

Abstract
Background and Purpose
Oxidative stress plays a key role in the vascular and metabolic abnormalities associated with obesity. Herein, we assessed whether obesity can increase coronary vasoconstriction induced by hydrogen peroxide (H2O2) and the signalling pathways involving COX‐2 and superoxide (O2 .‐) generation.
Experimental Approach
Contractile responses to H2O2 and O2 .‐ generation were measured in coronary arteries from genetically obese Zucker rats (OZR) and compared to lean Zucker rats (LZR).
Key Results
Both basal and H2O2‐stimulated O2 .‐ production were enhanced in coronary arteries from OZR, but H2O2‐induced vasoconstriction was unchanged. The selective COX‐2 inhibitor NS398 significantly reduced H2O2‐induced contractions in endothelium‐denuded arteries from LZR and OZR, but only in endothelium‐intact arteries from LZR. PGI2 (IP) receptor antagonism modestly reduced the vasoconstrictor action of H2O2 while antagonism of the PGE2 receptor 4 (EP4) enhanced H2O2 contractions in arteries from OZR but not LZR. Basal release of COX‐2‐derived PGE2 was higher in coronary arteries from OZR where the selective agonist of EP4 receptors TCS 2519 evoked potent relaxations. COX‐2 was up‐regulated after acute exposure to H2O2 in coronary endothelium and vascular smooth muscle (VSM) and inhibition of COX‐2 markedly reduced H2O2‐elicited O2 .‐ generation in coronary arteries and myocardium. Expression of Nox subunits in VSM and NADPH‐stimulated O2 .‐ generation was enhanced and contributed to H2O2 vasoconstriction in arteries from obese rats.
Conclusion and Implications
COX‐2 contributes to cardiac oxidative stress and to the endothelium‐independent O2 .‐‐mediated coronary vasoconstriction induced by H2O2 in obesity, which is offset by the release of COX‐2‐derived endothelial PGE2 acting on EP4 vasodilator receptors.
Abbreviations
- [Ca2+]i
intracellular Ca2 +
- EIA
enzyme immunoassay
- eNOS
endothelial NOS
- Fura2‐AM
fura‐2 acetoxymethyl ester
- K+30
30 mM K+ solution
- KPSS
high K+ solution
- LZR
lean Zucker rats
- Nox
NADPH oxidase cytosolic subunit
- O2.‐
superoxide anion
- OZR
obese Zucker rats
- VSM
vascular smooth muscle
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes b |
| EP4 receptor | COX‐2 |
| IP receptor | eNOS |
| TP receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Obesity is a major cardiovascular risk factor commonly associated with other metabolic and vascular abnormalities including dyslipidaemia, insulin resistance and hypertension, jointly referred to as metabolic syndrome (Grundy, 2012). Aetiological factors of cardiovascular disease in patients with metabolic syndrome include, among others, coronary atherosclerotic disease, arterial hypertension, left ventricular hypertrophy, endothelial dysfunction and coronary microvascular disease (Grundy, 2012; Bagi et al., 2014; Prieto et al., 2014). Obesity and metabolic syndrome greatly enhance the risk of heart disease and heart failure (Alexander et al., 2003). One common pathogenic factor in the development of cardiovascular disease in obesity, diabetes and other insulin resistant states is the increased oxidative stress in the heart (Boudina et al., 2009; Serpillon et al., 2009).
The high energy requirements of the heart largely rely on the mitochondrial aerobic metabolism that maintains a high ATP/ADP ratio and generates as byproducts ROS including O2 .‐ and the dismutated product of O2 .‐, H2O2 (Boudina et al., 2009). Despite the excessive formation or insufficient removal of ROS leading to oxidative tissue damage under pathological conditions, ROS are critically involved in physiological processes in the heart including regulation of excitation‐contraction coupling, differentiation and proliferation of cardiac myocytes and regulation of coronary blood flow (Shimokawa, 2010; Prosser et al., 2011; Burgoyne et al., 2012). H2O2, which plays a role along with NO and adenosine in the autoregulation of coronary blood flow (Yada et al., 2003), is released from the endothelium by shear stress in human coronary arterioles (Miura et al., 2003) and is involved in the pacing‐induced metabolic coronary vasodilatation coupling coronary blood flow to myocardial O2 consumption (Saitoh et al., 2006; Yada et al., 2007). H2O2 is an endogenous mediator of the endothelium‐derived hyperpolarization of coronary microvessels (Matoba et al., 2003; Shimokawa, 2010) and activates Ca2 +‐activated K+ channels (Barlow and White, 1998) and the Na+/K+ pump in coronary vascular smooth muscle (VSM) (Wong et al., 2014). We have recently demonstrated that H2O2 is also a coronary vasoconstrictor that activates COX and endothelial TXA2‐dependent contractions tightly coupled to Ca2 + entry through L‐type channels (Santiago et al., 2013). H2O2 can also regulate store‐operated Ca2 + entry not coupled to contraction in coronary VSM (Santiago et al., 2015).
Systemic, vascular and adipose tissue oxidative stress is increased in obese humans and in experimental models of obesity, and underlies the abnormal adipokine secretion and the development of endothelial dysfunction and insulin resistance (Furukawa et al., 2004; Katakam et al., 2005; Erdös et al., 2006; Marchesi et al., 2009; Sánchez et al., 2012; Bagi et al., 2014; Prieto et al., 2014). Enhanced H2O2 production has been found in right atrial appendages of type 2 diabetic patients (Anderson et al., 2009) and in the heart of db/db mice (Boudina et al., 2007), whereas increased NADPH oxidase‐ and mitochondria derived‐O2 .‐ generation in the heart was associated with aortic endothelial dysfunction and preceded cardiac dysfunction in genetically obese Zucker rats (OZR) (Serpillon et al., 2009). The pathophysiological implications of the higher levels of oxidative stress in redox signalling of coronary arteries under conditions of insulin resistance such as obesity and diabetes are unknown. Augmented vasoconstriction induced by H2O2 due to higher TXA2 production was found to be associated with oxidative stress and increased vascular production of H2O2 in systemic arteries in hypertension (Gao and Lee, 2001; García‐Redondo et al., 2009). Therefore, the aim of the present study was to determine whether obesity might exacerbate H2O2‐induced coronary vasoconstriction and impair the signalling pathways of the peroxide‐elicited contractions involving COX signalling and O2 .‐ generation (Santiago et al., 2013) in coronary arteries from the OZR. A role for COX‐2, recently involved in the increased vascular oxidative stress and endothelial dysfunction in hypertension (Tian et al., 2012; Virdis et al., 2013) and obesity (Muñoz et al., 2015), will specifically be assessed in coronary arteries from the OZR, a well‐established genetic model of obesity/insulin resistance caused by a dysfunctional gene of the leptin receptor.
Methods
Animal model and tissue preparation
All animal care and experimental protocols conformed to the European Union Guidelines for the Care and the Use of Laboratory Animals (European Union Directive 2010/63/EU) and were approved by the Institutional Animal Care and Use Committee at Complutense University (Madrid, Spain), and are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male OZR (fa/fa) and their control counterparts, lean Zucker rats (LZR) (fa/−), were purchased from Charles River Laboratories (Barcelona, Spain) at 8–10 weeks of age. Animals were housed at the Pharmacy School animal care facility and maintained on standard chow and water ad libitum, until they were used for the study, at 16–18 weeks of age.
Rats were anaesthetised with sodium pentobarbital (40 mg·kg−1, i.p.) and killed by cervical dislocation and exsanguination. The heart and the mesentery were removed and placed in a cold (4°C) physiological saline solution (PSS) of the following composition (mM): 119 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 1.5 CaCl2, 24.9 NaHCO3, 0.027 EDTA and 11 glucose; pH = 7.4. First‐ or second‐order branches of the left descending coronary artery and third‐order branches of mesenteric arteries were carefully dissected by removing the myocardium and adipose and connective tissue surrounding the arteries, respectively, and mounted in parallel in double microvascular myographs (Danish Myotechnology, Denmark) for isometric tension recording. The arteries were equilibrated for 30 min in PSS at 37°C continuously gassed with a mixture of 5% CO2/95% O2 to maintain pH, and then the relationship between passive wall tension and internal circumference was determined for each artery. From this, the internal circumference L100 corresponding to a transmural pressure of 100 mmHg for a relaxed vessel in situ was calculated. The arteries were set to an internal circumference L1 equal to 0.9 times L100 (L1 = 0.9 × L100), because force development is close to maximal at this internal circumference (Contreras et al., 2011).
Experimental procedure for the functional experiments
The vessel viability was tested at the beginning of each experiment by measuring the vasoconstrictor responses to a high K+ solution (KPSS), equivalent to PSS except that NaCl was exchanged for KCl on an equimolar basis, giving a final concentration of 123.7 mM K+. The vasoactive responses to H2O2 in coronary and mesenteric arteries were assessed by adding cumulative concentrations of this agent (1–300 μM) on arteries precontracted with a 30 mM K+ solution (K+30) in order to obtained maximal contractile responses to H2O2 (Santiago et al., 2013).
The contractile responses to H2O2 were assessed in the absence and the presence of the non‐selective inhibitor of COX, indomethacin (1 μM), the thromboxane receptor (TP) antagonist (ICI 192, 3 μM), the selective inhibitor of COX‐2 (NS398, 1 μM), the prostacyclin (PGI2) receptor (IP) antagonist (CAY 10441, 0.1 μM), the free radical scavenger, SOD mimetic tempol (30 μM) and the PGE2 receptor antagonist, L1613982 (0.1 μM). The relaxant effect of the selective agonist of the EP4 receptor TCS 2519 was assessed in endothelium‐intact coronary arteries precontracted with 5‐HT (1–2 μM). The effects of the selective inhibitor of NADPH oxidase (Nox) 2, Nox2ds‐tat (1 μM) and the dual inhibitor of Nox1‐Nox4 GKT137831 (0.1 μM) were also assessed on the vasoconstriction elicited by H2O2 in endothelium‐denuded coronary arteries from LZR and OZR. The endothelial integrity was tested in each artery by examining the relaxant effect of 10 μM ACh. The role of endothelial cells in the vasoactive response to H2O2 was tested in arteries in which the endothelium was mechanically removed by guiding a human hair inside the vessel lumen and gently moving it forward and back several times. The absence of a functional endothelium was confirmed by the lack of relaxation to ACh.
Simultaneous measurements of [Ca2 +]i and tension
Simultaneous measurements of intracellular Ca2 + ([Ca2 +]i) and tension were performed in intact arterial segments by fura‐2 acetoxymethyl ester (Fura2‐AM) fluorescence as previously described (Villalba et al., 2008; Santiago et al., 2013). Coronary arteries were incubated in the dark at 37°C in PSS containing the indicator 4 μM Fura‐2‐AM and 0.05% Cremophor EL for a 2 h period. They were washed three times in PSS to remove the remaining Fura‐2‐AM, and the solution was changed to PSS with fresh Fura‐2‐AM after 1 h. After Fura‐2‐AM loading, arteries were washed for 45 min in PSS. Experiments were performed in PSS (37°C) continuously gassed with a mixture of 5% CO2–95% O2 to maintain pH at 7.4. The myograph was mounted on an inverted microscope (Zeiss Axiovert S100 TV) equipped for dual excitation wavelength microfluorimetry (Deltascan, Photon Technology International).
The coronary artery was alternately illuminated at two different wavelengths, 340 and 380 nm light, using a monochromator‐based system. The intensity of emitted fluorescence was collected through a 510 nm filter using a photomultiplier and monitored together with the tension. Coronary arteries were stimulated with KPSS at the beginning of each experiment in order to test vessel viability. At the end of each experiment, Ca2 +‐insensitive signals were determined after quenching with Mn2 +, and the values obtained were subtracted from those recorded during the experiment. The ratio of fluorescence at 340 and 380 nm (F340/F380) corrected for autofluorescence was taken as a measure of [Ca2 +]i.
In the experiments aimed to assess whether the enzyme COX‐2 is involved in the [Ca2 +]i responses induced by H2O2 in coronary arteries, vasoconstriction and changes in [Ca2 +]i elicited by a single concentration of H2O2 (100 μM) were assessed in endothelium‐denuded arteries in the presence and absence of the selective COX‐2 inhibitor, NS398 (1 μM).
Measurement of superoxide production by chemiluminescence
The level of production of O2 .‐ in coronary arteries and myocardial tissue from control LZR and OZR under basal conditions and upon stimulation with H2O2 (100 μM) was detected by lucigenin‐enhanced chemiluminescence as previously described in intact small arteries (Prieto et al., 2010). Briefly, segments of coronary arteries and samples of myocardial tissue were dissected and equilibrated in PSS for 30 min at room temperature and then incubated in the absence (controls) or presence of tempol (30 μM), NS398 (1 μM), Nox2ds‐tat (1 μM) or GKT137831 (0.1 μM) at 37°C. Samples were then transferred to microtiter plate wells containing 5 μM lucigenin in air‐equilibrated Krebs solution buffered with 10 mM HEPES‐NaOH, in the absence and presence of tempol, NS398, Nox2ds‐tat or GKT137831, and H2O2 (100 μM) was then applied and the chemiluminescent signal was measured after 15 min. Chemiluminescence was measured in a luminometer (BMG Fluostar Optima). Baseline values were subtracted from the counting values under the different experimental conditions, and O2 .‐ production was normalized to tissue weight.
Measurement of NADPH oxidase activity
NADPH oxidase (Nox) activity was determined by lucigenin‐enhanced chemiluminescence in intact coronary arteries and samples of myocardium from LZR and OZR after addition of 0.1 mM NADPH, in the absence and presence of the selective Nox2 inhibitor Nox2ds‐tat (1 μM) and of the dual Nox1‐Nox4 inhibitor GKT137831 (0.1 μM). Samples were previously incubated with inhibitors for 30 min in PSS at 37°C before NADPH addition.
Immunohistochemistry
Tissue samples from the heart containing the left descending coronary artery from LZR and OZR were immersion‐fixed in 4% paraformaldehyde in 0.1 M sodium phosphate buffer (PB), cryoprotected in 30% sucrose in PB, and snap‐frozen in liquid nitrogen and stored at −80°C. Transversal sections 5 μm thick were obtained by means of a cryostat and preincubated in 10% normal goat serum in PB containing 0.3% Triton X‐100 for 2–3 h. Then, sections were incubated with a rabbit polyclonal anti‐COX‐2 (Santa Cruz Biotechnology, CA, USA) diluted at 1:50 or a mouse monoclonal anti‐eNOS (Chemicon International Inc) diluted at 1:500 for 48 h, washed, and allowed to react with a goat secondary serum (anti‐rabbit for the COX‐2 and anti‐mouse for the eNOS) diluted 1:200 for 2 h at room temperature. Some sections were incubated with a rabbit polyclonal anti‐COX‐1 (Santa Cruz Biotechnology, CA, USA) diluted at 1:50.
In order to determine the effects of acute exposure to H2O2 on COX‐2 expression in the coronary vascular wall, coronary arteries were incubated with 100 μM H2O2 for 45 min in oxygenated PSS at 37°C. Then the reaction was stopped and samples were snap‐frozen and processed for COX‐2 and eNOS staining, as described above.
Nox enzymes expression in the vascular wall of coronary arteries was determined by immunofluorescence by incubating coronary sections from LZR and OZR with rabbit monoclonal antibodies anti‐Nox1 (Santa Cruz Biotechnology Inc, CA, USA, 1:50 dilution), anti‐Nox2 (anti gp91‐phox, Santa Cruz Biotechnology, CA, USA, 1:50 dilution) or anti‐Nox4 (Santa Cruz Biotechnology, CA, USA, 1:50 dilution), along with the anti‐NOS antibody in order to colocalize Nox enzymes with eNOS in the coronary endothelium. Nox expression was also determined in sections from coronary arteries acutely exposed to H2O2.
Secondary antibodies used were Alexa Fluor 594 (red) and Alexa Fluor 488 (green). The slides were covered with a specific medium containing DAPI, which stains all cell nuclei. The observations were made with a fluorescence microscope (Olympus IX51). No immunoreactivity could be detected in sections incubated in the absence of the primary antisera (Fig. S1). Preadsorption with either COX‐2 or Nox proteins showed no cross‐reactivity to the antibodies.
Measurement of prostaglandin E2 by EIA
Basal and H2O2‐stimulated PGE2 levels were measured in intact coronary arteries by EIA (enzyme immune assay) (Arbor Assays). The left descending coronary artery and its branches were dissected out from the heart of LZR and OZR and incubated in 300 μL PSS at 37°C for 30 min, and the solution was collected to measure basal PGE2 levels. Arteries were thereafter stimulated with H2O2 (100 μM) for 30 min in fresh PSS in the absence and presence of the COX‐2 inhibitor NS398 (1 μM), and the supernatant was collected and kept at −80°C. Arterial tissue was removed, and the protein concentration was determined to standardize levels of PGE2.
Western blotting
The entire left descending coronary artery and its branches were dissected out from the heart of LZR (n = 12) and OZR (n = 11) and incubated in 1 mL PSS at 37°C for 30 min. LZR and OZR arteries were then divided in 2 groups: stimulated with H2O2 (100 μM) in PSS at 37°C or basal just left in PSS at 37°C for 1 h. Thereafter, arterial tissue was snap frozen in liquid nitrogen and kept at −80°C in order to determine basal and H2O2‐stimulated COX‐2 protein levels in intact coronary arteries by western blotting. Arterial tissue was homogenized in RIPA Buffer (ThermoFisher Scientific, IL, USA) with protease and phosphatase inhibitors (Roche) and centrifuged for 20 min at 12 000 g at 4°C. Protein content was determined by Bio‐Rad DC Protein Assay Kit (Bio Rad, Hercules, CA, USA) and equal amounts of proteins (20 μg) were loaded and subjected to electrophoresis on a SDS‐PAGE (7.5%) followed by transference to a PVDF membrane (Bio‐Rad). Protein expression was quantified using primary antibodies anti‐COX‐2 (Cayman, 1:150 dilution) or anti‐β‐actin as a loading control (Sigma Aldrich, Spain, 1:10 000 dilution) and horseradish peroxidase conjugated secondary goat‐anti mouse and anti‐rabbit antibodies (Santa Cruz Biotech, CA, USA, 1:2000 and 1:10 000 dilution). Proteins were detected using ECL Plus Western blotting reagents (Amersham, GE Healthcare, CT, USA) and analysed using Quantity One. Relative intensity for each protein was determined by comparison with the intensity of β‐actin staining.
Drugs
The sources of the compounds used were as follows: ACh, H2O2, 5‐HT, indomethacin, lucigenin and tempol were obtained from Sigma Aldrich (Spain); ICI 192, NS398, L1613982 and TCS 2519 were obtained from Tocris (Great Britain); Fura2‐AM, ionomycin, Alexa Fluor 594 goat‐antirabbit, Alexa Fluor 488 goat‐antimouse and DAPI were obtained from Invitrogen (Great Britain); CAY‐10441 and GKT137831 were obtained from Cayman chemical (USA); Nox2ds‐tat was obtained from bioNova Cientifica, s.l. (Spain); rabbit policlonal anti‐COX‐2, anti‐Nox1, anti‐Nox2 and anti‐Nox4 were obtained from Santa Cruz Biotechnology (USA); mouse monoclonal anti‐eNOS was obtained from Chemicon International Inc.
Data presentation and statistics analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Mechanical responses of the arteries were measured as force and expressed as active wall tension (ΔT), which is the increase in force (ΔF), divided by twice the segment length. The results are expressed as either Nm−1 of tension or as a percentage of the response to KPSS in each artery for the functional experiments, and in counts min‐1 mg‐1 of tissue for the measurement of O2 .‐ production, and as units of ratio (F340/F380) or as a percentage of the response to KPSS for the simultaneous measurements of [Ca2 +]i and tension. All the results are expressed as means ± SEM, n represents the number of arteries (1–2 per animal in the pharmacological and chemiluminescence experiments, and 1 per animal in the VSM [Ca2 +]i measurements, in Western blot analysis and in the PGE2 measurements). Statistical differences between means were analysed by Student's paired or unpaired t‐tests for comparison between two groups and by using one‐way anova followed by Bonferroni's post hoc test for comparisons involving more than two groups. Probability levels lower than 5% were considered significant. All calculations were made using a standard software package (Prism 5.0, GraphPad Software).
Results
General parameters
At 17–18 weeks of age, OZR were significantly heavier than LZR (494 ± 10 g vs 388 ± 7 g, P < 0.001 n = 40 and 39 respectively). We have reported that animals from OZR group exhibit mild hyperglycaemia, hyperinsulinaemia and dyslipidaemia with elevated total cholesterol and triglycerides levels (Villalba et al., 2009). The normalized internal lumen diameters, l1, of coronary arteries in the OZR group (310 ± 10 μm, n = 49 arteries, 1–2 per animal) were not significantly different from those in the LZR group (290 ± 10 μm, n = 48 arteries, 1–2 per animal), thus confirming that the structure is preserved in arteries from OZR compared to LZR (Villalba et al., 2009). Contractions elicited by depolarization with KPSS were not significantly different in coronary arteries of LZR and OZR (0.74 ± 0.10 Nm−1, n = 12 animals, in LZR and 0.85 ± 0.12 Nm−1, n = 13 animals, in OZR).
ROS levels in arterial and heart tissue and effects of H2O2 on vasoconstriction and [Ca2 +]i responses in coronary arteries from LZR and OZR
Basal O2 .‐ production was enhanced in coronary arteries and in the myocardium of OZR compared with their respective controls. Stimulation with H2O2 (10 and 100 μM) increased O2 .‐ generation in both LZR and OZR resulting in total higher levels of oxidative stress in coronary arteries from obese rats. After incubation with tempol (30 μM), O2 .‐ production was blunted in both coronary arteries and myocardium, and reduced to similar values in obese and lean animals (Figure 1A, B).
Figure 1.
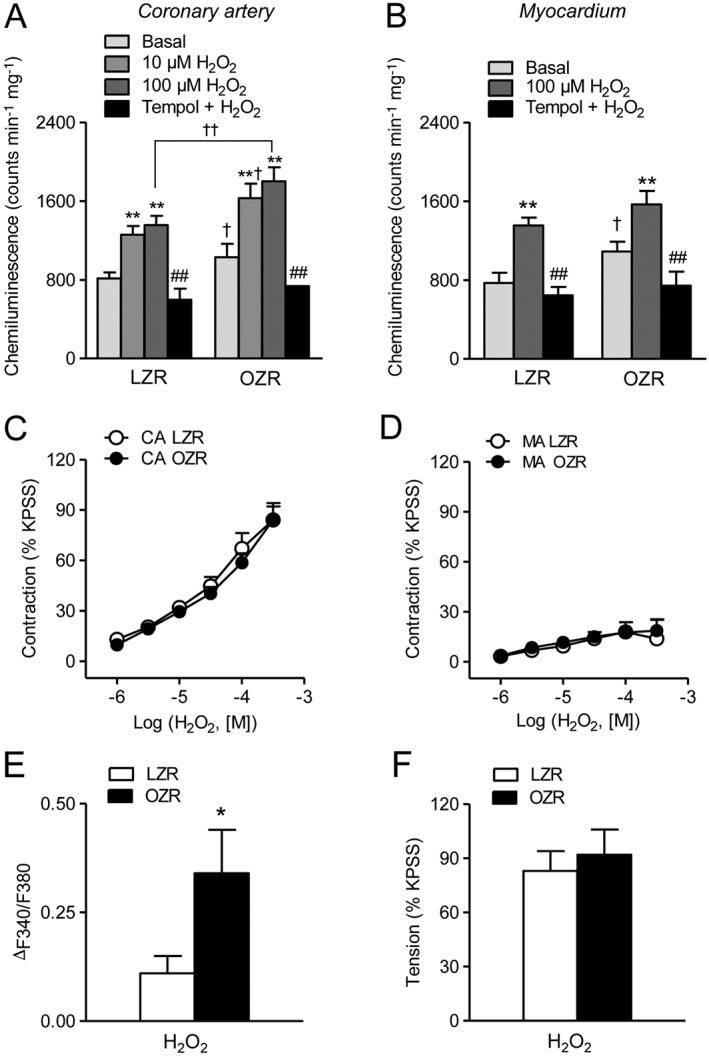
H2O2 increases O2 .‐ production and VSM [Ca2 +]i but not vasoconstriction in coronary arteries from genetically obese rats. (A, B) Effect of H2O2 (10 and 100 μM) and the free radical scavenger tempol (30 μM) on the production of O2 .‐ in coronary artery (A) and in the myocardium (B) of LZR and OZR. (C, D) H2O2 induced a contractile effect of similar magnitude in arteries from LZR and OZR in coronary (CA) (C) and mesenteric arteries (MA) (D) precontracted with K+30. (E, F) Summarized data showing the changes in [Ca2 +]i (E) and tension (F) in response to a single dose of H2O2 (100 μM) in endothelium‐intact coronary arteries from LZR and OZR. (A, B) Results are expressed as counts min‐1 mg‐1 of tissue and represent the mean ± SEM of 8–20 animals. **P < 0.01 versus control before treatment; ##P < 0.01 versus H2O2‐treated; †P < 0.05, ††P < 0.01 versus LZR. (C‐F) Results are expressed either as a percentage of the KPSS‐induced responses (C, D, F) or as absolute values of ratio (E) and represent the mean ± SEM of 5–13 arteries (1–2 per animal). *P < 0.05.
Increased ROS levels in coronary and myocardial tissue did not translate into differences in coronary vasoconstriction to H2O2 in obese rats. Thus, stimulation of intramyocardial coronary arteries precontracted with K+30 with increasing concentrations of H2O2 (1 μM–300 μM) elicited contractile responses that were not different in OZR compared with LZR (Emax 1.08 ± 0.15 Nm−1, n = 12 animals, and 1.26 ± 0.15 Nm−1, n = 13 animals, in LZR and OZR, respectively) (Figure 1C, F). No significant differences were observed either in the modest contraction induced by H2O2 in K+30‐depolarized mesenteric arteries from OZR and LZR (Figure 1D). However, stimulation of endothelium‐intact coronary arteries precontracted with K+30 with a single dose of H2O2 (100 μM) induced a significantly higher increase in [Ca2 +]i in OZR than in LZR (Figure 1E).
Effect of non‐selective inhibition of COX and a TP receptor antagonist on vasoconstrictor responses to H2O2
The involvement of prostanoids in the contractile response to H2O2 in obesity was assessed in coronary arteries after treatment with the nonselective COX inhibitor indomethacin and with the selective TP receptor antagonist ICI 192. Inhibition of COX with indomethacin (1 μM) reduced to a similar extent the contractile effects of H2O2 in coronary arteries from both OZR and LZR (Figure 2A, B). Likewise, ICI 192 (3 μM) produced a marked inhibition of the coronary vasoconstriction induced by H2O2 in both OZR and LZR (Figure 2C, D). These results suggest that the H2O2‐induced contractile effect is mediated by activation of COX and the release of contractile prostanoids acting on the TP receptor in coronary arteries from both lean and obese rats.
Figure 2.
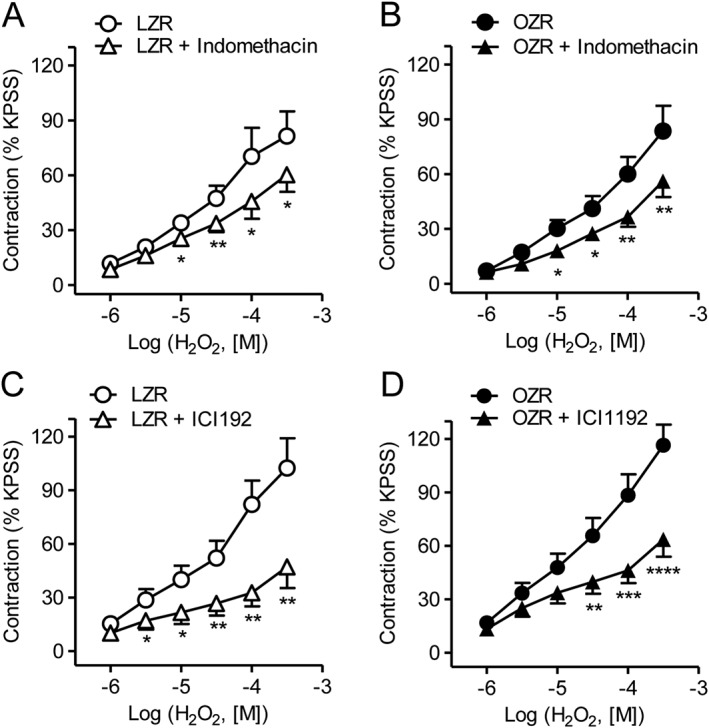
H2O2‐induced vasoconstriction is mediated via activation of TP receptor in both LZR and OZR. (A, B) The non‐selective COX inhibitor indomethacin (1 μM) significantly decreased the H2O2‐induced contractile response in coronary arteries from LZR and OZR. (C, D) The selective TP receptor inhibitor ICI192 (3 μM) significantly reduced the vasoconstrictor response to H2O2 in coronary arteries from LZR and OZR. Results are expressed as a percentage of KPSS‐induced contraction and represent the mean ± SEM from 6–8 arteries (1–2 per animal). *P < 0.05; **P < 0.01; ***P < 0.001.
Involvement of COX‐2 in the H2O2‐induced vasoconstriction of coronary arteries
Because COX‐2 is up‐regulated under inflammatory conditions in the vascular wall, the involvement of this inducible isoenzyme in the vascular actions of H2O2 was assessed. Treatment with the selective antagonist of COX‐2 NS398 (1 μM) significantly reduced the H2O2‐induced vasoconstriction in endothelium‐intact coronary arteries from LZR (Figure 3A), whereas it did not alter the contractile effect of H2O2 in coronary arteries from OZR (Figure 3B). However, removal of the endothelium increased vasoconstriction (Emax before and after endothelium removal being 84 ± 9, n = 13, and 88 ± 7 of KPSS, n = 14, in LZR, and 84 ± 8, n = 13, and 107 ± 7 of KPSS, n = 13, P < 0.05, n = 13, in OZR respectively) (Figure 3C, D) and unmasked a pronounced inhibitory effect of the COX‐2 antagonist on the contractile effect of H2O2 in coronary arteries from OZR of similar magnitude to the one in arteries from lean animals (Figure 3E, F). These results suggest that in obese rats, H2O2 stimulates the production of COX‐2‐derived contractile mediators in VSM of coronary arteries counterbalanced by the release of COX‐2‐derived relaxing prostanoids from the endothelium.
Figure 3.
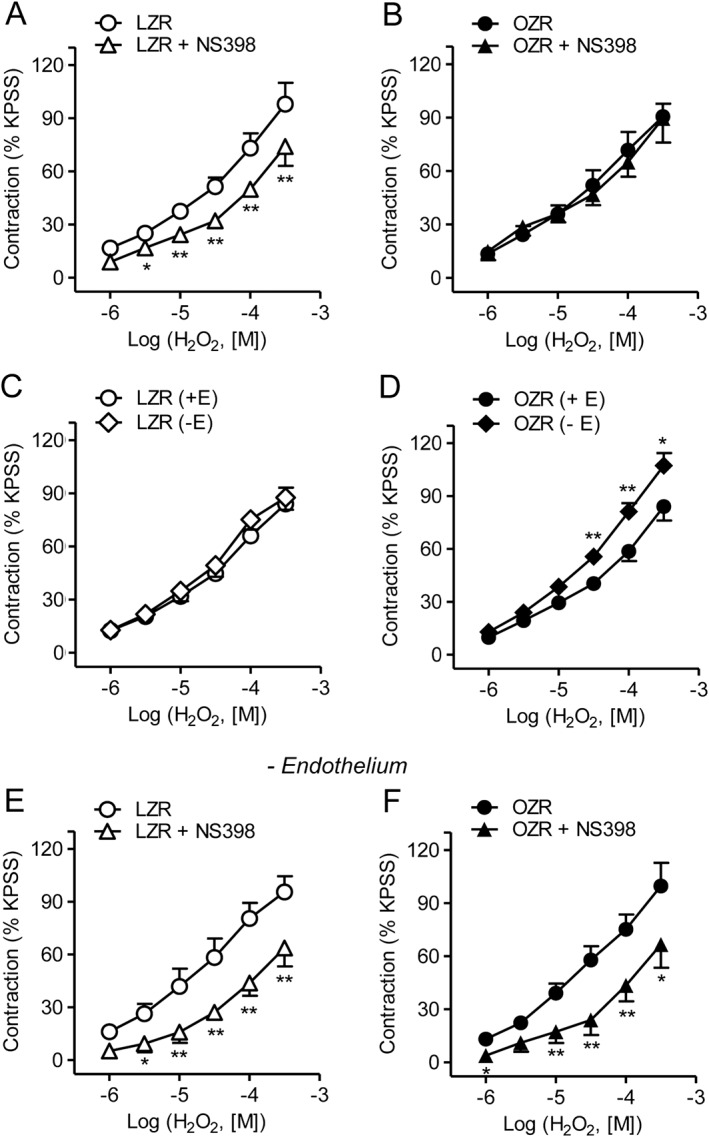
H2O2‐induced vasoconstriction is mediated via activation of COX‐2 in VSM of coronary arteries from both LZR and OZR and counterbalanced by COX‐2 derived endothelial mediators in OZR. (A, B) The selective inhibitor of COX‐2 NS398 (1 μM) significantly decreased the contractile response to H2O2 in endothelium‐intact coronary arteries from LZR (A), but not OZR (B). (C, D) Effect of endothelium removal on the average contractile responses elicited by H2O2 in coronary arteries from LZR and OZR. (E, F) In endothelium‐denuded coronary arteries, NS398 (1 μM) significantly reduced the contractile response to H2O2 in both OZR and LZR. Results are expressed as a percentage of KPSS‐induced contraction and represent the mean ± SEM from 6–14 arteries (1–2 per animal). *P < 0.05; **P < 0.01; ***P < 0.001.
Effect of COX‐2 inhibition on VSM [Ca2 +]i increase induced by H2O2
We further assessed whether COX‐2 is involved in the augmented increase in [Ca2 +]i elicited by peroxide in coronary VSM of obese rats (Figure 1E). In endothelium‐denuded coronary arteries from OZR, NS398 did not alter the increase in [Ca2 +]i (Figure 4A, B), but significantly reduced vasoconstriction induced by H2O2 (Figure 4A, C) suggesting Ca2 + sensitization mechanisms mediated by COX‐2.
Figure 4.
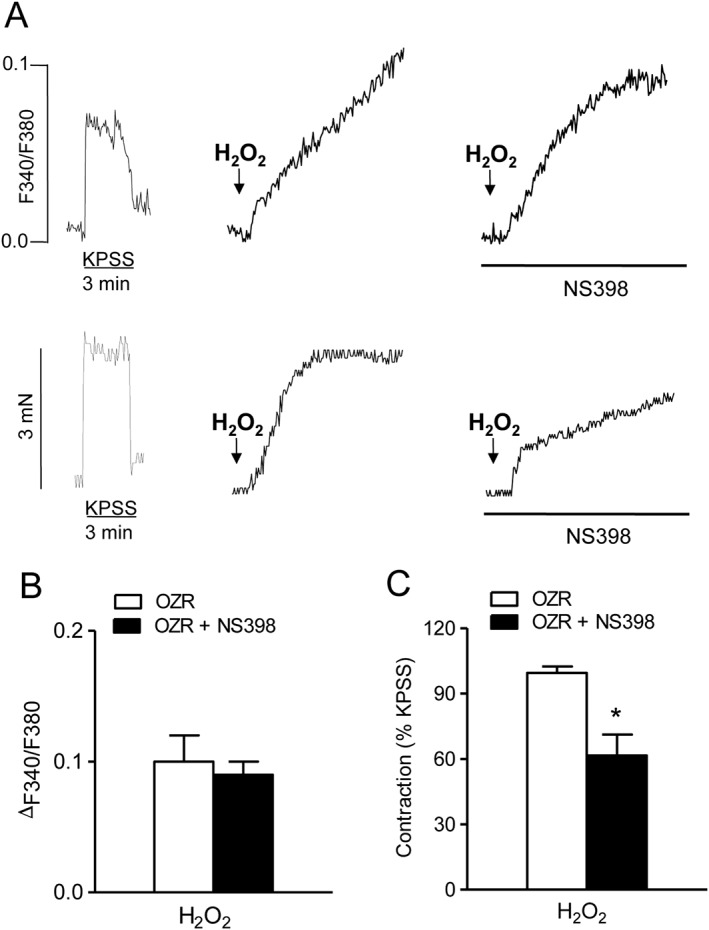
COX‐2 is involved in vasoconstriction but not in the increase in VSM [Ca2 +]i induced by H2O2 in coronary arteries from OZR. Simultaneous recordings showing changes in [Ca2 +]i (A, top) and tension (A, bottom) induced by H2O2 (100 μM) in the presence and absence of NS398 (1 μM) in endothelium‐denuded coronary arteries from OZR. (B, C) Average effects of NS398 on the H2O2‐induced rises in [Ca2 +]i (B) and tension (C). Data are expressed as absolute values of F340/F380 (B) or as % of the KPSS‐induced contraction (C) and represent the mean ± SEM from 5 arteries (1 per animal). *P < 0.05.
Effect of IP‐ and EP4 receptor antagonism on vasoconstrictor responses to H2O2 and levels of PGE2 in coronary arteries
In order to investigate the nature of the COX‐2 endothelial relaxant prostanoids that counterbalanced the H2O2 contractions in coronary arteries of obese rats, arteries were treated with selective IP‐ and EP4 receptor antagonists. Incubation with the IP receptor antagonist CAY 10441 (0.1 μM) modestly reduced the vasoconstrictor action of H2O2 in both LZR and OZR (Figure 5A, B), suggesting that PGI2 may act as a contractile mediator. In contrast, the antagonist of the EP4 receptors L1613982 (0.1 μM) significantly enhanced the contractions induced by the lowest concentrations of H2O2 in coronary arteries from OZR but not LZR (Figure 5C, D). Furthermore, the selective agonist of EP4 receptors TCS 2519 induced relaxations of higher potency and magnitude in coronary arteries from OZR (Figure 5E). Basal PGE2 secretion was significantly augmented in coronary arteries from obese rats and blunted by the COX‐2 inhibitor NS398 (1 μM), although PGE2 release in response to stimulation with H2O2 was not altered in arteries from either LZR or OZR (Figure 5F). These data suggest that basal increased production of PGE2 acting on EP4 receptors counterbalances H2O2‐induced coronary vasoconstriction in obese rats.
Figure 5.
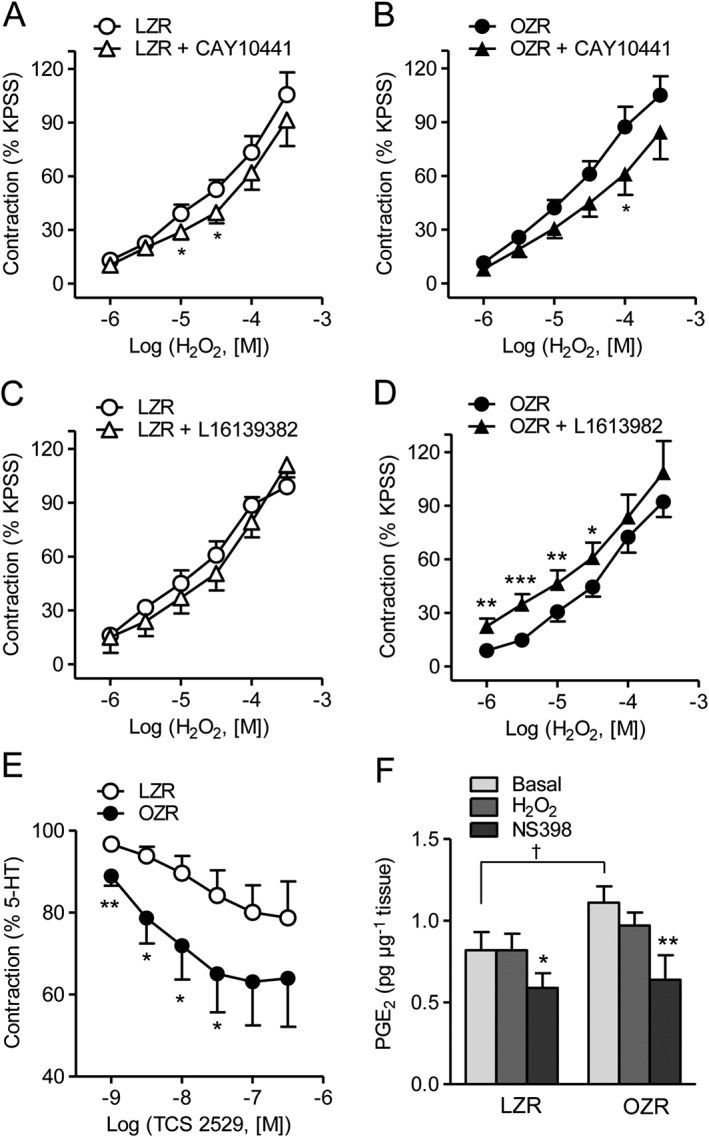
Effect of IP receptor and EP receptor antagonism on the contractile effect of H2O2 and PGE2 levels in coronary arteries of LZR and OZR. Effect of (A, B) the IP receptor antagonist CAY 10441 (0.1 μM) and (C, D) the EP4 receptor antagonist L1613982 (0.1 μM) on the contractile effect of H2O2 in endothelium‐intact coronary arteries from LZR (A,C) and OZR (B,D). (E) Relaxant effect of the selective EP4 receptor agonist TCS 2519 on coronary arteries from LZR and OZR precontracted with 5‐HT (1–2 μM). (F) Basal and H2O2‐stimulated PGE2 release in coronary arteries from LZR and OZR. Results are expressed as a percentage of the KPSS‐induced contraction and represent the mean ± SEM from 6–8 arteries (1–2 per animal) (A‐E) and from 5–6 arteries (1 per animal) (F). *P < 0.05; **P < 0.01; ***P < 0.001; †P < 0.05 versus LZR.
COX‐2 expression in coronary arteries and modulation by H2O2
Immunostaining of cross arterial sections of coronary arteries from LZR revealed that COX‐2 is discretely expressed in the coronary endothelium colocalized with eNOS, and to a minor extent in VSM (Figure 6A, left). Acute exposure to H2O2 for 45 min revealed a more intense COX‐2 immunostaining in the vascular wall and unmasked the induction of this enzyme primarily in the VSM, suggesting that H2O2 is able to induce COX‐2 expression (Figure 6A, right). In coronary arteries from obese rats, COX‐2 was expressed in both the endothelium and VSM, and the expression of this enzyme was higher than in arteries from LZR (Figure 6B, left) and also upon acute exposure to H2O2 (Figure 6B, right). Western blot analysis confirmed that COX‐2 expression was up‐regulated upon acute stimulation with H2O2 in coronary arteries of LZR. COX‐2 protein content was about threefold higher in coronary arteries from OZR compared with LZR, and incubation with H2O2 increased COX‐2 protein levels in arteries from LZR to the same levels as those in arteries from OZR (Figure 6C). COX‐1 protein was distributed throughout the endothelial lining but not in coronary VSM in arteries from both LZR and OZR, and its expression was not altered by H2O2 stimulation (Fig. S2).
Figure 6.
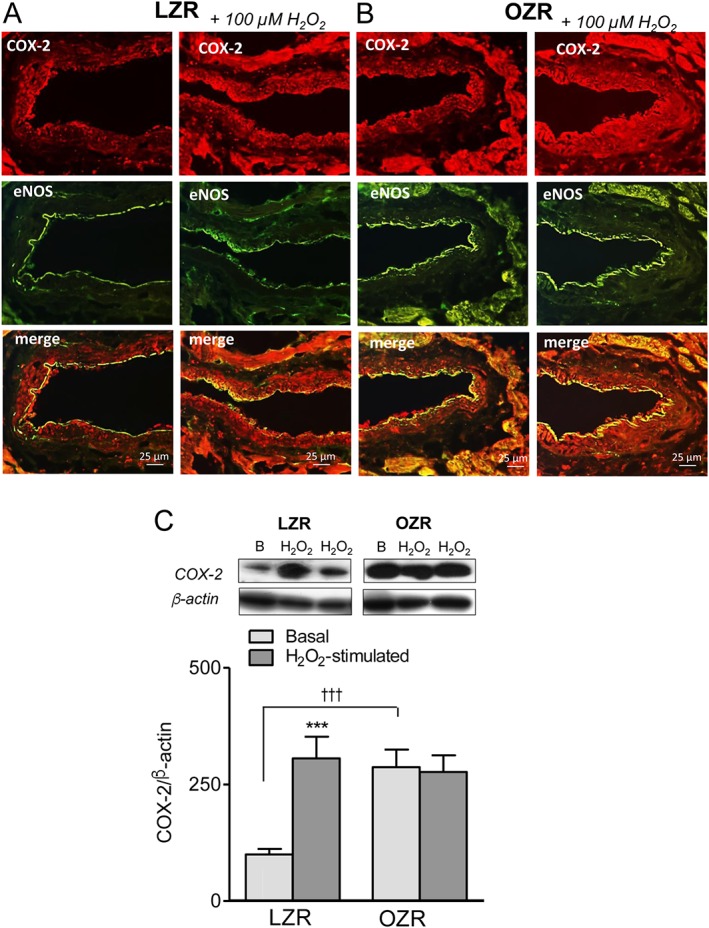
The up‐regulation of COX‐2 upon H2O2 stimulation in coronary arteries. (A, B) Immunohistochemical localization of COX‐2 in coronary arteries from LZR and OZR and effect of H2O2 stimulation. Immunofluorescence for COX‐2 protein (red areas) was modestly distributed in the endothelium of coronary arteries from LZR (A, left) and was more intense in both endothelium and VSM of coronary arteries from OZR (B, left). (A, B, right) Expression of COX‐2 was markedly increased in coronary VSM upon acute stimulation with H2O2 (100 μM, 45 min) in coronary arteries from both LZR (A, right) and OZR (B, right). The endothelium was visualized with anti‐eNOS antibodies (green areas), and double immunofluorescence shows the colocalization of eNOS and COX‐2 in the endothelium (yellow areas). The sections represent n = 3 animals. (C) H2O2 stimulation increases COX‐2 protein content in LZR. Western blot analysis of COX‐2 expression under basal conditions and after 1 h stimulation with 100 μM H2O2 in coronary arteries from LZR and OZR. Results were quantified by densitometry. Data are shown as means ± SEM of 5–6 animals. Significant differences from controls were analysed using one‐way ANOVA followed by a Bonferroni post test *** P < 0.01 versus LZR.
Involvement of COX‐2 in the H2O2‐induced ROS generation in coronary arteries
To assess whether enhanced COX‐2 expression contributes to oxidative stress in the heart of obese rats, ROS generation was measured in samples of coronary arteries and myocardium from LZR and OZR after selective COX‐2 inhibition. Treatment with NS398 significantly reduced both basal and H2O2‐elicited O2 .‐ generation (Figure 7A, B), suggesting that H2O2‐induced vasoactive effects are in part mediated by COX‐2‐derived O2 .‐, whose production is increased in obese rats. Involvement of O2 .‐ in the contractile effects of H2O2 was further confirmed by incubating endothelium‐denuded coronary arteries from LZR and OZR with the free radical scavenger tempol. Tempol reduced the vasoconstrictor effect of H2O2 to a larger extent in arteries from OZR, thus suggesting that the augmented generation of O2 .‐ radicals in VSM mediates the contraction induced by H2O2 in coronary arteries from obese rats (Figure 7C, D).
Figure 7.
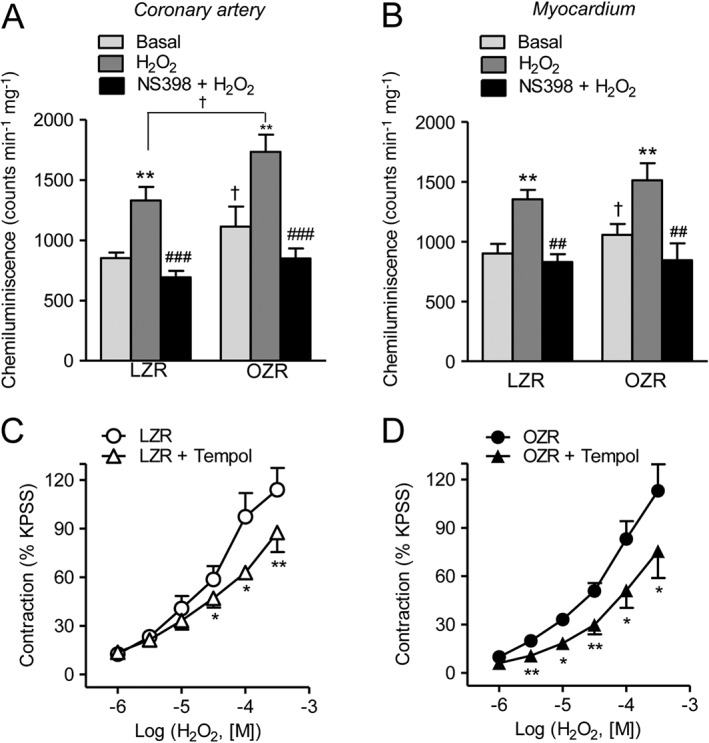
COX‐2 is involved in O2 .‐ generation and in the H2O2‐induced contraction in coronary VSM. (A, B) Effect of H2O2 (100 μM) and COX‐2 NS398 (1 μM) on O2 .‐ production in coronary artery (A) and in the myocardium (B) from OZR and LZR. (C, D) In endothelium‐denuded coronary arteries from LZR and OZR, the free radical scavenger tempol (30 μM) significantly reduced the contractile response to H2O2. (A, B) Results are expressed in counts min‐1 mg‐1 of tissue and represent the mean ± SEM of 8–20 animals. **P < 0.01 versus control before treatment; ##P < 0.01, ###P < 0.001 versus treated with H2O2; †P < 0.05, ††P < 0.01 versus LZR. (C, D) Results are expressed as a percentage of the KPSS‐induced contraction and represent the mean ± SEM from 5–7 arteries (1–2 per animal). *P < 0.05; **P < 0.01.
NADPH oxidase expression and activity in coronary arteries from LZR and OZR
Because NADPH oxidase is a major source of ROS generation in the vascular wall under conditions of insulin resistance, the expression and activity of NADPH isoforms (Nox1, Nox2 and Nox4) were assessed in coronary arteries from LZR and OZR, and their role in the H2O2‐induced vasoconstriction was determined. Immunoreaction for Nox1 (Figure 8A) and Nox2 (Figure 8B) was scarce in coronary arteries from lean rats but the expression of both isoenzymes was markedly enhanced in the endothelium and in particular in the VSM layer of arteries from OZR. A low immunoreaction for Nox4 could be detected in coronary arteries from LZR that was greatly augmented in VSM and endothelium of coronary arteries from obese rats (Figure 8C). An enhanced Nox immunoreactivity was not observed in lean animals upon acute stimulation with H2O2, and the up‐regulation of Nox1, Nox2 and Nox4 found in arteries of OZR was similar to that in the absence of H2O2 treatment (Fig. S3).
Figure 8.
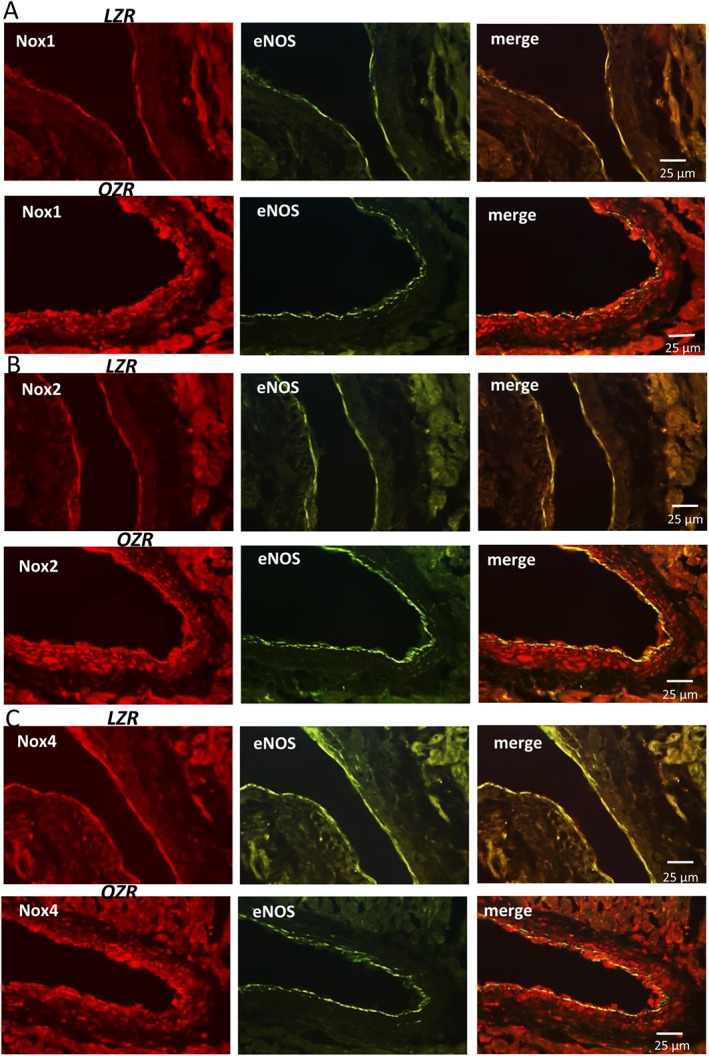
Immunohistochemical localization of Nox1, Nox2 and Nox4 in coronary arteries from LZR and OZR. Immunofluorescence for (A) Nox1 (red areas), (B) Nox2 (red areas) and (C) Nox4 (red areas) was absent or modest in coronary arteries from LZR (A,B,C left) but was markedly increased in both endothelium and VSM of coronary arteries from OZR (A,B,C, left). The endothelium was visualized with anti‐eNOS antibodies (green areas) and the double immunofluorescence shows colocalization of eNOS and Nox1 (A, right), Nox2 (B, right) and Nox4 (C, right) in the endothelium (yellow areas), but also a strong immunoreaction for all 3 Nox isoenzymes in coronary VSM (red areas) of obese rats. The sections represent n = 3 animals.
NADPH oxidase activity measured by the levels of NADPH‐stimulated O2 .‐ production was significantly higher in coronary arteries (Figure 9A) but not in myocardium (Figure 9B) from OZR compared with LZR. NADPH‐stimulated O2 .‐ generation was markedly reduced by the Nox‐2 inhibitor Nox2ds‐tat and by the dual Nox1‐Nox‐4 inhibitor GKT137831 in both coronary arteries (Figure 9A) and myocardium (Figure 9B) from LZR and OZR. Moreover, inhibition of Nox1‐Nox‐4 with GKT137831 significantly reduced NADPH‐stimulated O2 .‐ below basal levels in coronary arteries from obese rats (Figure 9A). The involvement of NADPH oxidase‐derived O2 .‐ from VSM in the contractile responses to H2O2 was further assessed in endothelium‐denuded coronary arteries. Treatment with either Nox2ds‐tat (Figure 9C, D) or GKT137831 (Figure 9E, F) markedly reduced H2O2‐induced coronary vasoconstriction in both LZR and OZR.
Figure 9.
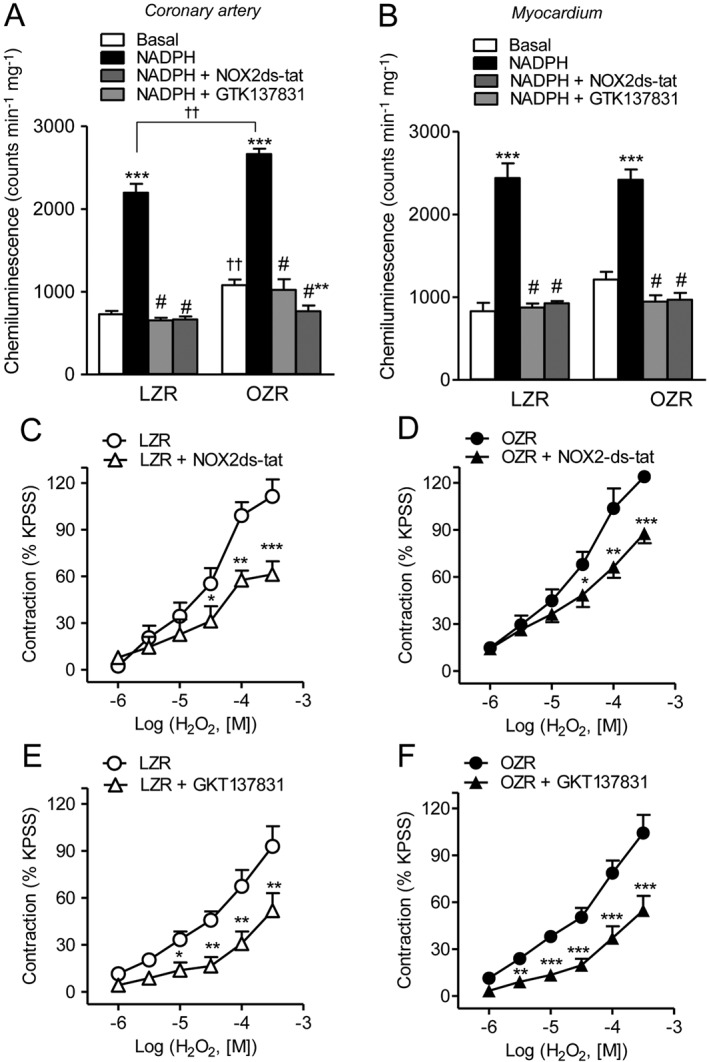
Nox1, Nox2 and Nox4‐derived O2 .‐ is involved in the H2O2‐induced contraction in coronary VSM. Effect of the selective Nox‐2 inhibitor Nox2ds‐tat (1 μM) and of the dual Nox1‐Nox4 inhibitor GKT137831 (0.1 μM) on (A,B) NADPH‐stimulated O2 .‐ generation of coronary arteries (A) and myocardium (B) from LZR and OZR, and on (C‐F) the contractions induced by H2O2 in endothelium‐denuded coronary arteries from LZR (C,E) and OZR (D,F). Results are expressed as a percentage of the KPSS‐induced contraction (C–F) and represent the mean ± SEM from 5–7 animals (A,B) and from 5–8 arteries (1–2 per animal) (C–F). *P < 0.05; **P < 0.01; ***P < 0.001 versus control; #P < 0.001 versus NADPH‐stimulated; ††P < 0.01 versus LZR.
Discussion
Oxidative stress plays a key role in the vascular and metabolic abnormalities associated with obesity and metabolic syndrome including atherosclerosis, hypertension, insulin resistance and type 2 diabetes, which suggests that it might be an early event rather than a consequence in the pathogenesis of these chronic diseases (Roberts and Sindhu, 2009). ROS like H2O2, have been demonstrated to contribute to the impaired vascular tone, augmented vasoconstriction and endothelial dysfunction in the insulin resistant states of hypertension (García‐Redondo et al., 2009, 2015) and obesity (Muñoz et al., 2015). The results of the present study demonstrate that cardiac and coronary vascular oxidative stress are associated with preserved H2O2 vasoconstriction in coronary and systemic arteries. COX‐2 expression and activity were up‐regulated by H2O2, and augmented O2 .‐ production derived from both COX‐2 and NADPH oxidase contributed to the H2O2‐induced VSM contractions but was counterbalanced by the increased release of COX‐2‐derived PGE2 acting on vasorelaxant EP4 receptors in coronary arteries from obese rats.
Obesity increases cardiac oxidation of free fatty acids and reduces glucose utilization regardless of diabetes in experimental models of metabolic syndrome, leading to enhanced mitochondrial ROS formation in the obese heart (Buchanan et al., 2005; Boudina et al., 2007). Increased NADPH oxidase‐derived O2 .‐ production and reduced activity of antioxidant enzymes such as cytosolic and mitochondrial SOD and glutathione peroxidase also importantly contribute to cardiac oxidative stress in obesity (Feillet‐Coudray et al., 2009; Serpillon et al., 2009; Ballal et al., 2010). The present results confirm the higher levels of oxidative stress in the myocardial tissue of OZR and further demonstrate that basal ROS production is increased in coronary arteries of obese rats. This, along with the feedback mechanism by which H2O2 is able to stimulate O2 .‐ production in the heart (Santiago et al., 2013), contributes to enhanced oxidative stress in arteries from OZR. However, the vasoconstrictor effects of H2O2 were not exacerbated either in coronary or in systemic arteries from obese rats, in contrast to that previously reported in arteries from hypertensive animals with elevated levels of vascular oxidative stress (Gao and Lee, 2001; García‐Redondo et al., 2009, 2015). In the latter studies, enhanced vasoconstrictor responses to H2O2 involved enhanced O2 .‐ generation, increased release and effects of TXA2 and higher VSM [Ca2 +]i mobilization. In healthy coronary arteries, the contractile effects of H2O2 are also mediated by activation of COX and TP receptors (Santiago et al., 2013) and this signalling pathway appears to be unchanged in arteries from obese rats, because selective inhibition of TP receptors reduced to a similar extent H2O2 coronary vasoconstriction in LZR and OZR, although H2O2‐induced VSM [Ca2 +]i mobilization was augmented in OZR. The lack of enhanced H2O2 contractile responses in arteries from obese rats compared with that reported in hypertension might be ascribed to the experimental model used. At 17–18 weeks OZR, a model of genetic obesity/metabolic syndrome, have not yet developed hypertension despite exhibiting hyperinsulinaemia and systemic endothelial dysfunction (Villalba et al., 2009; Muñoz et al., 2015). Moreover, we have recently shown that H2O2 activates stored‐operated Ca2 + entry not coupled to contraction that was augmented in coronary arteries of OZR (Santiago et al., 2013), which might explain the higher VSM [Ca2 +]i mobilization but preserved coronary vasoconstriction found for H2O2 in obese rats.
Interestingly, we demonstrated here that COX‐2 not only contributes to the H2O2‐induced VSM contractions in coronary arteries from both LZR and OZR rats but also mediates protective endothelial relaxant effects in arteries from OZR. COX‐2 is an inducible COX isoenzyme usually undetectable in healthy tissue but induced by mitogen, mechanical and inflammatory stimuli in the vascular wall, its expression being rapidly induced by cytokines, tumour promoters and growth factors (Eligini et al., 2005). A reciprocal relationship between ROS and COX‐2 in the vascular dysfunction associated with hypertension and diabetes is now well established, in which oxidative stress and ROS enhance COX‐2 expression, and COX‐2 is in turn a significant source of ROS generation (Shi and Vanhoutte, 2008; Martínez‐Revelles et al., 2012; Hernanz et al., 2014). COX‐2 is up‐regulated in coronary arteries under conditions of low‐grade vascular inflammation such as the insulin resistant states of diabetes (Szerafin et al., 2006) and obesity (Sánchez et al., 2010). Oxidative stress and ROS can increase the expression and activity of both COX‐1 and COX‐2, and specifically, H2O2 stimulates COX‐2 gene and protein expression in endothelial and VSM cells (Shi and Vanhoutte, 2008; Martín et al., 2012; Martínez‐Revelles et al., 2012; Tian et al., 2012; Muñoz et al., 2015). COX‐2 mRNA expression is rapid and in vitro studies have shown that it is maximally increased within the first hour in human coronary endothelial cells exposed to inflammatory stimuli, COX‐2 protein expression being detected within the first 2 h after initiation of stimulation (Tan et al., 2007). Also cytokines like IL‐1β and stimuli like angiotensin II have been reported to induce a fast COX‐2 protein expression as early as at 1 h in VSM cells (Aguado et al., 2015). Accordingly, the current data demonstrate that H2O2 up‐regulates COX‐2 expression and enhances COX‐2 activity in coronary arteries. COX‐2 protein was sparse in the endothelium of LZR coronary arteries and acute exposure to the oxidant H2O2 induced a rapid expression of the enzyme in endothelium and VSM and markedly increased COX‐2 protein content to levels similar to those in coronary arteries from OZR, where COX‐2 was already up‐regulated (Sánchez et al., 2010). H2O2 has also been demonstrated to induce early COX‐2 protein expression (90 min) in VSM from hypertensive rats through a mechanism involving the redox‐sensitive transcription factor NF‐kB (Martín et al., 2012).
H2O2‐induced COX‐2 expression was associated with COX‐2 involvement in H2O2 actions on vascular tone and ROS production in coronary arteries. Thus, selective COX‐2 inhibition significantly reduced H2O2 vasoconstriction in endothelium‐denuded coronary arteries from both LZR and OZR suggesting the involvement of COX‐2‐VSM‐derived contractile mediators in the H2O2‐induced coronary vasoconstriction. Endothelium‐independent contractions induced by H2O2 and O2 .‐ have been reported to be mediated by both Ca2 +‐dependent and Ca2 +‐sensitization mechanisms in VSM (Ardanaz and Pagano, 2006; Snetkov et al., 2011; Santiago et al., 2013). In the present study, an involvement of COX‐2 in the higher [Ca2 +]i mobilization induced by H2O2 in coronary VSM from OZR was however ruled out, because the selective COX‐2 inhibitor NS398 reduced vasoconstriction but not VSM [Ca2 +]i increases induced by H2O2 in coronary arteries of obese rats. This suggests that COX‐2‐mediated H2O2 contractions are mediated by Ca2 + sensitization of VSM probably involving kinases such as Rho kinase (Snetkov et al., 2011), also implicated in H2O2 coronary vasoconstriction under physiological conditions (Santiago et al., 2013).
On the other hand, the current data demonstrate that COX‐2 is associated with enhanced coronary vasodilatation in obese rats. The lack of effect of the COX‐2 inhibitor NS398 on the H2O2 contractile responses of endothelium‐intact coronary arteries from OZR compared with LZR, in contrast to the marked inhibitory effect in endothelium‐denuded vessels, suggests that COX‐2 mediates the production of endothelium‐derived relaxing prostanoids that mask the release of VSM contractile mediators in coronary arteries from OZR. These results support previous studies in our laboratory showing that an up‐regulation of COX‐2 was associated with the enhanced basal COX‐2‐mediated relaxation in coronary arteries from OZR (Sánchez et al., 2010). Also in type 2 diabetic patients (Szerafin et al., 2006) and mice (Przygodzki et al., 2015), COX‐2 inhibition reduced endothelium‐dependent relaxations of coronary arterioles and blunted basal coronary blood flow, respectively, suggesting an increased release of COX‐2‐derived vasodilator prostanoids likely to compensate for abnormal vascular function in the insulin resistant states of diabetes and obesity.
Although COX‐2 has traditionally been accepted as the main producer of PGI2 in the vascular endothelium, recent investigations support COX‐1 as the major isoform responsible for endothelial PGI2 production under both physiological (Kirkby et al., 2012) and pathophysiological conditions of vascular oxidative stress (Toniolo et al., 2013). In agreement with this, the involvement of PGI2 in the COX‐2‐mediated relaxing action activated by H2O2 in coronary arteries from obese rats may initially be ruled out, because the selective IP receptor antagonist did not enhance but rather decreased the vasoconstrictor effect of H2O2 in coronary arteries, therefore suggesting that PGI2 is involved in coronary vasoconstriction, as occasionally reported for PGI2 in cases of IP/TP receptor heterodimerization (Félétou et al., 2011). PGI2 and PGE2 are the main products of the COX metabolism in the coronary microvascular endothelium, and a reduction in the bioavailability of any of these endothelial protective factors can result in vascular dysfunction, as documented in patients with hypertension or diabetes (Hein et al., 2009). Inflammatory stimuli are able to stimulate COX‐2 gene expression associated with enhanced production of PGI2 and PGE2 in endothelial cells of human coronary artery (Tan et al., 2007). PGE2 is a potent COX‐2‐derived inflammatory mediator involved in cell infiltration, fibroblast proliferation and cardiac hypertrophy in the ischaemic heart (LaPointe et al., 2004), and in the protective effect of the endothelium during cardiac ischaemic preconditioning (Bouchard et al., 2000). Herein, we demonstrated that basal COX‐2‐dependent release of the vasodilator PGE2 acting on EP4 receptors counterbalances the ROS‐induced coronary vasoconstriction in obesity, based on the following findings. Firstly, the selective EP4 receptor antagonist L1613982 significantly increased the contractile effect of H2O2 in coronary arteries from obese but not from lean rats suggesting the involvement of PGE2 in the COX‐2‐mediated endothelial relaxant effects. This was further supported by the enhanced coronary vasodilator effect found for the selective EP4 receptor agonist TCS 2519 in OZR and confirmed by the augmented COX‐2‐mediated release of PGE2 in coronary arteries from obese rats compared with the lean controls, as reported in diabetic mice (Przygodzki et al., 2015). Hence, COX‐2 derived PGE2 acting on vasorelaxant EP4 receptors might have a protective role against oxidative damage induced by ROS in coronary arteries under conditions of obesity‐associated insulin resistance, consistent with the protective role recently ascribed to the EP4 receptor in obesity‐related inflammation (Yasui et al., 2015) and with reports showing that VSM‐specific EP4 receptor deletion exacerbates oxidative and renal injury induced by angiotensin II in mice (Thibodeau et al., 2016). Secondly, inducible COX‐2 was found in both endothelium and VSM of coronary arteries from obese rats, and further studies are needed to ascertain the source of PGE2 in the coronary vascular wall. Deletion of COX‐2 in both endothelial and VSM reduced PGE2 and PGI2 release, increased blood pressure and accelerated atherogenesis in mice fed a high fat diet (Tang et al., 2014) suggesting that VSM‐derived PGE2 may contribute to the protective effects of COX‐2 under conditions of vascular inflammation.
COX‐2 is a redox‐sensitive inducible enzyme associated with inflammation and recently confirmed as a source not only of prostanoids but also of ROS that importantly contribute to vascular oxidative stress (Shi and Vanhoutte, 2008; Martínez‐Revelles et al., 2012; Tian et al., 2012; Muñoz et al., 2015). Because COX‐2 is up‐regulated in coronary arteries from obese rats, a possible association of this enzyme with the increased ROS production and oxidative stress observed in coronary arteries was further assessed. Selective inhibition of COX‐2 induced a marked inhibitory effect on H2O2‐stimulated O2 .‐ production, and reduced the increased ROS generation in arteries from OZR to levels similar to those in controls, thus demonstrating that COX‐2 is partially responsible for the elevated levels of oxidative stress in the coronary arterial wall in obesity. Augmented COX‐2‐dependent ROS production is involved in the impaired endothelium‐dependent relaxations and endothelial dysfunction in patients with essential hypertension (Virdis et al., 2013), in experimental models of hypertension (Martínez‐Revelles et al., 2012; Tian et al., 2012) and diabetes (Shi and Vanhoutte, 2008), and also in renal arteries from insulin resistant OZR (Muñoz et al., 2015). Endothelial function is initially preserved in the 17–18 months old OZR used in the present study (Villalba et al., 2009; Contreras et al., 2011; Climent et al., 2014). However, the contribution of COX‐2‐derived O2 .‐ from VSM to the vasoconstrictor effect of H2O2 in coronary arteries was demonstrated in the present study by the inhibitory effect of the free radical scavenger tempol on the contractions elicited by H2O2 in endothelium‐denuded arteries. Such treatment produced a greater inhibition of the contractile effect in coronary arteries from OZR, consistent with the higher levels of O2 .‐ stimulated by H2O2 in these arteries, and indicative of an augmented vasoconstrictor action of H2O2 in the coronary VSM in part due to COX‐2‐ mediated oxidative stress in obese rats. The present data, along with the finding that H2O2 acutely up‐regulated COX‐2 expression in coronary arteries from lean but not obese animals, where COX‐2 expression was already enhanced, suggest that under conditions of cardiac oxidative stress and reduced activity of antioxidant enzymes such as glutathione peroxidase or catalase, as reported in obesity (Feillet‐Coudray et al., 2009; Serpillon et al., 2009; Ballal et al., 2010), augmented levels of ROS like H2O2 are responsible for the up‐regulation of vascular COX‐2, which in turn becomes an additional source of oxidative stress and increased coronary vasoconstriction.
The reciprocal relationship between COX‐2‐ and NADPH oxidase‐mediated vascular oxidative stress has recently been stablished in rodent models of hypertension, where NADPH‐derived ROS induced COX‐2 expression while COX‐2 inhibitors normalized augmented ROS generation, NADPH oxidase activity and Nox1 and Nox4 gene expression in arteries from hypertensive animals (Martín et al., 2012; Martínez‐Revelles et al., 2012). On the other hand, H2O2 can activate NADPH oxidase and O2 .‐ production in VSM in a feed‐forward mechanism that amplifies oxidant vascular injury (Li et al., 2001). We have demonstrated that NADPH oxidase‐derived O2 .‐ is involved in the H2O2 vasoconstriction of healthy coronary arteries (Santiago et al., 2013), and oxidative stress and augmented NADPH oxidase expression and activity have been reported in human coronary arteries from patients with coronary artery disease (Guzik et al., 2006). Interestingly, herein, we demonstrated that obesity markedly enhanced the expression of Nox1, Nox2 and Nox4 in both coronary endothelium and VSM, and that increased NADPH‐stimulated O2 .‐ production was inhibited by selective Nox2 and dual Nox1‐Nox4 inhibitors in coronary arteries from insulin resistant OZR. Moreover, we demonstrated the involvement of O2 .‐ derived from Nox1, Nox2 and Nox4 in the endothelium‐independent COX‐2 inhibitor‐sensitive contractions elicited by H2O2 in coronary arteries. Therefore, ROS derived from all three Nox subunits contribute to increased levels of vascular oxidative stress and mediate H2O2‐elicited coronary vasoconstriction in obese rats. Further studies are needed to elucidate the specific relationship between the up‐regulation of COX‐2 and enhanced NADPH expression and activity in coronary arteries. In addition, interaction of COX‐2 with sources of oxidative stress other than NADPH oxidase in coronary arteries from obese rats cannot be discarded. Under conditions of vascular inflammation like diabetes mellitus, oxidative stress and increased H2O2 generation have been associated with enhanced expression and activity of arginase, and the subsequent increase in the consumption of L‐arginine, eNOS uncoupling, reduced NO bioavailability and coronary endothelial dysfunction, (Beleznai et al., 2011; Pernow et al., 2015).
In summary, the present results demonstrate that increased oxidative stress is associated with preserved contractile responses to H2O2 and the up‐regulation of COX‐2 in coronary arteries from obese rats (Figure 10). H2O2 enhanced COX‐2 expression, which in turn contributed to augmented ROS generation and to endothelium‐independent O2 .‐‐mediated coronary vasoconstriction. Moreover, increased expression and activity of Nox1, Nox2 and Nox4 were also involved in the vascular oxidative stress and contraction in arteries from obese rats. Interestingly, vasoconstriction induced by H2O2 was counterbalanced by the release of COX‐2 derived endothelial PGE2 acting on vasodilator EP4 receptors, which suggests a protective mechanism to preserve coronary endothelial function against oxidative damage in obesity. The relevance of these findings relies on the dual beneficial/pathological role found for COX‐2 in coronary arteries from obese animals. On the one hand, COX‐2 represents a major source of oxidative stress and cardiovascular risk in the coronary arterial wall, but on the other, this isoenzyme is involved in protective vascular effects through the increased release of endothelial vasodilator PGE2, consistent with recent reports showing both protective and pro‐atherogenic/pro‐hypertensive effects for COX‐2 under conditions of hyperlipidaemia and vascular inflammation (Tang et al., 2014). The present findings further suggest that using selective drugs to target the pathway downstream of COX‐2, that is at the level of the EP4 receptor (Yasui et al., 2015), may shift the balance of cardiovascular efficacy and risk reported for non‐steroidal anti‐inflammatory drugs (NSAIDS) (Bhala et al., 2013).
Figure 10.
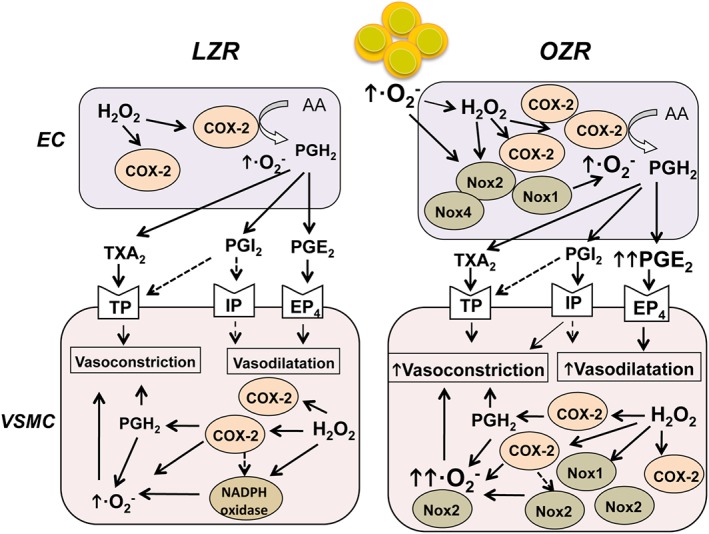
Proposed signal transduction mechanisms for COX‐2‐dependent actions elicited by H2O2 in coronary arteries. Oxidative stress and H2O2 up‐regulate COX‐2 expression in endothelium and VSM of coronary arteries. COX‐2 is already up‐regulated in arteries from obese rats. H2O2 activates TP receptor‐ and COX‐2‐dependent superoxide anion (O2 .‐)‐mediated vasoconstriction in VSM from both lean LZR and OZR. Nox1, Nox2 and Nox4 expression is enhanced in coronary arteries from OZR and contributes to the increased O2 .‐‐mediated H2O2‐induced vasoconstriction that is counterbalanced by vasodilator PGE2 acting through EP4 receptors. See Discussion for details. AA: arachidonic acid; EC, endothelial cell.
Author contributions
E.S., M.P.M, B.C., M.M., A.M.B. performed experiments and analysed data. D.P., E.S. designed and managed the research study. E.S.,B.C., A.M.B. , M.S., A.G.S, L.R., D.P., contributed to the Discussion and intellectual content. E.S. and D.P. wrote the manuscript. All authors contributed to manuscript preparation.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Control sections showing the absence of immunoreactivity in either VSM or endothelium of coronary arteries from LZR (A, B, upper panel) and OZR (C,D, lower panel) incubated just with the secondary antibodies (A, C, red or B, D, green) in the absence of the primary antibody anti‐COX‐2. Arrows indicate green autofluorescence of the internal elastic lamina. Cell nuclei stained with DAPI. The sections represent n = 3 animals.
Figure S2 H2O2 stimulation did not alter COX‐1 expression in coronary arteries. (A, B) Immunohistochemical localization of COX‐1 in coronary arteries from LZR and OZR and effect of H2O2 stimulation. Immunofluorescence for COX‐1 protein (red areas) was similarly distributed in the endothelium of coronary arteries (arrows) and myocardium (M) from LZR (A, left) and OZR (B, left). (A, B, right) Expression of COX‐1 was unaltered upon acute stimulation with H2O2 (100 μM, 45 min) in coronary arteries from both LZR (A, right) and OZR (B, right). The endothelium was visualized with anti‐endothelial nitric oxide synthase (eNOS) antibodies (green areas), and double immunofluorescence shows the colocalization of eNOS and COX‐1 in the endothelium (yellow areas). The sections represent n = 3 animals.
Figure S3 Immunohistochemical localization of Nox1, Nox2 and Nox4 in coronary arteries from LZR and OZR stimulated with H2O2. Immunofluorescence for (A) Nox1 (red areas), (B) Nox2 (red areas) and (C) Nox4 (red areas) was absent or modest in coronary arteries from LZR (A,B,C left) but was markedly increased in both endothelium and VSM of coronary arteries from OZR (A,B,C, left), after treatment with H2O2 (100 μM, 45 min). The endothelium was visualized with anti‐eNOS antibodies (green areas) and the double immunofluorescence shows colocalization of eNOS and Nox1 (A, right), Nox2 (B, right) and Nox4 (C, right) in the endothelium (yellow areas), but also a strong immunoreaction for all 3 Nox isoenzymes in coronary VSM (red areas) of obese rats. The sections represent n = 3 animals.
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This work was supported by grant SAF 2012‐31631 from MINECO‐Fondo Europeo de Desarrollo Regional (FEDER), Spain and grant GR3/14 from Universidad Complutense de Madrid. AMB was supported by the Ramón y Cajal Program (RYC‐2010‐06473) and by grant PI13/01488 from ISCIII‐FEDER. We thank Francisco Puente and Manuel Perales for their expert technical assistance.
Santiago, E. , Martínez, M. P. , Climent, B. , Muñoz, M. , Briones, A. M. , Salaices, M. , García‐Sacristán, A. , Rivera, L. , and Prieto, D. (2016) Augmented oxidative stress and preserved vasoconstriction induced by hydrogen peroxide in coronary arteries in obesity: role of COX‐2. British Journal of Pharmacology, 173: 3176–3195. doi: 10.1111/bph.13579.
References
- Aguado A, Rodríguez C, Martínez‐Revelles S, Avendaño MS, Zhenyukh O, Orriols M et al. (2015). HuR mediates the synergistic effects of angiotensin II and IL‐1β on vascular COX‐2 expression and cell migration. Br J Pharmacol 172: 3028–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander C, Landsman P, Teutsch S, Haffner S, (NHANES TNH and NES (NHANES I, Program NCE (NCEP) (2003). NCEP‐defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes 52: 1210–1214. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin C et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardanaz N, Pagano PJ (2006). Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med (Maywood) 231: 237–251. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Broskova Z, Feher A (2014). Obesity and coronary microvascular disease ‐ implications for adipose tissue‐mediated remote inflammatory response. Curr Vasc Pharmacol 12: 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballal K, Wilson C, Harmancey R, Taegtmeyer H (2010). Obesogenic high fat western diet induces oxidative stress and apoptosis in rat heart. Mol Cell Biochem 344: 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow R, White R (1998). Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. Am J Physiol 275: 1283–1289. [DOI] [PubMed] [Google Scholar]
- Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z (2011). Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol 300: H777–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA et al. (2013). Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. Lancet 382: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard J, Chouinard J, Lamontagne D (2000). Participation of prostaglandin E2 in the endothelial protective effect of ischaemic preconditioning in isolated rat heart. Cardiovasc Res 45: 418–427. [DOI] [PubMed] [Google Scholar]
- Boudina S, Sena S, Theobald H, Sheng X, Wright J, Hu X et al. (2007). Mitochondrial energetics in the heart in obesity‐related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466. [DOI] [PubMed] [Google Scholar]
- Boudina S, Bugger H, Sena S, O'Neill B, Zaha V, Ilkun O et al. (2009). Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119: 1272–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan J, Mazumder P, Hu P, Chakrabarti G, Roberts M, Yun U et al. (2005). Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146: 5341–5349. [DOI] [PubMed] [Google Scholar]
- Burgoyne J, Mongue‐Din H, Eaton P, Shah A (2012). Redox signaling in cardiac physiology and pathology. Circ Res 111: 1091–1106. [DOI] [PubMed] [Google Scholar]
- Climent B, Moreno L, Martínez P, Contreras C, Sánchez A, Pérez‐Vizcaíno F et al. (2014). Upregulation of SK3 and IK1 channels contributes to the enhanced endothelial calcium signaling and the preserved coronary relaxation in obese zucker rats. PLoS One 9: e109432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras C, Sánchez A, García‐Sacristán A, Martínez M, Andriantsitohaina R, Prieto D (2011). Preserved insulin vasorelaxation and up‐regulation of the Akt/eNOS pathway in coronary arteries from insulin resistant obese Zucker rats. Atherosclerosis 217: 331–339. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eligini S, Stella Barbieri S, Cavalca V, Camera M, Brambilla M, De Franceschi M et al. (2005). Diversity and similarity in signaling events leading to rapid Cox‐2 induction by tumor necrosis factor‐α and phorbol ester in human endothelial cells. Cardiovasc Res 65: 683–693. [DOI] [PubMed] [Google Scholar]
- Erdös B, Snipes J, Tulbert C, Katakam P, Miller A, Busija D (2006). Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase‐dependent superoxide production. Am J Physiol Heart Circ Physiol 290: H1264–H1270. [DOI] [PubMed] [Google Scholar]
- Feillet‐Coudray C, Sutra T, Fouret G, Ramos J, Wrutniak‐Cabello C, Cabello G et al. (2009). Oxidative stress in rats fed a high‐fat high‐sucrose diet and preventive effect of polyphenols: Involvement of mitochondrial and NAD(P)H oxidase systems. Free Radic Biol Med 46: 624–632. [DOI] [PubMed] [Google Scholar]
- Félétou M, Huang Y, Vanhoutte PM (2011). Endothelium‐mediated control of vascular tone: COX‐1 and COX‐2 products. Br J Pharmacol 164: 894–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y et al. (2004). Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Lee R (2001). Hydrogen peroxide induces a greater contraction in mesenteric arteries of spontaneously hypertensive rats through thromboxane A 2 production. Br J Pharmacol 134: 1639–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Redondo AB, Briones AM, Beltrán AE, Alonso MJ, Simonsen U, Salaices M (2009). Hypertension increases contractile responses to hydrogen peroxide in resistance arteries through increased thromboxane A2, Ca2+, and superoxide anion levels. J Pharmacol Exp Ther 328: 19–27. [DOI] [PubMed] [Google Scholar]
- García‐Redondo A, Briones A, Martínez‐Revelles S, Palao T, Vila L, Alonso M et al. (2015). c‐Src, ERK1/2 and Rho kinase mediate hydrogen peroxide‐induced vascular contraction in hypertension: role of TXA2, NAD(P)H oxidase and mitochondria. J Hypertens 33: 77–87. [DOI] [PubMed] [Google Scholar]
- Grundy S (2012). Pre‐diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol 59: 635–643. [DOI] [PubMed] [Google Scholar]
- Guzik T, Sadowski J, Guzik B, Jopek A, Kapelak B, Przybylowski P et al. (2006). Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler Thromb Vasc Biol 26: 333–339. [DOI] [PubMed] [Google Scholar]
- Hein T, Qamirani E, Ren Y, Kuo L (2009). C‐reactive protein impairs coronary arteriolar dilation to prostacyclin synthase activation: role of peroxynitrite. J Mol Cell Cardiol 47: 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernanz R, Briones AM, Salaices M, Alonso MJ (2014). New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo‐oxygenase in hypertension. Clin Sci (Lond) 126: 111–121. [DOI] [PubMed] [Google Scholar]
- Katakam P, Tulbert C, Snipes J, Erdös B, Miller A, Busija D (2005). Impaired insulin‐induced vasodilation in small coronary arteries of Zucker obese rats is mediated by reactive oxygen species. Am J Physiol Heart Circ Physiol 288: H854–H860. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby N, Lundberg M, Harrington L, Leadbeater P, Milne G, Potter C et al. (2012). Cyclooxygenase‐1, not cyclooxygenase‐2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A 109: 17597–17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe M, Mendez M, Leung A, Tao Z, Yang X (2004). Inhibition of cyclooxygenase‐2 improves cardiac function after myocardial infarction in the mouse. Am J Physiol Heart Circ Physiol 286: H1416–H1424. [DOI] [PubMed] [Google Scholar]
- Li W, Miller FJ, Zhang H, Spitz D, Oberley L, Weintraub N (2001). H2O2‐induced O2 production by a non‐phagocytic NAD(P)H oxidase causes oxidant injury. J Biol Chem 276: 29251–29256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin E (2009). Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension 54: 1384–1392. [DOI] [PubMed] [Google Scholar]
- Martín A, Pérez‐Girón J, Hernanz R, Palacios R, Briones A, Fortuño A et al. (2012). Peroxisome proliferator‐activated receptor‐γ activation reduces cyclooxygenase‐2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J Hypertens 30: 315–326. [DOI] [PubMed] [Google Scholar]
- Martínez‐Revelles S, Avendaño MS, García‐Redondo AB, Álvarez Y, Aguado A, Pérez‐Girón JV et al. (2012). Reciprocal relationship between reactive oxygen species and cyclooxygenase‐2 and vascular dysfunction in hypertension. Antioxid Redox Signal 18: 51–65. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Morikawa K, Kubota H, Kunihiro I, Urakami‐Harasawa L et al. (2003). Electron spin resonance detection of hydrogen peroxide as an endothelium‐derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol 23: 1224–1230. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Bosnjak J, Ning G, Saito T, Miura M, Gutterman D (2003). Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res 92: 31–40. [DOI] [PubMed] [Google Scholar]
- Muñoz M, Sánchez A, Martínez M, Benedito S, López‐Oliva M, García‐Sacristán A et al. (2015). COX‐2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic Biol Med 84: 77–90. [DOI] [PubMed] [Google Scholar]
- Pernow J, Kiss A, Tratsiakovich Y, Climent B (2015). Tissue‐specific up‐regulation of arginase I and II induced by p38 MAPK mediates endothelial dysfunction in type 1 diabetes mellitus. Br J Pharmacol 172: 4684–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto D, Kaminski P, Bagi Z, Ahmad M, Wolin M (2010). Hypoxic relaxation of penile arteries: involvement of endothelial nitric oxide and modulation by reactive oxygen species. Am J Physiol Heart Circ Physiol 299: H915–H924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto D, Contreras C, Sánchez A (2014). Endothelial dysfunction, obesity and insulin resistance. Curr Vasc Pharmacol 12: 412–426. [DOI] [PubMed] [Google Scholar]
- Prosser B, Ward C, Lederer W (2011). X‐ROS signaling: rapid mechano‐chemo transduction in heart. Science 333: 1440–1445. [DOI] [PubMed] [Google Scholar]
- Przygodzki T, Talar M, Przygodzka P, Watala C (2015). Inhibition of cyclooxygenase‐2 causes a decrease in coronary flow in diabetic mice. The possible role of PGE2 and dysfunctional vasodilation mediated by prostacyclin receptor. J Physiol Biochem 71: 71–78. [DOI] [PubMed] [Google Scholar]
- Roberts CK, Sindhu KK (2009). Oxidative stress and metabolic syndrome. Life Sci 84: 705–712. [DOI] [PubMed] [Google Scholar]
- Saitoh S, Zhang C, Tune J, Potter B, Kiyooka T, Rogers P et al. (2006). Hydrogen peroxide: a feed‐forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 26: 2614–2621. [DOI] [PubMed] [Google Scholar]
- Sánchez A, Contreras C, Martínez P, Villalba N, Benedito S, García‐Sacristán A et al. (2010). Enhanced cyclooxygenase 2‐mediated vasorelaxation in coronary arteries from insulin‐resistant obese Zucker rats. Atherosclerosis 213: 392–399. [DOI] [PubMed] [Google Scholar]
- Sánchez A, Contreras C, Martínez M, Climent B, Benedito S, García‐Sacristán A et al. (2012). Role of neural NO synthase (nNOS) uncoupling in the dysfunctional nitrergic vasorelaxation of penile arteries from insulin‐resistant obese Zucker rats. PLoS One 7: e36027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago E, Contreras C, García‐Sacristán A, Sánchez A, Rivera L, Climent B et al. (2013). Signaling pathways involved in the H2O2‐induced vasoconstriction of rat coronary arteries. Free Radic Biol Med 60: 136–146. [DOI] [PubMed] [Google Scholar]
- Santiago E, Climent B, Muñoz M, García‐Sacristán A, Rivera L, Prieto D (2015). Hydrogen peroxide activates store‐operated Ca2+ entry in coronary arteries. Br J Pharmacol 172: 5318–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpillon S, Floyd B, Gupte R, George S, Kozicky M, Neito V et al. (2009). Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose‐6‐phosphate dehydrogenase‐derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–H156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Vanhoutte PM (2008). Oxidative stress and COX cause hyper‐responsiveness in vascular smooth muscle of the femoral artery from diabetic rats. Br J Pharmacol 154: 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H (2010). Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pflugers Arch 459: 915–922. [DOI] [PubMed] [Google Scholar]
- Snetkov V, Smirnov S, Kua J, Aaronson P, Ward J, Knock G (2011). Superoxide differentially controls pulmonary and systemic vascular tone through multiple signalling pathways. Cardiovasc Res 89: 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerafin T, Erdei N, Fülöp T, Pasztor EET, Edes I, Koller A et al. (2006). Increased cyclooxygenase‐2 expression and prostaglandin‐mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ Res 99: e12–e17. [DOI] [PubMed] [Google Scholar]
- Tan X, Essengue S, Talreja J, Reese J, Stechschulte D, Dileepan K (2007). Histamine directly and synergistically with lipopolysaccharide stimulates cyclooxygenase‐2 expression and prostaglandin I(2) and E(2) production in human coronary artery endothelial cells. J Immunol 179: 7899–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SY, Monslow J, Todd L, Lawson J, Puré E, Fitz Gerald GA (2014). Cyclooxygenase‐2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation 129: 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeau JF, Holterman CE, He Y, Carter A, Gutsol A, Cron G et al. (2016). Vascular smooth muscle‐specific EP4 receptor deletion in mice exacerbates angiotensin II‐induced renal injury. Antioxid Redox Signal. (In press) [DOI] [PubMed] [Google Scholar]
- Tian XY, Wong WT, Leung FP, Zhang Y, Wang Y‐X, Lee HK et al. (2012). Oxidative stress‐dependent Cyclooxygenase‐derived Prostaglandin F2ɑ Impairs Endothelial Function in Renovascular Hypertensive Rats. Antioxid Redox Signal 16: 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toniolo A, Buccellati C, Pinna C, Gaion RM, Sala A, Bolego C (2013). Cyclooxygenase‐1 and Prostacyclin Production by Endothelial Cells in the Presence of Mild Oxidative Stress. PLoS One 8: 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba N, Stankevicius E, Simonsen U, Prieto D (2008). Rho kinase is involved in Ca2+ entry of rat penile small arteries. Am J Physiol Heart Circ Physiol 294: 1923–1932. [DOI] [PubMed] [Google Scholar]
- Villalba N, Martínez P, Bríones A, Sánchez A, Salaíces M, García‐Sacristán A et al. (2009). Differential structural and functional changes in penile and coronary arteries from obese Zucker rats. Am J Physiol Heart Circ Physiol 297: H696–H707. [DOI] [PubMed] [Google Scholar]
- Virdis A, Bacca A, Colucci R, Duranti E, Fornai M, Materazzi G et al. (2013). Endothelial dysfunction in small arteries of essential hypertensive patients: Role of cyclooxygenase‐2 in oxidative stress generation. Hypertension 62: 337–344. [DOI] [PubMed] [Google Scholar]
- Wong PS, Garle MJ, Alexander SPH, Randall MD, Roberts RE (2014). A role for the sodium pump in H2O2‐induced vasorelaxation in porcine isolated coronary arteries. Pharmacol Res 90: 25–35. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M et al. (2003). Hydrogen peroxide, an endogenous endothelium‐derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation 107: 1040–1045. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Hiramatsu O, Shinozaki Y, Mori H, Goto M et al. (2007). Important role of endogenous hydrogen peroxide in pacing‐induced metabolic coronary vasodilation in dogs in vivo. J Am Coll Cardiol 50: 1272–1278. [DOI] [PubMed] [Google Scholar]
- Yasui M, Tamura Y, Minami M, Higuchi S, Fujikawa R, Ikedo T et al. (2015). The Prostaglandin E2 receptor EP4 regulates obesity‐related inflammation and insulin sensitivity. PLoS One 10: e0136304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Control sections showing the absence of immunoreactivity in either VSM or endothelium of coronary arteries from LZR (A, B, upper panel) and OZR (C,D, lower panel) incubated just with the secondary antibodies (A, C, red or B, D, green) in the absence of the primary antibody anti‐COX‐2. Arrows indicate green autofluorescence of the internal elastic lamina. Cell nuclei stained with DAPI. The sections represent n = 3 animals.
Figure S2 H2O2 stimulation did not alter COX‐1 expression in coronary arteries. (A, B) Immunohistochemical localization of COX‐1 in coronary arteries from LZR and OZR and effect of H2O2 stimulation. Immunofluorescence for COX‐1 protein (red areas) was similarly distributed in the endothelium of coronary arteries (arrows) and myocardium (M) from LZR (A, left) and OZR (B, left). (A, B, right) Expression of COX‐1 was unaltered upon acute stimulation with H2O2 (100 μM, 45 min) in coronary arteries from both LZR (A, right) and OZR (B, right). The endothelium was visualized with anti‐endothelial nitric oxide synthase (eNOS) antibodies (green areas), and double immunofluorescence shows the colocalization of eNOS and COX‐1 in the endothelium (yellow areas). The sections represent n = 3 animals.
Figure S3 Immunohistochemical localization of Nox1, Nox2 and Nox4 in coronary arteries from LZR and OZR stimulated with H2O2. Immunofluorescence for (A) Nox1 (red areas), (B) Nox2 (red areas) and (C) Nox4 (red areas) was absent or modest in coronary arteries from LZR (A,B,C left) but was markedly increased in both endothelium and VSM of coronary arteries from OZR (A,B,C, left), after treatment with H2O2 (100 μM, 45 min). The endothelium was visualized with anti‐eNOS antibodies (green areas) and the double immunofluorescence shows colocalization of eNOS and Nox1 (A, right), Nox2 (B, right) and Nox4 (C, right) in the endothelium (yellow areas), but also a strong immunoreaction for all 3 Nox isoenzymes in coronary VSM (red areas) of obese rats. The sections represent n = 3 animals.
Supporting info item
Supporting info item
Supporting info item