

Abstract
Background and Purpose
Vascular inflammation is a major factor contributing to the development of vascular diseases. The aim of this study was to investigate the role of the nicotinic acetylcholine receptor α3 subtype (α3‐nAChR) in vascular inflammation.
Experimental Approach
Vascular inflammation was studied in apolipoprotein E knockout (ApoE−/−) mice fed a high‐fat diet. Inflammatory markers were measured in mouse aortic endothelial cells (MAECs) and macrophages after α3‐nAChRs were antagonized pharmacologically, or after the gene of α3‐nAChRs was silenced.
Key Results
Treatment with α‐conotoxin MII (MII; an α3‐nAChR antagonist) increased the number of inflammatory cells infiltrating the aortic walls and further impaired the endothelium‐dependent vasodilatations in the aorta of ApoE−/− mice. MII also increased the plasma levels of inflammatory cytokines. Furthermore, the infiltration of classical activated macrophages into the arterial wall of ApoE−/− mice was markedly elevated by MII but that of alternative activated macrophages was reduced. In MAECs, the lipopolysaccharide‐stimulated secretion of adhesion molecules and inflammatory cytokines was enhanced by MII, or by silencing the gene of α3‐nAChRs. This effect was reversed by inhibitors of the PI3K‐Akt‐IκKα/β‐IκBα‐NFκB pathways. In macrophages, the classical activation was enhanced, but the alternative activation was reduced when the gene of α3‐nACh receptors was silenced. These effects were prevented by inhibitors of the IκKα/β‐IκBα‐NFκB and JAK2‐STAT6‐PPARγ pathways respectively.
Conclusions and Implications
α3‐nAChRs play a pivotal role in regulating the inflammatory responses in endothelial cells and macrophages. The mechanisms involve the modulations of multiple cell signalling pathways.
Abbreviations
- ApoE
apolipoprotein E
- CRP
C reactive protein
- ECs
endothelial cells
- HE
hematoxylin and eosin
- HFD
High‐fat Diet
- ICAM‐1
intercellular adhesion molecule 1
- iNOS
inducible NOS
- MAECs
mouse aortic endothelial cells
- MCP‐1
monocyte chemotactic protein 1
- MII
α‐conotoxin MII
- nAChRs
Nicotinic acetylcholine receptors
- NO
nitric oxide
- PECAM‐1
platelet/endothelial cell adhesion molecule 1
- PIA
α‐Conotoxin PIA
- RgIA
α‐conotoxin RgIA
- siRNAs
Small interfering RNAs
- sICAM‐1
soluble intercellular adhesion molecule‐1
- VCAM‐1
vascular cell adhesion molecule 1
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Ligand‐gated ion channels c |
| α1D‐adrenoceptor | Nicotinic acetylcholine receptor α3 subunit |
| Enzymes b | Nicotinic acetylcholine receptor α4 subunit |
| eNOS | Nicotinic acetylcholine receptor α6 subunit |
| iNOS | Nicotinic acetylcholine receptor α9 subunit |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Advances in clinical and experimental studies have revealed a close relationship between the major cardiovascular diseases, such as atherosclerosis and hypertension, and a state of chronic inflammation in the vasculature (Hansson, 2005; Ghanem and Movahed, 2007). Vascular inflammation is initiated by the activation of endothelial cells (ECs), which can be induced by many different stimuli such as the accumulation of oxidized LDL (ox‐LDL) (Libby et al., 2002). Upon activation the ECs express adhesion molecules, such as selectins, vascular cell adhesion molecule 1 (VCAM‐1) and intercellular adhesion molecule 1 (ICAM‐1), and thereby facilitate both the tethering and rolling of monocytes onto the inflamed endothelial cell surface (Lusis, 2000). In addition, the activated ECs produce chemotactic factors, such as monocyte chemotactic protein 1 (MCP‐1, also known as CCL2) and IL‐8, which attract the migration of monocytes to subendothelial spaces and, finally, these monocytes differentiate into macrophages (Lusis, 2000). The activated macrophages in turn secrete different inflammatory cytokines that induce a broad range of effects during vascular inflammation (Mantovani et al., 2009). Therefore, ECs and macrophages are the main cells involved in triggering vascular inflammation.
Nicotinic acetylcholine receptors (nAChRs) are integral membrane proteins that belong to the pentameric ligand‐gated ion channel superfamily that mediate and/or modulate cellular signalling. Although widely expressed in the CNS, nAChRs are also found in non‐neuronal/non‐muscle cells, such as macrophages (Albuquerque et al., 2009). In mammals, nAChRs are formed by the assembly of specific combinations of five transmembrane subunits, selected from a pool of 16 homologous polypeptides (α1–7, α9–10, β1–4, δ, ϵ and γ). The individual subtypes can combine with each other with different stoichiometry, such as the (α7)5‐, (α4)2(β2)3‐ and (α3)2(β2)3‐nAChR (Albuquerque et al., 2009; Sambasivarao et al., 2014). nAChRs assembled with different subunits may mediate different physiological functions (Albuquerque et al., 2009).
A regulatory role of nAChRs in vascular inflammation was first suggested in the 1960s as its native ligand, nicotine, increased the extent of the aortic atherosclerotic lesions in rabbits fed an atherogenic diet (Stefanovich et al., 1969). However, it seems that different subunits of nAChRs play contradictory roles in the process of vascular inflammation. The α7 subunit of the nAChR plays a pivotal role in inhibiting the synthesis of inflammatory cytokines (Wang et al., 2003). However, the α1 subunit may be an atherogenic target as the gene silencing of α1‐nAChRs decreases the development of the aortic atherosclerotic plaque in mice (Zhang et al., 2011). Since the α3 subunit of nAChRs is expressed in the aorta (Zou et al., 2012) and macrophages, the present study was designed to verify the hypothesis that the nAChR α3 subtype (α3‐nAChR) may play a regulatory role in the process of vascular inflammation.
Methods
Mice treatment
Male apolipoprotein E knockout (ApoE−/−) mice (8 weeks old, backcrossed 10 times into a C57BL/6J background and originally obtained from Jackson Labs, ME) were purchased from the Laboratory Animal Centre of Yunnan Province (Kunming, China). All mice were maintained under barrier conditions. Water was available ad libitum. An atherogenic high fat diet (HFD), containing 5% (w w‐1) sucrose, 10% lard and 3% cholesterol, was prepared by Shuangshi Laboratory Animal Feed Science Co., Ltd (Suzhou, China). Mice were randomly divided into three groups (n = 5): (i) control group: ApoE−/− mice were fed the standard chow; (ii) HFD group: ApoE−/− mice were fed the HFD for 7 weeks; and (iii) HFD + MII group: ApoE−/− mice were fed the HFD and injected with MII (100 ng kg−1 day−1) i.p. for 7 weeks. All procedures involving animals were approved by the Committee on the Use of Live Animals in Teaching and Research of Yunnan Minzu University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Evaluation of autonomic activities and cytokine measurements in mice
The systolic blood pressure and heart rates of the conscious mice were recorded using a Biosignal Acquisition System (BL‐420F, Chengdu TME Technology, Chengdu, China). All recordings were made using non‐invasive techniques. Data were acquired and analysed using TM‐Wave 2.0 software (Chengdu TME Technology). Then, mice were anaesthetized by an i.p. injection of pentobarbitone sodium (50 mg·kg−1) before being killed. Plasma was prepared from EDTA‐treated blood. The concentrations of noradrenaline, C reactive protein (CRP), inducible NOS (iNOS), IL‐1β, IL‐12, TNF‐α and soluble ICAM‐1 (sICAM‐1) were measured using enzymatic assay kits (Cusabio Biotech Co., Ltd, Wuhan, China).
Isometric tension recordings
After the mice aortas had been dissected out, they were cut into rings (2 mm in length) and then suspended in organ chambers containing Krebs solution at 37°C, aerated with 5% CO2/95% O2, and connected to a force transducer (Powerlab model ML785 and ML119; ad Instruments Pty Ltd., NSW, Australia). The rings were then stretched progressively to their optimal resting tension (1.5 g; determined in preliminary experiments) and allowed to equilibrate for 60 min. The presence of a relaxation response to acetylcholine was taken as evidence that the blood vessel segments contained a functioning endothelium. All changes in tension were expressed as a percentage of the decrease in contraction to phenylephrine.
Histological characterization of aortic sections
For the morphological study, the thoracic aortic tissues were fixed in 8% formaldehyde, embedded in paraffin, sectioned (4 mm slices) and then stained with haematoxylin and eosin (HE) for microscopy.
Immunofluorescence for detecting activated macrophages in the arterial wall
Frozen sections of thoracic aorta were fixed in ice‐cold acetone for 15 min and treated with 0.1% hydrogen peroxide in Tris‐buffered saline (TBS) for another 15 min. After the nonspecific binding sites had been blocked with 3% BSA for 1 h, sections were incubated with rabbit anti‐mouse CD68 or CD206 antibodies (Cell Signaling Technology, Beverly, MA) overnight at 4°C. Following three washes in PBS, the sections were incubated with FITC‐conjugated goat anti‐rabbit antibodies (Cell Signaling Technology, 1:2000). After being rinsed, sections were stained with propidium iodide (PI, 5 mg·L−1, Sigma‐Aldrich). The stained sections were examined by use of a fluorescence microscope (Leica DMI3000B, Leica Microsystems Ltd., Wetzlar, Germany) using a mercury laser to excite PI and FITC at 535 and 488 nm respectively. Emissions were recorded simultaneously at 615 nm for PI and 525 nm for FITC using independent detectors. Figures were merged by using LAS AF software.
Cells
Mouse monocytes, WEHI‐274.1 (CRL‐1679), and macrophages, RAW 264.7 (TIB‐71), were purchased from the American Type Culture Collection (Manassas, VA) and maintained in DMEM supplemented with 10% fetal calf serum (FBS) and 4 mmol·L−1 glutamine. Primary cultured mouse aortic endothelial cells (MAECs) were purchased from PUHE Biotechnology (Wuxi, China) and maintained in DMEM supplemented with 20% FBS, 100 U·mL−1 penicillin‐G, 100 μg·mL−1 streptomycin, 2 mmol·L−1 L‐glutamine, 1× non‐essential amino acids, 1× sodium pyruvate, 25 mmol L−1 HEPES (pH 7.0–7.6), 100 μg·mL−1 heparin and 100 μg·mL−1 endothelial cell growth supplements. The identity of the MAECs was confirmed by immunofluorescence using goat anti‐rat Factor VIII (a stable endothelial antigen) and FITC conjugated rabbit anti‐goat IgG.
RNA interference
Small interfering RNAs (siRNAs) for mouse α3 and α4 subunits of nAChRs were obtained from Qiagen (Valencia, CA, USA). The cultured WEHI‐274.1, RAW 264.7 and MAECs were seeded onto six well plates (2 × 105 cells per well) in complete medium. After 24 h, the cells were serum‐deprived and 10 μL of 20 μmol·L−1 siRNA mixture, or siRNA control was transfected into the cells using HiPerFect transfection reagent (Qiagen). After 72 h of transfection, RT‐PCR was carried out to confirm that the mRNA expressions of the corresponding subunits of nAChRs had been knocked down in the cells.
Monocyte adhesion
MAECs were grown to confluence in 24 well plates and divided into 3 groups: (i) control group; (ii) LPS group: cells were treated with 5 ng·mL−1 LPS for 8 h; and (iii) LPS + Siα3 group: after the gene of α3‐nAChRs was silenced by siRNA, cells were incubated with 5 ng·mL−1 LPS for 8 h. The mouse monocytes WEHI‐274.1 cultured in DMEM were incubated with rabbit‐anti mouse CD136 polyclonal antibody (Cell Signaling Technology) overnight at 37°C. After being washed, the monocytes were added to the surface of the monolayer of MAECs and co‐cultured for 1 h. The co‐cultured cells were then fixed with 100% methanol for 20 min. After being washed with PBS, FITC‐conjugated goat‐anti rabbit antibodies were added to the cells. The stained cells were examined under a fluorescence microscope (Leica DMI3000B, Leica Microsystems Ltd., Wetzlar, Germany).
Transwell assay
The migration of monocytes was investigated in a Transwell chamber system (FluoroBlok, 3 μm pore size, BD Bioscience, Heidelberg, Germany). Briefly, MAECs, with or without the gene of α3‐nAChRs being knocked down, were seeded onto the membrane of the inner chamber and grown to 100% confluence. Culture medium was placed in the inner and outer chambers to reach the apical and basal surfaces of the cell monolayer. The MAECs were treated with 5 ng·mL−1 LPS for 8 h. Monocytes were then added onto the monolayer of MAECs. After 4 h, the cells on the upper surface of the membrane were removed mechanically, and the cells that had migrated into the lower compartment were fixed in 4% paraformaldehyde in PBS, stained with DAPI and counted under a fluorescence microscope.
Western blotting
Proteins from tissues or cells were collected using a commercial protein extraction kit (CWBIO, Beijing, China). The protein concentration was determined spectrophotometrically using the Bradford protein assay reagent (Bio‐rad Laboratories, Hercules, CA, USA) with serial dilution of BSA as the standard.
Twenty microgram of protein samples were loaded and electrophoresed on 7.5% SDS‐PAGEs at 200 V for 50 min. The proteins were then electrotransferred from the gels to PVDF membranes at 200 V for 45 min. After being blocked with 5% (w v‐1) dry milk in TBS for 1 h at room temperature, the membranes were incubated with the primary antibodies overnight at 4°C. Then, the membranes were incubated with HRP‐conjugated antibodies against different proteins of interest (Cell Signaling Technology). Monoclonal anti‐GAPDH antibody was used as an internal control. Bound secondary antibody was detected by chemiluminiscence (Amersham Biosciences, Piscataway, NJ, USA).
Total RNA extraction and quantitative real‐time PCR
Total RNA was extracted from the cells using TRIZOL reagent (Life Technologies, Inc., Grand Island, NY, USA), as directed by the manufacturer's protocol. Equal amounts of RNA were reverse‐transcribed into cDNA with the SuperRT cDNA Kit (CWbio. Co. Ltd., Beijing, China). Primers for all the subunits of nAChR tested and for β‐actin are listed in Table 1. The samples were processed using an ABI StepOne (ABI, Foster City, CA, USA). Real‐time PCR was performed using the UltraSYBR (CWbio., Beijing, China). The PCR amplifications were carried out with the following parameters: denaturation at 95°C for 1 min, annealing at 60°C for 2 min and extension at 72°C for 30 s. A total of 35 cycles was performed. The results of the log‐linear phase of the growth curve were analysed, and relative quantification was performed using the 2−ΔΔCT method with β‐actin as an internal standard.
Table 1.
The primers used in the present study
| Name | Sense | Antisense | Product length (bp) | Nucleotide sequence accession number |
|---|---|---|---|---|
| α1 | CTCTCGACTGTTCTCCTGCTG | GTAGACCCACGGTGACTTGTA | 160 | NM_007389.5 |
| α2 | TTATCTCTGGTGTCTGCTTCTGA | CCCAGCGATTGTAGCCTCC | 110 | NM_144803.2 |
| α3 | TCCAGTTTGAGGTGTCTATGTCT | TGGTAGTCAGAGGGTTTCCATTT | 127 | NM_145129.2 |
| α4 | CTAGCAGCCACATAGAGACCC | GACAAGCCAAAGCGGACAAG | 130 | NM_015730.5 |
| α5 | ATCCTCTGCTGCAAAACATGA | TCCACGTCCACTAACTGAGAT | 141 | NM_024354.1 |
| α6 | TAAAGGCAGTACAGGCTGTGA | AAAATGCACCGTGACGGGAT | 115 | NM_021369.2 |
| α7 | CACATTCCACACCAACGTCTT | AAAAGGGAACCAGCGTACATC | 106 | NM_007390.3 |
| α9 | GGAACCAGGTGGACATATTCAAT | GCAGCCGTAGGAGATGACG | 119 | NM_022930.1 |
| α10 | ATGGATGAACGGAACCAAGTG | GTCCCAATGTAGGTAGGCGT | 78 | NM_001081424.1 |
| β1 | CTCCAACTATGATAGCTCGGTGA | CAGGTCTAAGTACACCTTTGTGC | 139 | NM_027454.4 |
| β2 | AGGGGTTTTGGGTACTGACAC | AGCTTGTTATAGCGGGAAGGA | 72 | NM_009602.4 |
| β3 | CAGTGCCACTCTCTCAGGTTC | GGGCGGACACATTTCTGATAAC | 123 | NM_027454.4 |
| β4 | TGGATGATCTCCTGAACAAAACC | CAGGCGGTAGTCAGTCCATTC | 179 | NM_148944.4 |
| β‐actin | GACTACCTCATGAAGATCCTG | CAGCTCATAGCTCTTCTCCAG | 168 | NM_031144.3 |
| δ | GAATGAGGAACAAAGGCTGATCC | GGTGAGACTTAGGGCGACAT | 112 | NM_021600.3 |
| ε | ACCGCAGCTTTTACCGAGAA | CGACGGATGATGAGCGTGTA | 128 | NM_017194.1 |
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
In the western blot assay, changes in the protein expression levels are presented as the ratio to GAPDH. Statistical analyses were performed using spss 22.0 (spss Inc., Chicago, IL, USA). Data are presented as means ± SEM and n represents the number of experiments. Comparison between two groups was analysed using Student's t‐test. Comparisons among three or more groups were analysed using one‐way anova. Values of P < 0.05 were accepted to indicate statistically significant difference.
Drugs
LY294002 and LPS were purchased from Sigma‐Aldrich (St. Louis, MO, USA). IL‐4 was purchased from PeproTech, Inc. (Rocky Hill, NJ, USA). α‐conotoxin MII, α‐conotoxin RgIA and α‐conotoxin PIA were purchased from R&D Systems, Inc. (Minneapolis, MN, USA). The other reagents were of analytical grade.
Results
Regulatory effects of α3‐nAChRs on the morphology and function of the aorta
The histology and vascular tone of the aorta were studied since morphological and functional changes in blood vessels are detected in vascular wall inflammation (Blake and Ridker, 2001). Compared with the ApoE−/− control group fed a standard chow (Figure 1A), atherosclerotic lesions and an infiltration of macrophages were observed in ApoE−/− mice fed a HFD (Figure 1B). The number of inflammatory cells infiltrated into the aortic walls was further increased when the HFD‐fed ApoE−/− mice were treated with MII (an α3‐nAChR antagonist) (Figure 1C and D). The acetylcholine‐induced aortic relaxation was impaired (Figure 1E) in HFD‐fed ApoE−/− mice while the nitroprusside‐induced relaxation was not affected (Figure 1F). The impaired acetylcholine‐induced relaxation was exacerbated when the HFD‐fed ApoE−/− mice were treated with MII (Figure 1E).
Figure 1.
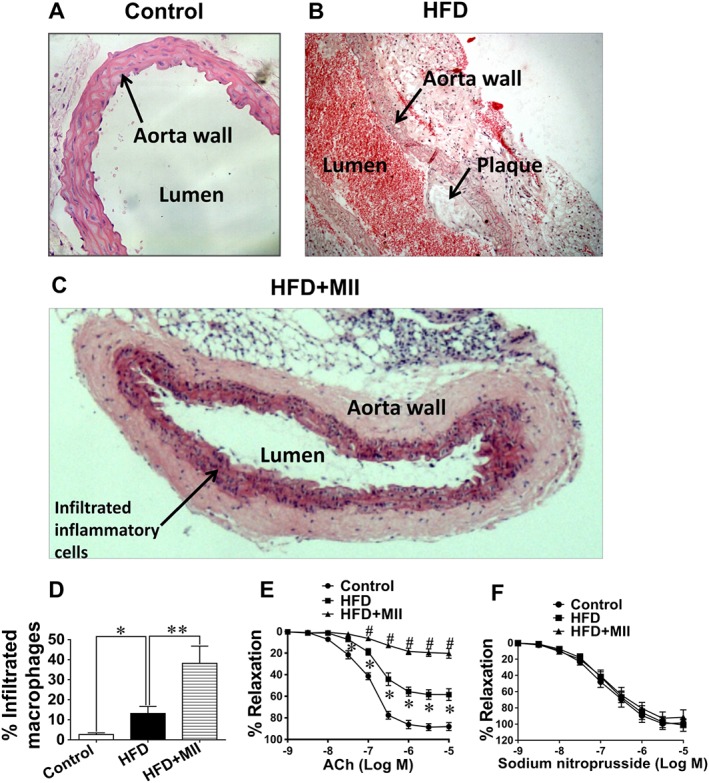
Effect of the α3‐nAChR antagonist α‐conotoxin MII on the morphology and function of aorta in ApoE−/− mice. (A–C) The haematoxylin and eosin staining of the thoracic aortic sections from the ApoE−/− mice (original magnification: 200×). (A) Control group: ApoE−/− mice were fed standard chow. (B) HFD group: ApoE−/− mice were fed a HFD only. (C) HFD + MII group: ApoE−/− mice were fed a HFD and injected with α‐conotoxin MII (MII, an antagonist of α3‐nAChRs, 100 ng·kg−1) i.p. (D) Quantitative evaluation of the infiltrated inflammatory cells as a percentage of the area of the vascular wall (* P < 0.05, ** P < 0.01). (E) The endothelium‐dependent vasodilatations induced by ACh (* P < 0.05, # P < 0.01). (F) The endothelium‐independent vasodilatations induced by sodium nitroprusside (n = 5). Values are means ± SEM. Data were obtained from five separate experiments (n = 5).
Roles of α3‐nAChRs in the production of inflammatory cytokines and autonomic activities in mice
The plasma levels of several markers of the inflammatory cascade were measured since they are elevated during vascular wall inflammation (Blake and Ridker, 2001). The amounts of the inflammatory cytokines including CRP, iNOS, IL‐1β, TNF‐α, IL‐2 and sICAM‐1 were markedly increased in HFD‐fed ApoE−/− mice after the treatment with MII (Figure 2A‐F). The changes in the autonomic functions after the administration of MII were also evaluated since α3‐nAChRs are essential for the normal functioning of the autonomic nervous system (Xu et al., 1999) and the autonomic activities may affect peripheral inflammation (Sorkin, 2015). The systolic blood pressures, heart rates and plasma noradrenaline concentrations in ApoE−/− mice were markedly increased in mice fed a HFD. However, MII did not significantly influence these parameters in HFD‐fed ApoE−/− mice (Figure 2G‐I).
Figure 2.
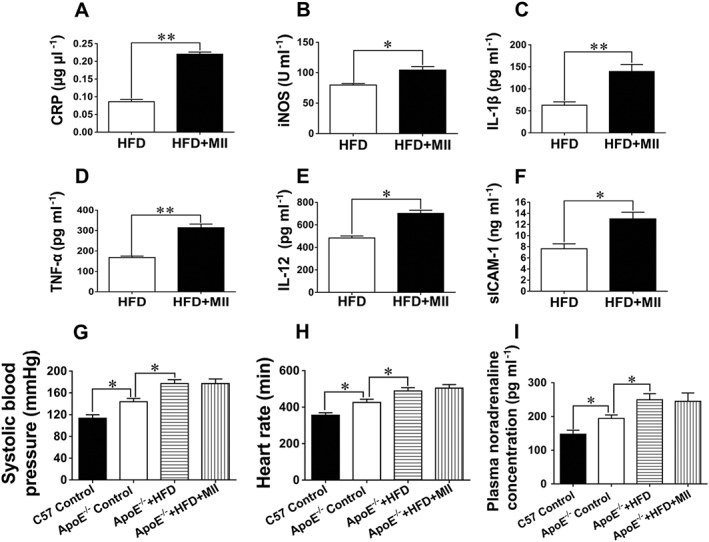
Effect of the α3‐nAChR antagonist α‐conotoxin MII on the production of inflammatory cytokines and the autonomic activities in mice. (A–F) Changes in the inflammatory cytokines in HFD‐fed ApoE−/− mice after the treatment with MII. (G) Changes in systolic blood pressures in mice. (H) Changes in heart rates in mice. (I) Changes in plasma noradrenaline concentrations in mice. Values are means ± SEM. Data were obtained from five separate experiments (n = 5). Significance of the difference between groups is indicated as follows: * P < 0.05, ** P < 0.01. C57: C57BL/6J mice.
Regulatory effect of α3‐nAChRs on the infiltration of macrophages into the arterial wall
Infiltration of macrophage into the arterial wall is one of the key characteristics of vascular inflammation (Hansson, 2005). So, the role of α3‐nAChRs in macrophage infiltration was further studied. As reflected from the immunofluorescent staining of aortic sections, the expression of CD68 (an indicator for the M1 type polarization of the macrophage) was notably elevated in HFD‐fed ApoE−/− mice after the treatment with MII (Figure 3A and C). In contrast, the expression of CD206 (an indicator for the M2 type polarization of the macrophage) was significantly decreased (Figure 3B and D).
Figure 3.
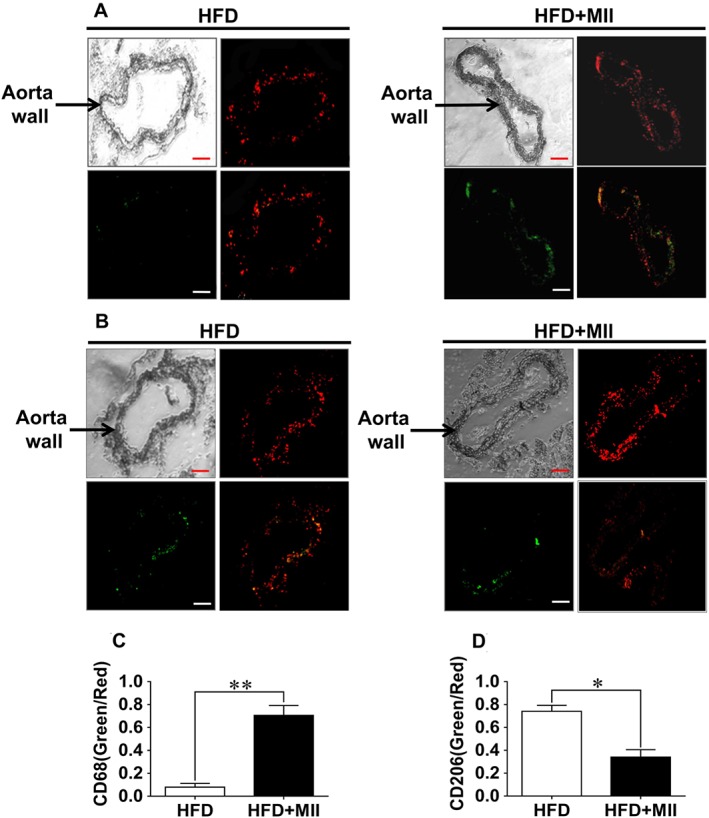
Effect of the α3‐nAChR antagonist α‐conotoxin MII on the infiltration of macrophages into the arterial wall. The infiltration of macrophages into the arterial walls was detected by performing immunofluorescence staining in the aortic sections from the ApoE−/− mice using antibodies to indicate the M1 or M2 type polarization of the macrophage. (A) The immunofluorescence staining of the aortic sections with an indicator for the M1 type polarization of the macrophage, CD68. (B) The immunofluorescence staining of the aortic sections with an indicator for the M2 type polarization of the macrophage, CD206. The upper left panels of each treatment group in (A) and (B) were the images of the aorta sections under the microscope. The upper right panels were the images of the aorta sections stained with PI for the detection of the cell nuclei. The lower left panels show the images of aorta sections stained with the indicator for the M1 or M2 type polarization of the macrophage under the immunofluorescence microscope. The lower right panels showed the merged images; (scale bar = 100 μm). (C and D) Quantification of the averaged fluorescence intensities of CD68 and CD206 with references to the fluorescence intensities of PI. Values are means ± SEM. Data were obtained from five separate experiments (n = 5). Significance of the difference between groups is indicated as follows: * P < 0.05; ** P < 0.01.
Regulatory effect of α3‐nAChRs on the inflammatory response in ECs
Stimulated ECs produce a number of pro‐inflammatory molecules, including adhesion molecules and chemotactic factors, which facilitate the activation and infiltration of macrophages (Lusis, 2000). The role of α3‐nAChRs in the production of pro‐inflammatory molecules by ECs was thus investigated. MAECs served as the endothelial cell model. Similar to the blood vessels, most of the α and β subtypes of nAChRs were expressed in MAECs (Figure 4A). After MAECs were treated with LPS (5 ng·mL−1) for 18 h, their production of MCP‐1, VCAM‐1, ICAM‐1, IL‐6, NO and TNFα by MAECs was significantly elevated (Figure 4B‐G). Pretreatment with MII (0.1 μmol·L−1) markedly augmented the inflammatory effect of LPS on MAECs. However, the LPS‐stimulated inflammatory responses were not significantly different in cells pretreated with α‐conotoxin PIA (PIA, 0.1 μmol·L−1), an α6‐nAChR antagonist, or α‐conotoxin RgIA (RgIA, 0.5 μmol·L−1), an α9‐nAChR antagonist. In contrast, when the cells were pre‐incubated with LY294002 (1 μmol·L−1), a selective PI3K inhibitor, the augmenting effect of MII on the LPS‐induced inflammatory response in MAECs was significantly attenuated. The protein expression levels of the platelet/endothelial cell adhesion molecule 1, COX‐2 and iNOS in MAECs were significantly elevated by LPS, and these levels were further augmented by MII or after knocking down the gene of α3‐nAChRs using siRNA (Figure 4H–M).The phosphorylation of endothelial NOS (p‐eNOS, Ser1177) was notably decreased by LPS (Figure 4N and O). Pretreatment with MII or knocking down the gene of α3‐nAChRs potentiated the inhibitory effect of LPS on the phosphorylation of eNOS.
Figure 4.
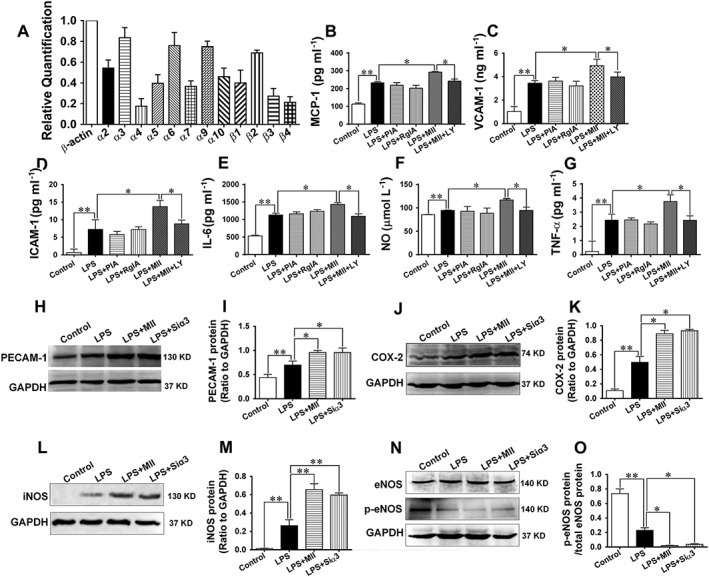
Effect of the α3‐nAChR antagonist α‐conotoxin MII on the inflammatory response in MAECs. (A) Expressions of the subunits of nAChRs in MAECs (relative to β‐actin). (B–G) The LPS (5 ng·mL−1)‐stimulated secretions of the inflammatory cytokines from MAECs in the absence or presence of α‐conotoxin MII (MII, 0.1 μmol·L−1, an α3‐nAChR antagonist), α‐conotoxin PIA (PIA, 0.1 μmol·L−1, an α6‐nAChR antagonist), α‐conotoxin RgIA (RgIA, 0.5 μmol·L−1, an α9‐nAChR antagonist), or LY294002 (1 μmol·L−1, a selective PI3K inhibitor). (H–O) The protein expressions of PECAM‐1, COX‐2, iNOS, eNOS and the p‐eNOS (Ser1177) after MAECs were stimulated with LPS in the absence or presence of MII or after cells were knocked down with the gene of α3‐nAChRs. Siα3: small interfering RNA targeting the α3‐nAChR gene. Values are means ± SEM. Data were obtained from six separate experiments (n = 6). Significance of the difference between groups is indicated as follows: * P < 0.05; ** P < 0.01.
The adhesion and migration of monocytes to MAECs were significantly elevated by LPS, and this situation was further exacerbated after the gene of α3‐nAChRs in MAECs was silenced (Figure 5A‐C). Moreover, the phosphorylations of Akt (Ser 473), IκB kinase‐α/β (IκKα/β, Ser176) and IκBα (Ser32), as well as the expression of NF‐κB were significantly increased by LPS, and these effects were augmented after the pretreatment with MII or after the gene of α3‐nAChRs was knocked down (Figure 5D–K).
Figure 5.
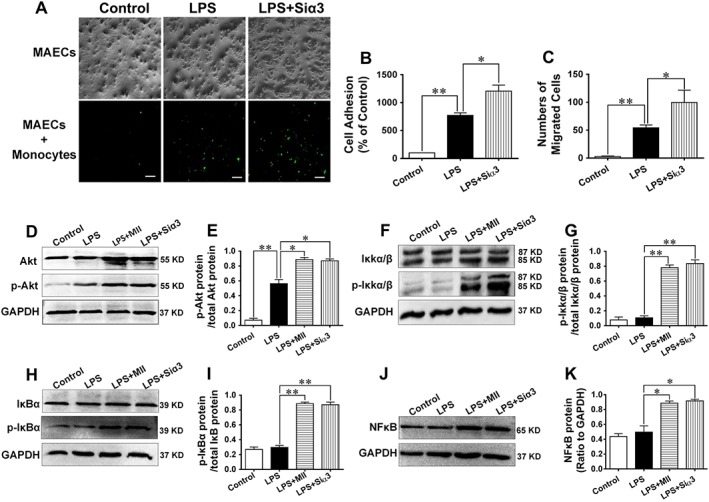
Adhesion and migration of monocytes to MAECs and mechanisms underlying the effect of the α3‐nAChR antagonist α‐conotoxin MII on the inflammatory response in MAECs. (A and B) Adhesion of monocytes to MAECs after the administration of LPS (5 ng·mL−1) in the absence or presence of α‐conotoxin MII (MII, 0.1 μmol·L−1). Monocytes were incubated with an anti‐CD136 polyclonal antibody and an FITC‐conjugated secondary antibody. Pictures were taken by a using a fluorescence microscope. (C) Numbers of monocytes migrating to the lower compartment of a Transwell system. (D–K) Expressions of Akt, p‐Akt (Ser473), IκKα/β, p‐IκKα/β (Ser176), IκBα, p‐IκBα (Ser32) and NFκB were detected by performing western blotting analysis after MAECs were stimulated with LPS in the absence or presence of MII or after cells were knocked down with the gene of the α3‐nAChR. Scale bar = 100 μm. Values are means ± SEM. Data were obtained from six separate experiments (n = 6). Significance of the difference between groups is indicated as follows: * P < 0.05; ** P < 0.01.
Regulatory effect of α3‐nAChRs on the inflammatory response in macrophages
After infiltrating the vascular wall, macrophages may undergo classical activation (or M1 type polarization) to promote inflammation or alternative activation (or M2 type polarization) to resolve inflammation in response to different environmental signals (Moore et al., 2013). Therefore, the roles of α3‐nAChRs in classical‐activated and alternative‐activated macrophages were further investigated. Different subunits of nAChRs were expressed in the mouse macrophage cell line (RAW 264.7, Figure 6A). After the treatment with LPS (100 ng·mL−1) for 24 h, the levels of the indicators of M1 type polarization of macrophages, including IL‐6, TNFα and NO were significantly elevated, and these levels were further augmented after the gene of α3‐nAChRs was knocked down (Figure 6B–D). IL‐10, an indicator for the M2 type polarization of macrophages, was also significantly increased after cells were treated with IL‐4 (25 ng·mL−1) for 24 h (Figure 6E), but the increase in IL‐10 was abolished after the gene of α3‐nAChRs was knocked down. The knock‐down of the gene of α4‐nAChRs did not influence the levles of the indicators of M1 or M2 type polarization of macrophages. The protein expression of CD206, another indicator of the M2 type polarization of macrophages, was markedly elevated by administration of IL‐4, and this effect was notably attenuated when cells were pretreated with MII (0.1 μmol·L−1) or after the gene of α3‐nAChRs was silenced (Figure 6F and G). Similarly, the protein expression of CD80 and iNOS, the indicators for the M1 type polarization of the macrophage, were markedly elevated by LPS, and was further increased in cells pretreated with MII or after the gene of α3‐nAChRs had been silenced (Figure 6H–K).
Figure 6.
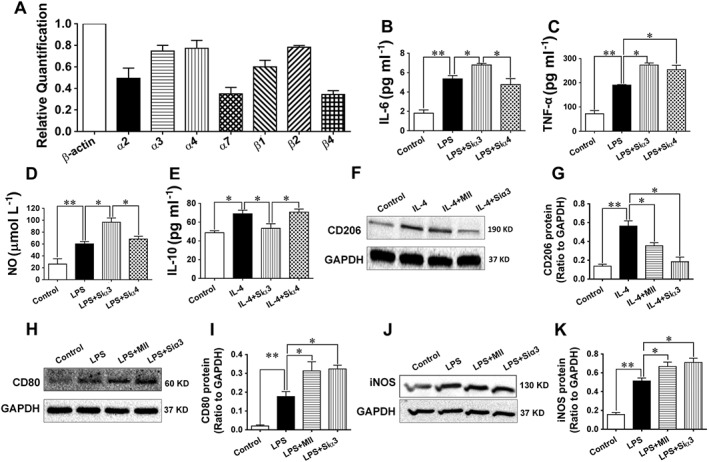
Effect of the α3‐nAChR antagonist α‐conotoxin MII on the inflammatory response in macrophages. (A) Expressions of the subunits of nAChRs in the mouse macrophage (RAW 264.7, relative to β‐actin). (B–E) The LPS (100 ng·mL−1)‐stimulated secretion of the indicators, IL‐6, NO and TNFα, for the M1 (classical) type polarization of the macrophages, and the IL‐4 (25 ng·mL−1)‐stimulated secretions of the indicator, IL‐10, for the M2 (alternative) type polarization of the macrophages after cells were knocked down with the gene of the α3‐nAChR or the α4‐nAChR. (F–K) Western blotting results of the IL‐4‐stimulated expression of the indicator, CD206, for the M2 (alternative) type polarization of the macrophages and the LPS‐stimulated expression of the indicators, CD80 and iNOS, for the M1 (classical) type polarization of the macrophages after cells were stimulated with IL‐4 or LPS in the presence of MII or after cells were knocked down with the gene of the α3‐nAChR. Values are means ± SEM. Data were obtained from six separate experiments (n = 6). Significance of the difference between groups is indicated as follows: * P < 0.05; ** P < 0.01.
The mechanisms involved in the regulatory effects of α3‐nAChRs on the M1 and M2 type polarizations of the macrophage were further studied. After the M1 type polarization of the macrophage was induced by LPS, the phosphorylations of IκKα/β (Ser176) and IκBα (Ser32) and the expression of NFκB were significantly increased, and were further augmented when cells were pretreated with MII or after the gene of α3‐nAChRs was knocked down (Figure 7A–F). After the M2 type polarization of the macrophage was induced by IL‐4, the protein expressions of phosphorylated‐JAK2 (Tyr1007), phosphorylated‐STAT6 (Tyr641) and PPARγ were markedly enhanced, but their expressions were significantly attenuated when cells were pre‐incubated with MII or after the gene of α3‐nAChRs was knocked down (Figure 7G–L).
Figure 7.
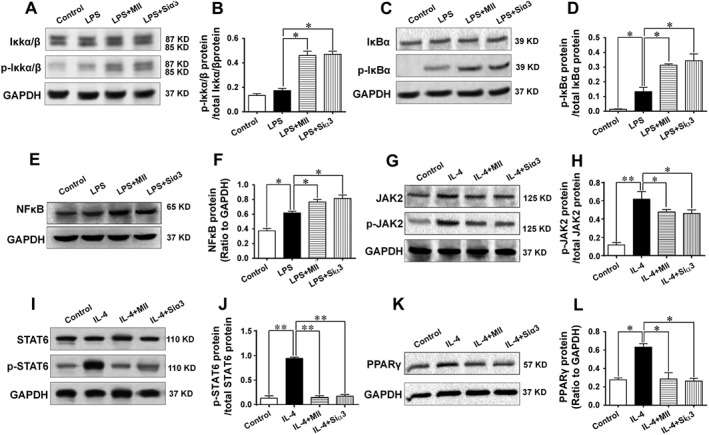
Mechanisms involved in the effect of the α3‐nAChR antagonist α‐conotoxin MII on the inflammatory response in macrophages. (A–F) Mechanisms underlying the effect of α3‐nAChRs on the M1 type polarization of macrophages. The protein expressions of the IκKα/β, p‐IκKα/β (Ser176), IκBα, p‐IκBα (Ser32) and NFκB were detected after macrophages were stimulated with LPS to induce the M1 type polarization in the presence of MII or after cells were knocked down with the gene of the α3‐nAChR. (G–L) Mechanisms involved in the effect of α3‐nAChRs on the M2 type polarization of macrophages. The protein expressions of JAK2, phosphorylated‐JAK2 (Tyr1007), STAT6, phosphorylated‐STAT6 (Tyr641) PPARγ were detected after macrophages were stimulated with IL‐4 to induce the M2 type polarization in the presence of MII or after cells were knocked down with the gene of the α3‐nAChR. Siα3: small interfering RNA targeting the gene of the α3‐nAChR. Values are means ± SEM. Data were obtained from five separate experiments (n = 5). Significance of the difference between groups is indicated as follows: * P < 0.05; ** P < 0.01.
In order to verify whether blocking or silencing the expression of α3‐nAChRs might lead to compensatory changes in the expressions of other nAChRs subunits, quantitative real‐time PCR was performed in MAECs and macrophages. Our results showed that MII induced compensatory increases in the expressions of α3 and/or α6 subunits in MAECs and macrophages. In contrast, the expression of the α3 subunit was abolished in MAECs and macrophages after the treatment with siRNA. The expressions of other subunits of nAChRs were not influenced by MII or siRNA of α3 (Fig. S1).
Discussion and conclusions
The purpose of the present study was to evaluate the regulatory roles of α3‐nAChRs in vascular inflammation in vitro and in vivo. It is well documented that mice with the null mutation for the gene of α3‐nAChRs always demonstrate impaired growth and increased mortality before and after weaning because of prominent inflammation (Xu et al., 1999). This finding suggests that the α3‐nAChRs have a crucial role in inflammatory responses in mice. Unfortunately, the high mortality of α3‐nAChR‐knockout mice also means it is extremely difficult to evaluate the regulatory roles of α3‐nAChRs in vivo. Hence, in the present study, MII was used to antagonize the α3‐nAChRs in vitro and in vivo, and the effects were investigated. Although MII is a highly effective antagonist of α3‐nAChRs, it fails to discriminate well between α3 and α6 subunits of nAChR since the α6 subunit is closely related to α3 subunit in terms of the structure (Dowell et al., 2003). Therefore, siRNAs were also used to further differentiate the functions of α3‐nAChRs from nAChRs with other α subunits. Our results clearly demonstrated that MII worsened the atherosclerotic lesion and further impaired the vasorelaxation responses in ApoE−/− mice. In addition, MII increased the plasma levels of inflammatory cytokines and the infiltration of macrophages into the blood vessels of ApoE−/− mice. These results suggest that α3‐nAChRs play a crucial role in regulating the inflammatory response in blood vessels.
ECs and macrophages are commonly accepted as the main cells involved in vascular inflammation. ECs, through the secretion of numerous mediators, act as one of the central players in cardiovascular homeostasis (Vanhoutte et al., 2009). Under pro‐inflammatory conditions such as the accumulation of ox‐LDL, the ECs are stimulated. Physiopathologically, during an inflammatory response ECs typically are stimulated to produce a number of molecules, which promote the adherence of monocytes onto their surface (Libby et al., 2002). Our present study has shown that the production of EC‐derived pro‐inflammatory cytokines was augmented by MII, but was abolished by a PI3K inhibitor. This indicates that α3‐nAChRs may play an anti‐inflammatory role in ECs, probably through the inhibition of the PI3K‐dependent mechanism. The down‐stream mechanisms might involve the Akt‐IκKα/β‐IκBα‐NFκB‐dependent pathways. Interestingly, LPS‐stimulated production of NO in MAECs was augmented by MII. This result seems to be paradoxical since NO decreases the activation of ECs and reduces the endothelial expression of adhesion molecules and pro‐inflammatory cytokines (De Caterina et al., 1995). However, the decreased phosphorylation of eNOS but the enhanced expression of iNOS may resolve this dichotomy, since it widely accepted that the pathological effects of NO in ECs are evoked by iNOS (Hickey et al., 2001). The larger amounts of NO produced by iNOS may result in the accumulation of the peroxynitrite (OONO−), which is commonly considered to be an oxygen‐ free radical and aggravates inflammatory responses (Heeba et al., 2009).
Macrophages also play a central role in the pathophysiological process of vascular inflammation. For instance, macrophages lodge in the intima and subintima of arteries, eventually leading to the generation of foam cells and formation of atherosclerotic plaques (Moore et al., 2013). Under inflammatory conditions, macrophages are very versatile cells with a high degree of plasticity in response to a range of environmental stimuli. It is generally accepted that the classically activated (or M1 type polarization) macrophages promote inflammation but the alternatively activated (or M2 type polarization) macrophages resolve inflammation (Moore et al., 2013). When the macrophages were pretreated with MII or siRNA of α3‐nAChRs, the production of the M1 type polarization cells was significantly elevated, whereas that of the M2 type was notably decreased. These findings indicate that after the inhibition of α3‐nAChRs or the silencing of their gene, M1 macrophages are predominant or M2 macrophages are switched to M1 macrophages. This theory is further strengthened by the results obtained after immunofluorescent staining of M1 or M2 type markers of the macrophages that had infiltrated into the vascular walls. With regard to the underlying mechanisms, we found that the suppressive effect of α3‐nAChRs on the M1 macrophages was due to the inhibition of the IκKα/β‐IκBα‐NFκB pathways, whereas the enhancement of the M2 macrophages was probably mediated through activation of the JAK2‐STAT6‐PPARγ pathways. Our findings are in line with the mechanisms previously reported for the activation of M1 and M2 types of macrophages (Ishii et al., 2009).
It is also well documented that another isoform of the nAChR, α7‐nAChR, has an anti‐inflammatory role (Wang et al., 2003; de Jonge and Ulloa, 2007). Therefore, it would be worth exploring the differences between the roles of α7‐nAChRs and α3‐nAChRs in inflammatory diseases. Of note, α7‐nAChRs have been shown to modulate the production of pro‐inflammatory cytokines from immune cells, such as macrophages, but have never been suggested to regulate inflammatory responses in ECs (Wang et al., 2003; de Jonge and Ulloa, 2007). In contrast, as demonstrated in our present study, α3‐nAChRs may function as an anti‐inflammatory target not only in immune cells but also in vascular cells. We propose that α3‐nAChRs, but not α7‐nAChRs, are the critical receptors involved in the inhibition of the inflammatory responses in ECs. Indeed, it has been suggested that ECs act as a target of anti‐inflammatory cholinergic mediators, since the cholinoceptor agonists, nicotine and CAP55, inhibit the generation of adhesion molecules from ECs, but mecamylamine, a non‐specific nAChR antagonist, reverses the activation of ECs (Saeed et al., 2005). Therefore, the findings of our present study extend the understandings of the cholinergic anti‐inflammatory pathway.
In conclusion, α3‐nAChRs may play a pivotal role in suppressing vascular inflammation through the regulation of the inflammatory responses in ECs and macrophages. In ECs, the activation of α3‐nAChRs may attenuate inflammatory responses by inhibiting the PI3K‐Akt‐IκKα/β‐IκBα‐NFκB pathways. Meanwhile, the α3‐nAChRs may depress the M1 (classical) activation of macrophages by inhibiting the IκKα/β‐IκBα‐NFκB pathway but stimulate the M2 (alternative) activation of macrophage by activating the JAK2‐STAT6‐PPARγ pathway. It is hoped that α3‐nAChRs might become a new therapeutic target for the treatment of diseases associated with vascular inflammation, such as atherosclerosis.
Author contributions
C.Y. and G.D. conceived and designed the study. C.Y., Z.L., G. L., S.Y., S.P. and J.Y. performed experiments, analysed data and wrote the paper. Y.H., R.D. and R.Y. performed some of the experimental work and data analysis.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Changes of the expressions of nAChRs subunits after antagonizing α3‐nAChRs or after silencing the gene of α3‐nAChRs. Quantitative real‐time PCR was performed in mouse aortic endothelial cells (MAECs) and macrophages. (A) Changes of the expressions of nAChRs subunits in MAECs after treated with α3‐nAChRs antagonist α‐Conotoxin MII (MII, 0.1 μmol L‐1). (B) Changes of the expressions of nAChRs subunits in macrophages after treated with α3‐nAChRs antagonist α‐Conotoxin MII (MII, 0.1 μmol L‐1). (C) Changes of the expressions of nAChRs subunits in MAECs after the gene of α3‐nAChRs was silenced with siRNA. (D) Changes of the expressions of nAChRs subunits in macrophages after the gene of α3‐nAChRs was silenced with siRNA. Relative quantification was performed using the 2‐ΔΔCT method with β‐actin as an internal standard. Values are means ±} S.E.M. Significance of the difference between groups is indicated as follows: *P < 0.05; **P < 0.01.
Supporting info item
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (no. 81160404, 81460553 and 11547233), and supported by Key Natural Scientific Fund of Yunnan Province (no. 2014FA036).
Yang, C. , Li, Z. , Yan, S. , He, Y. , Dai, R. , Leung, G. P. , Pan, S. , Yang, J. , Yan, R. , and Du, G. (2016) Role of the nicotinic acetylcholine receptor α3 subtype in vascular inflammation. British Journal of Pharmacology, 173: 3235–3247. doi: 10.1111/bph.13609.
Contributor Information
Cui Yang, Email: yangynni@163.com.
Guanhua Du, Email: dugh@imm.ac.cn.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89: 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake GJ, Ridker PM (2001). Novel clinical markers of vascular wall inflammation. Circ Res 89: 763–771. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr et al. (1995). Nitric oxide decreases cytokine‐induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 96: 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, Ulloa L (2007). The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151: 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A et al. (2003). Alpha‐conotoxin PIA is selective for alpha6 subunit‐containing nicotinic acetylcholine receptors. J Neurosci 23: 8445–8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem FA, Movahed A (2007). Inflammation in high blood pressure: a clinician perspective. J Am Soc Hypertens 1: 113–119. [DOI] [PubMed] [Google Scholar]
- Hansson GK (2005). Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352: 1685–1695. [DOI] [PubMed] [Google Scholar]
- Heeba G, Moselhy ME, Hassan M, Khalifa M, Gryglewski R, Malinski T (2009). Anti‐atherogenic effect of statins: role of nitric oxide, peroxynitrite and haem oxygenase‐1. Br J Pharmacol 156: 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MJ, Granger DN, Kubes P (2001). Inducible nitric oxide synthase (iNOS) and regulation of leucocyte/endothelial cell interactions: studies in iNOS‐deficient mice. Acta Physiol Scand 173: 119–126. [DOI] [PubMed] [Google Scholar]
- Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM et al. (2009). Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114: 3244–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A (2002). Inflammation and atherosclerosis. Circulation 105: 1135–1143. [DOI] [PubMed] [Google Scholar]
- Lusis AJ (2000). Atherosclerosis. Nature 407: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Garlanda C, Locati M (2009). Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 29: 1419–1423. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Sheedy FJ, Fisher EA (2013). Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13: 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed RW, Varma S, Peng‐Nemeroff T, Sherry B, Balakhaneh D, Huston J et al. (2005). Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med 201: 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivarao SV, Roberts J, Bharadwaj VS, Slingsby JG, Rohleder C, Mallory C et al. (2014). Acetylcholine promotes binding of alpha‐conotoxin MII at alpha3 beta2 nicotinic acetylcholine receptors. Chembiochem 15: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin LS (2015). Modulation of peripheral inflammation by the spinal cord. Handb Exp Pharmacol 227: 191–206. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovich V, Gore I, Kajiyama G, Iwanaga Y (1969). The effect of nicotine on dietary atherogenesis in rabbits. Exp Mol Pathol 11: 71–81. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H, Tang EH, Feletou M (2009). Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 196: 193–222. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S et al. (2003). Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421: 384–388. [DOI] [PubMed] [Google Scholar]
- Xu W, Gelber S, Orr‐Urtreger A, Armstrong D, Lewis RA, Ou CN et al. (1999). Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A 96: 5746–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Marshall AL, Thomas AL, Kernan KA, Su Y, LeBoeuf RC et al. (2011). In vivo knockdown of nicotinic acetylcholine receptor alpha1 diminishes aortic atherosclerosis. Atherosclerosis 215: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Q, Leung SW, Vanhoutte PM (2012). Activation of nicotinic receptors can contribute to endothelium‐dependent relaxations to acetylcholine in the rat aorta. J Pharmacol Exp Ther 341: 756–763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Changes of the expressions of nAChRs subunits after antagonizing α3‐nAChRs or after silencing the gene of α3‐nAChRs. Quantitative real‐time PCR was performed in mouse aortic endothelial cells (MAECs) and macrophages. (A) Changes of the expressions of nAChRs subunits in MAECs after treated with α3‐nAChRs antagonist α‐Conotoxin MII (MII, 0.1 μmol L‐1). (B) Changes of the expressions of nAChRs subunits in macrophages after treated with α3‐nAChRs antagonist α‐Conotoxin MII (MII, 0.1 μmol L‐1). (C) Changes of the expressions of nAChRs subunits in MAECs after the gene of α3‐nAChRs was silenced with siRNA. (D) Changes of the expressions of nAChRs subunits in macrophages after the gene of α3‐nAChRs was silenced with siRNA. Relative quantification was performed using the 2‐ΔΔCT method with β‐actin as an internal standard. Values are means ±} S.E.M. Significance of the difference between groups is indicated as follows: *P < 0.05; **P < 0.01.
Supporting info item