

Abstract
Background and Purpose
Chronic obstructive pulmonary disease (COPD) is a major cause of illness and death, often induced by cigarette smoking (CS). It is characterized by pulmonary inflammation and fibrosis that impairs lung function. Existing treatments aim to control symptoms but have low efficacy, and there are no broadly effective treatments. A new potential target is the ectoenzyme, semicarbazide‐sensitive mono‐amine oxidase (SSAO; also known as vascular adhesion protein‐1). SSAO is elevated in smokers' serum and is a pro‐inflammatory enzyme facilitating adhesion and transmigration of leukocytes from the vasculature to sites of inflammation.
Experimental Approach
PXS‐4728A was developed as a low MW inhibitor of SSAO. A model of COPD induced by CS in mice reproduces key aspects of human COPD, including chronic airway inflammation, fibrosis and impaired lung function. This model was used to assess suppression of SSAO activity and amelioration of inflammation and other characteristic features of COPD.
Key Results
Treatment with PXS‐4728A completely inhibited lung and systemic SSAO activity induced by acute and chronic CS‐exposure. Daily oral treatment inhibited airway inflammation (immune cell influx and inflammatory factors) induced by acute CS‐exposure. Therapeutic treatment during chronic CS‐exposure, when the key features of experimental COPD develop and progress, substantially suppressed inflammatory cell influx and fibrosis in the airways and improved lung function.
Conclusions and Implications
Treatment with a low MW inhibitor of SSAO, PXS‐4728A, suppressed airway inflammation and fibrosis and improved lung function in experimental COPD, demonstrating the therapeutic potential of PXS‐4728A for this debilitating disease.
Abbreviations
- BALF
bronchoalveolar lavage fluid
- COPD
chronic obstructive pulmonary disease
- CS
cigarette smoke
- EM
extracellular matrix
- G‐CSF
granulocyte‐colony stimulating factor
- MLI
mean linear intercept
- SSAO
semicarbazide‐sensitive amine oxidase
Tables of Links
| TARGETS |
|---|
| Enzymes |
| SSAO (VAP‐1), semicarbazide‐sensitive amine oxidase |
| Phosphodiesterase‐4 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of chronic morbidity and death worldwide, and its prevalence is increasing (Lozano et al., 2012). It is a heterogeneous disease variously comprising debilitating and complex pathological features including chronic pulmonary inflammation (bronchitis), fibrosis and emphysema (Keely et al., 2011; Fricker et al., 2014). These features combine to impair lung function and result in reduced oxygen transfer leading to breathlessness. Cigarette smoke (CS) exposure is the primary etiological agent in Western countries, accounting for more than 90% of cases. Once induced, the patients' condition often continues to deteriorate, even after cessation of smoking. Wood and cooking smoke and pollution are also important risk factors particularly in developing nations (Fabbri et al., 2004; Eisner et al., 2010; Ko and Hui, 2012).
There are no cures for COPD, and current treatments have substantial limitations. High doses of corticosteroids, long‐acting β‐adrenoceptor agonists, anti‐cholinergics, phosphodiesterase‐4 inhibitors (e.g. rolipram) are used in an attempt to control acute inflammation and reduce exacerbations (Yang et al., 2007; Calverley, 2014). However, these therapies have minimal effects on lung function, do not modify the underlying causes of disease, halt its progression or reverse pathological aspects. In addition, they are associated with deleterious side effects and can predispose patients to pneumonia, particularly when used long‐term (Suissa et al., 2013; Yang, 2012). Structural changes such as collagen deposition and fibrosis may also restrict the effectiveness of treatments. Vaccination protects against infections that cause exacerbations but is less effective in COPD (Nath et al., 2014). Thus, the development of novel, more effective, drugs that suppress symptoms and/or limit disease progression would be of considerable clinical benefit to patients with COPD.
The development of new therapies has been hampered by the lack of animal models that reproduce the characteristic features of human COPD, in a reasonable time frame (Vlahos and Bozinovski, 2014). To address this, we have generated a model induced by direct inhalation of CS that develops the characteristic features of COPD, including inflammation, emphysema‐like alveolar destruction, bronchitis and non‐responsiveness to corticosteroid treatment, in 8 weeks (Beckett et al., 2013; Franklin et al., 2014; Fricker et al., 2014; Hansbro et al., 2014; Chen‐Yu Hsu et al., 2015; Tay et al., 2015). The levels of CS‐exposure are representative of that of a one‐pack‐a‐day human smoker (Fricker et al., 2014).
Chronic CS‐exposure induces persistent inflammation dominated by macrophages, neutrophils and CD8+ lymphocytes, which drive the pathology and development of disease features (Maeno et al., 2007; Minematsu and Shapiro, 2007; Keely et al., 2011; Duan et al., 2012; Beckett et al., 2013; Fricker et al., 2014). We have recently shown that mast cells and the factors that they release may also play important pathogenetic roles by controlling macrophage responses (Beckett et al., 2013; Hansbro et al., 2014). The development, function and migration of these immune cells are controlled by stimulatory factors such as granulocyte‐macrophage colony‐stimulating factor (Vlahos et al., 2010) and granulocyte‐colony stimulating factor (G‐CSF) (Adachi et al., 2003; Yu et al., 2006; Kiss et al., 2008; Schilter et al., 2015). When they reach the airways and lungs, these cells release a range of inflammatory factors including cytokines, such as TNFα, and chemokines, such as CXCL1 (Keatings et al., 1996; Beckett et al., 2013). This leads to further inflammation and a feedback loop that results in chronic inflammatory responses and the further influx of cells to the site of injury. The repeated cycles of injury and repair result in collagen deposition and fibrosis of the airways, emphysema and reduced lung function (Keely et al., 2011; Fricker et al., 2014). However, the causal links between inflammation and fibrosis remain to be fully elucidated.
The development of fibrosis is a major problem in COPD with structural and compositional changes identified in most lung structures from the central airways down to the alveoli (Vignola et al., 1997). It results from the sub‐epithelial deposition of fibronectin, fibulin and tenascin and the accumulation of collagen (Dunsmore, 2008; Lau et al., 2010; Jaffar et al., 2014; Ge et al., 2015). The development of fibrosis in the small airways strongly correlates with the severity of COPD and results in narrowing of the airway lumen and airflow limitation (Bosken et al., 1990; Hogg et al., 2004). Fibrosis occurs initially in the small airways, as a result of persistent inflammation induced by CS, but is sustained as a consequence of reprogrammed immunity (Boorsma et al., 2013).
The enzyme semicarbazide‐sensitive amine oxidase (SSAO), also known as vascular adhesion protein‐1, is a copper dependent amine oxidase that is expressed in most tissues, and particularly in the lung (Singh et al., 2003). It has increased activity in numerous inflammation‐associated conditions including Alzheimer's disease (del Mar Hernandez et al., 2005), diabetes (Boomsma et al., 1995), stroke (Hernandez‐Guillamon et al., 2010), liver disease (Nemcsik et al., 2007), atherosclerosis (Karadi et al., 2002) and multiple sclerosis (Airas et al., 2006). A membrane bound form of the enzyme is predominantly expressed on the cell surface of adipocytes, smooth muscle and endothelial cells, and a soluble form is found in the bloodstream (Precious et al., 1988; Andres et al., 2001). The link with COPD has not been widely explored but in smokers, serum SSAO activity is positively correlated with the number of pack years (Wang et al., 2013). SSAO predominantly catalyses the deamination of primary amines, including methylamine, benzylamine and aminoacetone into toxic products, such as aldehydes, hydrogen peroxide (H2O2) and ammonia (Lizcano et al., 1994). Significantly, methylamine is a major component of CS and an end product of nicotine metabolism (US Dept Health, 1982; Yu, 1998). It is metabolized almost exclusively by SSAO and the aldehyde produced, formaldehyde, is a well‐recognized genotoxin and crosslinking agent (Boor et al., 1992). The production of the reactive oxygen species H2O2 results in oxidative stress which contributes to disease through the development and maintenance of chronic inflammation and tissue damage. SSAO also facilitates both acute and chronic inflammation by promoting leukocyte trafficking from blood vessels through the endothelium and into inflamed tissues (Merinen et al., 2005). Thus, treatments that inhibit both enzymic and cell adhesive functions associated with SSAO activity may ameliorate inflammation and the subsequent pulmonary fibrosis and impaired lung function, and could provide a potential therapeutic approach in COPD.
Here, we tested the ability of a selective, orally active low MW inhibitor of SSAO, PXS‐4728A (4‐[(E)‐2‐(aminomethyl)‐3‐fluoro‐allyloxy]‐N‐tert‐butyl‐benzamide hydro‐chloride; Schilter et al., 2015) to inhibit characteristic features of COPD in our mouse model that reproduces the human disease. Following acute CS‐exposure, we demonstrated that prophylactic and therapeutic administration of PXS‐4728A completely inhibited SSAO activity and reduced inflammatory cell influx into the airways, as well as decreasing cytokine and chemokine production. Importantly, therapeutic administration of PXS‐4728A during chronic CS‐exposure when COPD features have been established, reduced the influx of inflammatory cells, in particular neutrophils and macrophages, suppressed collagen deposition around small airways and improved lung function. Thus, targeting SSAO with PXS‐4728A may represent a viable therapeutic option in COPD.
Methods
Mice
All animal care and experimental procedures were conducted in accordance with the local Guidelines and were approved by the animal ethics committee of the University of Newcastle, NSW, Australia. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). Female wild‐type C57BL/6 mice were obtained from Australian BioResources Ltd (Mossvale, NSW, Australia) and group housed (5 mice per cage) under specific pathogen‐free conditions at the Animal Services Unit, Hunter Medical Research Institute, Newcastle, NSW, Australia. The light/dark cycle was 14/10 h; temperature range 20°–22°C and humidity range 55–70% relative humidity.
General procedures
Mice were randomly numbered and then separated into the experimental groups. All tissues and samples were processed using this number as the identifier. Assays were performed without knowing which number belonged to which group. Once the assay was completed, this number was used to identify the experimental group providing the sample.
Exposure to CS
Female C57BL/6 mice (6–8 weeks old) were exposed either to CS from 12 research‐grade cigarettes (3R4F, University of Kentucky, Lexington, KY, USA; each cigarette was smoked over 5 minutes) or to normal air for 75 min, twice per day. Female mice were used as they have a higher risk of developing small airway disease compared with male mice (Tam et al., 2016). CS was delivered to mice using an in‐house custom‐designed and purpose‐built nose‐only, directed‐flow inhalation and smoke‐exposure system (CH Technologies, Westwood, NJ, USA) with an air flow rate of 2.5L min−1, housed in a fume and laminar flow hood. Mice were either exposed each day for 4 days (acute exposure) or for 5 days a week for 12 weeks (chronic exposure) (Beckett et al., 2013; Franklin et al., 2014; Hansbro et al., 2014; Chen‐Yu Hsu et al., 2015). This model has been fully described earlier (Beckett et al., 2013).
Drug treatments
Mice were lightly anaesthetized by inhalation of 5% isoflurane. They were then treated with various doses of PXS‐4728A or rolipram (positive control, 3 mg⋅kg−1), resuspended in 0.5% w/v methylcellulose in sterile water (vehicle), or vehicle alone by oral gavage in a total volume of 200 μL immediately before CS‐exposure. For treatment in the acute 4 day exposure model, mice were treated before the first CS‐exposure of each day. To test the therapeutic potential of PXS‐4728A, mice were chronically exposed to CS for 12 weeks, with drug delivery prior to the first CS‐exposure of each day, commencing after 6 weeks, when features of COPD first emerge (Beckett et al., 2013). Previous studies have shown that the vehicle control has no effect on immune responses compared with PBS‐treated mice. Thus, in order to reduce animal use in these studies, PBS groups were omitted.
Assay of SSAO enzymic activity
To measure SSAO activity, animals were killed (overdose of pentobarbitone, i.p.) and abdominal fat and lungs were collected and snap frozen. Tissue samples were weighed and homogenized in ice‐cold HES buffer (20 mL per g tissue: composition; 20 mM HEPES, 1 mM EDTA, sucrose 250 mM, 1× proteases and phosphatases inhibitor, pH 7.4). Homogenates were centrifuged at 2000× g for 5 min at 4°C and the supernatants collected and diluted 1:5 in assay buffer (0.1 M sodium phosphate buffer, pH7.2) for the fluorimetric assay (fat tissue) and used undiluted for the radioactivity assay (lung tissue), of SSAO activity.
Fluorimetric assays of the enzymic activity were based on the production of H2O2 (Schilter et al., 2015). Briefly, tissue samples (1.25 mg lung; 0.25 mg fat) were incubated with 0.5 mM of pargyline, to inhibit any endogenous monoamine oxidase A and B activity. Samples (25 μL) were then incubated for 30 min at 37°C with or without the specific SSAO inhibitor mofegiline (1 μM; Palfreyman et al., 1994). Equal amounts of the reaction mixture containing Amplex Red (120 μM; Life Technologies, Australia), horseradish peroxidase (HRP; 1.5 U mL‐1; Sigma‐Aldrich, Sydney, Australia) and benzylamine (80 μM) were prepared in 0.1 M sodium phosphate buffer, pH 7.4 and 25 μL added into each well after the 30 min incubation. The relative fluorescence units were read every 2.5 min for 30 min at 37°C, excitation 565 nm and emission 590 on a BMG FLUOstar OPTIMA Microplate Reader (Optima, BMG labtech), and the slope of the kinetic curve for each sample was calculated using MARS data analysis software (BMG labtech) in relative fluorescence units⋅min−1. The signal obtained from the non‐treated samples was considered to be 100%. The difference between the signals obtained in the samples not treated (high‐signal control) or in the presence of mofegiline (low‐signal control) was considered to be the specific SSAO activity. The data from all treated samples were adjusted to yield a percentage response graph.
Radioactive enzymic activity (REA) assays were based on the oxidation of [14C]‐benzylamine to [14C]‐benzaldehyde by SSAO (Devlin et al., 1990). Assays were performed by a commercial service at Tetra‐Q ADME, The University of Queensland, Brisbane, Australia. Tissue samples were incubated with 3 μM pargyline and clorgyline to inhibit any endogenous monoamine oxidase A and B activity. Samples (150 μL) were incubated for 20 min at 37°C with or without the specific SSAO inhibitor PXS‐4728A (30 nM). [14C]‐benzylamine (50 μL of 915.5 μM stock) was added to each sample, which was incubated for 30 min at 37°C. Reactions were terminated by the addition of 50 μL of 2 M citric acid, and the products were extracted into 1 mL of toluene/ethyl acetate 1:1 (v/v) and assayed by liquid scintillation counting. The difference between the signals obtained in the absence of the inhibitor (high‐signal control) and in the presence of PXS‐4728A (low‐signal control) was considered to be the specific SSAO activity in the sample. The data are presented as a percentage of the signal to the non‐treated group (considered to be 100%).
In the REA assays, PXS‐4728A was used to selectively and specifically inhibit the SSAO component in order to measure the background, that is non‐SSAO mediated amine oxidase activity. As REA assays have a very high selectivity, we used the most selective inhibitor known to us, which is PXS‐4728A. Measurements in the presence of added PXS‐4728A were only used to determine the “low” signal for normalization while the “high” signal was that from non‐drug treated animals, not treated with the specific SSAO inhibitor. Thus, PXS‐4728A was not added to samples from drug‐treated animals and cannot interfere with the results in treatment groups. In contrast to REA in fluorometric measurements, SSAO concentrations in tissues are substantial, and selectivity and specificity were not critical. Therefore, the well‐characterized inhibitor mofegiline was used in these studies.
Airway inflammation
Airway inflammation was assessed by enumerating total leukocyte cell numbers as well as specific inflammatory cell types in bronchoalveolar lavage fluid (BALF) (Beckett et al., 2013; Hansbro et al., 2014; Chen‐Yu Hsu et al., 2015). BALF was obtained by lavaging lungs (after death) with two 400 μL aliquots of PBS at room temperature. Total cell numbers were assessed using Trypan Blue exclusion. Cytospins were performed on the remaining BALF cells, and differential cell counts were obtained based on morphology (Thorburn et al., 2010; Asquith et al., 2011; Beckett et al., 2013; Essilfie et al., 2015). Cytokine and chemokine levels were assessed in BALF. TNFα, CXCL1 and G‐CSF were assessed using a BD™ Cytometric Bead Array kit (BD Biosciences, San Jose, CA, USA), according to the manufacturer's instructions with a minimum detection limit of 0.274 pg⋅mL−1.
Small airway‐associated fibrosis
Small airway‐associated fibrosis was assessed in formalin‐fixed lung sections by measuring the deposition of Masson's Trichrome Blue‐stained collagen around the small airways (Hansbro et al., 2014). Stained lung sections on slides were photographed, and the amount of collagen was calculated using Image J software (version 1.47) by assessing the area of deposition around small airways of less than 1 mm diameter using the formula (Wct/Pbm), where Wct = Ao−Ai, inner collagen area = Ai, outer collagen area = Ao and basement membrane length = Pbm.
Soluble lung collagen
Snap frozen sections of lung (approximately 10 mg) were minced in ice‐cold PBS. After washing with PBS, acid‐soluble collagen was extracted by incubating overnight in 1 mL of 0.1 mg⋅kg−1 pepsin in 0.5 M acetic acid (Sigma Aldrich) at 4°C. Soluble collagen was concentrated and measured using Sircol Collagen Assay Kits according to the manufacturer's instructions (Biocolor, Carrickfergus, UK) (Chow et al., 2012).
Total lung collagen
Frozen lung tissues (10 mg) were weighed and incubated in HCl (6 N) for 7 h at 130°C. Solutions were then placed in a 100°C drying oven to evaporate the liquid. The resulting pure collagen was resuspended in a 96 well plate in 100 μL double‐distilled H2O per well. Plates were centrifuged (220× g for 15 s) and supernatants removed. Chloramine T solution (100 μL total; composition: 50 mM Chloramine T, 10% v/v n‐propanol, citrate/acetate buffer‐0.88 M sodium acetate, 0.24 M citric acid, 50 mM acetic acid, 0.85 M NaOH; pH 6‐6.5), was added to the samples and standards followed by incubation for 20 min at room temperature. Then Erhlich's solution (100 μL, Sigma Aldrich) was added and samples incubated for 18 min at 65°C. Total lung collagen was determined by measuring absorbance at 558 nm, compared with hydroxyproline standards (Sigma Aldrich, effective range 0.625–40 ug⋅mL−1).
Emphysema
Sections (4 μm thick) of paraffin‐embedded, formalin‐fixed inflated lung tissue were mounted on microscope slides and stained with haematoxylin and eosin. Emphysema‐like alveolar enlargement was assessed using the mean linear intercept (MLI) technique, which is a standard method for assessing alveolar size and emphysema in mice (Horvat et al., 2010; Beckett et al., 2013; Starkey et al., 2013; Hansbro et al., 2014; Chen‐Yu Hsu et al., 2015). A standardized template containing horizontal lines was laid over micrographs of lung sections. The number of alveolar intercepts was counted, and the resulting average number of intercepts was determined. Reduced numbers of intercepts are an indicator of increased alveolar size and of emphysema and tissue damage.
Lung function
Static lung compliance was obtained from quasistatic pressure–volume (PV) loops using Flexivent apparatus (Legacy System; SCIREQ, Montreal, Canada). Mice were anaesthetized with a mixture of xylazine (2 mg⋅mL−1, Troy Laboratories) and ketamine (40 mg⋅mL−1, Ceva), given i.p. in a volume of 50 μl per 10 g body weight. Cannulae were inserted into mouse tracheas. Animals were ventilated with a tidal volume of 8 mL⋅kg−1 at a rate of 450 breaths⋅min−1, with increasing airway pressure from 2–30 cm H2O into the lungs. The volume of air in the lungs at the end of maximal inspiration was determined. Static lung compliance was calculated as the volume change divided by applied pressure change, while hysteresis was assessed by measuring the difference between inspiratory and expiratory pressure volume loops. Each manoeuvre was performed at least three times and the average was calculated (Beckett et al., 2013; Hansbro et al., 2014; Chen‐Yu Hsu et al., 2015).
Data and statistical analysis
These studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data shown are means ± SEM (n = 6–8). All datasets were determined to be normally distributed based on previous experiments and were also tested using the Kolmogorov–Smirnov test (α = 0.05). Data were analysed using one‐way ANOVA with Tukey's multiple comparison test using PRISM software (V6.0d, GraphPad, La Jolla, CA, USA), with P < 0.05 considered as statistically significant.
Materials
The suppliers of the following compounds were as shown: mofegiline (Shanghai SynCores Technologies, Shanghai, P.R.China); clorgyline (Cayman Chemical Company, Ann Arbor, Michigan USA); PXS‐4728A (Pharmaxis Ltd, Frenchs Forest NSW, Australia. All other compounds were supplied by Sigma Chemicals, St Louis, MO, USA.
Results
Oral delivery of PXS‐4728A potently inhibits constitutive lung and peripheral SSAO activity
SSAO activity is found constitutively in the lungs and at higher levels in fat, in particularly in adipocytes. Thus, the effectiveness of PXS‐4728A treatment as an inhibitor was first assessed by measuring SSAO activity in the lungs and inguinal fat in mice after an acute, 4 day, exposure to CS. Acute CS‐exposure did not increase SSAO activity in the lungs when assessed 16 h after the last exposure, compared with normal air‐exposed controls (Figure 1). However, daily oral treatment with PXS‐4728A at either 2, 6 or 20 mg⋅kg−1 doses inhibited constitutive SSAO activity, compared with vehicle‐treated CS‐exposed mice. Additionally, SSAO activity in inguinal fat was completely inhibited with PXS‐4728A treatment (Figure 1). This shows that orally delivered PXS‐4728A is a potent in vivo inhibitor of SSAO activity both in the lungs and in peripheral tissue.
Figure 1.
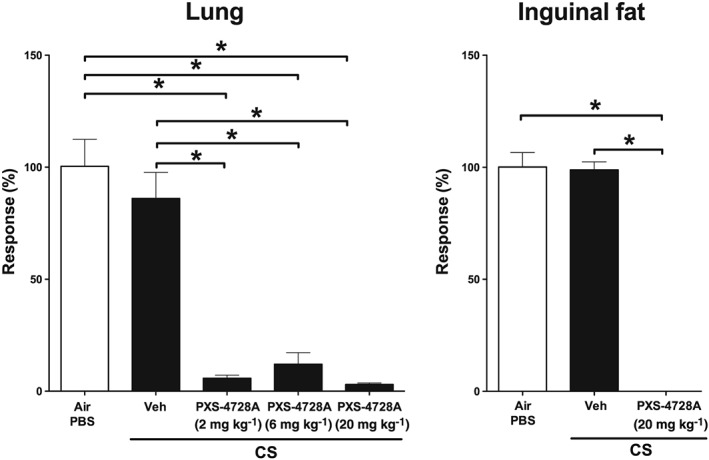
Oral treatment with PXS‐4728A inhibits constitutive lung and peripheral SSAO enzymic activity. Mice were exposed daily to CS or normal air for 4 days. Prior to each exposure, mice were treated by oral gavage with PXS‐4728A (2, 6 or 20 mg⋅kg−1). Lungs and inguinal fat were collected, and SSAO activity was measured using radiometric or fluorimetric enzymic assays respectively. Responses are represented as a percentage of activity relative to the normal, air‐exposed, group. Data represent the means ± SEM from six mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Oral delivery of PXS‐4728A reduces inflammatory cell influx into the airways following acute CS‐exposure
SSAO promotes inflammation largely by facilitating trafficking of inflammatory cells into inflamed tissues. Thus, we next assessed the effects of inhibiting SSAO with oral PXS‐4728A on the accumulation of total and differential leukocytes in the BALF of mice acutely exposed to CS for 4 days. Acute CS‐exposure increased the numbers of total leukocytes with significant increases in macrophages, neutrophils and lymphocytes compared with normal air‐exposed controls (Figure 2). Daily oral treatment with 12 or 20 mg⋅kg−1 of PXS‐4728A reduced the total number of leukocytes in the airways. With the lower dose, the numbers of macrophages and lymphocytes were reduced whereas, with the higher dose, neutrophil numbers were significantly suppressed.
Figure 2.
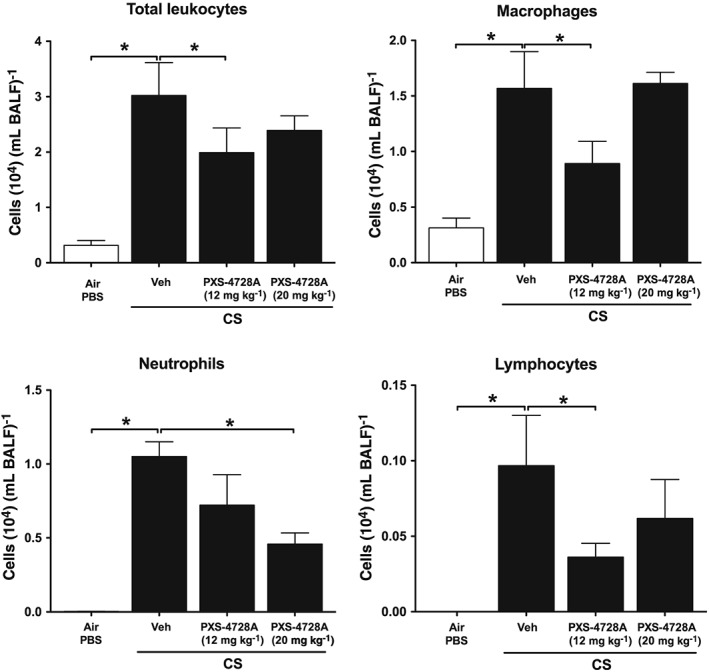
Oral treatment with PXS‐4728A reduces the numbers of inflammatory cells in the airways in response to acute CS‐exposure. Mice were exposed daily to CS for 4 days. Prior to each CS‐exposure, mice were treated by gavage with PXS‐4728A (12 or 20 mg⋅kg−1). BALF was collected, and total leukocytes, macrophages, neutrophils and lymphocytes were counted in stained cytospin slides. Data represent the means ± SEM from six mice per group. *P < 0.05, significantly different as indicated; one‐way ANOVA with Tukey's test.
Oral delivery of PXS‐4728A reduces pro‐inflammatory cytokine and chemokine levels in the airways following acute CS‐exposure
We then assessed the effects of PXS‐4728A treatment on pro‐inflammatory cytokine and chemokine levels induced by CS‐exposure. Acute CS‐exposure induced increases in TNFα and CXCL1 protein levels in the BALF compared with normal air‐exposed controls (Figure 3). Daily oral treatment with 12 or 20 mg⋅kg−1 of PXS‐4728A reduced the levels of TNFα protein in a dose‐dependent manner, with the higher dose reducing levels back to baseline levels in normal air‐exposed mice. CXCL1 levels were significantly reduced when treated with either dose.
Figure 3.
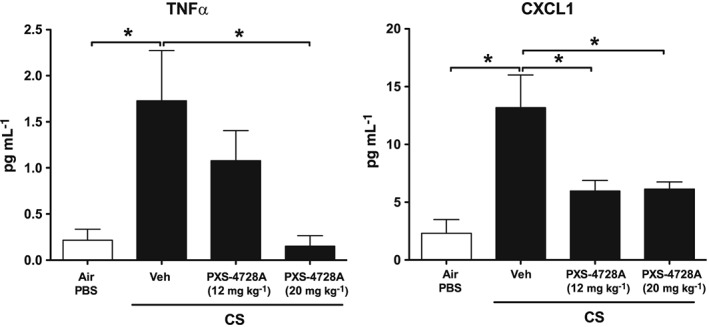
Oral treatment with PXS‐4728A reduces pro‐inflammatory cytokine and chemokine levels in the airways in response to acute CS‐exposure. Mice were exposed daily to CS or normal air 4 four days. Prior to each CS‐exposure, mice were treated by gavage with PXS‐4728A (12 or 20 mg⋅kg−1) or vehicle. BALF was collected, and the levels of TNFα and CXCL1 were measured by elisa. Data represent the means ± SEM from six mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Therapeutic treatment with PXS‐4728A potently inhibits SSAO activity in the lungs in experimental COPD
PXS‐4728A treatment reduced SSAO activity and inflammatory parameters during acute CS‐exposure. Thus, we determined whether therapeutic delivery would reduce disease features in a chronic model of COPD, which reproduces the characteristic features of the human disease. Our established model of experimental COPD developed the first signs of disease after 6 weeks of nose‐only CS‐exposure, which then progresses after 12 weeks of exposure. Disease features included chronic inflammation, fibrosis, emphysema and reduced lung function (Beckett et al., 2013; Franklin et al., 2014; Hansbro et al., 2014; Hsu et al., 2015, Tay et al., 2015). As the higher dose had greater effects in the acute model, mice were treated with PXS‐4728A (20 mg⋅kg−1) or, as a positive control, with the phosphodiesterase‐4 inhibitor rolipram (3 mg⋅kg−1) from 6 weeks of CS‐exposure. Rolipram is used here as a pharmacological comparator for the inhibition of COPD and comparator to the effects of inhibiting SSAO activity, as PDE4 inhibitors have been shown to be beneficial in other models. Rolipram has no known direct inhibitory activity on SSAO. We first assessed the effects on SSAO activity in the lung. The development and progression of experimental COPD was accompanied by a substantial increase in lung SSAO activity 16 h after the last exposure compared with normal air‐exposed controls (Figure 4). Daily oral treatment with PXS‐4728A completely inhibited SSAO activity induced by CS‐exposure. In comparison, treatment with rolipram reduced SSAO activity to levels found in normal air‐exposed controls. This is likely to be due to indirect effects that result from the suppression of inflammation.
Figure 4.
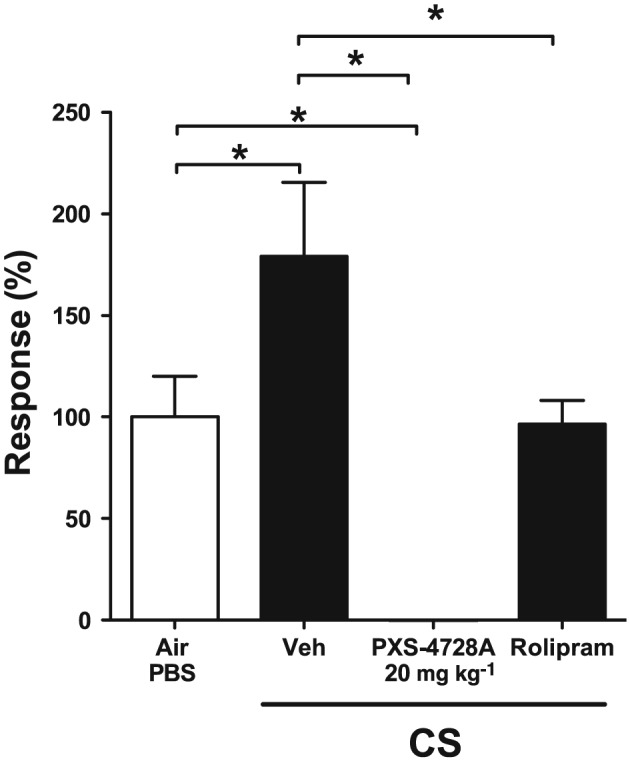
Therapeutic oral treatment with PXS‐4728A completely inhibits SSAO activity in the lung in experimental COPD. Mice were exposed to CS or normal air, on 5 days per week for 12 weeks. For the last 6 weeks, mice were treated by daily oral gavage, 1 h prior to CS‐exposure with PXS‐4728A, rolipram or vehicle. Lung tissue was collected, and SSAO activity was measured using REA. Activity is presented as the percentage of activity relative to the normal air‐exposed group. Data represent the means ± SEM from 6–8 mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Therapeutic treatment with PXS‐4728A reduces inflammatory cell influx into the airways in experimental COPD
We next assessed the effects of treatment on pulmonary inflammation in experimental COPD. Chronic CS‐exposure increased the numbers of total leukocytes and macrophages, neutrophils and lymphocytes in the BALF of mice, compared with normal air‐exposed controls (Figure 5). Treatment with PXS‐4728A significantly reduced the total numbers of leukocytes in the BALF. The numbers of macrophages and lymphocytes were reduced to those found in normal air‐exposed mice, and neutrophil numbers were also significantly decreased. Rolipram treatment had no effect on total leukocyte numbers. It did induce a shift in the proportion of the type of leukocytes and increased neutrophils but reduced lymphocytes. Interestingly, the reduction in leukocytes induced by PXS‐4728A treatment was not associated with changes in chemoattractants or growth factors (TNFα, CXCL1 and G‐CSF) associated with the influx into the airways and proliferation of inflammatory cells at this time‐point.
Figure 5.
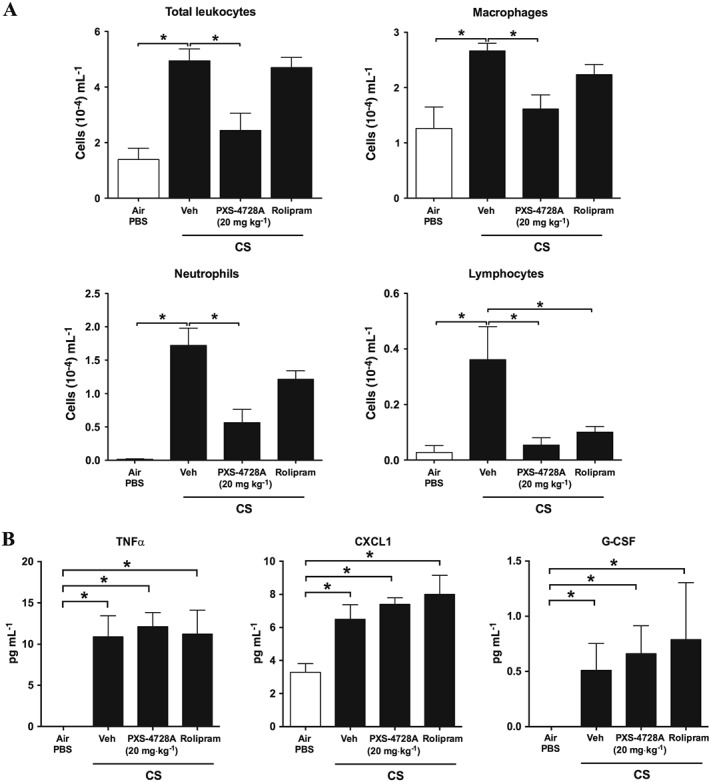
Therapeutic oral treatment with PXS‐4728A reduces the numbers of inflammatory cells in the airways in experimental COPD. Mice were exposed to CS or normal air on 5 days per week for 12 weeks. For the last 6 weeks, mice were treated by daily oral gavage, 1 h prior to CS‐exposure with PXS‐4728A, rolipram or vehicle. (A) BALF was collected, and total leukocytes, macrophages, neutrophils and lymphocytes were counted in stained cytospin slides. (B) The levels of TNFα, CXCL1 and G‐CSF were measured by elisa. Data represent the means ± SEM from 6–8 mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Therapeutic treatment with PXS‐4728A reduces small airway fibrosis in experimental COPD
We then assessed the effects of treatment on airway fibrosis and remodelling in our model. Exposure to CS for 12 weeks resulted in significantly increased peribronchiolar collagen deposition around the small airways compared with normal air‐exposed controls (Figure 6). Treatment with PXS‐4728A completely inhibited excess collagen deposition around the small airways with levels reduced to those seen in normal air‐exposed mice. In contrast, rolipram did not have any significant effects. Total lung collagen measured by hydroxyproline content was not altered by any treatment.
Figure 6.
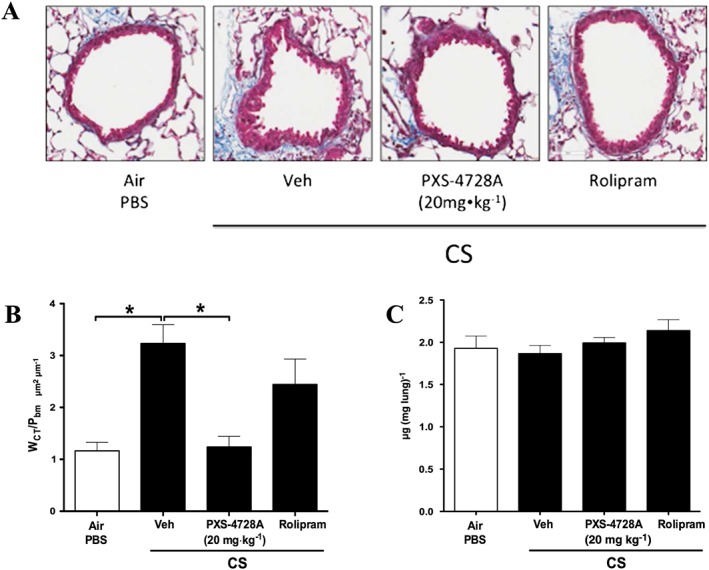
Therapeutic oral treatment with PXS‐4728A reduces airway fibrosis and remodelling in experimental COPD. Mice were exposed to CS or normal air on 5 days per week for 12 weeks. For the last 6 weeks, mice were treated daily by oral gavage prior to CS‐exposure with PXS‐4728A, rolipram or vehicle. Lungs were perfused, and inflated and airway associated fibrosis and remodelling were assessed by measuring the deposition of collagen around small airways in stained sections. (A) Representative micrographs of sections stained with Masson's Trichrome Blue. (B) Area of fibrosis around the small airways. (C) Hydroxyproline detected in lung lobes. Data represent the means ± SEM from 6–8 mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Therapeutic treatment with PXS‐4728A does not reverse emphysema‐like alveolar enlargement in experimental COPD
We then assessed the effects of treatment on emphysema‐like alveolar enlargement in our model. Chronic (12 weeks) CS‐exposure resulted in alveolar destruction with increases in mean linear intercept measurements that is representative of emphysema in human COPD, compared with normal air‐exposed controls (Figure 7). No treatment had any significant effect on alveolar enlargement.
Figure 7.
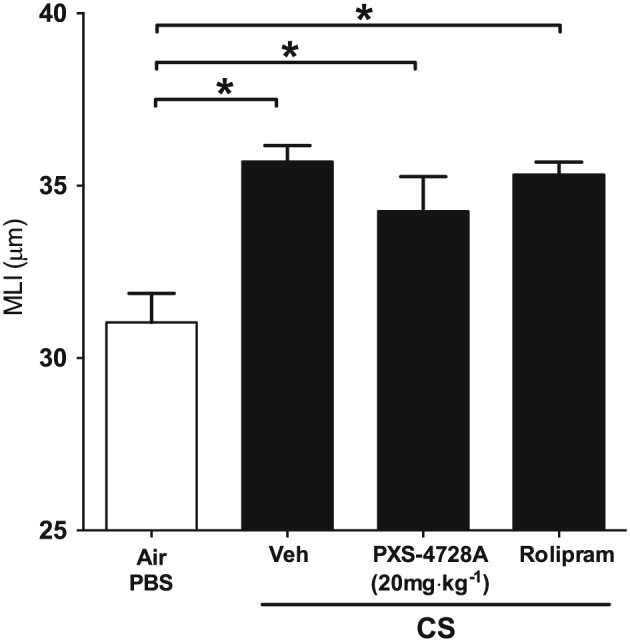
Therapeutic oral treatment with PXS‐4728A does not reduce emphysema. Mice were exposed to CS or normal air on 5 days per week for 12 weeks. For the last six weeks, mice were treated by daily oral gavage prior to CS‐exposure with PXS‐4728A, rolipram or vehicle. Lungs were perfused and emphysema‐like alveolar enlargement assessed by determining alveolar wall MLI in stained sections. Data represent the means ± SEM from 6–8 mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Therapeutic treatment with PXS‐4728A improves lung function in experimental chronic obstructive pulmonary disease
As chronic CS‐exposure induced pathological changes that result in impaired lung function, mouse lung function was assessed and analysis performed on PV loops, which represent the relationship between changes in pressure and lung volumes. Associated lung compliance (the lung's ability to stretch under a given pressure) and hysteresis (difference between compliance on inspiration vs. expiration) were also assessed. After 12 weeks of CS‐exposure, there was an increase in PV loops, lung compliance and hysteresis compared with normal air‐exposed controls (Figure 8). Treatment with either PXS‐4728A or rolipram improved lung function and completely inhibited changes in PV loops and lung compliance with levels equivalent to those in normal air‐exposed controls.
Figure 8.
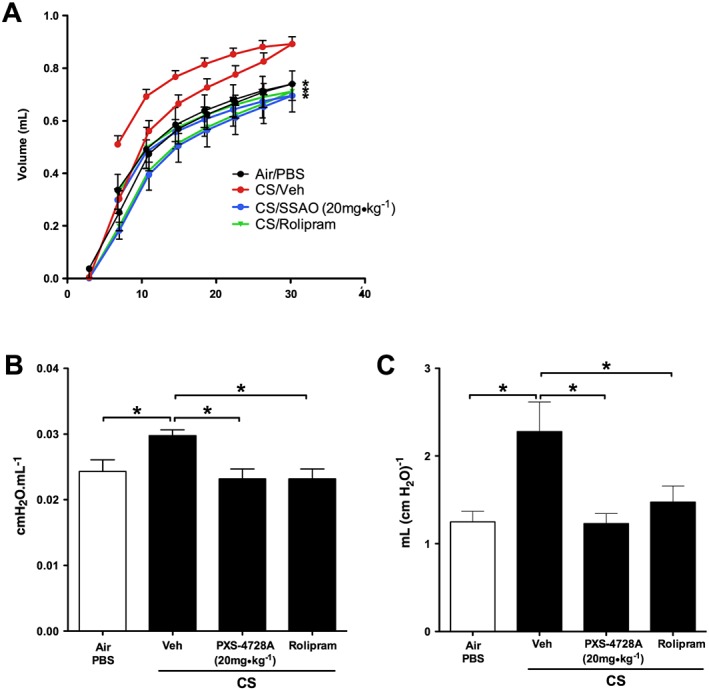
Therapeutic oral treatment with PXS‐4728A improves lung function. Mice were exposed to CS or normal air on 5 days per week for 12 weeks. For the last 6 weeks, mice were treated by daily oral gavage prior to CS‐exposure with PXS‐4728A, rolipram or vehicle. Mice were anaesthetized and cannulated. Pressure volume loops (A) lung static compliance (B) and hysteresis (C) were measured using flexiVent apparatus. Data represent the means ± SEM from 6–8 mice per group. *P < 0.05; significantly different as indicated; one‐way ANOVA with Tukey's test.
Discussion
COPD is a heterogeneous disease that results from a set of diverse pathologies and processes that can occur simultaneously, such as inflammation and fibrosis, resulting in poorer lung function, morbidity and mortality. There are no effective treatments that suppress the major disease features of COPD. SSAO levels are increased in the serum in smokers (Wang et al., 2013), and it may play an important role in COPD pathogenesis. Its enzymic activity results in the conversion of components of CS, such as methylamines, into toxic products, while its non‐enzymic activities induce pulmonary inflammation through facilitating leukocyte influx (Yu et al., 2006). When it becomes chronic, inflammation leads to the development of fibrosis and impaired lung function (Wong et al., 2014). Here, we have demonstrated that a low MW inhibitor of SSAO, PXS‐4728A, ameliorated important features of experimental COPD.
Initial assessment of lung SSAO demonstrated that its activity was not increased with acute CS‐exposure but required chronic exposure to induce significantly increased activity. Analysing activity in the lung has proven extremely difficult, as the Amplex Red/HRP assay available is not sufficiently sensitive to determine lung SSAO activity. To overcome this, a more sensitive radioactivity‐based assay was successfully employed, which for the first time enabled the demonstration of increased SSAO activity in lung homogenates in experimental COPD.
After both acute and chronic CS‐exposures, daily oral treatment of mice with PXS‐4728A was effective in completely inhibiting SSAO activity in the lung. The PXS‐4728A doses used were initially based on previous studies which defined the pharmacodynamic and pharmacokinetic properties of the inhibitor. In particular, its selectivity in targeting SSAO and its bioavailability and its lack of off‐target effects (Foot et al., 2013; Schilter et al., 2015). Oral treatment also inhibited systemic SSAO activity, as determined in inguinal fat after acute CS‐exposure. The adipose tissue was chosen as it contains high quantities of SSAO, allowing a large assay window to detect enzyme inhibition.
In addition to reducing SSAO activity, PXS‐4728A treatment also suppressed the airway leukocyte populations that dominate in the airways after acute and chronic CS‐exposure. Treatment reduced the increased numbers of macrophages, neutrophils and lymphocytes in the airways following acute CS‐exposure back to baseline levels (macrophages and lymphocytes). Suppression of these cell types was greatest with long‐term treatment in the chronic model, suggesting that longer treatments may be needed in humans. As there was no increased activity after exposure to acute CS, the reduction in activity to below control levels and the subsequent reduction in numbers of leukocyte subpopulations suggest a role for SSAO in cellular trafficking. We and others have previously demonstrated that macrophages play important roles in CS‐induced pathogenesis (Duan et al., 2012; Lenzo et al., 2012; Beckett et al., 2013), and their depletion prevented the development of experimental COPD (Beckett et al., 2013). While its effects on neutrophils has been well documented (Yu et al., 2006; Kiss et al., 2008; Foot et al., 2013), there has been no previously reported direct effect of alterations in SSAO activity on macrophages. The acute reduction in inflammatory cells was accompanied by significant decreases in the protein levels of the pro‐inflammatory cytokine TNFα. Among its other roles, TNFα contributes to the recruitment of inflammatory cells by increasing the expression of adhesion molecules, such as ICAM, VCAM and E‐selectin, on endothelial cells (Stangl et al., 2001; Mako et al., 2010; Kolaczkowska and Kubes, 2013; Schilter et al., 2015). SSAO activity can also induce the production of ROS products which can increase P‐selectin (Stangl et al., 2001; Mako et al., 2010). Leukocytes are able to tether to these molecules, which enables cellular attachment to endothelial surfaces and transmigration into sites of inflammation. Thus, PXS‐4728A treatment may reduce inflammatory cell influx into the lung by affecting adhesion molecules and endothelial tethering.
We previously showed that treatment with PXS‐4728A reduced the number of airway neutrophils that occurred in response to challenge with bacteria, viruses and allergens (Schilter et al., 2015), while others have demonstrated the effects of SSAO on neutrophils in pulmonary inflammation and ischaemic reperfusion injury (Yu et al., 2006; Kiss et al., 2008; Foot et al., 2013). Here, we used our models of CS‐exposure and experimental COPD to demonstrate for the first time that inhibition of SSAO reduces neutrophilic lung inflammation. TNFα can have pleiotropic effects on neutrophils depending on the environment. As alveolar macrophages are the major source of TNFα and other chemokines in the lung, even a partial suppression in the numbers of these cells would result in the reduction in surface molecule expression, a restricted immune response and subsequent cellular inflammation. TNFα contributes to the recruitment of these cells to the site of inflammation by inducing increases in the levels of surface molecules on neutrophils (e.g. CD44) and endothelial cells (e. g. ICAM, VCAM and E‐selectin) (Mako et al., 2010; Kolaczkowska and Kubes, 2013). It can also induce neutrophil apoptosis (Cross et al., 2008). PXS‐4728A treatment also reduced the levels of the neutrophil chemokine CXCL1, which can be induced by TNFα, and is produced by macrophages, neutrophils, epithelial and endothelial cells (Miyake et al., 2013; Hallstrand et al., 2014; Lo et al., 2014; Shieh et al., 2014). Phosphodiesterase‐4 inhibitors have also been shown to reduce CXCL1‐associated migration of neutrophils from COPD patients (Turner et al., 2011). Thus, inhibition of SSAO by PXS‐4728A may reduce the number of neutrophils in the lung by impairing transmigration and reducing the levels of TNFα that suppresses endothelial CXCL1 production and further restricts neutrophil chemoattraction to the lung.
In COPD, oxidative stress and inflammation become self‐sustaining even after the cessation of smoking. We considered that it would be more difficult to suppress disease with ongoing CS‐exposure. Thus, we used a more stringent test of the inhibitor by using it therapeutically during continued smoke exposure. Nevertheless, treatment is likely to be relevant to ex‐smokers also. Although we found that therapeutic treatment suppressed inflammatory cell influx into the airways in both the acute and chronic models, this was not associated with the suppression of TNFα or CXCL1, or of G‐CSF in the chronic model. Thus, the attenuation of inflammatory cell numbers is likely to occur through the reduction in endothelial proteins or other inflammatory factors described previously; this aspect requires further study. Nevertheless collectively, our data show that the inhibition of SSAO can suppress the cascade of inflammatory events and results in the reduction of acute CS‐induced inflammatory cell influx into the airways. Treatment had little effect on emphysema. PXS‐4728A was administered therapeutically after 6 weeks of CS‐exposure. At this time, conditions for the development of emphysema are likely to have already occurred. While PXS‐4728A treatment diminishes several important features of COPD such as inflammation and fibrosis, the treatment regime does not affect all the drivers of emphysema, such as the apoptosis of endothelial or epithelial cells. The inhibition of inflammation over time, however, is likely to prevent the progression of emphysema, although this has not been tested.
We demonstrate here for the first time that inhibition of SSAO inhibits fibrosis of the small airways in experimental COPD. Importantly, this was achieved by treating mice therapeutically when the first signs of COPD develop (Beckett et al., 2013). Other studies in different organs have examined the role of SSAO in the formation of fibrosis. SSAO activity positively correlated with hepatic liver disease including the influx of leukocytes, induction of oxidative stress through the production of toxic metabolites and the expression of fibrosis‐associated genes (Weston et al., 2014). Inhibition of SSAO with PXS‐4728A in an acute model of renal fibrosis reduced liver remodelling also through the suppression of leukocyte accumulation, oxidative stress and pro‐inflammatory and pro‐fibrotic gene expression (Wong et al., 2014). In COPD, the extracellular matrix and collagen that provide the structural integrity of the alveoli are often destroyed, whereas conversely these factors and thus fibrosis are increased around the small airways, causing stiffening and reducing lung function. Here, no change was observed in total lung tissue collagen levels, which is consistent with our emphysema data, which showed that there was no difference in alveolar enlargement after PXS‐4728A treatment. Others have shown that chronic CS‐exposure can simultaneously increase peribronchiolar fibrosis, resulting in restricted airflow, while at the same time causing the loss of parenchymal collagen, resulting in tissue destruction and emphysema (Chung and Adcock, 2008).
Treatment with PXS‐4728A and its associated protection against pathological changes, including reduced inflammation and fibrosis, resulted in a significant improvement in lung function. This involved reduction of experimental COPD‐associated increases in PV loops and improved static compliance and hysteresis. Many studies show that inflammation and airway remodelling are linked to decreased lung function. Indeed, persistent inflammation induces the development of fibrosis that particularly affects the function of the bronchioles and small airways (Wallace et al., 2006). Increased inflammation correlates with poorer lung function in patients with COPD (Turato et al., 2002; Donaldson et al., 2005; Aronson et al., 2006; Hancox et al., 2007), and small airway fibrosis contributes to airflow restriction through exaggerated tissue contraction, perhaps through the altered balance between fibrotic MMPs and their inhibitors (Hogg et al., 2004; Rennard, 2006; Salazar and Herrera, 2011; Churg et al., 2012; Jankowich and Rounds, 2012). Increases in the levels of alveolar macrophage‐associated MMPs negatively correlate with forced expiratory volume in 1 s/forced vital capacity in COPD patients (Ishii et al., 2013). We have shown that depletion of macrophages by clodronate improves transpulmonary and airway resistance as well as dynamic compliance in experimental COPD (Beckett et al., 2013), which possibly occurs through the reduction in MMPs. Thus, PXS‐4728A associated reductions in inflammation, particularly the number of immune cells and potentially their phenotypic features, as well as attenuated airway remodelling, reversed the decreases in lung function.
In summary, we show here, for the first time, that SSAO activity was significantly increased in the lungs in experimental COPD and that the inhibition of SSAO activity with PXS‐4728A resulted in suppression of inflammation and small airway remodelling and consequently improved lung function. These data in conjunction with other observations demonstrating the beneficial effect of inhibiting SSAO in pathologies associated with the liver and kidneys, provide strong evidence that PXS‐4728A may have broad efficacy in inflammatory and fibrotic diseases, with potential therapeutic benefits.
Author contributions
A.G.J. wrote the manuscript, planned and performed experiments and analysed data, H.S. planned and performed experiments and analysed data, T.T.Y. performed experiments and analysed data, A.E.T., G.L. and K.W. performed experiments, J.S.F., W.J. and P.M.H designed the study, planned the experiments, analysed data and revised the manuscript.
Conflict of interest
A.G.J. is a former employee of Pharmaxis P/L.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgments
This study was supported by grants from the Australian Federal Government Techvoucher and Research in Business schemes and the National Health and Medical Research Council (NHMRC) of Australia. A.G.J. was supported by The Australian Lung Foundation/Boehringer Ingelheim COPD Research Fellowship. P.M.H. is supported by an NHMRC Principal Research Fellowship and a Brawn Fellowship, Faculty of Health & Medicine, University of Newcastle.
Jarnicki, A. G. , Schilter, H. , Liu, G. , Wheeldon, K. , Essilfie, A. ‐T. , Foot, J. S. , Yow, T. T. , Jarolimek, W. , and Hansbro, P. M. (2016) The inhibitor of semicarbazide‐sensitive amine oxidase, PXS‐4728A, ameliorates key features of chronic obstructive pulmonary disease in a mouse model. British Journal of Pharmacology, 173: 3161–3175. doi: 10.1111/bph.13573.
References
- Adachi K, Suzuki M, Sugimoto T, Yorozu K, Takai H, Uetsuka K et al. (2003). Effects of Granulocyte Colony‐Stimulating Factor (G‐CSF) on Bleomycin‐Induced Lung Injury of Varying Severity. Toxicol Pathol 31: 665–673. [DOI] [PubMed] [Google Scholar]
- Airas L, Mikkola J, Vainio JM, Elovaara I, Smith DJ (2006). Elevated serum soluble vascular adhesion protein‐1 (VAP‐1) in patients with active relapsing remitting multiple sclerosis. J Neuroimmunol 177: 132–135. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres N, Lizcano JM, Rodriguez MJ, Romera M, Unzeta M, Mahy N (2001). Tissue Activity and Cellular Localization of Human Semicarbazide‐sensitive Amine Oxidase. J Histochem Cytochem 49: 209–217. [DOI] [PubMed] [Google Scholar]
- Aronson D, Roterman I, Yigla M, Kerner A, Avizohar O, Sella R et al. (2006). Inverse Association between Pulmonary Function and C‐Reactive Protein in Apparently Healthy Subjects. Am J Respir Crit Care Med 174: 626–632. [DOI] [PubMed] [Google Scholar]
- Asquith KL, Horvat JC, Kaiko GE, Carey AJ, Beagley KW, Hansbro PM et al. (2011). Interleukin‐13 Promotes Susceptibility to Chlamydial Infection of the Respiratory and Genital Tracts. PLoS Pathog 7: e1001339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett EL, Stevens RL, Jarnicki AG, Kim RY, Hanish I, Hansbro NG et al. (2013). A new short‐term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol 131: 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma F, Derkx FH, van den Meiracker AH, Man in ‘t Veld AJ, Schalekamp MA (1995). Plasma semicarbazide‐sensitive amine oxidase activity is elevated in diabetes mellitus and correlates with glycosylated haemoglobin. Clin Sci (Lond) 88: 675–679. [DOI] [PubMed] [Google Scholar]
- Boor PJ, Trent MB, Lyles GA, Tao M, Ansari GA (1992). Methylamine metabolism to formaldehyde by vascular semicarbazide‐sensitive amine oxidase. Toxicology 73: 251–258. [DOI] [PubMed] [Google Scholar]
- Boorsma CE, Draijer C, Melgert BN (2013). Macrophage Heterogeneity in Respiratory Diseases. Mediators Inflamm 2013: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosken CH, Wiggs BR, Paré PD, Hogg JC (1990). Small Airway Dimensions in Smokers with Obstruction to Airflow. Am Rev Respir Dis 142: 563–570. [DOI] [PubMed] [Google Scholar]
- Calverley P (2014). Current Drug Treatment, Chronic and Acute. Clin Chest Med 35: 177–189. [DOI] [PubMed] [Google Scholar]
- Chen‐Yu Hsu A, Starkey MR, Hanish I, Parsons K, Haw TJ, Howland LJ et al. (2015). Targeting PI3K‐p110alpha Suppresses Influenza Virus Infection in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 191: 1012–1023. [DOI] [PubMed] [Google Scholar]
- Chow M‐J, Mondonedo JR, Johnson VM, Zhang Y (2012). Progressive structural and biomechanical changes in elastin degraded aorta. Biomech Model Mechanobiol 12: 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KF, Adcock IM (2008). Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J 31: 1334–1356. [DOI] [PubMed] [Google Scholar]
- Churg A, Zhou S, Wright JL (2012). Series ‘matrix metalloproteinases in lung health and disease’: Matrix metalloproteinases in COPD. Eur Respir J 39: 197–209. [DOI] [PubMed] [Google Scholar]
- Cross A, Moots RJ, Edwards SW (2008). The dual effects of TNFalpha on neutrophil apoptosis are mediated via differential effects on expression of Mcl‐1 and Bfl‐1. Blood 111: 878–884. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Mar Hernandez M, Esteban M, Szabo P, Boada M, Unzeta M (2005). Human plasma semicarbazide sensitive amine oxidase (SSAO), β‐amyloid protein and aging. Neurosci Lett 384: 183–187. [DOI] [PubMed] [Google Scholar]
- Devlin AJ, Bhatti AR, Williams AC, Ramsden DB (1990). Inhibition of human benzylamine oxidase (BzAO) by analogues of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). Toxicol Lett 54: 135–142. [DOI] [PubMed] [Google Scholar]
- Donaldson GC, Seemungal TAR, Patel IS, Bhowmik A, Wilkinson TMA, Hurst JR et al. (2005). Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 128: 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML (2012). Distinct Macrophage Subpopulations Characterize Acute Infection and Chronic Inflammatory Lung Disease. J Immunol 189: 946–955. [DOI] [PubMed] [Google Scholar]
- Dunsmore SE (2008). Treatment of COPD: A matrix perspective. COPD 3: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner MD, Anthonisen N, Coultas D, Kuenzli N, Perez‐Padilla R, Postma D et al. (2010). An official American Thoracic Society public policy statement: Novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 182: 693–718. [DOI] [PubMed] [Google Scholar]
- Essilfie A‐T, Horvat JC, Kim RY, Mayall JR, Pinkerton JW, Beckett EL et al. (2015). Macrolide therapy suppresses key features of experimental steroid‐sensitive and steroid‐insensitive asthma. Thorax 70: 458–467. [DOI] [PubMed] [Google Scholar]
- Fabbri L, Pauwels RA, Hurd SS (2004). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary updated 2003. COPD 1: 105–41– discussion 103–4 [DOI] [PubMed] [Google Scholar]
- Foot JS, Yow TT, Schilter H, Buson A, Deodhar M, Findlay AD et al. (2013). PXS‐4681 A, a potent and selective mechanism‐based inhibitor of SSAO/VAP‐1 with anti‐inflammatory effects in vivo . J Pharmacol Exp Ther 347: 365–374. [DOI] [PubMed] [Google Scholar]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G et al. (2014). The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15: 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, Deane A, Hansbro PM (2014). Animal models of chronic obstructive pulmonary disease. Expert Opin Drug Discovery 9: 629–645. [DOI] [PubMed] [Google Scholar]
- Ge Q, Chen L, Jaffar J, Argraves WS, Twal WO, Hansbro P et al. (2015). Fibulin1C peptide induces cell attachment and extracellular matrix deposition in lung fibroblasts. Sci Rep 5: 9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrand TS, Hackett TL, Altemeier WA, Matute‐Bello G, Hansbro PM, Knight DA (2014). Airway epithelial regulation of pulmonary immune homeostasis and inflammation. Clin Immunol 151: 1–15. [DOI] [PubMed] [Google Scholar]
- Hancox RJ, Poulton R, Greene JM, Filsell S, McLachlan CR, Rasmussen F et al. (2007). Systemic inflammation and lung function in young adults. Thorax 62: 1064–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansbro PM, Hamilton MJ, Fricker M, Gellatly SL, Jarnicki AG, Zheng D et al. (2014). Importance of mast cell Prss31/transmembrane tryptase/tryptase‐γ in lung function and experimental chronic obstructive pulmonary disease and colitis. J Biol Chem 289: 18214–18227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Guillamon M, Garcia‐Bonilla L, Sole M, Sosti V, Pares M, Campos M et al. (2010). Plasma VAP‐1/SSAO activity predicts intracranial hemorrhages and adverse neurological outcome after tissue plasminogen activator treatment in stroke. Stroke 41: 1528–1535. [DOI] [PubMed] [Google Scholar]
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L et al. (2004). The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350: 2645–2653. [DOI] [PubMed] [Google Scholar]
- Horvat JC, Starkey MR, Kim RY, Beagley KW, Preston JA, Gibson PG et al. (2010). Chlamydial Respiratory Infection during Allergen Sensitization Drives Neutrophilic Allergic Airways Disease. J Immunol 184: 4159–4169. [DOI] [PubMed] [Google Scholar]
- Hsu A, Starkey M, Hanish I, Parsons K, Haw T, Howland L et al. (2015). Targeting PI3K-p110α suppresses influenza viral infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 191: 1012–1023. [DOI] [PubMed] [Google Scholar]
- Ishii T, Abboud RT, Wallace AM, English JC, Coxson HO, Finley RJ et al. (2013). Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur Respir J 43: 82–91. [DOI] [PubMed] [Google Scholar]
- Jaffar J, Unger S, Corte TJ, Keller M, Wolters PJ, Richeldi L et al. (2014). Fibulin‐1 predicts disease progression in patients with idiopathic pulmonary fibrosis. Chest 146: 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowich MD, Rounds SIS (2012). Combined Pulmonary Fibrosis and Emphysema Syndrome. Chest 141: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadi I, Meszaros Z, Csanyi A, Szombathy T, Hosszufalusi N, Romics L et al. (2002). Serum semicarbazide‐sensitive amine oxidase (SSAO) activity is an independent marker of carotid atherosclerosis. Clin Chim Acta 323: 139–146. [DOI] [PubMed] [Google Scholar]
- Keatings VM, Collins PD, Scott DM, Barnes PJ (1996). Differences in interleukin‐8 and tumor necrosis factor‐alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 153: 530–534. [DOI] [PubMed] [Google Scholar]
- Keely S, Talley NJ, Hansbro PM (2011). Pulmonary‐intestinal cross‐talk in mucosal inflammatory disease. Mucosal Immunol 5: 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss J, Jalkanen S, Fülöp F, Savunen T, Salmi M (2008). Ischemia–reperfusion injury is attenuated in VAP‐1‐deficient mice and by VAP‐1 inhibitors. Eur J Immunol 38: 3041–3049. [DOI] [PubMed] [Google Scholar]
- Ko FWS, Hui DSC (2012). Air pollution and chronic obstructive pulmonary disease. Respirology 17: 395–401. [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P (2013). Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–175. [DOI] [PubMed] [Google Scholar]
- Lau JY, Oliver BG, Baraket M, Beckett EL, Hansbro NG, Moir LM et al. (2010). Fibulin‐1 is increased in asthma‐‐a novel mediator of airway remodeling? PLoS One 5: e13360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzo JC, Turner AL, Cook AD, Vlahos R, Anderson GP, Reynolds EC et al. (2012). Control of macrophage lineage populations by CSF‐1 receptor and GM‐CSF in homeostasis and inflammation. Immunol Cell Biol 90: 429–440. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Fernandez de Arriba A, Lyles GA, Unzeta M (1994). Several aspects on the amine oxidation by semicarbazide‐sensitive amine oxidase (SSAO) from bovine lung. J Neural Transm Suppl 41: 415–420. [DOI] [PubMed] [Google Scholar]
- Lo H‐M, Lai T‐H, Li C‐H, Wu W‐B (2014). TNF‐α induces CXCL1 chemokine expression and release in human vascular endothelial cells in vitro via two distinct signaling pathways. Nat Publ Group 35: 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V et al. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD (2007). CD8+ T Cells Are Required for Inflammation and Destruction in Cigarette Smoke‐Induced Emphysema in Mice. J Immunol 178: 8090–8096. [DOI] [PubMed] [Google Scholar]
- Mako V, Czucz J, Weiszhar Z, Herczenik E, Matko J, Prohaszka Z et al. (2010). Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL‐1beta, TNF‐alpha, and LPS. Cytometry A 77: 962–970. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merinen M, Irjala H, Salmi M, Jaakkola I, Hänninen A, Jalkanen S (2005). Vascular adhesion protein‐1 is involved in both acute and chronic inflammation in the mouse. Am J Pathol 166: 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minematsu N, Shapiro SD (2007). To live and die in the LA (lung airway): mode of neutrophil death and progression of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 37: 129–130. [DOI] [PubMed] [Google Scholar]
- Miyake M, Goodison S, Urquidi V, Gomes Giacoia E, Rosser CJ (2013). Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab Invest 93: 768–778. [DOI] [PubMed] [Google Scholar]
- Nath KD, Burel JG, Shankar V, Pritchard AL, Towers M, Looke D et al. (2014). Clinical factors associated with the humoral immune response to influenza vaccination in chronic obstructive pulmonary disease. COPD 9: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemcsik J, Szoko E, Soltesz Z, Fodor E, Toth L, Egresits J et al. (2007). Alteration of serum semicarbazide‐sensitive amine oxidase activity in chronic renal failure. J Neural Transm 114: 841–843. [DOI] [PubMed] [Google Scholar]
- Palfreyman MG, McDonald IA, Bey P, Danzin C, Zreika M, Cremer G (1994). Haloallylamine inhibitors of MAO and SSAO and their therapeutic potential. J Neural Transm Suppl. 41: 407–414. [DOI] [PubMed] [Google Scholar]
- Precious E, Gunn CE, Lyles GA (1988). Deamination of methylamine by semicarbazide‐sensitive amine oxidase in human umbilical artery and rat aorta. Biochem Pharmacol 37: 707–713. [DOI] [PubMed] [Google Scholar]
- Rennard SI (2006). Chronic obstructive pulmonary disease: linking outcomes and pathobiology of disease modification. Proc Am Thorac Soc 3: 276–280. [DOI] [PubMed] [Google Scholar]
- Salazar LM, Herrera AM (2011). Fibrotic Response of Tissue Remodeling in COPD. Lung 189: 101–109. [DOI] [PubMed] [Google Scholar]
- Schilter HC, Collison A, Russo RC, Foot JS, Yow TT, Vieira AT et al. (2015). Effects of an anti‐inflammatory VAP‐1/SSAO inhibitor, PXS‐4728A, on pulmonary neutrophil migration. Respir Res 16: 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh J‐M, Tsai Y‐J, Tsou C‐J, Wu W‐B (2014). CXCL1 Regulation in Human Pulmonary Epithelial Cells by Tumor Necrosis Factor. Cell Physiol Biochem 34: 1373–1384. [DOI] [PubMed] [Google Scholar]
- Singh B, Tschernig T, van Griensven M, Fieguth A, Pabst R (2003). Expression of vascular adhesion protein‐1 in normal and inflamed mice lungs and normal human lungs. Virchows Arch 442: 491–495. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res: 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangl V, Günther C, Jarrin A, Bramlage P, Moobed M, Staudt A et al. (2001). Homocysteine inhibits TNF‐alpha‐induced endothelial adhesion molecule expression and monocyte adhesion via nuclear factor‐kappaB dependent pathway. Biochem Biophys Res Commun 280: 1093–1100. [DOI] [PubMed] [Google Scholar]
- Starkey MR, Nguyen DH, Essilfie AT, Kim RY, Hatchwell LM, Collison AM et al. (2013). Tumor necrosis factor‐related apoptosis‐inducing ligand translates neonatal respiratory infection into chronic lung disease. Mucosal Immunol 7: 478–488. [DOI] [PubMed] [Google Scholar]
- Suissa S, Patenaude V, Lapi F, Ernst P (2013). Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax 68: 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam A, Churg A, Wright JL, Zhou S, Kirby M, Coxson HO et al. (2016). Sex differences in airway remodeling in a mouse model of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 193: 825–834. [DOI] [PubMed] [Google Scholar]
- Tay HL, Kaiko GE, Plank M, Li J, Maltby S, Essilfie A‐T et al. (2015). Antagonism of miR‐328 increases the antimicrobial function of macrophages and neutrophils and rapid clearance of non‐typeable Haemophilus influenzae (NTHi) from infected lung. PLoS Pathog 11: e1004549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn AN, O'Sullivan BJ, Thomas R, Kumar RK, Foster PS, Gibson PG et al. (2010). Pneumococcal conjugate vaccine‐induced regulatory T cells suppress the development of allergic airways disease. Thorax 65: 1053–1060. [DOI] [PubMed] [Google Scholar]
- Turato G, Zuin R, Miniati M, Baraldo S, Rea F, Beghé B et al. (2002). Airway Inflammation in Severe Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 166: 105–110. [DOI] [PubMed] [Google Scholar]
- Turner A, Fenwick P, Barnes P, Donnelly L (2011). Roflumilast potently inhibits CXCL1 mediated neutrophil migration in COPD patients. Eur Respir J 38: 3826. [Google Scholar]
- US Dept. Health and Human Services (1982). Constituents of Tobacco Smoke. USPHS publication No. 82‐50179, 322.
- Vignola AM, Chanez P, Chiappara G, Merendino A, Pace E, Rizzo A et al. (1997). Transforming growth factor‐beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med 156: 591–599. [DOI] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S (2014). Recent advances in pre‐clinical mouse models of COPD. Clin Sci 126: 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S, Chan SPJ, Ivanov S, Linden A, Hamilton JA et al. (2010). Neutralizing granulocyte/macrophage colony‐stimulating factor inhibits cigarette smoke‐induced lung inflammation. Am J Respir Crit Care Med 182: 34–40. [DOI] [PubMed] [Google Scholar]
- Wallace WAH, Fitch PM, Simpson AJ, Howie SEM (2006). Inflammation‐associated remodelling and fibrosis in the lung ‐ a process and an end point. Int J Exp Pathol 88: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li H, Wei J, Lin M, Shih S, Hua C et al. (2013). Serum vascular adhesion protein‐1 level is higher in smokers than non‐smokers. Ann Hum Biol 40: 413–418. [DOI] [PubMed] [Google Scholar]
- Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW et al. (2014). Vascular adhesion protein‐1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest 125: 501–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Saad S, Zhang J, Gross S, Jarolimek W, Schilter H et al. (2014). Semicarbazide‐sensitive amine oxidase (SSAO) inhibition ameliorates kidney fibrosis in a unilateral ureteral obstruction murine model. Am J Physiol Renal Physiol 307: F908–F916. [DOI] [PubMed] [Google Scholar]
- Yang IA, Fong KM, Sim EHA, Black PN, Lasserson TJ (2007). Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2: 1–126. [DOI] [PubMed] [Google Scholar]
- Yang IA, Clarke MS, Sim EHA, Fong KM (2012). Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane (Rev) 10:1–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu PH (1998). Increase of formation of methylamine and formaldehyde in vivo after administration of nicotine and the potential cytotoxicity. Neurochem Res 23: 1205–1210. [DOI] [PubMed] [Google Scholar]
- Yu PH, Lu L‐X, Fan H, Kazachkov M, Jiang Z‐J, Jalkanen S et al. (2006). Involvement of Semicarbazide‐Sensitive Amine Oxidase‐Mediated Deamination in Lipopolysaccharide‐Induced Pulmonary Inflammation. Am J Pathol 168: 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]