmid RD. P450 in biotechnology: zinc driven omega-hydroxylation of p-nitrophenoxydodecanoic acid using P450BM-3 F87A as a catalyst. J Biotechnol. 2000;84:249–257. doi: 10.1016/s0168-1656(00)00357-6. [DOI] [PubMed] [Google Scholar]
- 30.Nazor J, Dannenmann S, Adjei RO, Fordjour YB, Ghampson IT, Blanusa M, Roccatano D, Schwaneberg U. Laboratory evolution of P450BM3 for mediated electron transfer yielding an activity-improved and reductase-independent variant. Prot Eng Des Select. 2008;21:29–35. doi: 10.1093/protein/gzm074. [DOI] [PubMed] [Google Scholar]
- 31.Sadeghi SJ, Fantuzzi A, Gilardi G. Breakthrough in P450 bioelectrochemistry and future perspectives. Biochim Biophys Acta -Proteins Proteom. 2011;1814:237–248. doi: 10.1016/j.bbapap.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Park JH, Lee SH, Cha GS, Choi DS, Nam DH, Lee JH, Lee JK, Yun CH, Jeong KJ, Park CB. Cofactor-Free Light-Driven Whole-Cell Cytochrome P450 Catalysis. Angew Chem Int Ed. 2015;54:969–973. doi: 10.1002/anie.201410059. [DOI] [PubMed] [Google Scholar]
- 33.Qian J, Zhu W, Mi L, Xu X, Yu JC, Cui DM, Xue YC, Liu SQ. Nanohybrids of quantum dots and cytochrome P450 for light-driven drug metabolism. J Electroanal Chem. 2014;733:27–32. [Google Scholar]
- 34.Girhard M, Kunigk E, Tihovsky S, Shumyantseva VV, Urlacher VB. Light-driven biocatalysis with cytochrome P450 peroxygenases. Biotechnol Appl Bio. 2013;60:111–118. doi: 10.1002/bab.1063. [DOI] [PubMed] [Google Scholar]
- 35.Jensen K, Jensen PE, Moller BL. Light-Driven Cytochrome P450 Hydroxylations. ACS Chem Biol. 2011;6:533–539. doi: 10.1021/cb100393j. [DOI] [PubMed] [Google Scholar]
- 36.Zilly FE, Taglieber A, Schulz F, Hollmann F, Reetz MT. Deazaflavins as mediators in light-driven cytochrome P450 catalyzed hydroxylations. Chem Commun. 2009:7152–7154. doi: 10.1039/b913863c. [DOI] [PubMed] [Google Scholar]
- 37.O’Reilly E, Kohler V, Flitsch SL, Turner NJ. Cytochromes P450 as useful biocatalysts: addressing the limitations. Chem Commun. 2011;47:2490–2501. doi: 10.1039/c0cc03165h. [DOI] [PubMed] [Google Scholar]
- 38.Renault H, Bassard JE, Hamberger B, Werck-Reichhart D. Cytochrome P450-mediated metabolic engineering: current progress and future challenges. Curr Opin Plant Biol. 2014;19:27–34. doi: 10.1016/j.pbi.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Tripathi S, Li HY, Poulos TL. Structural Basis for Effector Control and Redox Partner Recognition in Cytochrome P450. Science. 2013;340:1227–1230. doi: 10.1126/science.1235797. [DOI] [PubMed] [Google Scholar]
- 40.Sevrioukova IF, Li HY, Zhang H, Peterson JA, Poulos TL. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc Natl Acad Sci USA. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiruma Y, Hass MAS, Kikui Y, Liu WM, Olmez B, Skinner SP, Blok A, Kloosterman A, Koteishi H, Lohr F, Schwalbe H, Nojiri M, Ubbink M. The Structure of the Cytochrome P450cam-Putidaredoxin Complex Determined by Paramagnetic NMR Spectroscopy and Crystallography. J Mol Biol. 2013;425:4353–4365. doi: 10.1016/j.jmb.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Roccatano D. Structure, dynamics, and function of the monooxygenase P450 BM-3: insights from computer simulations studies. J Phys Condens Matter. 2015;27:273102. doi: 10.1088/0953-8984/27/27/273102. [DOI] [PubMed] [Google Scholar]
- 43.Verma R, Schwaneberg U, Roccatano D. Insight into the redox partner interaction mechanism in cytochrome P450BM-3 using molecular dynamics simulations. Biopolymers. 2014;101:197–209. doi: 10.1002/bip.22301. [DOI] [PubMed] [Google Scholar]
- 44.Winkler JR, Gray HB. Electron Flow through Metalloproteins. Chem Rev. 2014;114:3369–3380. doi: 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winkler JR, Gray HB. Long-range electron tunneling. J Am Chem Soc. 2014;136:2930–2939. doi: 10.1021/ja500215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishio M, Umezawa Y, Fantini J, Weiss MS, Chakrabarti P. CH-pi hydrogen bonds in biological macromolecules. Phys Chem Chem Phys. 2014;16:12648–12683. doi: 10.1039/c4cp00099d. [DOI] [PubMed] [Google Scholar]
- 47.Farver O, Pecht I. Elucidation of Electron-Transfer Pathways in Copper and Iron Proteins by Pulse Radiolysis Experiments. Prog Inorg Chem. 2007;55:1–78. [Google Scholar]
- 48.Marcus RA, Sutin N. Electron Transfers in Chemistry and Biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 49.Whitehouse CJC, Yang W, Yorke JA, Tufton HG, Ogilvie LCI, Bell SG, Zhou WH, Bartlam M, Rao ZH, Wong LL. Structure, electronic properties and catalytic behaviour of an activity-enhancing CYP102A1 (P450(BM3)) variant. Dalton Trans. 2011;40:10383–10396. doi: 10.1039/c1dt10098j. [DOI] [PubMed] [Google Scholar]
- 50.Whitehouse CJC, Bell SG, Yang W, Yorke JA, Blanford CF, Strong AJF, Morse EJ, Bartlam M, Rao ZH, Wong LL. A Highly Active Single-Mutation Variant of P450(BM3) (CYP102A1) Chembiochem. 2009;10:1654–1656. doi: 10.1002/cbic.200900279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.