

Abstract
Background and Purpose
Liver fibrosis is one of the leading causes of morbidity and mortality worldwide with very limited therapeutic options. Given the pivotal role of activated hepatic stellate cells in liver fibrosis, attention has been directed towards the signalling pathways underlying their activation and fibrogenic functions. Recently, the hedgehog (Hh) signalling pathway has been identified as a potentially important therapeutic target in liver fibrosis. The present study was designed to explore the antifibrotic effects of the potent Hh signalling inhibitor, forskolin, and the possible molecular mechanisms underlying these effects.
Experimental Approach
Male Sprague‐Dawley rats were treated with either CCl4 and/or forskolin for 6 consecutive weeks. Serum hepatotoxicity markers were determined, and histopathological evaluation was performed. Hepatic fibrosis was assessed by measuring α‐SMA expression and collagen deposition by Masson's trichrome staining and hydroxyproline content. The effects of forskolin on oxidative stress markers (GSH, GPx, lipid peroxides), inflammatory markers (NF‐κB, TNF‐α, COX‐2, IL‐1β), TGF‐β1 and Hh signalling markers (Ptch‐1, Smo, Gli‐2) were also assessed.
Key Results
Hepatic fibrosis induced by CCl4 was significantly reduced by forskolin, as indicated by decreased α‐SMA expression and collagen deposition. Forskolin co‐treatment significantly attenuated oxidative stress and inflammation, reduced TGF‐β1 levels and down‐regulated mRNA expression of Ptch‐1, Smo and Gli‐2 through cAMP‐dependent PKA activation.
Conclusion and Implications
In our model, forskolin exerted promising antifibrotic effects which could be partly attributed to its antioxidant and anti‐inflammatory effects, as well as to its inhibition of Hh signalling, mediated by cAMP–dependent activation of PKA.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ECM
extracellular matrix
- Gli‐2
glioblastoma transcription factor‐2
- HCC
hepatocellular carcinoma
- Hh
hedgehog
- HSCs
hepatic stellate cells
- P‐CREB
phosphorylated cAMP response element‐binding protein
- Ptch‐1
patched‐1 receptor
- α‐SMA
α‐smooth muscle actin
- Smo
smoothened receptor
- TC
total cholesterol
- TG
triglycerides
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| Smo, Smoothened |
| Enzymes b |
| COX‐2, cyclooxygenase‐2 |
| PKA, protein kinase A |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Liver fibrosis is a reversible wound healing process characterized by excessive accumulation of extracellular matrix (ECM) or scar in response to chronic liver injury (Friedman, 2003). If left untreated, fibrosis can progress into cirrhosis, hepatocellular carcinoma (HCC) and ultimately liver failure. The main cellular source of ECM is activated hepatic stellate cells (HSCs). Following liver injury, HSCs undergo activation and transdifferentiation from a quiescent form into proliferative, fibrogenic, pro‐inflammatory and contractile myofibroblast‐like cells (Mormone et al., 2011). Many factors play key pathogenic roles in the activation of HSCs including ROS, lipid peroxidation products, NF‐κB and many growth factors such as TGF‐β1 and pro‐inflammatory cytokines such as TNF‐α (Gressner et al., 2002; Elsharkawy and Mann, 2007; Mormone et al., 2011). The activation of HSCs and hepatic accumulation of myofibroblasts represent the central pathophysiological convergence point of multiple signalling pathways that ultimately lead to liver fibrosis (He and Dai, 2015). A signalling pathway that has recently been involved in the pathogenesis of liver fibrosis is the hedgehog (Hh) signalling pathway (Yang et al., 2014).
The Hh pathway plays an important role in embryogenesis, tissue homeostasis, cell differentiation and proliferation. In healthy adult liver, this pathway is quiescent or silenced where Hh ligands are absent (Omenetti et al., 2011). In this case, the free Hh cell surface receptor patched‐1 (Ptch‐1) inhibits the 7‐transmembrane protein smoothened (Smo) signalling which in turn causes the glioblastoma (Gli) family transcription factors (Gli‐1, Gli‐2 and Gli‐3) to undergo phosphorylation by various intracellular kinases such as PKA. Finally, these Gli‐phosphorylated forms become a target for proteasomal degradation (Pan et al., 2006; Pan et al., 2009). However, there is aberrant activation of Hh signalling in chronic liver injury. The pathway is initiated when Hh ligands, produced from injured hepatocytes and cholangiocytes, interact with Ptch‐1, expressed on Hh‐responsive target cells (HSCs, hepatic myofibroblasts, liver progenitors, cholangiocytes and liver lymphocytes) (Choi et al., 2011). This interaction de‐represses the activity of Smo, which leads to the inhibition of PKA and other intracellular kinases, thus permitting the nuclear translocation of Gli family transcription factors that induce the expression of Gli target genes including several components of the Hh pathway itself as well as genes that regulate cell viability, proliferation and differentiation (Hooper and Scott, 2005).
Activation of the Hh pathway increases hepatic recruitment of inflammatory cells (Syn et al., 2010). It also promotes hepatic accumulation of myofibroblasts by stimulating activation of HSCs and cholangiocytes to undergo epithelial–mesenchymal transition (Choi et al., 2009; Omenetti and Diehl, 2011). Furthermore, various growth factors and cytokines produced during fibrosis such as TGF‐β1 stimulate HSCs to produce Hh ligands (Javelaud et al., 2012). The latter promotes the proliferation and viability of myofibroblasts as well as the neighbouring Hh responsive cells including liver progenitors, the initiators of HCC (Yang et al., 2008).
Recent studies have reported that Hh pathway inhibitors effectively attenuated liver fibrosis and promoted myofibroblasts to reacquire a more quiescent phenotype (Pratap et al., 2012; Hirsova et al., 2013). Several Hh pathway antagonists have been identified (Mas and Ruiz i Altaba, 2010). Forskolin, a labdane diterpenoid isolated from the Indian plant Coleus forskohlii, functions as an Hh pathway antagonist (Yamanaka et al., 2010). It is known for its antioxidant, anti‐inflammatory and anti‐metastatic effects (Agarwal and Parks, 1983; Niaz and Singh, 1999; Irie et al., 2001). Forskolin increases the level of cAMP through the activation of adenylyl cyclase, resulting in PKA activation. This subsequently leads to Gli phosphorylation and their proteasomal degradation (Makinodan and Marneros, 2012). In addition, forskolin suppresses cell proliferation and tumour growth in basal cell carcinoma and paediatric malignancies including rhabdomyosarcoma, hepatoblastoma and neuroblastoma through inhibition of Hh signalling (Yamanaka et al., 2010; Makinodan and Marneros, 2012).
Based on these earlier findings, the present study was designed to assess the antifibrotic effects of forskolin in carbon tetrachloride (CCl4)‐induced liver fibrosis as well as the possible molecular mechanisms underlying these effects, including its effect on oxidative stress, inflammation, NF‐κB, fibrosis markers and, more importantly, its effect on the Hh signalling pathway.
Methods
Animals
All animal care and experimental procedures complied with the guidelines of the Ethical Committee of Faculty of Pharmacy, Ain Shams University, Egypt and were approved by this Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Male Sprague‐Dawley rats (8 weeks old; 150–190 g) were obtained from El‐Nile Company for Pharmaceutical and Chemical industries, Cairo, Egypt. The animals were housed in controlled environmental conditions (24 ± 2°C temperature, 60–70% relative humidity, 12 h light/dark cycle) and provided with a standard pellet diet (containing not less than 20% protein, 5% fibre, 3.5% fat, 6.5% ash and a vitamin mixture) and water ad libitum.
General procedures
Chronic administration of CCl4 in rats has been widely used for experimental induction of hepatic fibrosis (Choi et al., 2009; Shen et al., 2014). Randomisation was carried out as follows. On arrival from El‐Nile Company, animals were assigned a group designation and weighed. A total number of 32 animals were divided into four different weight groups (eight animals per group). Each animal was assigned a temporary random number within the weight range group. On the basis of their position on the rack, cages were given a numerical designation. For each group, a cage was selected randomly from the pool of all cages. Two animals were removed from each weight range group and given their permanent numerical designation in the cages. Then, the cages were randomized within the exposure group. The experimenters were blinded to the treatments given to the animals and to the biochemical and histological analyses and the data analyses.
Experimental design
After acclimatization for 2 weeks, animals were randomly divided into four groups of eight rats each and treated for six consecutive weeks as follows: The first group was treated with CCl4 (50% CCl4/corn oil; 0.5 mL·kg−1, i.p.) twice a week to induce liver fibrosis. The second group was given forskolin only at a dose of 10 mg·kg−1, i.p., dissolved in a DMSO/saline solution (1:49) five times a week. The third group was given both CCl4 and forskolin. The dose of forskolin used here was based on the results of our preliminary study (Supporting Information). The fourth group served as the normal control, receiving vehicles only. At 24 h after the last injection, blood samples were collected from the retro‐orbital plexus after light anaesthesia with sodium pentobarbital (50 mg·kg−1, i.p.). Serum was separated by centrifugation at 3000× g for 10 min and was used for the assessment of liver functions. Rats were killed by cervical dislocation, and livers were removed and weighed. A portion of liver tissue was washed and homogenized to obtain a 20% (w·v−1) homogenate, which was used for assessment of oxidative stress, inflammatory and fibrogenic markers. Another portion was placed in formalin for immunohistochemical and histopathological analyses. The remainder was stored at −80°C, together with the 20% homogenate, until needed.
Serum biochemistry and liver index
Serum concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), total cholesterol (TC), triglycerides (TG) and albumin were determined colourimetrically using available commercial kits (Spectrum diagnostics, Cairo, Egypt). Liver index was calculated according to the formula: (liver weight/body weight) × 100.
Histopathological examination
Liver samples taken from rats in the different experimental groups were fixed in 10% formol saline for 24 h. Tissue washing was done by tap water; then serial dilutions of alcohol (methyl, ethyl and absolute ethyl) were used for dehydration. Specimens were cleared in xylene and embedded in paraffin at 56°C in a hot air oven for 24 h. Sections were embedded in paraffin and sliced into 4‐μm‐thick sections by a sledge microtome. The tissue sections were collected on glass slides, deparaffinized, stained with haematoxylin & eosin (H&E) and then examined by light microscopy (Banchroft et al., 1996).
Quantitative immunohistochemical analysis of NF‐κB (p65), COX‐2 and α‐SMA
Paraffin‐embedded tissue sections of 3 μm thickness were rehydrated first in xylene and then in graded ethanol solutions. The slides were blocked with 5% BSA in tris buffered saline (TBS) for 2 h. Immunohistochemical analyses were performed by a standard streptavidin–biotin–peroxidase procedure. The sections were immunostained with one of the following primary antibodies: rabbit polyclonal anti‐rat NF‐κB (p65) (Thermoscientific, Rockford, IL, USA; NF‐κB Cat. #RB‐9034‐R7), rabbit polyclonal antibody to rat COX‐2 (COX‐2 Cat. #RB‐9072‐R7; Thermoscientific) or mouse monoclonal antibody to rat α‐SMA (α‐SMA Cat.#MS‐113‐R7; Thermoscientific) at a concentration of 1 μg·mL−1 and incubated overnight at 4°C. After washing the slides with TBS, the sections were incubated with the corresponding biotinylated secondary antibody for 10–15 min. After that, the horseradish peroxidase‐conjugated streptavidin solution was added and incubated at room temperature for 10–15 min. Sections were then washed with TBS and incubated for 5–10 min in a solution of 0.02% diaminobenzidine containing 0.01% hydrogen peroxide. Counterstaining was performed using haematoxylin, and the slides were visualized under the light microscope. Immunohistochemical quantification was performed by measuring the percentage of immunopositive area using image analysis software (ImageJ, 1.46a, NIH, Bethesda, Maryland, USA).
elisa for TGF‐β1, TNF‐α, IL‐1β, NF‐κB (p65) and P‐CREB
Liver tissues were washed and homogenized in ice‐cold PBS (pH = 7.4) to obtain a 20% homogenate (w·v−1), which was then centrifuged for 15 min at 3000 x g and 4°C. The supernatant obtained was used for measuring TGF‐β1, TNF‐α (R&D Systems, Minneapolis, MN, USA) and IL‐1β (Cell Biolabs, San Diego, USA) using sandwich elisa kits according to the manufacturer's instructions. They were expressed as pg.mg−1 protein. Protein content was also determined in the supernatant using a commercially available kit (Biodiagnostic, Cairo, Egypt). NF‐κB (p65) and phosphorylated cAMP response element‐binding protein (P‐CREB; Ser133), a marker for cAMP‐dependent PKA activation, were measured in nuclear protein extracts using sandwich elisa kits from Cloud‐Clone Corp., Houston, TX, USA, and R&D Systems, Minneapolis, MN, USA, respectively. Nuclear protein extract was prepared from liver tissues using NE‐PER nuclear and cytoplasmic extraction reagent kit according to the manufacturer's protocol (Thermoscientific). Protein concentration was determined in the nuclear extract as well.
Hepatic oxidative stress markers
As described above, the supernatant obtained by centrifugation of the 20% homogenate was used for the assessment of oxidative stress markers. Lipid peroxidation was determined by estimating the level of thiobarbituric acid reactive substances measured as malondialdehyde (MDA), according to the method of Mihara (Mihara and Uchiyama, 1978). MDA is a decomposition product of the process of lipid peroxidation; thus, it is used as an indicator of this process. Briefly, 0.5 mL of the supernatant was added to 2.5 mL of 20% trichloroacetic acid and 1.0 mL of 0.6% thiobarbituric acid; then the mixture was heated for 20 min in a boiling water bath. After cooling, 4 mL of n‐butanol was added with shaking. The alcohol layer was then separated by centrifugation at 500 x g for 10 min, and the absorbance was measured at 535 nm. The results were expressed as nmol of MDA per g of wet tissue using 1,1,3,3‐tetraethoxypropane as standard. Reduced GSH was measured using a commercial kit (Biodiagnostic, Cairo, Egypt). In addition, GSH peroxidase (GPx) activity was determined by a kinetic method using a commercial kit (RANSEL, Randox Laboratories, Antrim, UK).
Markers of hepatic fibrosis
Besides TGF‐β1 and α‐SMA, hepatic collagen content was assessed histologically by visualizing the blue colour of collagen fibres using Masson's trichrome staining as well as biochemically through the determination of hydroxyproline concentration using the simplified method of Reddy (Reddy and Enwemeka, 1996). Briefly, 25 μL of 20% liver homogenate was hydrolyzed in 25 μL of 2N NaOH at 120°C for 30 min. Chloramine T solution (450 μL) was then added to the hydrolysate and left at room temperature for 25 min. After that, 500 μL of Ehrlich's solution was added and the mixture was incubated at 65°C for 20 min. After cooling for 10 min, the colour developed was measured spectrophotometrically at 550 nm. Results were expressed as μg·g−1 of wet tissue.
qRT‐PCR analysis of Ptch‐1, Smo and Gli‐2 gene expression
Total RNA was isolated from rat liver samples using Qiagen RNeasy Mini Kit. Reverse transcription and quantitative real‐time PCR (qRT‐PCR) were performed using QuantiTect SYBR® Green PCR Kit according to the manufacturer's protocol. The cycling conditions were as follows: reverse transcription at 50°C for 30 min, initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 55°C (β‐actin, Ptch‐1 and Smo) or 50°C (Gli‐2) for 30 s and extension at 72°C for 30 s. The relative mRNA level of the target genes was calculated by the comparative threshold cycle (Ct) method using β‐actin as an internal control for normalization. The fold change in the expression of each target gene was calculated by the following formula: relative quantification (RQ) = 2−ΔΔCt . The following primer sequences were used: β‐actin forward primer: 5′‐TCA CCC ACA CTG TGC CCA TCT ATG A‐3′, β‐actin reverse primer: 5′‐CAT CGG AAC CGC TCA TTG CCG ATA G‐3′, Ptch‐1 forward primer: 5′‐ACG CTC CTT TCC TCT TGA GAC‐3′, Ptch‐1 reverse primer: 5′‐TGA ACT GGG CAG CTA TGA AGT C‐3′, Smo forward primer: 5′‐GCC TGG TGC TTA TTG TGG‐3′, Smo reverse primer: 5′‐GGT GGT TGC TCT TGA TGG‐3′, Gli‐2 forward primer: 5′‐ATA AGC GGA GCA AGG TCA AG‐3′, and Gli‐2 reverse primer: 5′‐CAG TGG CAG TTG GTC TCG TA‐3′.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are expressed as mean ± SD. Multiple comparisons were performed using one‐way ANOVA followed by Tukey–Kramer as a post hoc test. P < 0.05 was considered statistically significant. All analyses were performed using Instat software package (version 3.06). Graphs were sketched using GraphPad Prism software (version 5).
Materials
Forskolin, CCl4, DMSO, chloramine‐T, p‐dimethylaminobenzaldehyde (Ehlrich reagent), hydroxyproline, 1,1,3,3‐tetraethoxypropane and thiobarbituric acid were purchased from Sigma Chemical Co. (St Louis, MO, USA). n‐Butanol and trichloroacetic acid were purchased from El‐Nasr Chemical Co., Cairo, Egypt. All other chemicals used were of highest grade commercially available.
Results
Forskolin treatment inhibited histopathological deterioration induced by CCl4
As shown in Figure 1A, liver sections obtained from both control and forskolin‐only treated group exhibited normal hepatic architecture of the central vein and surrounding hepatocytes, while those taken from CCl4 group involved ballooning degeneration and fatty change in the hepatocytes associated with strands of fibrous connective tissue, as well as severe inflammatory cell infiltration in between and in the portal area. Nevertheless, forskolin co‐treatment effectively restored the hepatic architecture with little inflammatory cell infiltration and mild ballooning degeneration.
Figure 1.
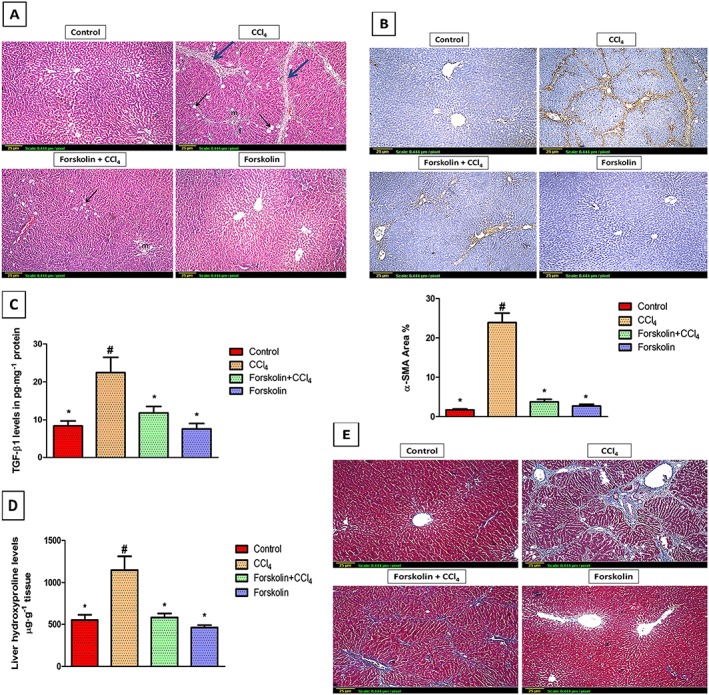
Forskolin treatment decreased histopathological deterioration and hepatic fibrogenesis induced by CCl4. (A) Histopathological analysis of rat liver sections using H&E staining (×100). Control and forskolin‐only treated groups showing normal histological structure of the central vein and surrounding hepatocytes. CCl4 group showing severe ballooning degeneration (black arrow), inflammatory cell infiltration (m) and fatty change (f) associated with strands of fibrous connective tissue (blue arrow). Forskolin + CCl4 group showing reduction in histological abnormalities with only few inflammatory cell infiltration (m) and mild ballooning degeneration (black arrow). (B) Immunohistochemical detection of α‐SMA (×100). Control and forskolin‐only treated groups showing minimal α‐SMA expression. CCl4 group showing extensive α‐SMA expression (brown stain). Forskolin + CCl4 group showing less α‐SMA expression. Quantitative image analysis for immunohistochemical staining of α‐SMA expressed as percentage of stained area averaged across 6 different fields for each rat of at least five rats. Each column represents mean ± SD. (C,D) Effect of forskolin on hepatic (C) TGF‐β1 and (D) hydroxyproline levels. Each column represents mean ± SD (n = 7). # P <0.05; significantly different from control group; * P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test. (E) Masson's trichrome staining of rat liver sections (×100). Control and forskolin‐only treated groups showing absence of collagen fibres (stained blue). CCl4 group showing extensive collagen fibres deposition (blue stain) with pseudolobules formation (bridging fibrosis). The forskolin + CCl4 group showed fewer fibres than the CCl4 group.
Forskolin treatment mitigated CCl4‐induced hepatic fibrogenesis
Immunohistochemical detection of α‐SMA revealed extensive brown staining in the CCl4 only group, amounting to 24% of the immunopositive area. However, in forskolin co‐treatment group, the positive stained area of α‐SMA was markedly reduced (Figure 1B). elisa analysis revealed that TGF‐β1 levels were higher in the CCl4 group than in the control group. Forskolin treatment caused a significant decrease in TGF‐β1 levels, compared with the CCl4 group (Figure 1C). Moreover, the antifibrotic effect of forskolin was further confirmed by biochemical determination of hydroxyproline levels. As seen in Figure 1D, the increase in hydroxyproline levels induced by CCl4 was completely prevented by forskolin co‐treatment. These results confirmed those obtained from the histological visualization of collagen fibres by Masson's trichrome stain (Figure 1E).
Forskolin treatment amended CCl4‐induced hepatic damage
As compared with the control group, ALT and AST serum levels were significantly elevated in the CCl4 group (Table 1). On the other hand, rats co‐treated with forskolin showed a significant decrease in the ALT and AST serum levels, compared with the CCl4 group but remained significantly higher than the control values. The CCl4 group also showed a significant rise in TC and TG, compared to the control. Notably, TC and TG serum levels were normalized in forskolin co‐treated group. Albumin levels were reduced in the CCl4 group, compared with the control but forskolin treatment returned albumin levels nearly to the normal value. Regarding liver index, the increase observed in the CCl4 group, compared with the control group, was significantly reduced by concurrent administration of forskolin with CCl4.. No significant changes were observed in the forskolin‐only treated group in all hepatotoxicity markers when compared with the values from the control group (Table 1).
Table 1.
Effect of forskolin on hepatotoxicity indices and liver index in CCl4‐intoxicated rats
| Group | ALT (U·L−1) | AST (U·L−1) | TC (g·L−1) | TG (g·L−1) | Albumin (g·L−1) | Liver index (%) |
|---|---|---|---|---|---|---|
| Control | 9.1 ± 1.18b | 16.6 ± 2.84b | 0.50 ± 0.12b | 0.99 ± 0.10b | 42.1 ± 1.91b | 2.86 ± 0.25b |
| CCl4 | 38.7 ± 3.64a | 45.5 ± 2.95a | 1.08 ± 0.15a | 1.56 ± 0.20a | 31.6 ± 3.10a | 4.63 ± 0.55a |
| Forskolin + CCl4 | 22.5 ± 4.67a , b | 31.3 ± 5.87a , b | 0. 62 ± 0.13b | 0. 86 ± 0.19b | 39.1 ± 3.83b | 3.24 ± 0.18b |
| Forskolin | 11.4 ± 2.55b | 18.4 ± 4.74b | 0. 46 ± 0.12b | 0. 95 ± 0.18b | 39.2 ± 2.63b | 3.23 ± 0.39b |
Data presented are means ± SD (n = 8).
P <0.05; significantly different from control group;
P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Forskolin treatment attenuated CCl4‐induced oxidative stress
GSH levels and GPx activity were markedly reduced while levels of lipid peroxides were significantly elevated in the CCl4 group, compared with the control group. Interestingly, forskolin co‐treatment returned both GSH and MDA to the normal levels and restored GPx activity, thus protecting against CCl4‐induced oxidative liver damage. Besides, rats given forskolin only did not show any change in GPx activity, GSH and MDA tissue levels when compared with the control values (Table 2).
Table 2.
Effect of forskolin on oxidative stress markers in CCl4‐intoxicated rats
| Group | GSH (μmol·g−1 tissue) | GPx (U·g−1 tissue) | MDA (nmol·g−1 tissue) |
|---|---|---|---|
| Control | 3.47 ± 0.8b | 99.73 ± 15.78b | 27.8 ± 0.69b |
| CCl4 | 0.94 ± 0.21a | 51.5 ± 3.65a | 35.1 ± 3.25a |
| Forskolin + CCl4 | 3.36 ± 0.75b | 87.01 ± 8.98b | 27.01 ± 2.27b |
| Forskolin | 2.98 ± 0.29b | 94.83 ± 12.36b | 28.2 ± 0.88b |
Data presented are means ± SD (n = 8).
P <0.05; significantly different from control group;
P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Forskolin treatment alleviated CCl4‐induced hepatic inflammation
As shown in Figure 2A, the marked rise in TNF‐α levels observed in the CCl4 group was completely blocked by forskolin treatment. Similarly, IL‐1β tissue levels were elevated in the CCl4 group, compared with the control group. Treatment with forskolin significantly reduced IL‐1β levels, compared with the CCl4 group (Figure 2B). In addition, the immunohistochemical detection of COX‐2 revealed a significant increase in its expression in the CCl4 group as shown by the intense brown staining, whereas minimum expression was observed in both the control and forskolin‐only treated groups. Forskolin co‐treatment significantly decreased COX‐2 expression to near control levels of the immunopositive area (Figure 2C).
Figure 2.
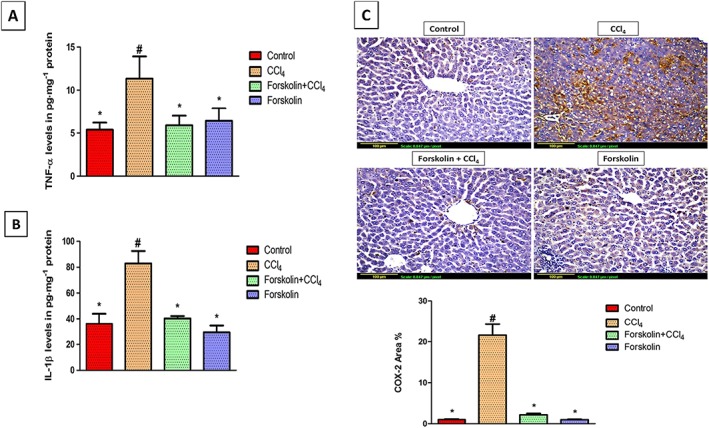
Forskolin treatment reduced inflammation associated with CCl4‐induced liver fibrosis. (A,B) Effect of forskolin on hepatic (A) TNF‐α levels and (B) IL‐1β levels. Each column represents mean ± SD (n = 7). (C) Immunohistochemical detection of COX‐2 (×200). Control and forskolin‐only treated groups showing minimal COX‐2 expression. CCl4 group showing extensive COX‐2 expression (brown stain). Forskolin + CCl4 group showing less COX‐2 expression. Quantitative image analysis for immunohistochemical staining of COX‐2 expressed as percentage of stained area averaged across six different fields for each rat of at least five rats. Each column represents mean ± SD. # P <0.05; significantly different from control group; * P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Forskolin treatment reduced NF‐κB expression in hepatic tissue
elisa analysis showed that chronic CCl4 administration caused a substantial rise in NF‐κB tissue levels, compared with the control group. Forskolin treatment successfully reduced NF‐κB levels, compared with the CCl4 group; however, it remained significantly higher than the control group (Figure 3A). These results matched those obtained from immunohistochemistry. CCl4‐intoxicated rats showed a significant increase in hepatic NF‐κB expression and treatment with forskolin successfully attenuated this effect (Figure 3B).
Figure 3.
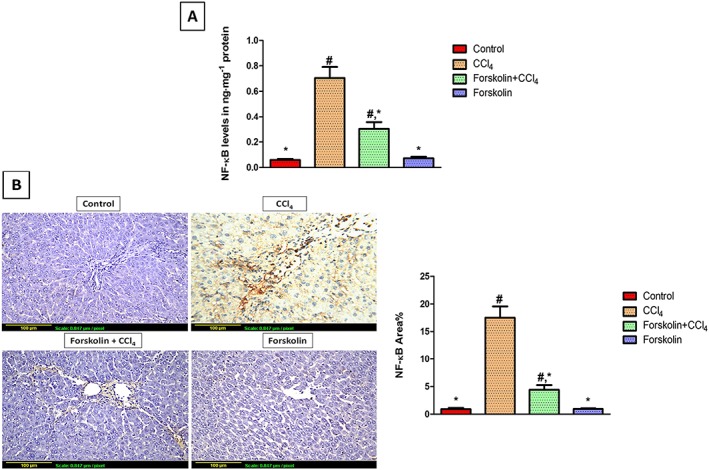
Forskolin treatment suppressed NF‐κB expression in liver tissue. (A) NF‐κB tissue levels determined by elisa. Each column represents mean ± SD (n = 6). (B) Immunohistochemical detection of NF‐κB (×200). Control and forskolin‐only treated groups showing minimal NF‐κB expression. CCl4 group showing extensive NF‐κB expression (brown stain). Forskolin + CCl4 group showing less NF‐κB expression. Quantitative image analysis for immunohistochemical staining of NF‐κB expressed as percentage of stained area averaged across six different fields for each rat of at least five rats. Each column represents mean ± SD. # P <0.05; significantly different from control group; * P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Forskolin treatment activated PKA and suppressed the extensive activation of Hh signalling induced by CCl4
The effect of forskolin on PKA was determined by measuring its downstream target, P‐CREB. elisa analysis revealed that P‐CREB tissue levels were significantly decreased in the CCl4 group, compared with the control group. However, a significant increase in P‐CREB levels was observed in the forskolin co‐treatment group, compared with the CCl4 group, confirming the activation of PKA (Figure 3A). Regarding Hh signalling, qRT‐PCR analysis of Ptch‐1, Smo and Gli‐2 mRNA expression in the forskolin‐only treated group showed no significant changes from the control group. Strikingly, CCl4 significantly increased Ptch‐1, Smo and Gli‐2 mRNA expression, compared with the control group. Co‐treatment with forskolin significantly down‐regulated the genetic expression of Ptch‐1, Smo and Gli‐2, compared with the CCl4 group (Figure 4B–D), confirming the inhibitory effect of forskolin on the Hh signalling pathway.
Figure 4.
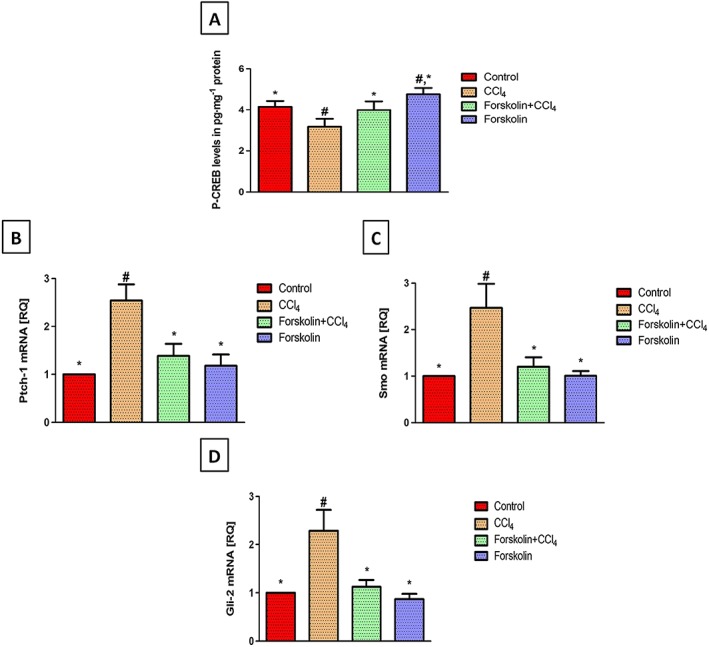
Forskolin treatment activated PKA and blocked Hh signalling induced by chronic CCl4 administration. (A) Effect of forskolin on tissue levels of P‐CREB (marker for PKA activation). Each column represents mean ± SD (n = 6). (B–D) Effect of forskolin on mRNA expression of (B) Ptch‐1, (C) Smo and (D) Gli‐2 expressed as relative quantification (RQ) compared with the control group, which was assigned a value of 1. Each column represents mean ± SD (n = 5). # P <0.05; significantly different from control group; * P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Discussion
Liver fibrosis is associated with high morbidity and mortality worldwide and has very limited therapeutic options. Attention has been directed towards the emerging signalling pathways underlying fibrosis progression and regression in order to develop new therapies that either arrest or reverse the fibrotic process, as even advanced fibrosis could be reversed (Soriano et al., 2006). Recent studies have focused on the Hh pathway as an important target in the treatment of liver fibrosis (Choi et al., 2011; Philips et al., 2011). Accordingly, the current study aimed to investigate the potential antifibrotic effect of the Hh pathway inhibitor, forskolin, as well as to elucidate the possible underlying mechanisms by studying its effect on key events involved in hepatic fibrosis including oxidative stress, inflammation, NF‐κB, fibrogenesis and Hh signalling.
CCl4 has been widely used for experimental induction of hepatic fibrosis in rats (Hernandez‐Munoz et al., 1990) and this compound is metabolised by cytochrome P450 2E1, causing the production of free radicals. These free radicals cause lipid peroxidation of the hepatocellular membrane, followed by release of inflammatory cytokines and, eventually, hepatocellular damage (Weber et al., 2003). The latter was shown, in our model, by the elevation in serum ALT, AST, TC and TG in the CCl4 group, while significantly decreased serum albumin reflected the negative effect of chronic CCl4 administration on hepatic synthetic function. Co‐treatment with forskolin significantly attenuated all of these hepatic changes, indicating improved functional status of hepatocytes as well as restored hepatocellular architecture, shown by the H&E staining. These results confirm the hepatoprotective effect of forskolin that was reported previously (Ji et al., 2012).
Liver fibrosis of any aetiology is characterized by excessive accumulation of ECM components especially collagen, the predominant component of this ECM (Friedman, 2003). Chronic CCl4 administration induced liver fibrosis and collagen deposition as indicated by Masson's trichrome staining of collagen fibres as well as increased liver hydroxyproline content, a marker of collagen deposition. The present study demonstrates that forskolin possesses a potent antifibrotic activity and prevents CCl4‐induced fibrogenesis. This was confirmed by the significant decrease in liver hydroxyproline levels that coincided histologically with the reduction of collagen deposition, as measured with Masson's stain.
Activation of HSCs and their transdifferentiation to myofibroblasts represent the main pathophysiological event in liver fibrosis. Activated HSCs are the major fibrogenic cells in injured liver and are characterized by enhanced proliferation, overproduction of ECM and de novo expression of α‐SMA, which is a marker for activated HSCs (Friedman, 2000). Therefore, antifibrotic strategies target activated HSCs as an approach for the prevention and treatment of liver fibrosis. The antifibrotic effect of forskolin was further confirmed by the significant reduction in α‐SMA expression, indicating that forskolin successfully inhibited HSC activation and the subsequent fibrogenic process. This result is consistent with previous in vitro studies showing that forskolin decreased collagen synthesis and suppressed the proliferation and differentiation in human pulmonary fibroblasts and activated HSCs (Mallat et al., 1996; Liu et al., 2004).
In order to determine the possible mechanisms underlying this antifibrotic effect and inhibition of HSC activation, we examined some of the key events inducing this activation. Oxidative stress plays an important role in hepatocellular damage and fibrogenesis (Parola and Robino, 2001). Damaged hepatocytes release lipid peroxidation products and ROS that cause death of the hepatocytes, amplify the hepatic inflammatory response as well as inducing HSC activation (Nieto et al., 2002; Nieto, 2006). The endogenous GSH antioxidant defence system provides the first line of defence against lipid peroxidation and ROS. GSH is an important non‐enzymatic part of this system that can directly scavenge free radicals or act as a substrate for the antioxidant enzyme GPx during detoxification of lipid peroxides and ROS (Szymonik‐Lesiuk et al., 2003; Masella et al., 2005).
CCl4 toxicity contributes to oxidative stress through production of free radicals. This was supported by the finding that chronic CCl4 administration significantly increased lipid peroxides, as shown by raised MDA levels in our model, accompanied by a significant depletion in liver GSH levels and GPx activity (Colak et al., 2016). In agreement with previous studies, our results showed that forskolin indeed has a potent antioxidant activity. Forskolin effectively inhibited lipid peroxidation and restored the hepatic antioxidant defence system as shown by the normalized levels of lipid peroxides, GSH and GPx activity (Kamata et al., 1996; Niaz and Singh, 1999).
Another main event involved in the fibrogenic process is the inflammatory process initiated by Kupffer cell activation (Racanelli and Rehermann, 2006). Following liver injury, NF‐κB and its downstream signalling are activated in both Kupffer cells and HSCs. NF‐κB is a transcription factor that up‐regulates the expression of various pro‐inflammatory cytokines such as TNF‐α and IL‐1β as well as pro‐inflammatory enzymes such as COX‐2. TNF‐α and IL‐1β cause further activation of NF‐κB, hence perpetuating the inflammatory cascade. Besides promoting inflammation, NF‐κB signalling is an important contributor to the activation and survival of HSCs (Elsharkawy and Mann, 2007; Luedde and Schwabe, 2011). Consistent with previous studies, our study showed that CCl4 increased NF‐κB expression with a subsequent rise in liver TNF‐α, IL‐1β levels as well as hepatic COX‐2 expression, thus worsening hepatic inflammation and fibrosis (Orfila et al., 2005).
Forskolin is known to suppress TNF‐α production in rat Kupffer cells and in a mouse model of lipopolysaccharide‐induced inflammation (Irie et al., 2001; Dahle et al., 2005). Notably, forskolin inhibited macrophage infiltration and suppressed TNF‐α and IL‐1β production in a mouse model of acute pyelonephritis (Wei et al., 2015). Moreover, Ji and his co‐workers showed that forskolin diminished neutrophil and macrophage infiltration or activation as well as reducing hepatocyte necrosis and apoptosis – in vivo and in vitro – through inhibition of NF‐κB and its downstream signalling leading to suppression of multiple pro‐inflammatory cytokines including TNF‐α and IL‐1β (Ji et al., 2012). In this context, cAMP/PKA activation by forskolin inhibited NF‐κB either directly or indirectly through phosphorylation of CREB. These data were reflected in our findings that forskolin induced an elevation in P‐CREB levels, suppressed NF‐κB expression and inhibited the subsequent inflammatory cascade shown by the decreased hepatic content of COX‐2, TNF‐α and IL‐1β.
Indeed, NF‐κB promotes fibrogenesis by up‐regulating the expression of several growth factors including TGF‐β1, the most potent fibrogenic stimuli to HSCs (Friedman, 1999). TGF‐β1 produced from autocrine and paracrine sources is a strong inducer of HSC activation, epithelial–mesenchymal transition and collagen deposition (Inagaki and Okazaki, 2007). Notably, forskolin co‐treatment significantly decreased liver TGF‐β1 levels which may be explained by the inhibitory effect of forskolin on NF‐κB.
The Hh signalling pathway is now identified as a novel key player in liver fibrosis. Several types of resident liver cells produce Hh ligands in response to chronic liver injury. These Hh ligands activate the pathway in a range of Hh‐responsive cells including HSCs (Choi et al., 2011; Omenetti et al., 2011). Activation of Hh signalling contributes to the pathogenesis of liver fibrosis by promoting recruitment of inflammatory cells and capillarization of the sinusoids (Omenetti et al., 2009; Syn et al., 2010; Xie et al., 2013). More importantly, several in vitro and in vivo studies confirmed that activity of the Hh pathway is required for activation and transdifferentiation of quiescent HSCs into myofibroblasts (Sicklick et al., 2005; Choi et al., 2009; Choi et al., 2010). Hh signalling activation also promotes the up‐regulation of myofibroblast‐associated genes including α‐SMA, collagen 1α1 and TGF‐β1, a potent inducer of Gli‐2 transcription and Hh signalling (Choi et al., 2009; Choi et al., 2010). Studies also revealed that inhibiting the Hh pathway down‐regulated myofibroblast‐associated genes and caused the reversal of myofibroblasts to a quiescent phenotype, thus reducing hepatic content of myofibroblasts and attenuating fibrosis (Choi et al., 2009; Pratap et al., 2012). Interestingly, Philips and his co‐workers proved that inhibition of Hh pathway with vismodegib, a Smo antagonist, promoted the regression of both advanced liver fibrosis and HCC (Philips et al., 2011).
In agreement with previous studies, our results showed that liver fibrosis induced by CCl4 is associated with excessive activation of Hh signalling as shown by increased expression of the Hh pathway components, Ptch‐1, Smo and the transcriptional activator Gli‐2, where the level of Hh pathway activation paralleled the increase in hepatic collagen content (Fleig et al., 2007; Choi et al., 2009; Shen et al., 2014). Activation of the Hh pathway results in Smo‐dependent inhibition of PKA. The observed reduction in P‐CREB levels in fibrotic rat livers indicates the inhibition of PKA. Recent in vitro and in vivo studies have shown that activation of PKA by the cAMP stimulant forskolin is sufficient to suppress Hh signalling by indirectly antagonizing Gli‐1 and Gli‐2 (Cohen et al., 2010; Yamanaka et al., 2010, Yamanaka et al., 2011; Makinodan and Marneros, 2012). In addition, activation of PKA by cAMP results in phosphorylation of its main downstream target CREB at Ser133; thus, P‐CREB can be used as a marker for activation of the cAMP/PKA system (Gonzalez and Montminy, 1989; Makinodan and Marneros, 2012; Lee et al., 2013).
Our findings confirming that forskolin is a potent Hh signalling inhibitor, demonstrated by the significant decrease in Ptch‐1, Smo and Gli‐2 expression are consistent with earlier studies. Forskolin indirectly inhibited Hh signalling through increasing cAMP levels. The latter is known to induce the activation of PKA which results in phosphorylation and proteasomal degradation of Gli‐1 and Gli‐2. Forskolin‐induced cAMP‐dependent PKA activation was confirmed by the elevation in tissue levels of P‐CREB(Ser133). Such inhibition of the Hh pathway would explain the observed reduction in hepatic TGF‐β1, α‐SMA and collagen deposition.
In light of all the previous findings, the current study demonstrated for the first time that the Hh pathway inhibitor, forskolin, has a promising antifibrotic effect. Forskolin markedly attenuated liver fibrosis through suppressing HSC activation. The molecular mechanisms underlying this effect involve attenuating oxidative stress and inflammation, inhibiting NF‐κB, reducing fibrogenesis and, more importantly, inhibiting the Hh signalling pathway through cAMP‐mediated activation of PKA. The latter, in turn, caused further attenuation of inflammation, repression of HSC activation and, consequently, decreased liver fibrogenesis.
Because forskolin acts through increasing cAMP, a highly versatile second messenger that regulates tissue fibrosis through activating many downstream effectors other than PKA, such as Epac (exchange protein activated by cAMP) which are also involved in cAMP‐mediated inhibition of tissue fibrosis (Insel et al., 2012), there is a strong possibility that the antifibrotic effect of forskolin could be also mediated through other cAMP‐binding proteins and signalling pathways involved in the fibrotic process. Therefore, further studies are needed to elucidate the other mechanisms underlying the antifibrotic effects of forskolin.
Author contributions
R.N.E.‐N. and E.E.‐D. designed the research study. N.N.E.‐A. performed the research. N.N.E.‐A. and R.N.E.‐N. performed the analysis and interpretation of data. N.N.E.‐A. wrote the article. N.N.E.‐A., R.N.E.‐N., R.A.E.‐R. and E.E.‐D. revised the article critically for important intellectual content.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of different doses of forskolin on serum levels of (A) ALT and (B) AST. Each column represents mean ± SD (n = 6). # P <0.05; significantly different from control group; * P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Figure S2 Histopathological analysis of rat liver sections using H&E staining (x100). A: Control group showing normal histological structure of the central vein and surrounding hepatocytes. B: CCl4 group showing centrilobular necrosis (n) with ballooning degeneration in the hepatocytes (arrow) associated with dilatation in the central vein (CV). C: Forskolin (5 mg.kg−1) + CCl4 group showing centrilobular necrosis (n) with ballooning degeneration (arrow) in diffuse manner all over the hepatocytes in association with congestion in the portal vein (PV). D: Forskolin (10 mg.kg−1) + CCl4 group showing reduction in histological abnormalities with only ballooning degeneration (arrow) in some of the hepatocytes and dilatation in the central vein (CV). E: Forskolin (20 mg.kg−1) + CCl4 group showing centrilobular necrosis (n) with ballooning degeneration (arrow) associated with congestion in the portal vein (PV).
Supporting info item
El‐Agroudy, N. N. , El‐Naga, R. N. , El‐Razeq, R. A. , and El‐Demerdash, E. (2016) Forskolin, a hedgehog signalling inhibitor, attenuates carbon tetrachloride‐induced liver fibrosis in rats. British Journal of Pharmacology, 173: 3248–3260. doi: 10.1111/bph.13611.
References
- Agarwal KC, Parks RE Jr (1983). Forskolin: a potential antimetastatic agent. Int J Cancer 32: 801–804. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchroft JD, Stevens A, Turner DR (1996). Theory and Practice of Histological Techniques, Fourth Edition edn. New York, London, San Francisco, Tokyo: Churchil Livingstone. [Google Scholar]
- Choi SS, Omenetti A, Syn WK, Diehl AM (2011). The role of Hedgehog signaling in fibrogenic liver repair. Int J Biochem Cell Biol 43: 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Omenetti A, Witek RP, Moylan CA, Syn WK, Jung Y et al. (2009). Hedgehog pathway activation and epithelial‐to‐mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol 297: G1093–G1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Witek RP, Yang L, Omenetti A, Syn WK, Moylan CA et al. (2010). Activation of Rac1 promotes hedgehog‐mediated acquisition of the myofibroblastic phenotype in rat and human hepatic stellate cells. Hepatology 52: 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JR, Resnick DZ, Niewiadomski P, Dong H, Liau LM, Waschek JA (2010). Pituitary adenylyl cyclase activating polypeptide inhibits Gli1 gene expression and proliferation in primary medulloblastoma derived tumorsphere cultures. BMC Cancer 10: 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak E, Ustuner MC, Tekin N, Burukoglu D, Degirmenci I, Gunes HV (2016). The hepatocurative effects of Cynara scolymus L. leaf extract on carbon tetrachloride‐induced oxidative stress and hepatic injury in rats. Springerplus 5: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahle MK, Myhre AE, Aasen AO, Wang JE (2005). Effects of forskolin on Kupffer cell production of interleukin‐10 and tumor necrosis factor alpha differ from those of endogenous adenylyl cyclase activators: possible role for adenylyl cyclase 9. Infect Immun 73: 7290–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsharkawy AM, Mann DA (2007). Nuclear factor‐kappaB and the hepatic inflammation–fibrosis–cancer axis. Hepatology 46: 590–597. [DOI] [PubMed] [Google Scholar]
- Fleig SV, Choi SS, Yang L, Jung Y, Omenetti A, VanDongen HM et al. (2007). Hepatic accumulation of Hedgehog‐reactive progenitors increases with severity of fatty liver damage in mice. Lab Invest 87: 1227–1239. [DOI] [PubMed] [Google Scholar]
- Friedman SL (1999). Cytokines and fibrogenesis. Semin Liver Dis 19: 129–140. [DOI] [PubMed] [Google Scholar]
- Friedman SL (2000). Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275: 2247–2250. [DOI] [PubMed] [Google Scholar]
- Friedman SL (2003). Liver fibrosis – from bench to bedside. J Hepatol 38 (Suppl. 1): S38–S53. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR (1989). Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59: 675–680. [DOI] [PubMed] [Google Scholar]
- Gressner AM, Weiskirchen R, Breitkopf K, Dooley S (2002). Roles of TGF‐beta in hepatic fibrosis. Front Biosci 7: d793–d807. [DOI] [PubMed] [Google Scholar]
- He W, Dai C (2015). Key fibrogenic signaling. Curr Pathobiol Rep 3: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Munoz R, Diaz‐Munoz M, Suarez J, Chagoya de Sanchez V (1990). Adenosine partially prevents cirrhosis induced by carbon tetrachloride in rats. Hepatology 12: 242–248. [DOI] [PubMed] [Google Scholar]
- Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ (2013). Vismodegib suppresses TRAIL‐mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS One 8: e70599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JE, Scott MP (2005). Communicating with Hedgehogs. Nat Rev Mol Cell Biol 6: 306–317. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Okazaki I (2007). Emerging insights into transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 56: 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Murray F, Yokoyama U, Romano S, Yun H, Brown L et al. (2012). cAMP and Epac in the regulation of tissue fibrosis. Br J Pharmacol 166: 447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie K, Fujii E, Ishida H, Wada K, Suganuma T, Nishikori T et al. (2001). Inhibitory effects of cyclic AMP elevating agents on lipopolysaccharide (LPS)‐induced microvascular permeability change in mouse skin. Br J Pharmacol 133: 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javelaud D, Pierrat MJ, Mauviel A (2012). Crosstalk between TGF‐beta and hedgehog signaling in cancer. FEBS Lett 586: 2016–2025. [DOI] [PubMed] [Google Scholar]
- Ji H, Shen XD, Zhang Y, Gao F, Huang CY, Chang WW et al. (2012). Activation of cyclic adenosine monophosphate‐dependent protein kinase a signaling prevents liver ischemia/reperfusion injury in mice. Liver Transpl 18: 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Tanaka C, Yagisawa H, Hirata H (1996). Nerve growth factor and forskolin prevent H2O2‐induced apoptosis in PC12 cells by glutathione independent mechanism. Neurosci Lett 212: 179–182. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YY, Moujalled D, Doerflinger M, Gangoda L, Weston R, Rahimi A et al. (2013). CREB‐binding protein (CBP) regulates beta‐adrenoceptor (beta‐AR)‐mediated apoptosis. Cell Death Differ 20: 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ostrom RS, Insel PA (2004). cAMP‐elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol 286: C1089–C1099. [DOI] [PubMed] [Google Scholar]
- Luedde T, Schwabe RF (2011). NF‐kappaB in the liver – linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 8: 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan E, Marneros AG (2012). Protein kinase A activation inhibits oncogenic Sonic hedgehog signalling and suppresses basal cell carcinoma of the skin. Exp Dermatol 21: 847–852. [DOI] [PubMed] [Google Scholar]
- Mallat A, Preaux AM, Serradeil‐Le Gal C, Raufaste D, Gallois C, Brenner DA et al. (1996). Growth inhibitory properties of endothelin‐1 in activated human hepatic stellate cells: a cyclic adenosine monophosphate‐mediated pathway. Inhibition of both extracellular signal‐regulated kinase and c‐Jun kinase and upregulation of endothelin B receptors. J Clin Invest 98: 2771–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas C, Ruiz i Altaba A (2010). Small molecule modulation of HH‐GLI signaling: current leads, trials and tribulations. Biochem Pharmacol 80: 712–723. [DOI] [PubMed] [Google Scholar]
- Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C (2005). Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione‐related enzymes. J Nutr Biochem 16: 577–586. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Uchiyama M (1978). Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem 86: 271–278. [DOI] [PubMed] [Google Scholar]
- Mormone E, George J, Nieto N (2011). Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact 193: 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niaz MA, Singh RB (1999). Modulation of free radical stress in human red blood cell membrane by forskolin and the prospects for treatment of cardiovascular disease and diabetes. Cell Mol Biol (Noisy‐le‐Grand) 45: 1203–1207. [PubMed] [Google Scholar]
- Nieto N (2006). Oxidative‐stress and IL‐6 mediate the fibrogenic effects of rodent Kupffer cells on stellate cells. Hepatology 44: 1487–1501. [DOI] [PubMed] [Google Scholar]
- Nieto N, Friedman SL, Cederbaum AI (2002). Cytochrome P450 2E1‐derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem 277: 9853–9864. [DOI] [PubMed] [Google Scholar]
- Omenetti A, Diehl AM (2011). Hedgehog signaling in cholangiocytes. Curr Opin Gastroenterol 27: 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenetti A, Choi S, Michelotti G, Diehl AM (2011). Hedgehog signaling in the liver. J Hepatol 54: 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenetti A, Syn WK, Jung Y, Francis H, Porrello A, Witek RP et al. (2009). Repair‐related activation of hedgehog signaling promotes cholangiocyte chemokine production. Hepatology 50: 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orfila C, Lepert JC, Alric L, Carrera G, Beraud M, Pipy B (2005). Immunohistochemical distribution of activated nuclear factor kappaB and peroxisome proliferator‐activated receptors in carbon tetrachloride‐induced chronic liver injury in rats. Histochem Cell Biol 123: 585–593. [DOI] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B (2006). Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol 26: 3365–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Wang C, Wang B (2009). Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog‐regulated mouse development. Dev Biol 326: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola M, Robino G (2001). Oxidative stress‐related molecules and liver fibrosis. J Hepatol 35: 297–306. [DOI] [PubMed] [Google Scholar]
- Philips GM, Chan IS, Swiderska M, Schroder VT, Guy C, Karaca GF et al. (2011). Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS One 6: e23943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap A, Singh S, Mundra V, Yang N, Panakanti R, Eason JD et al. (2012). Attenuation of early liver fibrosis by pharmacological inhibition of smoothened receptor signaling. J Drug Target 20: 770–782. [DOI] [PubMed] [Google Scholar]
- Racanelli V, Rehermann B (2006). The liver as an immunological organ. Hepatology 43: S54–S62. [DOI] [PubMed] [Google Scholar]
- Reddy GK, Enwemeka CS (1996). A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 29: 225–229. [DOI] [PubMed] [Google Scholar]
- Shen X, Cheng S, Peng Y, Song H, Li H (2014). Attenuation of early liver fibrosis by herbal compound "Diwu Yanggan" through modulating the balance between epithelial‐to‐mesenchymal transition and mesenchymal‐to‐epithelial transition. BMC Complement Altern Med 14: 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicklick JK, Li YX, Choi SS, Qi Y, Chen W, Bustamante M et al. (2005). Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab Invest 85: 1368–1380. [DOI] [PubMed] [Google Scholar]
- Soriano V, Labarga P, Ruiz‐Sancho A, Garcia‐Samaniego J, Barreiro P (2006). Regression of liver fibrosis in hepatitis C virus/HIV‐co‐infected patients after treatment with pegylated interferon plus ribavirin. AIDS 20: 2225–2227. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A et al. (2010). Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 51: 1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymonik‐Lesiuk S, Czechowska G, Stryjecka‐Zimmer M, Slomka M, Madro A, Celinski K et al. (2003). Catalase, superoxide dismutase, and glutathione peroxidase activities in various rat tissues after carbon tetrachloride intoxication. J Hepatobiliary Pancreat Surg 10: 309–315. [DOI] [PubMed] [Google Scholar]
- Weber LW, Boll M, Stampfl A (2003). Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol 33: 105–136. [DOI] [PubMed] [Google Scholar]
- Wei Y, Li K, Wang N, Cai GD, Zhang T, Lin Y et al. (2015). Activation of endogenous anti‐inflammatory mediator cyclic AMP attenuates acute pyelonephritis in mice induced by uropathogenic Escherichia coli . Am J Pathol 185: 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Choi SS, Syn WK, Michelotti GA, Swiderska M, Karaca G et al. (2013). Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 62: 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Oue T, Uehara S, Fukuzawa M (2010). Forskolin, a Hedgehog signal inhibitor, inhibits cell proliferation and induces apoptosis in pediatric tumor cell lines. Mol Med Rep 3: 133–139. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Oue T, Uehara S, Fukuzawa M (2011). Hedgehog signal inhibitor forskolin suppresses cell proliferation and tumor growth of human rhabdomyosarcoma xenograft. J Pediatr Surg 46: 320–325. [DOI] [PubMed] [Google Scholar]
- Yang JJ, Tao H, Li J (2014). Hedgehog signaling pathway as key player in liver fibrosis: new insights and perspectives. Expert Opin Ther Targets 18: 1011–1021. [DOI] [PubMed] [Google Scholar]
- Yang L, Wang Y, Mao H, Fleig S, Omenetti A, Brown KD et al. (2008). Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J Hepatol 48: 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of different doses of forskolin on serum levels of (A) ALT and (B) AST. Each column represents mean ± SD (n = 6). # P <0.05; significantly different from control group; * P <0.05; significantly different from CCl4 group; ANOVA followed by Tukey–Kramer post hoc test.
Figure S2 Histopathological analysis of rat liver sections using H&E staining (x100). A: Control group showing normal histological structure of the central vein and surrounding hepatocytes. B: CCl4 group showing centrilobular necrosis (n) with ballooning degeneration in the hepatocytes (arrow) associated with dilatation in the central vein (CV). C: Forskolin (5 mg.kg−1) + CCl4 group showing centrilobular necrosis (n) with ballooning degeneration (arrow) in diffuse manner all over the hepatocytes in association with congestion in the portal vein (PV). D: Forskolin (10 mg.kg−1) + CCl4 group showing reduction in histological abnormalities with only ballooning degeneration (arrow) in some of the hepatocytes and dilatation in the central vein (CV). E: Forskolin (20 mg.kg−1) + CCl4 group showing centrilobular necrosis (n) with ballooning degeneration (arrow) associated with congestion in the portal vein (PV).
Supporting info item